
The Molecular Mechanisms and Therapeutic Prospects of Alternative Lengthening of Telomeres (ALT)


Abstract
Simple Summary
Abstract
1. Introduction
Telomere Maintenance Mechanisms
2. Main Body
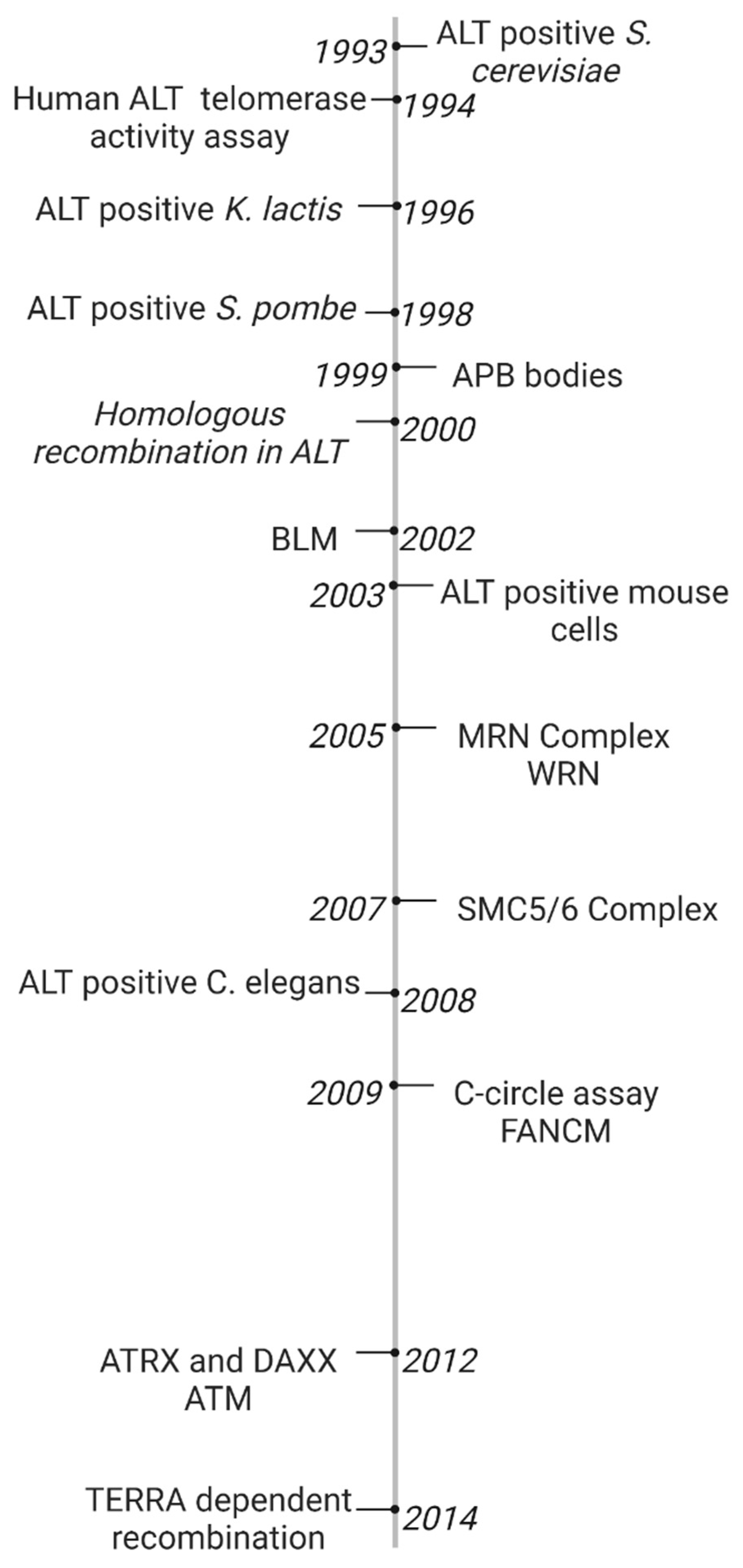
2.1. Timeline of Early ALT Discoveries
2.2. ALT Cancer Hallmarks
2.3. Replication Stress
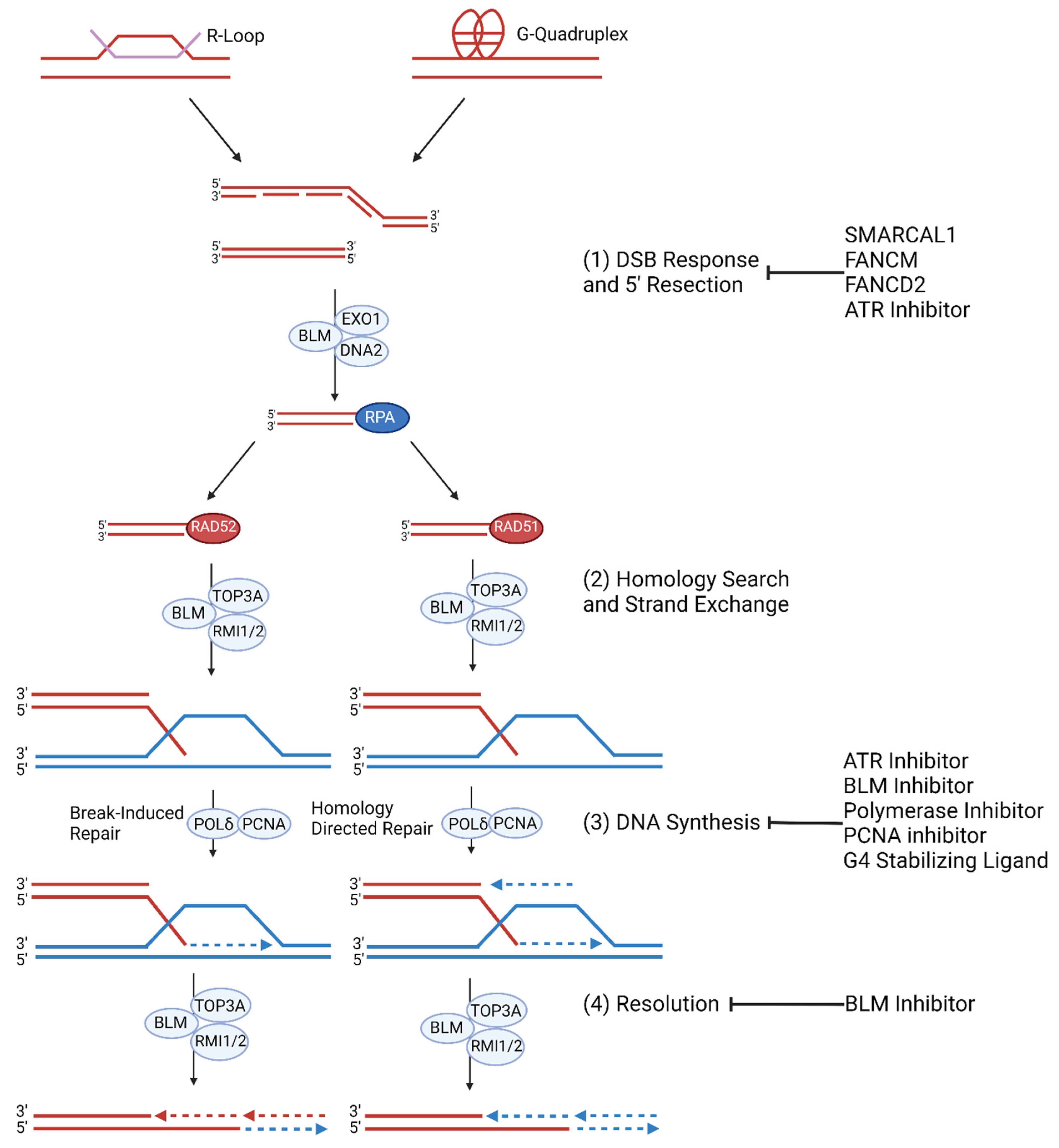
2.4. ALT Molecular Mechanism
2.5. APB Formation and Telomere Instability
2.6. Homologous Recombination and Telomeric MiDAS
2.7. Hyperactive ALT Pathway
3. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, V.; Lingner, J. Replication of telomeres and the regulation of telomerase. Cold Spring Harb. Perspect. Biol. 2013, 5, a010405. [Google Scholar] [CrossRef]
- Muraki, K.; Nyhan, K.; Han, L.; Murnane, J.P. Mechanisms of telomere loss and their consequences for chromosome instability. Front. Oncol. 2012, 2, 135. [Google Scholar] [CrossRef]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef]
- Erdel, F.; Kratz, K.; Willcox, S.; Griffith, J.D.; Greene, E.C.; de Lange, T. Telomere Recognition and Assembly Mechanism of Mammalian Shelterin. Cell Rep. 2017, 18, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Falconer, E.; Chavez, E.A.; Henderson, A.; Poon, S.S.; McKinney, S.; Brown, L.; Huntsman, D.G.; Lansdorp, P.M. Identification of sister chromatids by DNA template strand sequences. Nature 2009, 463, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Dunham, M.A.; Neumann, A.A.; Fasching, C.L.; Reddel, R.R. Telomere maintenance by recombination in human cells. Nat. Genet. 2000, 26, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence: Figure 1. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [PubMed]
- Letsolo, B.T.; Rowson, J.; Baird, D.M. Fusion of short telomeres in human cells is characterized by extensive deletion and microhomology, and can result in complex rearrangements. Nucleic Acids Res. 2010, 38, 1841–1852. [Google Scholar] [CrossRef] [PubMed]
- Capper, R.; Britt-Compton, B.; Tankimanova, M.; Rowson, J.; Letsolo, B.; Man, S.; Haughton, M.; Baird, D.M. The nature of telomere fusion and a definition of the critical telomere length in human cells. Genes Dev. 2007, 21, 2495–2508. [Google Scholar] [CrossRef]
- Lafferty-Whyte, K.; Cairney, C.J.; Will, M.B.; Serakinci, N.; Daidone, M.G.; Zaffaroni, N.; Bilsland, A.; Keith, W.N. A gene expression signature classifying telomerase and ALT immortalization reveals an hTERT regulatory network and suggests a mesenchymal stem cell origin for ALT. Oncogene 2009, 28, 3765–3774. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.L.; Batista, L.F.; Freund, A.; Pech, M.F.; Venteicher, A.S.; Artandi, S.E. TPP1 OB-fold domain controls telomere maintenance by recruiting telomerase to chromosome ends. Cell 2012, 150, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, J.; Bell, C.F.; Weidenfeld, I.; Zaug, A.J.; Leinwand, L.A.; Cech, T.R. The TEL patch of telomere protein TPP1 mediates telomerase recruitment and processivity. Nature 2012, 492, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Y.; Redon, S.; Lingner, J. The human CST complex is a terminator of telomerase activity. Nature 2012, 488, 540–544. [Google Scholar] [CrossRef]
- Latrick, C.M.; Cech, T.R. POT1-TPP1 enhances telomerase processivity by slowing primer dissociation and aiding translocation. EMBO J. 2010, 29, 924–933. [Google Scholar] [CrossRef]
- Smogorzewska, A.; de Lange, T. Regulation of telomerase by telomeric proteins. Annu. Rev. Biochem. 2004, 73, 177–208. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, C.; Kurth, I.; Lingner, J. Human protection of telomeres 1 (POT1) is a negative regulator of telomerase activity in vitro. Mol. Cell. Biol. 2005, 25, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Silva, B.; Pentz, R.; Figueira, A.M.; Arora, R.; Lee, Y.W.; Hodson, C.; Wischnewski, H.; Deans, A.J.; Azzalin, C.M. FANCM limits ALT activity by restricting telomeric replication stress induced by deregulated BLM and R-loops. Nat. Commun. 2019, 10, 2253. [Google Scholar] [CrossRef] [PubMed]
- Hakin-Smith, V.; Jellinek, D.A.; Levy, D.; Carroll, T.; Teo, M.; Timperley, W.R.; McKay, M.J.; Reddel, R.R.; Royds, J.A. Alternative lengthening of telomeres and survival in patients with glioblastoma multiforme. Lancet 2003, 361, 836–838. [Google Scholar] [CrossRef] [PubMed]
- Henson, J.D.; Hannay, J.A.; McCarthy, S.W.; Royds, J.A.; Yeager, T.R.; Robinson, R.A.; Wharton, S.B.; Jellinek, D.A.; Arbuckle, S.M.; Yoo, J.; et al. A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin. Cancer Res. 2005, 11, 217–225. [Google Scholar] [CrossRef]
- Costa, A.; Daidone, M.G.; Daprai, L.; Villa, R.; Cantù, S.; Pilotti, S.; Mariani, L.; Gronchi, A.; Henson, J.D.; Reddel, R.R.; et al. Telomere maintenance mechanisms in liposarcomas: Association with histologic subtypes and disease progression. Cancer Res. 2006, 66, 8918–8924. [Google Scholar] [CrossRef]
- Villa, R.; Daidone, M.G.; Motta, R.; Venturini, L.; De Marco, C.; Vannelli, A.; Kusamura, S.; Baratti, D.; Deraco, M.; Costa, A.; et al. Multiple mechanisms of telomere maintenance exist and differentially affect clinical outcome in diffuse malignant peritoneal mesothelioma. Clin. Cancer Res. 2008, 14, 4134–4140. [Google Scholar] [CrossRef] [PubMed]
- Subhawong, A.P.; Heaphy, C.M.; Argani, P.; Konishi, Y.; Kouprina, N.; Nassar, H.; Vang, R.; Meeker, A.K. The alternative lengthening of telomeres phenotype in breast carcinoma is associated with HER-2 overexpression. Mod. Pathol. 2009, 22, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Jeyapalan, J.N.; Mendez-Bermudez, A.; Zaffaroni, N.; Dubrova, Y.E.; Royle, N.J. Evidence for alternative lengthening of telomeres in liposarcomas in the absence of ALT-associated PML bodies. Int. J. Cancer 2008, 122, 2414–2421. [Google Scholar] [CrossRef] [PubMed]
- Yeager, T.R.; Neumann, A.A.; Englezou, A.; Huschtscha, L.I.; Noble, J.R.; Reddel, R.R. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999, 59, 4175–4179. [Google Scholar]
- Onitake, Y.; Hiyama, E.; Kamei, N.; Yamaoka, H.; Sueda, T.; Hiyama, K. Telomere biology in neuroblastoma: Telomere binding proteins and alternative strengthening of telomeres. J. Pediatr. Surg. 2009, 44, 2258–2266. [Google Scholar] [CrossRef] [PubMed]
- Pezzolo, A.; Pistorio, A.; Gambini, C.; Haupt, R.; Ferraro, M.; Erminio, G.; De Bernardi, B.; Garaventa, A.; Pistoia, V. Intratumoral diversity of telomere length in individual neuroblastoma tumors. Oncotarget 2015, 6, 7493–7503. [Google Scholar] [CrossRef]
- Matsuo, T.; Shay, J.W.; Wright, W.E.; Hiyama, E.; Shimose, S.; Kubo, T.; Sugita, T.; Yasunaga, Y.; Ochi, M. Telomere-maintenance mechanisms in soft-tissue malignant fibrous histiocytomas. J. Bone Jt. Surg. Am. 2009, 91, 928–937. [Google Scholar] [CrossRef]
- Matsuo, T.; Shimose, S.; Kubo, T.; Fujimori, J.; Yasunaga, Y.; Ochi, M. Alternative lengthening of telomeres as a prognostic factor in malignant fibrous histiocytomas of bone. Anticancer. Res. 2010, 30, 4959–4962. [Google Scholar]
- Venturini, L.; Motta, R.; Gronchi, A.; Daidone, M.; Zaffaroni, N. Prognostic relevance of ALT-associated markers in liposarcoma: A comparative analysis. BMC Cancer 2010, 10, 254. [Google Scholar] [CrossRef]
- McDonald, K.L.; McDonnell, J.; Muntoni, A.; Henson, J.D.; Hegi, M.E.; von Deimling, A.; Wheeler, H.R.; Cook, R.J.; Biggs, M.T.; Little, N.S.; et al. Presence of alternative lengthening of telomeres mechanism in patients with glioblastoma identifies a less aggressive tumor type with longer survival. J. NeuroPathol. Exp. Neurol. 2010, 69, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Wright, W.E.; Shay, J.W. Alternative lengthening of telomeres can be maintained by preferential elongation of lagging strands. Nucleic Acids Res. 2017, 45, 2615–2628. [Google Scholar] [CrossRef]
- Perrem, K.; Colgin, L.M.; Neumann, A.A.; Yeager, T.R.; Reddel, R.R. Coexistence of alternative lengthening of telomeres and telomerase in hTERT-transfected GM847 cells. Mol. Cell Biol. 2001, 21, 3862–3875. [Google Scholar] [CrossRef] [PubMed]
- Lundblad, V.; Blackburn, E.H. An alternative pathway for yeast telomere maintenance rescues est1- senescence. Cell 1993, 73, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.M.; Cooper, J.P.; Cech, T.R. Two modes of survival of fission yeast without telomerase. Science 1998, 282, 493–496. [Google Scholar] [CrossRef] [PubMed]
- McEachern, M.J.; Blackburn, E.H. Cap-prevented recombination between terminal telomeric repeat arrays (telomere CPR) maintains telomeres in Kluyveromyces lactis lacking telomerase. Genes Dev. 1996, 10, 1822–1834. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.Y.; Perez-Martin, J.; Holloman, W.K.; Lue, N.F. Mre11 and Blm-Dependent Formation of ALT-Like Telomeres in Ku-Deficient Ustilago maydis. PLoS Genet. 2015, 11, e1005570. [Google Scholar] [CrossRef]
- Blasco, M.A.; Lee, H.W.; Hande, M.P.; Samper, E.; Lansdorp, P.M.; DePinho, R.A.; Greider, C.W. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 1997, 91, 25–34. [Google Scholar] [CrossRef]
- Chang, S.; Khoo, C.M.; Naylor, M.L.; Maser, R.S.; DePinho, R.A. Telomere-based crisis: Functional differences between telomerase activation and ALT in tumor progression. Genes Dev. 2003, 17, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Raices, M.; Verdun, R.E.; Compton, S.A.; Haggblom, C.I.; Griffith, J.D.; Dillin, A.; Karlseder, J.C. elegans telomeres contain G-strand and C-strand overhangs that are bound by distinct proteins. Cell 2008, 132, 745–757. [Google Scholar] [CrossRef]
- Seo, B.; Kim, C.; Hills, M.; Sung, S.; Kim, H.; Kim, E.; Lim, D.S.; Oh, H.S.; Choi, R.M.J.; Chun, J.; et al. Telomere maintenance through recruitment of internal genomic regions. Nat. Commun. 2015, 6, 8189. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Quantitation of the frequency of immortalization of normal human diploid fibroblasts by SV40 large T-antigen. Exp. Cell Res. 1989, 184, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Bryan, T.M.; Englezou, A.; Gupta, J.; Bacchetti, S.; Reddel, R.R. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 1995, 14, 4240–4248. [Google Scholar] [CrossRef]
- Bryan, T.M.; Englezou, A.; Dalla-Pozza, L.; Dunham, M.A.; Reddel, R.R. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med. 1997, 3, 1271–1274. [Google Scholar] [CrossRef] [PubMed]
- Henson, J.D.; Cao, Y.; Huschtscha, L.I.; Chang, A.C.; Au, A.Y.; Pickett, H.A.; Reddel, R.R. DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat. Biotechnol. 2009, 27, 1181–1185. [Google Scholar] [CrossRef] [PubMed]
- Teng, S.C.; Zakian, V.A. Telomere-telomere recombination is an efficient bypass pathway for telomere maintenance in Saccharomyces cerevisiae. Mol. Cell. Biol. 1999, 19, 8083–8093. [Google Scholar] [CrossRef]
- Jiang, W.Q.; Zhong, Z.H.; Henson, J.D.; Neumann, A.A.; Chang, A.C.; Reddel, R.R. Suppression of alternative lengthening of telomeres by Sp100-mediated sequestration of the MRE11/RAD50/NBS1 complex. Mol. Cell Biol. 2005, 25, 2708–2721. [Google Scholar] [CrossRef]
- Potts, P.R.; Yu, H. The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat. Struct. Mol. Biol. 2007, 14, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Zhang, F.; Barrett, B.; Ren, K.; Andreassen, P.R. A role for monoubiquitinated FANCD2 at telomeres in ALT cells. Nucleic Acids Res. 2009, 37, 1740–1754. [Google Scholar] [CrossRef] [PubMed]
- Stavropoulos, D.J.; Bradshaw, P.S.; Li, X.; Pasic, I.; Truong, K.; Ikura, M.; Ungrin, M.; Meyn, M.S. The Bloom syndrome helicase BLM interacts with TRF2 in ALT cells and promotes telomeric DNA synthesis. Hum. Mol. Genet. 2002, 11, 3135–3144. [Google Scholar] [CrossRef] [PubMed]
- Laud, P.R.; Multani, A.S.; Bailey, S.M.; Wu, L.; Ma, J.; Kingsley, C.; Lebel, M.; Pathak, S.; DePinho, R.A.; Chang, S. Elevated telomere-telomere recombination in WRN-deficient, telomere dysfunctional cells promotes escape from senescence and engagement of the ALT pathway. Genes Dev. 2005, 19, 2560–2570. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Hwang, S.S.; Liesa, M.; Gan, B.; Sahin, E.; Jaskelioff, M.; Ding, Z.; Ying, H.; Boutin, A.T.; Zhang, H.; et al. Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell 2012, 148, 651–663. [Google Scholar] [CrossRef]
- Lovejoy, C.A.; Li, W.; Reisenweber, S.; Thongthip, S.; Bruno, J.; de Lange, T.; De, S.; Petrini, J.H.; Sung, P.A.; Jasin, M.; et al. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 2012, 8, e1002772. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Lee, Y.; Wischnewski, H.; Brun, C.M.; Schwarz, T.; Azzalin, C.M. RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat. Commun. 2014, 5, 5220. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Griffith, J.D. Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops. Mol. Cell. Biol. 2004, 24, 9948–9957. [Google Scholar] [CrossRef] [PubMed]
- Nabetani, A.; Ishikawa, F. Unusual telomeric DNAs in human telomerase-negative immortalized cells. Mol. Cell. Biol. 2009, 29, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Mazzucco, G.; Huda, A.; Galli, M.; Piccini, D.; Giannattasio, M.; Pessina, F.; Doksani, Y. Telomere damage induces internal loops that generate telomeric circles. Nat. Commun. 2020, 11, 5297. [Google Scholar] [CrossRef]
- Chung, I.; Osterwald, S.; Deeg, K.I.; Rippe, K. PML body meets telomere: The beginning of an ALTernate ending? Nucleus 2012, 3, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Loe, T.K.; Li, J.S.Z.; Zhang, Y.; Azeroglu, B.; Boddy, M.N.; Denchi, E.L. Telomere length heterogeneity in ALT cells is maintained by PML-dependent localization of the BTR complex to telomeres. Genes Dev. 2020, 34, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Wright, W.E.; Shay, J.W. Clustered telomeres in phase-separated nuclear condensates engage mitotic DNA synthesis through BLM and RAD52. Genes Dev. 2019, 33, 814–827. [Google Scholar] [CrossRef]
- Blagoev, K.B.; Goodwin, E.H. Telomere exchange and asymmetric segregation of chromosomes can account for the unlimited proliferative potential of ALT cell populations. DNA Repair 2008, 7, 199–204. [Google Scholar] [CrossRef]
- Wilson, D.M., 3rd; Thompson, L.H. Molecular mechanisms of sister-chromatid exchange. Mutat. Res. 2007, 616, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.M.; Brenneman, M.A.; Goodwin, E.H. Frequent recombination in telomeric DNA may extend the proliferative life of telomerase-negative cells. Nucleic Acids Res. 2004, 32, 3743–3751. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, A.; Reddel, R.R. The first molecular details of ALT in human tumor cells. Hum. Mol. Genet. 2005, 14, R191–R196. [Google Scholar] [CrossRef]
- Varley, H.; Pickett, H.A.; Foxon, J.L.; Reddel, R.R.; Royle, N.J. Molecular characterization of inter-telomere and intra-telomere mutations in human ALT cells. Nat. Genet. 2002, 30, 301–305. [Google Scholar] [CrossRef]
- Onozawa, M.; Zhang, Z.; Kim, Y.J.; Goldberg, L.; Varga, T.; Bergsagel, P.L.; Kuehl, W.M.; Aplan, P.D. Repair of DNA double-strand breaks by templated nucleotide sequence insertions derived from distant regions of the genome. Proc. Natl. Acad. Sci. USA 2014, 111, 7729–7734. [Google Scholar] [CrossRef]
- Feretzaki, M.; Pospisilova, M.; Valador Fernandes, R.; Lunardi, T.; Krejci, L.; Lingner, J. RAD51-dependent recruitment of TERRA lncRNA to telomeres through R-loops. Nature 2020, 587, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, Z.; Shengzhao, G.; Li, X.; Liu, H.; Zhao, Y. Strand break-induced replication fork collapse leads to C-circles, C-overhangs and telomeric recombination. PLoS Genet. 2019, 15, e1007925. [Google Scholar] [CrossRef]
- Silva, B.; Arora, R.; Bione, S.; Azzalin, C.M. TERRA transcription destabilizes telomere integrity to initiate break-induced replication in human ALT cells. Nat. Commun. 2021, 12, 3760. [Google Scholar] [CrossRef]
- Clynes, D.; Jelinska, C.; Xella, B.; Ayyub, H.; Scott, C.; Mitson, M.; Taylor, S.; Higgs, D.R.; Gibbons, R.J. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun. 2015, 6, 7538. [Google Scholar] [CrossRef] [PubMed]
- Law, M.J.; Lower, K.M.; Voon, H.P.; Hughes, J.R.; Garrick, D.; Viprakasit, V.; Mitson, M.; De Gobbi, M.; Marra, M.; Morris, A.; et al. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell 2010, 143, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Sakellariou, D.; Chiourea, M.; Raftopoulou, C.; Gagos, S. Alternative lengthening of telomeres: Recurrent cytogenetic aberrations and chromosome stability under extreme telomere dysfunction. Neoplasia 2013, 15, 1301–1313. [Google Scholar] [CrossRef]
- Rippe, K.; Luke, B. TERRA and the state of the telomere. Nat. Struct. Mol. Biol. 2015, 22, 853–858. [Google Scholar] [CrossRef]
- Hasegawa, D.; Okabe, S.; Okamoto, K.; Nakano, I.; Shin-ya, K.; Seimiya, H. G-quadruplex ligand-induced DNA damage response coupled with telomere dysfunction and replication stress in glioma stem cells. Biochem. Biophys. Res. Commun. 2016, 471, 75–81. [Google Scholar] [CrossRef]
- Min, J.; Wright, W.E.; Shay, J.W. Alternative Lengthening of Telomeres Mediated by Mitotic DNA Synthesis Engages Break-Induced Replication Processes. Mol. Cell. Biol. 2017, 37, e00226-17. [Google Scholar] [CrossRef] [PubMed]
- Tarsounas, M.; Sung, P. The antitumorigenic roles of BRCA1-BARD1 in DNA repair and replication. Nat. Rev. Mol. Cell Biol. 2020, 21, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S.; Gautier, J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef]
- Napier, C.E.; Huschtscha, L.I.; Harvey, A.; Bower, K.; Noble, J.R.; Hendrickson, E.A.; Reddel, R.R. ATRX represses alternative lengthening of telomeres. Oncotarget 2015, 6, 16543–16558. [Google Scholar] [CrossRef] [PubMed]
- Episkopou, H.; Draskovic, I.; Van Beneden, A.; Tilman, G.; Mattiussi, M.; Gobin, M.; Arnoult, N.; Londoño-Vallejo, A.; Decottignies, A. Alternative Lengthening of Telomeres is characterized by reduced compaction of telomeric chromatin. Nucleic Acids Res. 2014, 42, 4391–4405. [Google Scholar] [CrossRef]
- Goldberg, A.D.; Banaszynski, L.A.; Noh, K.M.; Lewis, P.W.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef]
- Lewis, P.W.; Elsaesser, S.J.; Noh, K.M.; Stadler, S.C.; Allis, C.D. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA 2010, 107, 14075–14080. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Deng, Z.; Zhang, L.; Wu, C.; Jin, Y.; Hwang, I.; Vladimirova, O.; Xu, L.; Yang, L.; Lu, B.; et al. ATRX loss induces telomere dysfunction and necessitates induction of alternative lengthening of telomeres during human cell immortalization. EMBO J. 2019, 38, e96659. [Google Scholar] [CrossRef] [PubMed]
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, J.; Wild, A.T.; Wu, W.H.; Shah, R.; Danussi, C.; Riggins, G.J.; Kannan, K.; Sulman, E.P.; Chan, T.A.; et al. G-quadruplex DNA drives genomic instability and represents a targetable molecular abnormality in ATRX-deficient malignant glioma. Nat. Commun. 2019, 10, 943. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, E.; Matsusaka, T.; Morrison, C.; Vagnarelli, P.; Hoshi, O.; Ushiki, T.; Nojima, K.; Fukagawa, T.; Waizenegger, I.C.; Peters, J.M.; et al. Scc1/Rad21/Mcd1 is required for sister chromatid cohesion and kinetochore function in vertebrate cells. Dev. Cell 2001, 1, 759–770. [Google Scholar] [CrossRef]
- Hartsuiker, E.; Vaessen, E.; Carr, A.M.; Kohli, J. Fission yeast Rad50 stimulates sister chromatid recombination and links cohesion with repair. EMBO J. 2001, 20, 6660–6671. [Google Scholar] [CrossRef] [PubMed]
- González-Barrera, S.; Cortés-Ledesma, F.; Wellinger, R.E.; Aguilera, A. Equal sister chromatid exchange is a major mechanism of double-strand break repair in yeast. Mol. Cell 2003, 11, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Horiuchi, T.; Tongaonkar, P.; Vu, L.; Nomura, M. SIR2 regulates recombination between different rDNA repeats, but not recombination within individual rRNA genes in yeast. Cell 2004, 117, 441–453. [Google Scholar] [CrossRef]
- Potts, P.R.; Porteus, M.H.; Yu, H. Human SMC5/6 complex promotes sister chromatid homologous recombination by recruiting the SMC1/3 cohesin complex to double-strand breaks. EMBO J. 2006, 25, 3377–3388. [Google Scholar] [CrossRef]
- Covo, S.; Westmoreland, J.W.; Gordenin, D.A.; Resnick, M.A. Cohesin Is limiting for the suppression of DNA damage-induced recombination between homologous chromosomes. PLoS Genet. 2010, 6, e1001006. [Google Scholar] [CrossRef] [PubMed]
- Gelot, C.; Guirouilh-Barbat, J.; Le Guen, T.; Dardillac, E.; Chailleux, C.; Canitrot, Y.; Lopez, B.S. The Cohesin Complex Prevents the End Joining of Distant DNA Double-Strand Ends. Mol. Cell 2016, 61, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, C.A.; Takai, K.; Huh, M.S.; Picketts, D.J.; de Lange, T. ATRX affects the repair of telomeric DSBs by promoting cohesion and a DAXX-dependent activity. PLoS Biol. 2020, 18, e3000594. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, M.; Smith, S. Loss of ATRX Suppresses Resolution of Telomere Cohesion to Control Recombination in ALT Cancer Cells. Cancer Cell 2015, 28, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Teber, E.T.; Holmes, O.; Nones, K.; Patch, A.M.; Dagg, R.A.; Lau, L.M.S.; Lee, J.H.; Napier, C.E.; Arthur, J.W.; et al. Telomere sequence content can be used to determine ALT activity in tumours. Nucleic Acids Res. 2018, 46, 4903–4918. [Google Scholar] [CrossRef]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef]
- De Nonneville, A.; Salas, S.; Bertucci, F.; Sobinoff, A.P.; Adelaide, J.; Guille, A.; Finetti, P.; Noble, J.R.; Churikov, D.; Chaffanet, M.; et al. TOP3A amplification and ATRX inactivation are mutually exclusive events in pediatric osteosarcomas using ALT. EMBO Mol. Med. 2022, 14, e15859. [Google Scholar] [CrossRef] [PubMed]
- Brosnan-Cashman, J.A.; Davis, C.M.; Diplas, B.H.; Meeker, A.K.; Rodriguez, F.J.; Heaphy, C.M. SMARCAL1 loss and alternative lengthening of telomeres (ALT) are enriched in giant cell glioblastoma. Mod. Pathol. 2021, 34, 1810–1819. [Google Scholar] [CrossRef]
- Sieverling, L.; Hong, C.; Koser, S.D.; Ginsbach, P.; Kleinheinz, K.; Hutter, B.; Braun, D.M.; Cortes-Ciriano, I.; Xi, R.; Kabbe, R.; et al. Genomic footprints of activated telomere maintenance mechanisms in cancer. Nat. Commun. 2020, 11, 733. [Google Scholar] [CrossRef] [PubMed]
- De Nonneville, A.; Reddel, R.R. Alternative lengthening of telomeres is not synonymous with mutations in ATRX/DAXX. Nat. Commun. 2021, 12, 1552. [Google Scholar] [CrossRef]
- Feuerbach, L. Formal reply to “Alternative lengthening of telomeres is not synonymous with mutations in ATRX/DAXX”. Nat. Commun. 2021, 12, 1551. [Google Scholar] [CrossRef]
- Conomos, D.; Reddel, R.R.; Pickett, H.A. NuRD-ZNF827 recruitment to telomeres creates a molecular scaffold for homologous recombination. Nat. Struct. Mol. Biol. 2014, 21, 760–770. [Google Scholar] [CrossRef]
- Gauchier, M.; Kan, S.; Barral, A.; Sauzet, S.; Agirre, E.; Bonnell, E.; Saksouk, N.; Barth, T.K.; Ide, S.; Urbach, S.; et al. SETDB1-dependent heterochromatin stimulates alternative lengthening of telomeres. Sci. Adv. 2019, 5, eaav3673. [Google Scholar] [CrossRef]
- Murnane, J.P. Telomere dysfunction and chromosome instability. Mutat. Res. Mol. Mech. Mutagen. 2012, 730, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Marzec, P.; Armenise, C.; Perot, G.; Roumelioti, F.M.; Basyuk, E.; Gagos, S.; Chibon, F.; Dejardin, J. Nuclear-receptor-mediated telomere insertion leads to genome instability in ALT cancers. Cell 2015, 160, 913–927. [Google Scholar] [CrossRef] [PubMed]
- Schwab, R.A.; Nieminuszczy, J.; Shah, F.; Langton, J.; Lopez Martinez, D.; Liang, C.C.; Cohn, M.A.; Gibbons, R.J.; Deans, A.J.; Niedzwiedz, W. The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol. Cell 2015, 60, 351–361. [Google Scholar] [CrossRef]
- Lafuente-Barquero, J.; Luke-Glaser, S.; Graf, M.; Silva, S.; Gomez-Gonzalez, B.; Lockhart, A.; Lisby, M.; Aguilera, A.; Luke, B. The Smc5/6 complex regulates the yeast Mph1 helicase at RNA-DNA hybrid-mediated DNA damage. PLoS Genet. 2017, 13, e1007136. [Google Scholar] [CrossRef]
- Deans, A.J.; West, S.C. FANCM connects the genome instability disorders Bloom’s Syndrome and Fanconi Anemia. Mol. Cell 2009, 36, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liu, S.; Bellani, M.A.; Thazhathveetil, A.K.; Ling, C.; de Winter, J.P.; Wang, Y.; Wang, W.; Seidman, M.M. The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol. Cell 2013, 52, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Rohleder, F.; Huang, J.; Xue, Y.; Kuper, J.; Round, A.; Seidman, M.; Wang, W.; Kisker, C. FANCM interacts with PCNA to promote replication traverse of DNA interstrand crosslinks. Nucleic Acids Res. 2016, 44, 3219–3232. [Google Scholar] [CrossRef]
- Bétous, R.; Mason, A.C.; Rambo, R.P.; Bansbach, C.E.; Badu-Nkansah, A.; Sirbu, B.M.; Eichman, B.F.; Cortez, D. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012, 26, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.A.; Zhao, R.; Glick, G.G.; Lovejoy, C.A.; Eischen, C.M.; Cortez, D. SMARCAL1 maintains telomere integrity during DNA replication. Proc. Natl. Acad. Sci. USA 2015, 112, 14864–14869. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.E.; Marechal, A.; Flynn, R.L. SMARCAL1 Resolves Replication Stress at ALT Telomeres. Cell Rep. 2016, 14, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; O’Rourke, J.J.; Sobinoff, A.P.; Allen, J.A.M.; Nelson, C.B.; Tomlinson, C.G.; Lee, M.; Reddel, R.R.; Deans, A.J.; Pickett, H.A. The FANCM-BLM-TOP3A-RMI complex suppresses alternative lengthening of telomeres (ALT). Nat. Commun. 2019, 10, 2252. [Google Scholar] [CrossRef]
- Root, H.; Larsen, A.; Komosa, M.; Al-Azri, F.; Li, R.; Bazett-Jones, D.P.; Stephen Meyn, M. FANCD2 limits BLM-dependent telomere instability in the alternative lengthening of telomeres pathway. Hum. Mol. Genet. 2016, 25, 3255–3268. [Google Scholar] [CrossRef]
- Pan, X.; Chen, Y.; Biju, B.; Ahmed, N.; Kong, J.; Goldenberg, M.; Huang, J.; Mohan, N.; Klosek, S.; Parsa, K.; et al. FANCM suppresses DNA replication stress at ALT telomeres by disrupting TERRA R-loops. Sci. Rep. 2019, 9, 19110. [Google Scholar] [CrossRef]
- Roumelioti, F.M.; Sotiriou, S.K.; Katsini, V.; Chiourea, M.; Halazonetis, T.D.; Gagos, S. Alternative lengthening of human telomeres is a conservative DNA replication process with features of break-induced replication. EMBO Rep. 2016, 17, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Cejka, P.; Symington, L.S. DNA End Resection: Mechanism and Control. Annu. Rev. Genet. 2021, 55, 285–307. [Google Scholar] [CrossRef]
- Zhang, J.M.; Yadav, T.; Ouyang, J.; Lan, L.; Zou, L. Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cell Rep. 2019, 26, 955–968.e3. [Google Scholar] [CrossRef]
- Yadav, T.; Zhang, J.M.; Ouyang, J.; Leung, W.; Simoneau, A.; Zou, L. TERRA and RAD51AP1 promote alternative lengthening of telomeres through an R- to D-loop switch. Mol. Cell 2022, 82, 3985–4000.e4. [Google Scholar] [CrossRef]
- Costantino, L.; Sotiriou, S.K.; Rantala, J.K.; Magin, S.; Mladenov, E.; Helleday, T.; Haber, J.E.; Iliakis, G.; Kallioniemi, O.P.; Halazonetis, T.D. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 2014, 343, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Drosopoulos, W.C.; Sethi, L.; Madireddy, A.; Schildkraut, C.L.; Zhang, D. FANCM, BRCA1, and BLM cooperatively resolve the replication stress at the ALT telomeres. Proc. Natl. Acad. Sci. USA 2017, 114, E5940–E5949. [Google Scholar] [CrossRef] [PubMed]
- Özer, Ö.; Bhowmick, R.; Liu, Y.; Hickson, I.D. Human cancer cells utilize mitotic DNA synthesis to resist replication stress at telomeres regardless of their telomere maintenance mechanism. Oncotarget 2018, 9, 15836–15846. [Google Scholar] [CrossRef]
- Sobinoff, A.P.; Allen, J.A.; Neumann, A.A.; Yang, S.F.; Walsh, M.E.; Henson, J.D.; Reddel, R.R.; Pickett, H.A. BLM and SLX4 play opposing roles in recombination-dependent replication at human telomeres. EMBO J. 2017, 36, 2907–2919. [Google Scholar] [CrossRef]
- Zhang, J.M.; Genois, M.M.; Ouyang, J.; Lan, L.; Zou, L. Alternative lengthening of telomeres is a self-perpetuating process in ALT-associated PML bodies. Mol. Cell 2021, 81, 1027–1042.e4. [Google Scholar] [CrossRef] [PubMed]
- Bakhos-Douaihy, D.; Desmaze, C.; Jeitany, M.; Gauthier, L.R.; Biard, D.; Junier, M.P.; Chneiweiss, H.; Boussin, F.D. ALT cancer cells are specifically sensitive to lysine acetyl transferase inhibition. Oncotarget 2019, 10, 773–784. [Google Scholar] [CrossRef]
- Yang, C.W.; Hsieh, M.H.; Sun, H.J.; Teng, S.C. Nuclear envelope tethering inhibits the formation of ALT-associated PML bodies in ALT cells. Aging 2021, 13, 10490–10516. [Google Scholar] [CrossRef]
- Episkopou, H.; Diman, A.; Claude, E.; Viceconte, N.; Decottignies, A. TSPYL5 Depletion Induces Specific Death of ALT Cells through USP7-Dependent Proteasomal Degradation of POT1. Mol. Cell 2019, 75, 469–482 e466. [Google Scholar] [CrossRef]
- Mukherjee, J.; Johannessen, T.C.; Ohba, S.; Chow, T.T.; Jones, L.; Pandita, A.; Pieper, R.O. Mutant IDH1 Cooperates with ATRX Loss to Drive the Alternative Lengthening of Telomere Phenotype in Glioma. Cancer Res. 2018, 78, 2966–2977. [Google Scholar] [CrossRef]
- Diotti, R.; Loayza, D. Shelterin complex and associated factors at human telomeres. Nucleus 2011, 2, 119–135. [Google Scholar] [CrossRef]
- Wilson, F.R.; Ho, A.; Walker, J.R.; Zhu, X.D. Cdk-dependent phosphorylation regulates TRF1 recruitment to PML bodies and promotes C-circle production in ALT cells. J. Cell Sci. 2016, 129, 2559–2572. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, L.; Bosso, G.; Louzame, J.; Serrano, R.; Gómez-Casero, E.; Martínez-Torrecuadrada, J.; Martínez, S.; Blanco-Aparicio, C.; Pastor, J.; Blasco, M.A. Multiple cancer pathways regulate telomere protection. EMBO Mol. Med. 2019, 11, e10292. [Google Scholar] [CrossRef]
- Bejarano, L.; Schuhmacher, A.J.; Méndez, M.; Megías, D.; Blanco-Aparicio, C.; Martínez, S.; Pastor, J.; Squatrito, M.; Blasco, M.A. Inhibition of TRF1 Telomere Protein Impairs Tumor Initiation and Progression in Glioblastoma Mouse Models and Patient-Derived Xenografts. Cancer Cell 2017, 32, 590–607.e4. [Google Scholar] [CrossRef] [PubMed]
- García-Beccaria, M.; Martínez, P.; Méndez-Pertuz, M.; Martínez, S.; Blanco-Aparicio, C.; Cañamero, M.; Mulero, F.; Ambrogio, C.; Flores, J.M.; Megias, D.; et al. Therapeutic inhibition of TRF1 impairs the growth of p53-deficient K-RasG12V-induced lung cancer by induction of telomeric DNA damage. EMBO Mol. Med. 2015, 7, 930–949. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Pertuz, M.; Martínez, P.; Blanco-Aparicio, C.; Gómez-Casero, E.; Belen García, A.; Martínez-Torrecuadrada, J.; Palafox, M.; Cortés, J.; Serra, V.; Pastor, J.; et al. Modulation of telomere protection by the PI3K/AKT pathway. Nat. Commun. 2017, 8, 1278. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, L.; Chen, Y.; Yang, Y.; Yang, C.Y.; Guo, T.; Lei, M.; Sun, H.; Wang, S. Cyclic Peptidic Mimetics of Apollo Peptides Targeting Telomeric Repeat Binding Factor 2 (TRF2) and Apollo Interaction. ACS Med. Chem. Lett. 2018, 9, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Di Maro, S.; Zizza, P.; Salvati, E.; De Luca, V.; Capasso, C.; Fotticchia, I.; Pagano, B.; Marinelli, L.; Gilson, E.; Novellino, E.; et al. Shading the TRF2 recruiting function: A new horizon in drug development. J. Am. Chem. Soc. 2014, 136, 16708–16711. [Google Scholar] [CrossRef]
- Benarroch-Popivker, D.; Pisano, S.; Mendez-Bermudez, A.; Lototska, L.; Kaur, P.; Bauwens, S.; Djerbi, N.; Latrick, C.M.; Fraisier, V.; Pei, B.; et al. TRF2-Mediated Control of Telomere DNA Topology as a Mechanism for Chromosome-End Protection. Mol. Cell 2016, 61, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Chen, Y.; Lei, M.; Chang, S. TRF2-RAP1 is required to protect telomeres from engaging in homologous recombination-mediated deletions and fusions. Nat. Commun. 2016, 7, 10881. [Google Scholar] [CrossRef] [PubMed]
- Henson, J.D.; Neumann, A.A.; Yeager, T.R.; Reddel, R.R. Alternative lengthening of telomeres in mammalian cells. Oncogene 2002, 21, 598–610. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef]
- Hopkins, T.A.; Ainsworth, W.B.; Ellis, P.A.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Abraham, V.C.; Algire, M.A.; Shi, Y.; Olson, A.M.; et al. PARP1 Trapping by PARP Inhibitors Drives Cytotoxicity in Both Cancer Cells and Healthy Bone Marrow. Mol. Cancer Res. 2019, 17, 409–419. [Google Scholar] [CrossRef]
- Zimmermann, M.; Murina, O.; Reijns, M.A.M.; Agathanggelou, A.; Challis, R.; Tarnauskaite, Z.; Muir, M.; Fluteau, A.; Aregger, M.; McEwan, A.; et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 2018, 559, 285–289. [Google Scholar] [CrossRef]
- Demeny, M.A.; Virag, L. The PARP Enzyme Family and the Hallmarks of Cancer Part 2: Hallmarks Related to Cancer Host Interactions. Cancers 2021, 13, 2057. [Google Scholar] [CrossRef]
- Chen, Q.; Ijpma, A.; Greider, C.W. Two survivor pathways that allow growth in the absence of telomerase are generated by distinct telomere recombination events. Mol. Cell. Biol. 2001, 21, 1819–1827. [Google Scholar] [CrossRef]
- Kockler, Z.W.; Comeron, J.M.; Malkova, A. A unified alternative telomere-lengthening pathway in yeast survivor cells. Mol. Cell 2021, 81, 1816–1829.e5. [Google Scholar] [CrossRef] [PubMed]
- Bugreev, D.V.; Huang, F.; Mazina, O.M.; Pezza, R.J.; Voloshin, O.N.; Camerini-Otero, R.D.; Mazin, A.V. HOP2-MND1 modulates RAD51 binding to nucleotides and DNA. Nat. Commun. 2014, 5, 4198. [Google Scholar] [CrossRef] [PubMed]
- Barroso-Gonzalez, J.; Garcia-Exposito, L.; Hoang, S.M.; Lynskey, M.L.; Roncaioli, J.L.; Ghosh, A.; Wallace, C.T.; de Vitis, M.; Modesti, M.; Bernstein, K.A.; et al. RAD51AP1 Is an Essential Mediator of Alternative Lengthening of Telomeres. Mol. Cell 2019, 76, 11–26.e17. [Google Scholar] [CrossRef]
- Tauchi, H.; Kobayashi, J.; Morishima, K.; van Gent, D.C.; Shiraishi, T.; Verkaik, N.S.; van Heems, D.; Ito, E.; Nakamura, A.; Sonoda, E.; et al. Nbs1 is essential for DNA repair by homologous recombination in higher vertebrate cells. Nature 2002, 420, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.H.; Jiang, W.Q.; Cesare, A.J.; Neumann, A.A.; Wadhwa, R.; Reddel, R.R. Disruption of telomere maintenance by depletion of the MRE11/RAD50/NBS1 complex in cells that use alternative lengthening of telomeres. J. Biol. Chem. 2007, 282, 29314–29322. [Google Scholar] [CrossRef] [PubMed]
- Dupre, A.; Boyer-Chatenet, L.; Sattler, R.M.; Modi, A.P.; Lee, J.H.; Nicolette, M.L.; Kopelovich, L.; Jasin, M.; Baer, R.; Paull, T.T.; et al. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat. Chem. Biol. 2008, 4, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Bakr, A.; Oing, C.; Kocher, S.; Borgmann, K.; Dornreiter, I.; Petersen, C.; Dikomey, E.; Mansour, W.Y. Involvement of ATM in homologous recombination after end resection and RAD51 nucleofilament formation. Nucleic Acids Res. 2015, 43, 3154–3166. [Google Scholar] [CrossRef]
- ATM Inhibition Sensitizes ALT Neuroblastomas to Chemotherapy. Cancer Discov. 2021, 11, 2368. [CrossRef] [PubMed]
- Macha, S.J.; Koneru, B.; Burrow, T.A.; Zhu, C.; Savitski, D.; Rahman, R.L.; Ronaghan, C.A.; Nance, J.; McCoy, K.; Eslinger, C.; et al. Alternative Lengthening of Telomeres in Cancer Confers a Vulnerability to Reactivation of p53 Function. Cancer Res. 2022, 82, 3345–3358. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suva, M.L.; Benes, C.H.; et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015, 347, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Schoppy, D.W.; Ragland, R.L.; Gilad, O.; Shastri, N.; Peters, A.A.; Murga, M.; Fernandez-Capetillo, O.; Diehl, J.A.; Brown, E.J. Oncogenic stress sensitizes murine cancers to hypomorphic suppression of ATR. J. Clin. Investig. 2012, 122, 241–252. [Google Scholar] [CrossRef]
- Grudic, A.; Jul-Larsen, A.; Haring, S.J.; Wold, M.S.; Lonning, P.E.; Bjerkvig, R.; Boe, S.O. Replication protein A prevents accumulation of single-stranded telomeric DNA in cells that use alternative lengthening of telomeres. Nucleic Acids Res. 2007, 35, 7267–7278. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Kaul, Z.; Gocha, A.S.; Martinez, A.R.; Harris, J.; Parvin, J.D.; Groden, J. Association of BLM and BRCA1 during Telomere Maintenance in ALT Cells. PLoS ONE 2014, 9, e103819. [Google Scholar] [CrossRef] [PubMed]
- Gocha, A.R.; Acharya, S.; Groden, J. WRN loss induces switching of telomerase-independent mechanisms of telomere elongation. PLoS ONE 2014, 9, e93991. [Google Scholar] [CrossRef]
- Kargaran, P.K.; Yasaei, H.; Anjomani-Virmouni, S.; Mangiapane, G.; Slijepcevic, P. Analysis of alternative lengthening of telomere markers in BRCA1 defective cells. Genes Chromosomes Cancer 2016, 55, 864–876. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wang, F.; Liu, L. Alternative Lengthening of Telomeres (ALT) in Tumors and Pluripotent Stem Cells. Genes 2019, 10, 1030. [Google Scholar] [CrossRef] [PubMed]
- Makarova, A.V.; Stodola, J.L.; Burgers, P.M. A four-subunit DNA polymerase zeta complex containing Pol delta accessory subunits is essential for PCNA-mediated mutagenesis. Nucleic Acids Res. 2012, 40, 11618–11626. [Google Scholar] [CrossRef] [PubMed]
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.W.; Shen, H.; Liu, Y.; Hickson, I.D. Replication stress activates DNA repair synthesis in mitosis. Nature 2015, 528, 286–290. [Google Scholar] [CrossRef]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Minocherhomji, S.; Chan, K.L.; Palmai-Pallag, T.; Chu, W.K.; Wass, T.; Mankouri, H.W.; Liu, Y.; Hickson, I.D. MUS81 promotes common fragile site expression. Nat. Cell Biol. 2013, 15, 1001–1007. [Google Scholar] [CrossRef]
- Naim, V.; Wilhelm, T.; Debatisse, M.; Rosselli, F. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 2013, 15, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Guh, C.Y.; Shen, H.J.; Chen, L.W.; Chiu, P.C.; Liao, I.H.; Lo, C.C.; Chen, Y.; Hsieh, Y.H.; Chang, T.C.; Yen, C.P.; et al. XPF activates break-induced telomere synthesis. Nat. Commun. 2022, 13, 5781. [Google Scholar] [CrossRef]
- Higa, M.; Fujita, M.; Yoshida, K. DNA Replication Origins and Fork Progression at Mammalian Telomeres. Genes 2017, 8, 112. [Google Scholar] [CrossRef]
- Lezaja, A.; Panagopoulos, A.; Wen, Y.; Carvalho, E.; Imhof, R.; Altmeyer, M. RPA shields inherited DNA lesions for post-mitotic DNA synthesis. Nat. Commun. 2021, 12, 3827. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Pickett, H.A. Telomeric replication stress: The beginning and the end for alternative lengthening of telomeres cancers. Open Biol. 2022, 12, 220011. [Google Scholar] [CrossRef]
- Ghelli Luserna di Rora, A.; Cerchione, C.; Martinelli, G.; Simonetti, G. A WEE1 family business: Regulation of mitosis, cancer progression, and therapeutic target. J. Hematol Oncol. 2020, 13, 126. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zhao, H.; Diplas, B.H.; Liu, S.; Liu, J.; Wang, D.; Lu, Y.; Zhu, Q.; Wu, J.; Wang, W.; et al. Genome-Wide CRISPR-Cas9 Screen Reveals Selective Vulnerability of ATRX-Mutant Cancers to WEE1 Inhibition. Cancer Res. 2020, 80, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Koneru, B.; Farooqi, A.; Nguyen, T.H.; Chen, W.H.; Hindle, A.; Eslinger, C.; Makena, M.R.; Burrow, T.A.; Wilson, J.; Smith, A.; et al. ALT neuroblastoma chemoresistance due to telomere dysfunction-induced ATM activation is reversible with ATM inhibitor AZD0156. Sci. Transl. Med. 2021, 13, 5750. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Schwab, R.A.; Nieminuszczy, J.; Deans, A.J.; West, S.C.; Niedzwiedz, W. The DNA translocase activity of FANCM protects stalled replication forks. Hum. Mol. Genet. 2012, 21, 2005–2016. [Google Scholar] [CrossRef]
- Forment, J.V.; O’Connor, M.J. Targeting the replication stress response in cancer. Pharmacol. Ther. 2018, 188, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Byun, T.S.; Pacek, M.; Yee, M.C.; Walter, J.C.; Cimprich, K.A. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005, 19, 1040–1052. [Google Scholar] [CrossRef]
- Delacroix, S.; Wagner, J.M.; Kobayashi, M.; Yamamoto, K.; Karnitz, L.M. The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 2007, 21, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Gorecki, L.; Andrs, M.; Rezacova, M.; Korabecny, J. Discovery of ATR kinase inhibitor berzosertib (VX-970, M6620): Clinical candidate for cancer therapy. Pharmacol. Ther. 2020, 210, 107518. [Google Scholar] [CrossRef]
- O’Rourke, J.J.; Bythell-Douglas, R.; Dunn, E.A.; Deans, A.J. ALT control, delete: FANCM as an anti-cancer target in Alternative Lengthening of Telomeres. Nucleus 2019, 10, 221–230. [Google Scholar] [CrossRef]
- Voter, A.F.; Manthei, K.A.; Keck, J.L. A High-Throughput Screening Strategy to Identify Protein-Protein Interaction Inhibitors That Block the Fanconi Anemia DNA Repair Pathway. SLAS Discov. Adv. Sci. Drug Discov. 2016, 21, 626–633. [Google Scholar] [CrossRef]
- Kent, T.; Gracias, D.; Shepherd, S.; Clynes, D. Alternative Lengthening of Telomeres in Pediatric Cancer: Mechanisms to Therapies. Front. Oncol. 2020, 9, 1518. [Google Scholar] [CrossRef]
- Bryan, T.M. G-Quadruplexes at Telomeres: Friend or Foe? Molecules 2020, 25, 3686. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Qi, H.; Lin, C.P.; Lin, R.K.; Kerrigan, J.E.; Rzuczek, S.G.; LaVoie, E.J.; Rice, J.E.; Pilch, D.S.; Lyu, Y.L.; et al. A G-quadruplex stabilizer induces M-phase cell cycle arrest. J. Biol. Chem. 2009, 284, 22535–22543. [Google Scholar] [CrossRef]
- Amato, R.; Valenzuela, M.; Berardinelli, F.; Salvati, E.; Maresca, C.; Leone, S.; Antoccia, A.; Sgura, A. G-quadruplex Stabilization Fuels the ALT Pathway in ALT-positive Osteosarcoma Cells. Genes 2020, 11, 304. [Google Scholar] [CrossRef]
- Pennarun, G.; Granotier, C.; Gauthier, L.R.; Gomez, D.; Hoffschir, F.; Mandine, E.; Riou, J.F.; Mergny, J.L.; Mailliet, P.; Boussin, F.D. Apoptosis related to telomere instability and cell cycle alterations in human glioma cells treated by new highly selective G-quadruplex ligands. Oncogene 2005, 24, 2917–2928. [Google Scholar] [CrossRef]
- Castillo Bosch, P.; Segura-Bayona, S.; Koole, W.; van Heteren, J.T.; Dewar, J.M.; Tijsterman, M.; Knipscheer, P. FANCJ promotes DNA synthesis through G-quadruplex structures. EMBO J. 2014, 33, 2521–2533. [Google Scholar] [CrossRef]
- Zhang, J.M.; Zou, L. Alternative lengthening of telomeres: From molecular mechanisms to therapeutic outlooks. Cell Biosci. 2020, 10, 30. [Google Scholar] [CrossRef]
- MacKenzie, D., Jr.; Watters, A.K.; To, J.T.; Young, M.W.; Muratori, J.; Wilkoff, M.H.; Abraham, R.G.; Plummer, M.M.; Zhang, D. ALT Positivity in Human Cancers: Prevalence and Clinical Insights. Cancers 2021, 13, 2384. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.; Royle, N.J. ALT: A Multi-Faceted Phenomenon. Genes 2020, 11, 133. [Google Scholar] [CrossRef] [PubMed]
- Dilley, R.L.; Greenberg, R.A. ALTernative Telomere Maintenance and Cancer. Trends Cancer 2015, 1, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Pompili, L.; Leonetti, C.; Biroccio, A.; Salvati, E. Diagnosis and treatment of ALT tumors: Is Trabectedin a new therapeutic option? J. Exp. Clin. Cancer Res. 2017, 36, 189. [Google Scholar] [CrossRef] [PubMed]
- Kent, T.; Clynes, D. Alternative Lengthening of Telomeres: Lessons to Be Learned from Telomeric DNA Double-Strand Break Repair. Genes 2021, 12, 1734. [Google Scholar] [CrossRef]
- Domingues-Silva, B.; Silva, B.; Azzalin, C.M. ALTernative Functions for Human FANCM at Telomeres. Front. Mol. Biosci. 2019, 6, 84. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef]
- Gao, J.; Pickett, H.A. Targeting telomeres: Advances in telomere maintenance mechanism-specific cancer therapies. Nat. Rev. Cancer 2022, 22, 515–532. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Therapeutic Target | Molecular Mechanism | Relevant Drugs |
|---|---|---|
| SP100 | MRN sequestering and APB inhibition | |
| TRF1 (T271) | APB formation | |
| TRF1 (T371 phosphorylation) | APB formation | |
| MMS21 SUMO ligase | SMC5/6 complex maintenance interference | |
| TSPYL5 | USP7 repression | |
| FANCM/FAAP24 | Replication stress suppression | |
| FANCM/BTR | D-loop branch migration | PIP-199 |
| ATRX/DAXX | Chromatin decompaction and telomere cohesion | |
| NRSC/F | NuRD and ZNF827 recruitment | |
| SMARCAL1 | Replication fork reversal and re-initiation | |
| FA core complex | FANCD2 monoubiquitination and BRCA1/2 localization | |
| TRF2 | APOLLO exonuclease recruitment and 5′ resection | |
| TRF2 | SLX4 repression | |
| PARP | BRCA1/2 interaction | |
| HOP2-MND1 heterodimer | Recombinase activity | |
| DMC1 | DNA strand exchange | |
| RAD51AP1 | Telomere dysfunction and fragmentation | |
| NBS1 | T-SCEs | |
| MRN | ATM signaling | Mirin |
| POLD3/4 | BIR and CFS-MiDAS | |
| XPF | DDR pathway activation | |
| WEE1 | CDK phosphorylation | MK-1775 |
| PKMYT1 | CDK phosphorylation | RP-6306 |
| ATM | Replication form regression and stabilization | AZD0156 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sohn, E.J.; Goralsky, J.A.; Shay, J.W.; Min, J. The Molecular Mechanisms and Therapeutic Prospects of Alternative Lengthening of Telomeres (ALT). Cancers 2023, 15, 1945. https://doi.org/10.3390/cancers15071945
Sohn EJ, Goralsky JA, Shay JW, Min J. The Molecular Mechanisms and Therapeutic Prospects of Alternative Lengthening of Telomeres (ALT). Cancers. 2023; 15(7):1945. https://doi.org/10.3390/cancers15071945
Chicago/Turabian StyleSohn, Eric J., Julia A. Goralsky, Jerry W. Shay, and Jaewon Min. 2023. "The Molecular Mechanisms and Therapeutic Prospects of Alternative Lengthening of Telomeres (ALT)" Cancers 15, no. 7: 1945. https://doi.org/10.3390/cancers15071945
APA StyleSohn, E. J., Goralsky, J. A., Shay, J. W., & Min, J. (2023). The Molecular Mechanisms and Therapeutic Prospects of Alternative Lengthening of Telomeres (ALT). Cancers, 15(7), 1945. https://doi.org/10.3390/cancers15071945