Simple Summary
Mitochondria, often called “the powerhouse of the cell”, generate most of the energy required for all cellular processes. Mitochondria also provide a platform for multiple functions, including building essential macromolecules (nucleotides, proteins, and lipids), cell signaling, detoxification, and cell fate control. Studies have demonstrated that mitochondrial function is widely reprogrammed in breast cancers to promote tumor progression. Therefore, targeting mitochondria is currently one of the focal points in breast cancer research. Over 1000 proteins are involved with mitochondrial functions. The mitochondrial protease called ClpP plays a central role in mitochondrial protein quality control. Recently, ClpP agonists have emerged as a novel class of mitochondria-targeting drugs. Hyperactivating ClpP induces uncontrolled, but selective, degradation of ClpP substrates and disrupts mitochondrial functions, leading to growth inhibition of breast cancer cells, without adverse effect in non-malignant cells. The unique characteristics and mechanism of action of ClpP agonists provide new opportunities in breast cancer treatment.
Abstract
Breast cancer is the most frequently diagnosed malignancy worldwide and the leading cause of cancer mortality in women. Despite the recent development of new therapeutics including targeted therapies and immunotherapy, triple-negative breast cancer remains an aggressive form of breast cancer, and thus improved treatments are needed. In recent decades, it has become increasingly clear that breast cancers harbor metabolic plasticity that is controlled by mitochondria. A myriad of studies provide evidence that mitochondria are essential to breast cancer progression. Mitochondria in breast cancers are widely reprogrammed to enhance energy production and biosynthesis of macromolecules required for tumor growth. In this review, we will discuss the current understanding of mitochondrial roles in breast cancers and elucidate why mitochondria are a rational therapeutic target. We will then outline the status of the use of mitochondria-targeting drugs in breast cancers, and highlight ClpP agonists as emerging mitochondria-targeting drugs with a unique mechanism of action. We also illustrate possible drug combination strategies and challenges in the future breast cancer clinic.
1. Introduction
Breast cancer is the most diagnosed cancer worldwide (excluding skin cancer), accounting for 12% of all new cancer cases annually [1]. For women in the US, breast cancer is the most commonly diagnosed malignancy (excluding skin cancer) (31%), and the second leading cause of cancer mortality overall after lung cancer [2]. Breast cancer is the leading cause of cancer death among African American and Hispanic women [3,4]. Incidence rates of breast cancer continue to rise by ~0.5% per year, although the cancer mortality rates have declined steadily since their peak in 1989, albeit at a slower pace in recent years (1.3% annually from 2011 to 2020) [3]. Early diagnosed breast cancers are often effectively treated using surgery, radiation, chemotherapy, endocrine therapy, and targeted therapies [5]. Breast cancers can be classified into distinct subtypes: estrogen receptor positive (ER+), human epidermal growth factor receptor 2 (HER2) amplified, and triple-negative breast cancer (TNBC) defined by the absence of ER and progesterone receptor (PR) expression and HER2 amplification [6]. TNBC lacks specific molecular markers and is usually more aggressive compared with other subtypes. Endocrine therapies for ER+, and HER2-targeted drug therapies for HER2 amplified subtypes have led to significant improvement of the survival rate for both early stage and advanced cancers [7]. TNBC therapy remains challenging with high rates of relapse and metastatic disease, despite significant improvements in chemotherapy and targeted therapies (e.g., trop2-targeted therapy and immune checkpoint inhibitors [ICIs]). There is an urgent need to explore more effective and targeted strategies to overcome such drug resistance and metastasis in current therapy, especially in TNBC [8,9,10,11,12].
Much effort has been made to uncover underlying mechanisms that drive the proliferation and metastasis of cancers. As a result, it has now become clear that cancer is not only a genetic disease but also a metabolic disease, where oncogenic signaling pathways dysregulate (or co-opt) energy regulation and anabolism to support rapidly spreading tumors [13,14,15,16]. Metabolic reprogramming has recently emerged as one of the major hallmarks of cancer [17,18,19]. Metabolism is rewired in cancer cells to meet the high demand of energy to promote proliferation. For instance, uptake of glucose and amino acids is increased to fulfill the biosynthetic demands; the demand for carbon and nitrogen is upregulated for biosynthesis of macromolecules such as nucleotides, proteins, and lipids; nicotinamide adenine dinucleotide phosphate (NADPH) production is elevated to support reductive metabolic reactions and to maintain redox balance [20]. These complex metabolic changes are intricately orchestrated by the mitochondria, a master regulator of cell metabolism.
In this review, we will first discuss the recent understanding of the role of mitochondria in breast cancers and analyze a wealth of evidence supporting the rationale that mitochondria are an emerging therapeutic target in breast cancers. We then examine the status of the development of mitochondria-targeting drugs, with special highlight of the caseinolytic mitochondrial matrix peptidase proteolytic subunit (ClpP) as an emerging drug target in breast cancers.
2. Mitochondria and Breast Cancer—An Overview
The mitochondrion is a double-membrane-bound organelle found in the cytoplasm of a majority of eukaryotic cells [21]. Mitochondria contain their own genome called mitochondrial DNA (mtDNA), a circular DNA of 16,569 bp in human, which encodes 13 subunits of the electron transport chain (ETC) complexes, 2 ribosomal RNAs (12S and 16S), and 22 transfer RNAs. Mitochondria are often called the ‘powerhouse’ of the cell, as they generate chemical energy in the form of adenosine triphosphate (ATP) via oxidative phosphorylation (OxPhos) at the ETC. Mitochondria also play pleiotropic roles as a center of macromolecule synthesis, such as nucleic acids, amino acids, and lipids. Mitochondria also regulate apoptosis, redox homeostasis, calcium homeostasis, innate immunity, cell signaling, and epigenetic modification of the nuclear genome [22,23,24,25,26,27,28,29,30].
Outside of OxPhos in mitochondria, ATP is generated via glycolysis, in which glucose is used and metabolized to pyruvate and lactate. From an energy efficiency perspective, OxPhos is a more efficient way to produce ATP (36 mol ATP/mol glucose) compared with glycolysis (2 mol ATP/mol glucose). Normal mammalian cells generate ATP from either OxPhos or glycolysis, in the presence or absence of O2, respectively. On the other hand, it has long been viewed that cancer cells primarily rely on glycolysis even in the presence of O2, due to “defective” mitochondria, called the “Warburg effect” [31,32]. In accordance with this view, some studies indicated that downregulation of OxPhos genes is associated with poor clinical outcomes in multiple cancer types [33], and that impaired OxPhos is linked to reduced or mutated mtDNA in cancers [34,35].
In recent years, however, our understanding of the “Warburg effect” has evolved to a more complex cognition of tumor metabolism. The recent development of quantitative omics tools such as RNAseq, proteomics, and metabolomics has provided compelling evidence to support that tumors, including breast cancers, are metabolically heterogeneous and flexible, depending on multiple factors such as cell lineage, differentiation state, and microenvironment [36,37]. In addition, cancer cells have a metabolic demand beyond ATP, such as nucleic acids, amino acids, and lipids to support growth [32,38]. A large body of evidence shows that mitochondria are not defective, but rather essential to cancer cells [39,40]. Herein, we discuss the recent findings with regard to how mitochondria contribute to breast cancer progression.
3. Evidence That Mitochondria Are a Target in Breast Cancer
3.1. OxPhos Is Upregulated in Breast Cancers
A study which analyzed ~2000 breast cancer patients showed that an OxPhos gene signature is significantly upregulated in human breast tumors (p < 1 × 10−20) relative to normal breast tissue, in both ER+ and ER- breast cancers [41]. An analysis from TCGA also showed that OxPhos genes are significantly increased in breast tumor samples compared to normal samples (p = 3.11 × 10−7) [42]. In ER-negative breast cancers, the OxPhos gene signature was associated with a ~30% increase in metastasis over a 10-year follow-up period (50% or 21.4% developed metastasis with or without the signature, respectively) [41]. Moreover, a recent study identified OxPhos as the top pathway upregulated in micrometastases of breast cancers, and pharmacological inhibition of OxPhos attenuated lung metastasis, highlighting the potential of OxPhos as a therapeutic target to prevent metastasis in breast cancers [43].
3.2. Breast Cancer Stem Cells (CSCs) and Circulating Tumor Cells (CTCs) Are Mitochondria-Dependent
CSCs and circulating tumor cells CTCs are responsible for tumorigenesis, chemoresistance, metastasis, and relapse [44,45]. In primary tumors, the majority of cancer cells are highly proliferative, differentiated, non-stem cells. In contrast, CSCs are a rare population (<1%) of cancer cells harboring an undifferentiated, slow-cycling phenotype, with self-renewal and tumor-initiating capacities [46,47,48,49]. These cells show distinct metabolic preferences; the bulk breast cancer cells rely on glycolysis, while quiescent breast CSCs depend on OxPhos, and proliferating CSCs utilize both OxPhos and glycolysis. CSCs also utilize metabolites generated from tumor stromal cells in what is called a “Reverse Warburg effect” [37,50,51,52,53] (Figure 1, left). OxPhos is shown to be critical for breast CSCs function [54,55,56,57,58,59,60,61,62,63]. For instance, inhibition of OxPhos by depleting mtDNA reduced an anchorage-independent survival and propagation of breast CSC-like cells [55,56,57,58]. MtDNA in exosomes mediates endocrine therapy resistance [59]. Samples from chemoresistant TNBC showed high dependency on OxPhos [60,61].
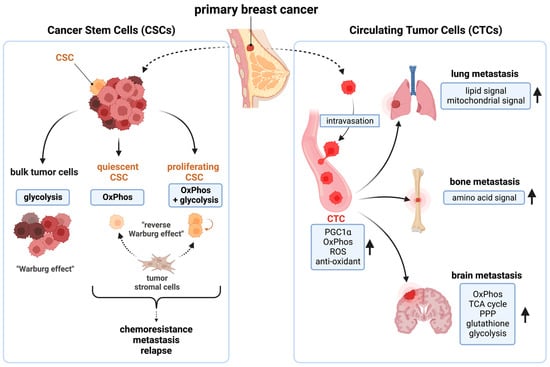
Figure 1.
The function of breast cancer stem cells (CSCs) and circulating tumor cells (CTCs) are mitochondria dependent. (Left): while bulk cancer cells are glycolysis dependent, CSCs depend on OxPhos for survival. (Right): CTC-mediated distant metastases are also dependent on multiple mitochondrial functions. OxPhos: oxidative phosphorylation; PGC1α: peroxisome proliferator-activated receptor gamma coactivator 1-alpha, ROS: reactive oxygen species; TCA cycle: the tricarboxylic acid cycle; PPP: pentose phosphate pathway. This figure was generated by BioRender.com (19 January 2023).
CTCs are shed from the primary tumor and intravasate into the blood circulation and mediate tumor metastasis [64]. CSCs and CTCs share similar phenotypes in breast cancers [65]. Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α), the master regulator of mitochondrial biogenesis, is overexpressed in breast CTCs [66]. OxPhos is elevated in breast CTCs, along with purine/pyrimidine metabolism, ROS, and antioxidant mechanisms [66,67,68]. Lung/bone-metastatic breast cancer cells show high PGC1α expression and mitochondrial metabolic activity [66,69,70]. In brain-metastatic breast cancers, OxPhos, the TCA cycle, and the pentose phosphate pathway (PPP), glutathione, and glycolysis pathways were all elevated [71] (Figure 1, right). A recent clinical study also provided the evidence that OxPhos contributes to breast cancer metastasis. The copper chelating agent tetrathiomolybdate (TM) inhibited lung metastasis and improved survival benefit in TNBC patients (NCT00195091) [72]. TM’s anti-tumor effect is shown to be an inhibition of OxPhos by copper depletion [73]. Thus, mitochondria are critical for breast CSCs/CTCs functions [50,52,74].
3.3. Mitochondrial Metabolism in Breast Cancers Is Widely Reprogrammed to Promote Their Growth
In addition to high demands of ATP, breast cancers heavily rely on multiple mitochondrial metabolic pathways. Many mitochondrial enzymes are overexpressed as discussed below and indicated shown in Supplementary Figure S1.
ETC: The de novo pyrimidine synthesis is coupled to the respiratory chain and mediated by dihydroorotate dehydrogenase (DHODH) [75]. DHODH is overexpressed in human breast cancer tissues [76]. Ubiquinol-cytochrome C reductase core protein II (UQCRC2), a subunit of complex III, is also elevated in breast cancers [77].
Glutamine metabolism: Glutamine serves as a nitrogen donor for protein and nucleotide synthesis and maintains redox homeostasis and detoxication [78]. Glutaminase (GLS) is an enzyme that converts glutamine to glutamate. Glutamate is further converted to alpha-ketoglutaric acid (α-KG) via multiple enzymes, including glutamate dehydrogenase 1 (GLUD/GDH1). GLUD is upregulated in breast cancers [79]. α-KG is a key metabolite that drives the TCA cycle. The TCA cycle produces reducing power of nicotinamide adenine dinucleotide (NADH), which is utilized at the ETC to generate ATP. The TCA cycle also contributes to the biosynthesis of macromolecules, such as amino-acids, nucleotides, and lipids [80,81]. GLS1/GLS2 is pro-tumorigenic [82]. Glutamine metabolism is promoted by PGC1α in HER2+ breast cancer [83]. TNBC are known to be “glutamine-addicted”, with higher expression of key enzymes mediators of glutamine metabolism, including GLS, amino acid transporter-2 (ASCT2) [84,85,86,87,88,89]. Glutamate is enriched in breast cancer tissue [90]. The expression of glutamine synthetase (GS) and asparagine synthetase (ASNS) is correlated with poor prognosis in breast cancers [91,92].
The TCA cycle: High expression of dihydrolipoamide S-succinyltransferase (DLST/2-oxoglutarate dehydrogenase complex [OGDC]) and malic enzyme (ME2) is associated with poor prognosis in breast cancers [93,94].
Proline metabolism: Proline is derived from glutamate, and contributes to synthesis of arginine, glutamate polyamines through the urea cycle. Proline and its precursor delta-1-pyrroline-5-carboxylate (P5C) shuttle between the mitochondria and the cytoplasm, forming the “proline cycle”, thereby contributing to nucleotide synthesis, collagen synthesis, redox homeostasis by coupling with PPP. Proline biosynthesis is elevated in breast cancers [95,96], and pyrroline-5-carboxylate reductase (PYCR) 1, which mediates proline synthesis from P5C, is elevated in invasive breast cancers [97,98]. Proteomic analysis showed that PYCR1, PYCR2 and P5C synthase (P5CS/ALDH18A1) are all elevated in breast cancer patients’ samples [99]. Proline dehydrogenase (PRODH), which converts proline to P5C, is overexpressed in breast cancer metastases compared with primary tumors [100].
Serine, glycine and folate-mediated one carbon cycle: Serine and glycine are essential metabolites for tumor growth, and the related enzymes are upregulated in breast cancers [101,102,103,104]. Phosphoglycerate dehydrogenase (PHGDH), the first rate-limiting enzyme in the serine synthesis pathway, is elevated in ~70% of ER- breast cancers, and PHGDH inhibition showed an anti-proliferative effect [103]. PHGDH expression was elevated in 90% of brain metastases of breast cancer [105]. Serine serves as a major donor of one-carbon units to the folate cycle, which is required for de novo purine/pyrimidine synthesis and production of NADPH [106,107]. Serine hydroxymethyltransferase 2 (SHMT2), an enzyme that converts serine to glycine, is shown as a prognostic marker and therapeutic target for breast cancers [108]. In the folate cycle, methylenetetrahydrofolate dehydrogenase (MTHFD) produces purines. High MTHFD2 is correlated with worse prognosis in breast cancers [109,110]. Thymidylate synthase (TYMS/TS), the target of 5′-fluorouracil, is a rate-limiting enzyme in thymidylate biosynthesis [111]. TYMS expression was elevated in breast cancer compared with normal tissues, and was greater in TNBC than non-TNBC, and higher TYMS was associated with poor prognosis [112]. Consistent with this, purine/pyrimidine metabolism enzyme gene sets are elevated in TNBC [89]. Dihydrofolate reductase (DHFR), another enzyme in folate metabolism, is essential to purine synthesis and contribute to mitochondrial thymidylate biosynthesis. DHFR is a target of methotrexate (MTX). The DHFR activity is elevated in MTX-resistant breast cancer cells [113].
Fatty acid synthesis (FAS): Elevated FAS is one of the most common metabolic traits in cancers [114,115]. FAS occurs in the cytoplasm via lipogenic enzymes, such as acetyl-CoA carboxylase 1 (ACC1/ACACA) and fatty acid synthase (FASN), using acetyl-CoA as the substrate. ACC1 is involved with breast cancer metastasis [116]. FASN is overexpressed in 70% of operable TNBC and is associated with poor prognosis [117,118]. FASN regulates Bcl-2 family proteins and controls breast cancer cell survival [119]. Acetyl-CoA synthetase 2 (ACSS2) is amplified in breast cancers and targeting ACSS2 impaired tumor growth in TNBC [120,121]. ATP citrate lyase (ACLY), an enzyme involved with de novo synthesis of cholesterol and FA, is overexpressed in breast cancer [122].
Fatty acid oxidation (FAO): FAO is the mitochondrial aerobic process of breaking down fatty acid (FA) into acetyl-CoA-units [123]. Overexpression of carnitine palmitoyl transferase I (CPTI), a rate-limiting enzyme of FAO, is associated with breast cancer progression [124,125,126]. FAO contributes to elevated ATP in TNBC and metastasis [127,128], and promotes apoptosis-resistance of TNBC by increasing mitochondrial membrane lipids [129]. FAO is also critical for breast CSCs function [130], and CD36, a FA receptor, is linked to breast cancer metastasis [131].
The mevalonate pathway: The mevalonate pathway synthesizes cholesterol from acetyl-CoA. High cholesterol is associated with breast cancer metastasis, and cholesterol biosynthesis is elevated in breast CSCs [132,133,134,135]. Statin use is associated with lower mortality in breast cancers [136]. The mevalonate precursor enzyme 3-hydroxy-3-methylglutaryl-CoA synthase 1 (HMGCS1) is overexpressed in breast cancer, and a key mediator of enrichment of breast CSCs [137,138]. High expression of HMGCS1 and 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) is correlated with poor prognosis in breast cancer [139]. Together, these data suggest that many metabolic pathways within or linked to the mitochondria are widely dysregulated in breast cancers, and those alterations collectively promote tumor growth and metastasis.
3.4. Myc, TP53, PIK3CA, Bcl-2 Family Proteins Control Mitochondrial Metabolism in Breast Cancers
The Myc proto-oncogene regulates multiple cellular processes. It is well established that Myc promotes breast tumorigenesis and progression [140,141,142,143]. Myc is amplified in breast cancers (~25% of all breast cancers, ~50% in TNBC) and is associated with poor prognosis [144,145,146,147]. Myc is a major driver of mitochondrial biogenesis [148,149]. Among ~1000 nuclear-encoded human mitochondrial genes, over 500 genes are Myc targets [150,151]. Myc also indirectly regulates mitochondrial gene expression via repression of microRNAs [152,153]. Myc globally participates mitochondrial metabolism [154]. Myc promotes glutamine metabolism [152,155,156], proline biosynthesis [96,157], serine biosynthesis [158] and FAO [159,160].
The tumor-suppressor gene TP53 regulates gene expression involved in cell cycle arrest, apoptosis, and senescence [161]. TP53 mutations (TP53mut) are observed in 25–30% of all breast cancers, ~80% in TNBC and 72% in HER2 amplified subtypes [162]. TP53mut participate metabolic changes via numerous mechanisms [163,164]. For instance, TP53mut enhances OxPhos [165,166] and FAS [167]. It also elevates the transcriptional activity of sterol regulatory element-binding proteins (SREBP) family, supporting the mevalonate pathway and cell growth [168].
Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) mutation (PIK3CAmut) leads to increased phosphatidylinositol 3-kinase (PI3K) activity. PIK3CAmut is observed in ~40% of ER+/HER2-breast cancer patients [169]. PIK3CAmut potentiates de novo lipid synthesis, pyruvate entry into the TCA cycle, and glutamine metabolism via the PI3K-AKT serine/threonine kinase (AKT) signaling pathway in breast epithelial cells [170,171,172].
The Bcl-2 family proteins are apoptosis regulatory proteins functioning at the mitochondria [173]. The anti-apoptotic Bcl-2 proteins (Bcl-2, Bcl-xL, Bcl-w, and Mcl-1) restrain pro-apoptotic proteins (Bax, Bak), thus preserving mitochondrial outer membrane integrity, preventing release of pro-apoptotic proteins, thereby inhibiting apoptosis. Bcl-2 is overexpressed in breast cancers (~75%) [174]. Bcl-xL is also overexpressed (43%) and associated with metastasis and higher tumor grade [175,176]. Overexpression of Mcl-1 is even more frequent and is associated with poor prognosis, and TNBC are dependent on Mcl-1 [177]. The Bcl-2 family proteins mediate therapy resistance in breast cancers [178]. Bcl-2 and Bcl-xL also regulate mitochondrial dynamics [179] and support metabolic robustness by elevating NAD(P)H and ATP in breast cancer cells [180,181].
3.5. Reactive Oxygen Species (ROS) Plays a Dual, Complex Role in Breast Cancer
Mitochondria are the major source of ROS, and also provide ROS-scavenging systems, such as glutathione, superoxide dismutase, and NADPH [182,183]. Thus, mitochondria play a major role in redox homeostasis in a cell [25,184]. ROS has a dual, complex role in cancers; either pro- or anti-tumorigenic, depending on the context [185,186]. High ROS is often observed in aggressive, drug-resistant cancers, but it can be lethal; therefore, cancer cells rewire the ROS-scavenging systems [115,187,188,189,190,191,192]. TNBC exhibit higher ROS and higher antioxidant capacity than non-TNBC and are dependent on ROS for cell survival [193,194]. High ROS causes mitochondrial damage and further generates ROS as a “vicious cycle” [195,196]. High ROS stimulates the PI3K-AKT-mammalian target of the rapamycin (mTOR) pathway, leading to the activation of Myc, Ras-mitogen-activated protein kinase (MAPK), the NF-kB pathway, and anti-apoptotic Bcl-2 family proteins, promoting cell survival and metastasis [188]. ROS also triggers DNA mutations, leading to cellular senescence, increased inflammatory cytokine release, and activation of the innate immune system [29,197,198,199,200]. In addition, low levels of ROS are critical for CSCs to maintain their self-renewal ability [25,187,201,202,203]. Breast CSCs have a lower level of ROS compared with bulk tumors, and pharmacologic inhibition of ROS scavengers in breast CSCs decreased their clonogenicity [204]. Similarly, exposure to H2O2 treatment impaired tumor formation ability in breast CSCs and led to senescence [205]. Thus, either a high or low level of ROS can enhance breast cancer growth via different mechanisms.
3.6. Additional Mechanisms by Which Mitochondria Contribute to Breast Cancer Progression
The tumor microenvironment (TME) consists of tumor cells and their neighbor cells, such as cancer-associated fibroblasts (CAFs), immune cells, and the extra cellular matrix. Interactions between these components of the TME promote tumor progression [206]. While understanding the complex nature of breast TME is challenging, a few studies indicate the role of mitochondria in breast TME. Breast cancer cells hijack the mitochondria from immune cells via nanotubes, and inhibition of the nanotube assembly and an ICIs showed the anti-tumor effect [207]. PYCR1 is elevated in the stroma and CAFs in breast cancers, and inhibition of PYCR1 reduced collagen synthesis, tumor growth, and metastasis [208]. Free fatty acids produced by tumor-surrounding adipocytes are used by breast cancer cells to promote FAO and cancer invasion [209].
Dysregulated epigenetic processes contribute to therapy resistance in breast cancers [210]. Mitochondrial metabolites drive epigenetic changes, such as histone acetylation, methylation, and DNA methylation [211]. A study showed that oncometabolite 2-hydroxyglutarate (2-HG) is elevated in a subset of breast cancers, and high 2-HG and a distinct DNA methylation pattern were associated with poor prognosis [212]. Mechanistically, Myc stimulates GLS1 activity, and the increased glutamine consumption promotes aberrant 2-HG accumulation, leading to DNA methylation in breast tumors [212].
The ATP-driven efflux transporters contribute to chemoresistance [213]. ATP-binding cassette (ABC) transporters utilize mitochondria derived-ATP as a source of energy to efflux drugs out of cancer cells, leading to chemo-resistance [214].
Mitochondrial undergo continuous cycles of fission and fusion, and these dynamics are closely related to its function [215,216,217]. Studies indicate a role of mitochondrial dynamics in breast cancer, although there are contradictory findings. Mitochondrial fission is associated with either poor outcomes [218,219] or anti-tumor activity [220,221].
In summary, mitochondria and breast cancer cells maintain a symbiotic relationship via multiple mechanisms that are intricately interconnected to each other to further promote breast cancer survival and growth (Figure 2). Additional mitochondria-associated factors not discussed in this review, such as mitophagy and ROS-induced senescence [222,223], may also support breast cancer progression. Thus, targeting mitochondria is reasonable in breast cancers, and numerous drugs have been developed [224,225]. Next, we will discuss the status of ClpP agonists, a novel class of mitochondrial drugs.
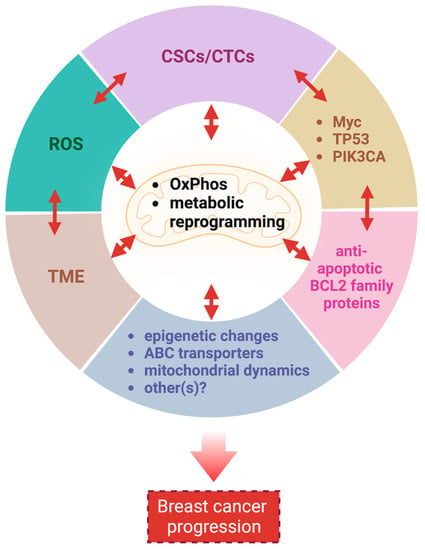
Figure 2.
Mitochondria are critical for breast cancer progression. Mitochondria support breast cancer progression via multiple interrelated mechanisms. CSCs: cancer stem cells; CTCs: circulating tumor cells; ROS: reactive oxygen species; TME: tumor microenvironment; PIK3CA: phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; ABC: ATP-binding cassette. This figure was generated by BioRender.com (19 January 2023).
4. ClpP
Because of the interest in targeting mitochondria, a number of drugs targeting different aspects of mitochondria have been developed. In the rest of the review, we will focus on ClpP agonists. A summary of other drugs targeting other mitochondrial pathways are discussed in the supplemental materials and supplemental Table S1.
4.1. ClpP—The Structure and Functions
In humans, mitochondria contain over ~1000 proteins that fulfil multiple functions [226,227]. According to the latest version of MitoCarta 3.0, an inventory of mammalian mitochondrial proteins, a total of 1136 human genes, encode mitochondrial proteins [150]. Among them, 99% proteins are encoded by nuclear DNA, and 13 proteins are encoded by mtDNA. The sub-mitochondrial localizations of these proteins are the mitochondria matrix (46%), the mitochondrial inner membrane (IMM, 32%), the mitochondrial outer membrane (OMM, 10%), the mitochondrial intermembrane space (IMS, 5%), the mitochondrial membrane (3%), and unknown (5%). These proteins participate in multiple mitochondrial pathways including metabolism (40.6%), mtDNA maintenance, mtRNA metabolism and translation (20%), OxPhos (14.9%), protein import, sorting and homeostasis (7.6%), mitochondrial dynamics (9.1%), small-molecule transport (7.5%), and signaling (4.2%) [150].
Mitochondrial proteins undergo processing events, form functional assemblies, and localize to correct locations. During this process, unfolded and misfolded proteins are cleared to maintain mitochondrial protein homeostasis, which is critical for mitochondrial function [228]. Misregulation of mitochondrial proteostasis leads to human diseases, including cancers [229,230,231,232], and specialized molecular chaperones and proteases conduct this maintenance [233]. At least 45 proteases are present in the different compartments of a mitochondrion [234]. Among these proteases, the evolutionally conserved ATP-dependent proteases (AAA+ [ATPases Associated with various cellular Activities] superfamily) represent core components of the mitochondrial proteolytic system performing mitochondrial protein quality control in a mammalian cell. The members of this AAA+ proteases family are the Lon protease 1 and the ClpXP complex in the mitochondria matrix, and the i-AAA and m-AAA proteases in the inner membrane [234,235,236] (Figure 3). Among the four AAA+ proteases, ClpXP is most extensively studied in terms of its biochemical and genetic features, crystal structures, and the molecular basis of protein substrate recognition [237].
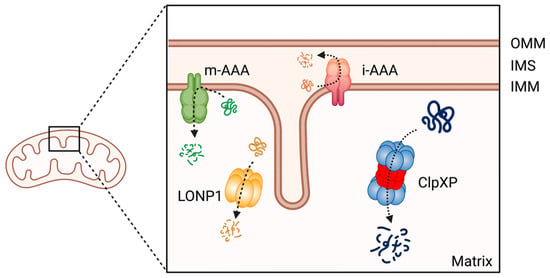
Figure 3.
The member of AAA+ protease family in human mitochondria. ClpXP and LONP1 are located in the mitochondrial matrix, the i-AAA and m-AAA are located in the inner mitochondrial membrane. OMM: Outer mitochondrial membrane; IMS: intermembrane space; IMM: inner mitochondrial membrane. This figure was generated by BioRender.com (19 January 2023).
ClpXP consists of the caseinolytic mitochondrial matrix peptidase chaperone subunit X (ClpX; AAA+ ATPase) and ClpP (a tetradecameric peptidase). ClpX is a hexametric ATP-dependent protein unfoldase and translocase [238]. ClpP is a barrel-like peptidase assembled from two stacked heptameric rings, which enclose a roughly spherical proteolytic chamber [239] (Figure 4A). The proteolytic activity of ClpP is usually tightly regulated by ClpX [240]. ClpX recognizes degrons on proteins to be degraded and provides energy to unfold and translocate linearized protein into the barrel of ClpP [241]. Substrate specificity is not defined by the amino acid sequence, rather ClpX recognizes specific degradation signals and interacts with certain adaptor proteins [242]. ClpX then feeds the unfolded substrate through the axial pore to the proteolytic chamber of ClpP, and the substrate is cleaved into small peptide fragments [242,243] (Figure 4B). ClpX binds to one or both ends of ClpP and this allows for both single- and double-capped ClpXP complexes [237]. As with bacterial ClpP, human ClpP cylinders switches dynamically between extended and compact forms (Figure 4C). The active, extended form is required for substrate degradation, while an inactive compact state allows peptide product release from the ClpP inner chamber [243,244,245,246].
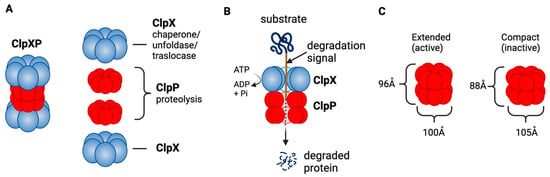
Figure 4.
ClpXP structure and function as a mitochondrial protease. (A) ClpXP consists of ClpX (ATPase) and ClpP. (B) ClpX recognizes degrons of target protein, unfolds the proteins and threads them into ClpP for degradation. (C) Extended (active) and compact structure (inactive) of Staphylococcus aureus ClpP [243]. This figure was generated by BioRender.com (19 January 2023).
The role of ClpP as a master regulator of mitochondrial protein quality control and homeostasis is well established. Many mitochondrial proteins are identified as substrates of ClpXP, including proteins involved in ETC, the TCA cycle, mitochondrial ribosome, mitochondrial gene transcription and translation, glutamine metabolism, and folate metabolism [58,247,248,249]. By degrading misfolded or damaged proteins, ClpXP maintains the integrity of mitochondrial functions.
ClpP also serves as a central mediator of the mitochondrial unfolded protein response (UPRmt) [250,251], a conserved transcriptional response activated by multiple forms of mitochondrial dysfunction and regulated by mitochondrial-nuclear communication. The UPRmt supports the recovery of mitochondria from damage, restores ETC function, eliminates excess amounts of ROS, and promotes cell survival [252,253]. Recent studies indicated the prolonged UPRmt contributes to organismal deterioration including cancer [196,253,254]. In breast cancers, elevated expression of UPRmt-related genes is significantly associated with poor overall and metastasis-free survival [255]. Activated UPRmt was observed in HER2 amplified breast cancer tissues [256].
ClpP is predominantly expressed in non-malignant tissues with high mitochondrial content such as skeletal muscle, liver, and heart, suggesting its critical role in normal tissue [257]. Despite the expression in such critical organs, mice with homozygous deletion of the CLPP gene are viable, but infertile, and exhibit hearing loss and growth retardation with accumulation of ClpX and mtDNA [258]. Similarly, recessive CLPP mutations in the human CLPP genes are linked to a rare genetic disease Perrault syndrome, which shows infertility and hearing loss [259,260].
4.2. ClpP Activation Impairs Breast Cancer Cell Viability
While the exact roles of ClpP in tumorigenesis are not clearly established, emerging evidence demonstrate that ClpP is overexpressed in multiple malignancies including breast cancers [261,262,263]. Recent analysis of TCGA database also confirmed that ClpP is overexpressed in breast and other cancers [264]. In addition, a high level of ClpP expression is associated with the disease stage, metastasis, poor prognosis and is proposed as a prognostic marker in breast cancers [261,265,266]. Thus, ClpP is considered an emerging target for breast cancer.
Interestingly, both inhibition and hyperactivation of ClpP can lead to impaired OxPhos, resulting in cancer cell death [267]. Inhibition of ClpP induces the accumulation of damaged and misfolded mitochondrial proteins, whereas hyperactivation of ClpP leads to unregulated degradation of ClpP substrates, and both mechanisms can elicit defective cellular respiration and eventual cell death [233]. Importantly, whether inhibition or activation of ClpP induces cell death appears to be context dependent. CLPP is an essential gene for cell survival in leukemia cells and ClpP inhibition is lethal in acute myelogenous leukemia (AML), chronic myelogenous leukemia, and osteosarcoma [262]. In contrast, ClpP activators show anti-tumor effect in breast, ovary, colorectal, glioblastoma, and other cancers [58,268,269,270,271,272]. In breast cancer cells, transient knockdown of CLPP by siRNA induced apoptosis and inhibited cell viability, migration, invasion in breast cancer cells [265], while CRISPR/Cas9-mediated deletion of CLPP gene did not affect cell viability presumably due to cellular adaptation [58,270]. In contrast, activation of ClpP by ClpP agonists exerts an anti-tumor effect in breast cancer cells [58,270]. This suggests that ClpP activation, but not ClpP inhibition, is the better targeting approach in breast cancers.
4.3. ClpP Activating Drugs
Since ClpP activators, but not ClpP inhibitors, show cytotoxicity in breast cancers, we will focus on ClpP activators in this review (for ClpP inhibitors, we refer readers to other excellent reviews [233,264,266]. There are three classes of ClpP agonists: acyldepsipeptide antibiotics (ADEPs), ONC201 and its analogs (imipridones), and TR compounds. The representative ClpP agonists and structures are illustrated in Figure 5.
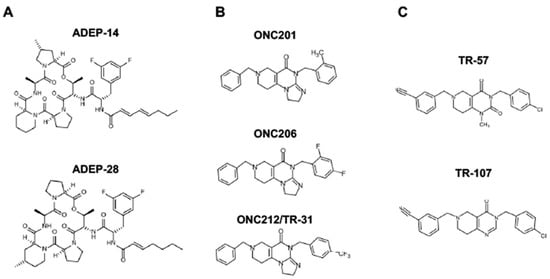
Figure 5.
Representative ClpP agonists and structures. (A) Acyldepsipeptide antibiotics (ADEPs), (B) ONC201 and its analogs (imipridones), and (C) TR compounds.
ADEPs: ClpP has emerged as an anti-bacterial target during the mechanism of action studies on acyldepsipeptide antibiotics (ADEPs) in last two decades. ADEPs were first reported as antibiotics [273]. Identification of the resistance-mediating mutation within an ADEP-resistant Escherichia coli mutant and affinity chromatography with an immobilized ADEP analog led to identification of ClpP as the direct target of ADEPs [274]. ADEPs increase the activity of the bacterial ClpP, leading to bacterial cell death. As ClpP plays important roles in the bacterial virulence due to the broad range of substrates, extensive effort has been made to develop ADEPs as novel therapeutics for drug-resistant bacteria [242,274,275,276,277]. Medicinal chemistry studies revealed the structure–activity relationship and yielded many derivatives with enhanced in vitro potency and stability [278,279,280,281]. The anti-bacterial efficacy of ADEPs has been demonstrated in lethal bacterial infections in rodent models [274,278,282]. Wong et al. were the first to demonstrate that ADEPs binds to human ClpP (HsClpP) using crystallographic structural analysis [283]. ADEPs induce caspase-dependent apoptosis, mitochondrial fragmentation, and OxPhos inhibition in HEK293 cells. At present, ADEP analogues are being tested in preclinical models in breast cancers, but the findings are limited.
ONC201 and its analogs (Imipridones): ONC201 (a.k.a., TIC10) is the first-in-class small molecule of the imipridone family [284]. ONC201 was identified in a chemical library screen to find a small molecule that transcriptionally induces tumor necrosis factor-alpha related apoptosis-inducing ligand (TRAIL), leading to an autocrine induction of apoptosis in the cancer cell. The proposed mechanism of action of ONC201 was that it inhibits extracellular signal-regulated kinase (ERK) and AKT, leading to the translocation of Foxo3a into the nucleus, where it activates transcription of the TRAIL gene [284]. Subsequent studies evaluated ONC201 as an antagonist of dopamine receptors (DRD)2/3 [285,286,287]. There were some preclinical data supportive of DRD2 antagonistic activity [286], and the clinical activity seen in diffuse intrinsic pontine gliomas (DIPG) has been suggested to be mediated through DRD2 inhibition [288,289]. However, this remains controversial. No direct evidence of ONC201 binding to DRD2/3 has been shown, gene suppression or deletion of DRD2/3 did not abrogate the ONC201’s cytotoxic effect [286], and DRD2 transcript is not detectable in many breast cancer cell lines that are sensitive to ONC201 [269]. This requires further evaluation to determine if any activity of ONC201 or other ClpP agonists is due to DRD2 antagonism.
While ONC201 was shown to increase Poly (ADP-ribose) polymerase 1 (PARP) cleavage and apoptosis in some cancer models [284,290], apoptosis was not observed in breast cancers [269,270]. Our group was the first to report that the cytotoxicity of ONC201 is due to targeting mitochondria and is independent of TRAIL-mediated apoptosis [269]. We found that ONC201 inhibits OxPhos, depletes ATP, mtDNA, multiple mitochondrial proteins, such as mitochondrial transcription factor A (TFAM), ETC proteins, accompanied with mitochondrial structural damage and an integrated stress response (ISR) indicated by ATF4 and CHOP induction [269]. We also found that breast cancer cells that lack functional mitochondria (e.g., rho0 cells) were ONC201-resistant. Our finding was confirmed and extended by two studies which demonstrated that ONC201 binds to and activates ClpP using crystallography structural analysis, mass-spectrometry, and affinity chromatography/drug competition assays [268,271]. Another group identified ClpP as the target of ONC201 using a genome wide CRISPR KO library screen [291]. Later, ONC206 and ONC212 were developed by Chimerix, Inc. (https://www.chimerix.com/, accessed on 2 January 2023) as more potent imipridones compared with ONC201. Studies confirmed that these imipridones impair OxPhos, induce mitochondrial damage, ROS, and ISR [271,292]. ONC201 has been shown to penetrate the brain-blood barrier [284].
TR compounds: TR compounds are a novel series of imipridone-derived compounds and related chemicals being developed by Madera Therapeutics, LLC (https://maderat.com/, accessed on 2 January 2023). Multiple TR compounds, including TR-57, TR-31 (=ONC212), were found to be ∼50–100-fold more potent compared with ONC201 in cytotoxicity in breast cancer cells. TR compounds induced an ISR at nanomolar concentrations, significantly lower compared with those induced by ONC201 at micromolar concentrations [268]. Affinity chromatography/drug competition assays demonstrated that the TR compounds bind to ClpP with ∼10-fold higher affinity compared to ONC201 [268]. Similar to ONC201, TR compounds reduce target protein levels (e.g., TFAM) and impair OxPhos [58,268,269].
The unique feature of all ClpP agonists described above is that they activate ClpP in the absence of ClpX (Figure 6 and Figure 7). The X-ray crystallography structure demonstrated that ADEP binds to the hydrophobic pockets of ClpP and dissociates the ClpXP complex at substoichiometric concentrations, leading to ClpP activation independent of regulatory subunit ClpX [283] (Figure 6A). Similar to ADEPs, imipridones directly binds to ClpP, and displace ClpX, and the bindings triggers the opening of channel-like pore of ClpP, and thereby increase its protease activity without the ClpX [233,271,293] (Figure 6B). Recently, X-ray crystallography demonstrated that TR compounds bind to ClpP, with enhanced binding affinities due to their greater shape and charge complementarity with the surface hydrophobic pockets of ClpP [294] (Figure 7C). Moreover, N-terminome profiling of MDA-MB-231 cell line upon treatment with one of TR compounds revealed the global proteomic changes and characterized the sequence and structural properties for protein cleavage generated upon TR compound-induced ClpP-dependent proteolysis of cellular proteins [294].
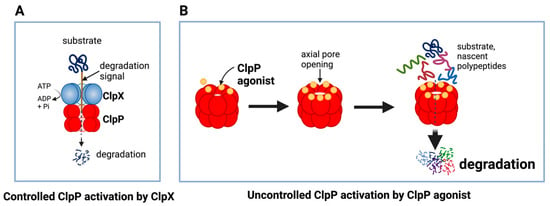
Figure 6.
Controlled and uncontrolled proteolysis by ClpP. (A) In controlled proteolysis, target substrates are specifically recognized and unfolded by ClpX. The unfolded substrate is transferred into the barrel-like chamber of the ClpP, and proteolysis is carried out by active proteases at the inner surface of the chamber. (B) Uncontrolled proteolysis triggered by binding of ClpP agonists to ClpP. Binding of ClpP agonists (yellow spheres) to ClpP subunits displaces ClpX and triggers an opening of the entrance pore of the ClpP barrel, leading to unregulated degradation of nascent polypeptides and unfolded proteins. This uncontrolled ClpP activation does not require ClpX. This figure was generated by BioRender.com (20 January 2023).
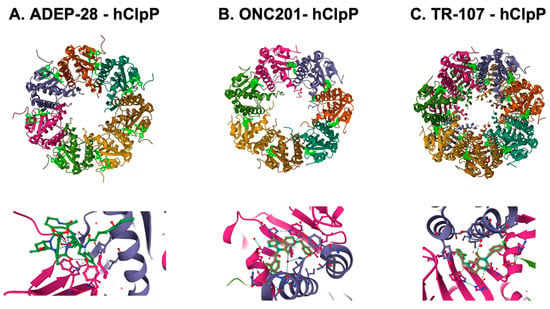
Figure 7.
Crystal structures of human mitochondrial ClpP complex with ClpP ligands. (A) ADEP-28-hClpP binding [283] (PDB DOI: 10.2210/pdb6BBA/pdb), (B) ONC201-hClpP [271] (PDB DOI: 10.2210/pdb6DL7/pdb), (C) TR-107-hClpP [294]. (PDB DOI: 10.2210/pdb7UVU/pdb). Top panels show that each ClpP ligand (bright green) bound to ClpP. One heptamer ring for ADEP-28 and ONC201, or two heptamer rings for TR-107 are shown. Each subunit of ClpP is shown in a different color. Bottom panels show a magnified view of ligands (bright green) bound to the pocket between two ClpP subunits.
5. Preclinical Studies Using ClpP Agonists in Breast Cancers
Many preclinical studies have demonstrated the efficacy of ClpP agonists in breast cancer models (Table 1). Among ClpP agonists, ONC201 has been most extensively tested in various preclinical models, including breast cancers. TR compounds have also been tested recently in breast and other cancer cells. At present, data on ADEPs as a cancer therapeutic are limited. ADEP1 and ADEP2 showed anti-tumor activities in multiple renal cancer cells [295]. ADEP41 showed cytotoxicity in HeLa (cervical), U2OS (osteosarcoma), SH-SY5Y (neuroblastoma) at IC50 0.5–0.9 uM, but the effect on breast cancers is not yet reported [283]. Several critical findings from the preclinical studies using ClpP agonists in breast cancer models are discussed below.

Table 1.
Preclinical studies using ClpP agonists in breast cancers.
5.1. Findings from In Vitro Studies
5.1.1. ClpP Agonists Uniquely and Broadly Impair Mitochondrial Function
ClpP agonists induce degradation of mitochondrial proteins (e.g., TFAM, mitochondrial elongation factor Tu [TUFM], and ETC complex proteins), depletion of mtDNA copy number, downregulation of OxPhos, loss of ATP, mitochondrial structural damage, and elevation of ROS level in multiple breast cancer cell lines [58,269,270,304]. Moreover, ClpP agonists dysregulate metabolic pathways not affected by other mitochondrial drugs such as metformin, oligomycin, CPI-613 [58]. ClpP agonists downregulate multiple enzymes; GLS, P5CS/ALDH18A1, PYCR1/2, NADK2/mitochondrial NAD+ kinase, ME2, SHMT2, MTHFD2, TYMS and HMGCS1. All of these enzymes are associated with metabolic rewiring that drives breast cancers as described above. The proline cycle, ME2, and folate cycle are involved with production of NAD(P)+/NAD(P)H; therefore, ClpP agonists also deplete NAD(P)+/NAD(P)H, leading to inhibition of macromolecule biosynthesis and ROS production [58].
5.1.2. ClpP Agonists Inhibit Breast Cell Growth, Proliferation, Viability, but Do Not Induce Apoptosis
Studies testing ONC201 and/or TR compounds in breast cancer models collectively show that ClpP agonists exert a significant anti-proliferative effect accompanied with ISR and cell cycle arrest, rather than inducing apoptosis [58,268,270,298,299,301,302,304,306]. These observations are distinct from what has been reported as apoptosis observed in other malignancies, such as colon cancers and hematological malignancies [307,308,309]. Recent studies revealed that the anti-proliferative effect of ClpP agonists observed in breast cancers represent a senescence-like phenotype [304,310]. These findings suggests that the impact of ClpP targeting in cancers is context dependent.
5.1.3. Non-Malignant Cells Are Resistant with ClpP Agonists
One notable finding from ClpP agonist studies is that ClpP agonists do not induce cytotoxicity in non-malignant cells. While ONC201 induces mitochondrial structural damage and depletes mtDNA in human foreskin fibroblast (HFF) cells, it does not impair cell viability [269]. Other studies also reported that ONC201 and its analogs do not exert cytotoxicity in normal human fibroblasts, such as MRC5, WI38, HFF1, 18Co, 19Lu [269,305], normal lung epithelial cells [311], and normal bone marrow cells [312]. The mechanisms of the resistance to ClpP agonists in normal cells remains elusive but intriguing, suggesting a beneficial feature of ClpP targeting therapies.
5.1.4. The Subtype-Specific Effect of ClpP Agonists in Breast Cancer Remains Unclear
Several studies investigated ONC201 sensitivity with a specific focus on breast cancer subtypes. We tested ONC201 in 17 breast cancer cell lines consisting of different subtypes (4 ER+, 1 ER+/HER2+, 3 HER2+, 4 TNBC Basal A, and 5 TNBC Basal B), and the IC50 range was from 0.78 to 4.83 micromolar without any subtype-specific difference [269]. A study tested ONC201 in 10 TNBC cell lines and found that the IC50 range varied from 0.032 to 10.12 micro molar; however, mesenchymal-like TNBC (e.g., SUM159, SUM149, MDA-MB-157, MDA-MB-231, and BT549) and epithelial-like TNBC (e.g., HCC70, BT20, HCC1806, HCC1937, and MDA-MB-468) were equally sensitive to ONC201 [298]. Another study tested 17 TNBC cell lines and reported that the ONC201 IC50 ranged from 2.05 to 43.39 micromolar, and that HCC70 (Basal-like 1), MDA-MB-157 and SUM159 (Mesenchymal) were relatively resistant compared with MDA-MB-468 (Basal-like 1), CAL51 (Basal-like2) and that higher ClpP protein expression is correlated with a lower ONC201 IC50 in TNBC cell lines [302]. This proposed link between ClpP protein expression and ClpP agonist sensitivity needs to be further investigated, as the ClpP protein expression level does not appear to be correlated with sensitivity to ClpP agonists in 17 breast cancer cell lines with different subtypes (3 ER+, 1 ER+/HER2+, 3 HER2+, 5 TNBC Basal A, and 4 TNBC Basal B) in our study [58].
5.1.5. Comparison of ClpP Agonist Potency in Representative Breast Cancer Cell Lines
The IC50 of ONC201 is in the micromolar range, while that of ONC206 is several folds lower, and ONC212 is more potent in the nanomolar range in breast cancer cells. Compared with imipridones, TR compounds show significantly higher potency; IC50 of TR-57 and TR-107 exert anti-proliferative effects in the 10–25 nanomolar in TNBC cells (Table 2). The superior potency of TR compounds over other ClpP agonists is attributed to the better binding properties to ClpP [294].
Table 2.
Comparison of IC50 (nM) of ClpP agonists in representative breast cancer cell lines.
5.2. Findings from In Vivo Studies
5.2.1. ClpP Agonists Showed an Anti-Tumor Effect in Animal Models of Breast Cancer
The anti-tumor activity of ClpP agonists in breast tumors has been demonstrated in multiple in vivo studies. In these studies, ONC201 was used at doses of 25–100 mg/kg, weekly, orally, or intraperitoneally (i.p.) (Table 2). ONC201 treatment (i.p. at 100 mg/kg, weekly) had an anti-tumor growth effect in MDA-MB-231 xenograft model [284]. Another study also showed that ONC201 ablated MDA-MB-231 xenograft tumors, and that dose intensification of ONC201 inhibited invasion, migration, and metastasis [300]. Anti-tumor growth effects of TR-107 in MDA-MB-231 xenograft models was also reported. TR-107 treatment (oral, 4 or 8 mg/kg, twice a day, twice a week) induced a ~50% reduction in tumor volume and extended the animal median survival ~35% compared to control. TR-107 treatment was well tolerated with less than 5% weight loss observed even at the highest dosing regimen. Importantly, the on-target in vivo activity of TR-107 was confirmed, as mtDNA copy number was reduced and ClpP substrate proteins such as TFAM, PYCR2, and HMGCS1 were all downregulated in tumor samples from the TR-107-treated group [270]. The inhibitory effect of ClpP on breast CSC function has also been shown in vivo. MDA-MB-231 cells pre-treated with either ONC201 or TR-57 failed to form tumor in mice, indicating that ClpP agonists impair CSC function [58]. Collectively, these studies demonstrated that ClpP agonists induce cytotoxicity in TNBC and inhibit breast CSC functions.
5.2.2. Pharmacokinetics
The pharmacokinetics (PK) property of select ClpP agonists in mice from three independent studies is summarized in Table 3 [270,284,305]. In the initial study, the half-life of ONC201 was reported as 6.42 h (25 mg/kg, oral) [284], and that of ONC212 was 4.31 h (125 mg/kg, oral) [305], whereas a recent study showed the shorter half-life [270]; ONC201 was 0.31 h (10 mg/kg, oral), and that of ONC212 was 1.49 h (10 mg/kg, oral). The half-life of TR-57 was 1.4 h or 1.52 h (10 mg/kg, oral or 2 mg/kg, i.v., respectively), that of TR-107 was 0.9 h (10 mg/kg, oral). Despite similar half-life in mice models, the exposure (AUC) of TR-57 and TR-107 (10 mg/kg, oral) was 750–900-fold higher than that of ONC201 (10 mg/kg, oral). TR-57 showed high rapid absorption rate (F% of 61%) after oral administration. The protein-binding studies for TR-107 show a serum-free fraction of 10%. Collectively, both TR-57 and TR-107 show favorable PK characteristics compared with ONC201 and ONC212.
Table 3.
Pharmacokinetic analysis of select ClpP agonists.
5.2.3. ClpP Agonists Induce Natural Killer (NK) Cell Recruitment and Activation of Immune Cells
As discussed above, mitochondria participate in the modulation of TME in breast cancers. Studies have shown that ONC201 promotes NK cell recruitment to tumors and NK cells mediated killing of breast cancer cells [300,301]. It is also shown that ONC201 induces accumulation of CD4+/CD8+CD3+ T cells in syngeneic MC38 colorectal tumors [300]. Similarly, ONC206 treatment increased NK cells and activated CD4+ cells within ovarian tumors in a mouse model [313]. The impact of ClpP agonists on immune cells needs to be further investigated.
6. Clinical Trials of ClpP Agonists in Breast Cancers
ClpP agonists have been actively exploited in multiple clinical trials with various types of malignancies; however, clinical studies of ClpP agonists in breast cancers have not reported results. Among multiple ClpP agonists, ONC201 is the only ClpP agonist entered in clinical trials for breast cancers as of January 2023.
In the first-in-human phase 1/2 study of ONC201 in patients with advanced cancers (no breast cancer patients were included), ONC201 was orally administered once every 3 weeks defined as one cycle, with a dose escalation from 125 to 625 mg in 10 patients. ONC201 was well-tolerated, and the recommended phase 2 dose (RP2D) was determined as 625 mg, weekly. An additional 18 patients were treated at the RP2D in an expansion phase to collect additional safety, PK/PD information. PK analysis revealed a Cmax of 1.5 to 7.5 μg/mL (~3.9–19.4 micromolar), the mean half-life ONC201 was 11.3 h and mean AUC was 37.7 h·μg/L [285,314]. No objective responses by RECIST were achieved; however, radiographic regression of several individual metastatic lesions was observed along with prolonged stable disease (>9 cycles) in prostate and endometrial cancer patients.
Table 4 summaries trials targeted to breast cancer patients. The phase 2 clinical trial of ONC201 in metastatic, previously treated ER+ and TNBC breast cancer patient was recently completed but has not yet published the results (NCT03394027). The dosing schedule in this study was 625 mg of the ONC201 taken orally, every 7 days in 28-day cycles. Unfortunately, ONC201 did not show clinical efficacy as reported at Clinicaltrials.gov (accessed on 20 January 2023).
Table 4.
Status of clinical trials using ClpP agonists in breast cancer patients.
ONC206, ONC212, and TR compounds are not yet tested in breast cancers. Another study intended to determine the objective response rate to ONC201 with a methionine-restricted diet in patients with metastatic TNBC, but was terminated due to the low number of patients enrolled (NCT03733119).
7. Challenges to Achieve Better Clinical Outcomes of ClpP Agonists Therapy
While ClpP agonist-induced mitochondria targeting therapy is an emerging and attractive approach, there are many questions to be addressed. Establishing predictive biomarkers that can identify patients who will be most effectively treated with ClpP agonists will be significantly beneficial to improving therapeutic outcomes. Pharmacodynamic (PD) biomarkers are also required to monitor the activity of ClpP agonists in patients. To further enhance the therapeutic efficacy, the combination of ClpP agonists with other drugs needs to be explored. In this section, we will discuss the research status in these challenges.
7.1. Predictive Biomarkers
Gene Expression: As discussed above, a higher ClpP expression level is correlated with a lower IC50 of ONC201 in TNBC [302]; however, this needs to be further investigated, as other studies did not find this correlation [58,269]. Increased protein expression of four genes, HER2_pY1248, PLK1, Rb_pS807/811, and EMA (MUC1), has been shown to be correlated with ONC201 resistance in TNBC, suggesting that inhibiting these genes may improve ONC201 efficacy [302]. The expression of CSC-related genes (e.g., ID1, CD44, HES7, and TCF3) correlates with ONC201 efficacy, and combining the expression of multiple genes is shown to be a potential predictive as well as PD biomarker [315]. In non-breast cancer settings, a high level of the c-myc protein is reported as a predictive biomarker for ClpP agonists in glioblastoma [316], and mutation of succinate dehydrogenase is proposed as a potential biomarker in neuroendocrine tumors [317].
Mitochondrial activity: Basal mitochondrial ATP% (e.g., the fraction OxPhos-derived ATP, as opposed to glycolysis-derived ATP) is proposed as a predictive biomarker of ONC212 in pancreatic cancer models [318], but not yet tested in breast cancers.
7.2. PD Biomarkers
Detecting mitochondrial dysfunction caused specifically by ClpP agonists in patients is needed to evaluate the drug efficacy. Detecting impaired OxPhos is feasible; however, specificity and sensitivity remain as major challenges. For instance, quantification of lactate/pyruvate can be used to detect OxPhos dysfunction [269,270]. However, absence of elevated lactate/pyruvate does not exclude the presence of OxPhos impairment, and the elevation of those metabolites may be condition dependent (e.g., exercise and acute crisis) [319]. Cytokines, such as fibroblast growth factor 21, growth differentiation factor 15 are stress response factors linked to mitochondrial function, and are proposed as diagnostic biomarkers of mitochondrial diseases; however, the specificity remains controversial [319,320,321].
Mitochondrial damage caused by ClpP agonists was successfully detected in vivo. Downregulation of TFAM, TUFM, PYCR2, HMGCS1, and mtDNA copy number was detected in breast tumors in mice treated with TR-107 [270]. However, mitochondrial dysfunction may trigger a compensatory protective response, making the assessment of specificity of ClpP agonists difficult [319]. Moreover, performing multiple tumor biopsies is not practical and often not available in clinic. Minimally or non-invasive monitoring of mitochondrial function in tumors is needed. The ClpP agonist increased NK cells in peripheral blood of prostate cancer patients, suggesting that NK cell activation might be useful as an indirect biomarker of drug activity [300,313].
7.3. Drug Combination
Combination therapy with ClpP agonists is likely to be needed to improve the therapeutic efficacy in breast cancer. Several combination strategies are currently being proposed and investigated as discussed below.
TRAIL: Most breast cancers are highly resistant to TRAIL, except for a subset of TNBC [322,323]. A synergistic effect of TRAIL and the ClpP agonist has been reported in breast and other cancer types. ONC201 downregulates multiple anti-apoptotic proteins, such as Mcl-1, Bcl-xL, XIAP [324], which modulate the TRAIL-mediated extrinsic apoptosis pathway [325,326]. Combining TRAIL with ONC201 converted the response from anti-proliferative to apoptosis in TRAIL-resistant non-TNBC cells and in vivo breast cancer models [301]. In addition, ClpP agonist-induced senescent TNBC cells are sensitized to TRAIL-mediated apoptosis [310].
Chemotherapy agents: In a study using a panel of human cancer cell lines including breast cancer cell lines, ONC201 was shown to be synergistic with multiple FDA-approved chemotherapeutic and targeted agents, such as azacitidine, bortezomib, dacarbazine, hydroxyurea, pralatrexate, sorafenib, topotecan, and vismodegib [296]. Taxanes (e.g., docetaxel and paclitaxel) have also been shown to synergize with ONC201 in a subset of TNBC cells [299].
PARP inhibitors: PARP inhibitors are relatively new class of agents showing an efficacy in BRCA-mutated breast cancer and TNBC [327]. The synergistic effect of PARP inhibitors (e.g., olaparib and rucaparib) with imipridone has been shown in vitro, but not yet in vivo [328].
Mitogen-activated protein kinase kinase (MEK) inhibitor: The combination of ONC201 and trametinib, a MEK inhibitor, showed a synergistic anti-tumor effect and increased in caspase 3/7 activity in TNBC [302].
The Enhancer of Zeste Homolog 2 inhibitors (EZH2i): EZH2 mediates gene silencing via H3K27 tri-methylation. Inhibition of EZH2 catalytic activity is shown to target a metastatic subpopulation in TNBC [329]. The combination of EZH2i and ONC201 showed a synergistic apoptosis in multiple breast cancer cell lines [303].
Histone deacetylase inhibitor (HDACi): HDACi are promising anti-cancer agents that inhibit cell proliferation of many types of cancer cells, and have been actively tested in clinical trials including breast cancers (see a review [330]). The combination of vorinostat (FDA-approved) and ONC201 had a synergistic effect on cell viability, and induced apoptosis in MCF7 cells [303]. Several HDACi have been exploited in combination with imipridones in CNS malignancies, and a synthetic lethality of combination of ClpP activation and HDACi is reported in glioblastoma [272,331].
Venetoclax/ABT-199: Synergistic anti-tumor effect of the combination of venetoclax and imipridones has been reported in AML models [312,332], but not yet in breast cancers.
Immunotherapy: Immunotherapies are revolutionizing the treatment of many cancers, including breast cancer. The clinical activity of programmed cell death-1/programmed death ligand-1 (PD-1/PD-L1) blockade by ICIs is shown with early- [12,333] and late-stage breast cancer patients [9]. The ICIs are currently being tested in combination with multiple therapies (e.g., chemotherapy, HER2-directed therapy, and CDK4/6 inhibitor) [334]. ONC201 has been shown to induce CD4/CD8+CD3+ T cell accumulation in colon cancer models, suggesting a that the combination of ClpP agonists with ICIs may be useful [300]. The combination of atezolizumab, an anti-PD-L1 antibody, and ONC201 is currently being tested in a clinical trial in endometrial cancer (NCT05542407). The combination of ClpP agonists with ICIs needs to be explored in breast cancers.
NK cells are anti-tumorigenic, innate cytotoxic lymphocytes, and are being investigated for use in breast cancers [335,336]. Peritumoral abundance of NK cells is shown to correlate with better pathological complete response (pCR) rate after neoadjuvant chemotherapy in large and locally advanced breast cancer [337]. ClpP agonists recruit and activate NK cells in breast and other cancer models [300,301], and the cytotoxicity of ClpP-agonist was enhanced by NK cells in breast cancer cell line [304].
While the combination of immunotherapy with a ClpP agonist is appealing, the impact of ClpP agonists on immune cells needs to be considered. For instance, T cell metabolism changes dynamically depending on the activation state [338]; Naïve T cells (quiescent) rely on OxPhos, and activated effector T cells primarily use glycolysis, while memory T cells and Treg are dependent on OxPhos and FAO [339,340]. Thus, breast cancer cells and T cells both rely on mitochondria. Therefore, defining similarities and differences between the cancer metabolism and immune cells is critical to determine potential metabolic crosstalk or competition within the TME. Such mechanistic insight may reveal novel strategies to inhibit tumor growth while maximizing anti-tumor immunity [341]. The function of NK cells also depends on mitochondrial dynamics and OxPhos [342], and is modulated by various metabolic conditions [343]. The presence of lactate in the TME is a major obstacle for cell-based immunotherapies as it impairs the cytotoxic abilities of T cells and NK cells. [344,345]. At present, our knowledge on the impact on ClpP agonists on immune system is limited. Understanding the impact of ClpP agonists on immune cells and TME is needed in future research.
8. Conclusions and Future Directions
Recent studies established that mitochondria are essential for breast cancer progression, and numerous efforts have been made to target mitochondria. ClpP agonists are an emerging class of mitochondria-targeting drugs featuring unique mechanism of action. Hyperactivation of ClpP leads to uncontrolled protein degradation in mitochondria. The impact of ClpP agonists goes beyond OxPhos inhibition; they broadly disrupt multiple mitochondrial functions (e.g., OxPhos, the TCA cycle, glutamine-proline metabolism, one-carbon metabolism, nucleotide biosynthesis, and the mevalonate pathway). ClpP agonist-induced mitochondrial dysfunction effectively halts cell growth and induces senescence, but not apoptosis in breast cancers. ClpP agonists show cytotoxicity in cancer models but not in non-malignant cells. More studies are required to understand the impact of ClpP agonists on immune cells and TME. The development of predictive biomarkers, PD biomarkers, and effective drug combination strategies is also needed. Regardless, further development of ClpP agonists is warranted.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cancers15071936/s1, Figure S1: Mitochondrial metabolic pathways dysregulated in breast cancers; Table S1: The status of development of mitochondria-targeting drugs in breast cancers (not including ClpP agonists). References [346,347,348,349,350,351,352,353,354,355,356,357,358,359,360,361,362,363,364,365,366,367,368,369,370,371,372,373,374,375,376,377,378,379,380,381,382,383,384,385,386,387,388,389,390,391,392,393,394,395,396,397] are cited in the supplementary materials.
Author Contributions
Conceptualization, R.W., Y.E.G., D.J.W., S.W., M.K., D.V. and S.L. Methodology, R.W. and Y.E.G. Resources, S.L. Writing—original draft preparation, R.W. Writing—review and editing, R.W., Y.E.G., D.J.W. and S.L. Visualization, R.W. and Y.E.G. Supervision, S.L. Project administration, S.L. Funding acquisition, S.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research (ZIA SC 007263).
Acknowledgments
This work was supported by the Center for Cancer Research Intramural Research Program of the National Cancer Institute at the NIH.
Conflicts of Interest
The authors have no conflict of interest.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Giaquinto, A.N.; Sung, H.; Miller, K.D.; Kramer, J.L.; Newman, L.A.; Minihan, A.; Jemal, A.; Siegel, R.L. Breast Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 524–541. [Google Scholar] [CrossRef]
- Miller, K.D.; Ortiz, A.P.; Pinheiro, P.S.; Bandi, P.; Minihan, A.; Fuchs, H.E.; Martinez Tyson, D.; Tortolero-Luna, G.; Fedewa, S.A.; Jemal, A.M.; et al. Cancer statistics for the US Hispanic/Latino population, 2021. CA Cancer J. Clin. 2021, 71, 466–487. [Google Scholar] [CrossRef]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Fragomeni, S.M.; Sciallis, A.; Jeruss, J.S. Molecular Subtypes and Local-Regional Control of Breast Cancer. Surg. Oncol. Clin. N. Am. 2018, 27, 95–120. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Chang-Qing, Y.; Jie, L.; Shi-Qi, Z.; Kun, Z.; Zi-Qian, G.; Ran, X.; Hui-Meng, L.; Ren-Bin, Z.; Gang, Z.; Da-Chuan, Y.; et al. Recent treatment progress of triple negative breast cancer. Prog. Biophys. Mol. Biol. 2020, 151, 40–53. [Google Scholar] [CrossRef]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Bardia, A.; Hurvitz, S.A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Brufsky, A.; Sardesai, S.D.; Kalinsky, K.; Zelnak, A.B.; et al. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2021, 384, 1529–1541. [Google Scholar] [CrossRef]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e813. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Dent, R.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; et al. Event-free Survival with Pembrolizumab in Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2022, 386, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Coller, H.A. Is cancer a metabolic disease? Am. J. Pathol. 2014, 184, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Gyamfi, J.; Kim, J.; Choi, J. Cancer as a Metabolic Disorder. Int. J. Mol. Sci. 2022, 23, 1155. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D’Agostino, D.P. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis 2014, 35, 515–527. [Google Scholar] [CrossRef]
- Wishart, D.S. Is Cancer a Genetic Disease or a Metabolic Disease? EBioMedicine 2015, 2, 478–479. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef]
- Ju, H.Q.; Lin, J.F.; Tian, T.; Xie, D.; Xu, R.H. NADPH homeostasis in cancer: Functions, mechanisms and therapeutic implications. Signal Transduct. Target. Ther. 2020, 5, 231. [Google Scholar] [CrossRef]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef]
- Chakrabarty, R.P.; Chandel, N.S. Mitochondria as Signaling Organelles Control Mammalian Stem Cell Fate. Cell Stem Cell 2021, 28, 394–408. [Google Scholar] [CrossRef]
- Tan, J.X.; Finkel, T. Mitochondria as intracellular signaling platforms in health and disease. J. Cell Biol. 2020, 219, e202002179. [Google Scholar] [CrossRef]
- Missiroli, S.; Perrone, M.; Genovese, I.; Pinton, P.; Giorgi, C. Cancer metabolism and mitochondria: Finding novel mechanisms to fight tumours. EBioMedicine 2020, 59, 102943. [Google Scholar] [CrossRef] [PubMed]
- Matilainen, O.; Quirós, P.M.; Auwerx, J. Mitochondria and Epigenetics—Crosstalk in Homeostasis and Stress. Trends Cell Biol. 2017, 27, 453–463. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef]
- Sandhir, R.; Halder, A.; Sunkaria, A. Mitochondria as a centrally positioned hub in the innate immune response. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1090–1097. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Gaude, E.; Frezza, C. Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat. Commun. 2016, 7, 13041. [Google Scholar] [CrossRef] [PubMed]
- Weerts, M.J.A.; Sleijfer, S.; Martens, J.W.M. The role of mitochondrial DNA in breast tumors. Drug Discov. Today 2019, 24, 1202–1208. [Google Scholar] [CrossRef]
- Yu, M. Generation, function and diagnostic value of mitochondrial DNA copy number alterations in human cancers. Life Sci. 2011, 89, 65–71. [Google Scholar] [CrossRef]
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef]
- Patra, S.; Elahi, N.; Armorer, A.; Arunachalam, S.; Omala, J.; Hamid, I.; Ashton, A.W.; Joyce, D.; Jiao, X.; Pestell, R.G. Mechanisms Governing Metabolic Heterogeneity in Breast Cancer and Other Tumors. Front. Oncol. 2021, 11, 700629. [Google Scholar] [CrossRef]
- Kubicka, A.; Matczak, K.; Łabieniec-Watała, M. More Than Meets the Eye Regarding Cancer Metabolism. Int. J. Mol. Sci. 2021, 22, 9507. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, Y. Mitochondria as a target in cancer treatment. MedComm (2020) 2020, 1, 129–139. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Flomenberg, N.; Birbe, R.C.; Witkiewicz, A.K.; Howell, A.; Pavlides, S.; Tsirigos, A.; Ertel, A.; Pestell, R.G.; et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: Visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle 2011, 10, 4047–4064. [Google Scholar] [CrossRef]
- Xu, Y.; Xue, D.; Bankhead, A.; Neamati, N. Why All the Fuss about Oxidative Phosphorylation (OXPHOS)? J. Med. Chem. 2020, 63, 14276–14307. [Google Scholar] [CrossRef]
- Davis, R.T.; Blake, K.; Ma, D.; Gabra, M.B.I.; Hernandez, G.A.; Phung, A.T.; Yang, Y.; Maurer, D.; Lefebvre, A.E.Y.T.; Alshetaiwi, H.; et al. Transcriptional diversity and bioenergetic shift in human breast cancer metastasis revealed by single-cell RNA sequencing. Nat. Cell Biol. 2020, 22, 310–320. [Google Scholar] [CrossRef]
- Marzagalli, M.; Fontana, F.; Raimondi, M.; Limonta, P. Cancer Stem Cells-Key Players in Tumor Relapse. Cancers 2021, 13, 376. [Google Scholar] [CrossRef]
- Lin, D.; Shen, L.; Luo, M.; Zhang, K.; Li, J.; Yang, Q.; Zhu, F.; Zhou, D.; Zheng, S.; Chen, Y.; et al. Circulating tumor cells: Biology and clinical significance. Signal Transduct. Target. Ther. 2021, 6, 404. [Google Scholar] [CrossRef]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef]
- Bai, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Cancer stem cell in breast cancer therapeutic resistance. Cancer Treat. Rev. 2018, 69, 152–163. [Google Scholar] [CrossRef]
- Chang, J.C. Cancer stem cells: Role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine 2016, 95, S20–S25. [Google Scholar] [CrossRef]
- Zeng, X.; Liu, C.; Yao, J.; Wan, H.; Wan, G.; Li, Y.; Chen, N. Breast cancer stem cells, heterogeneity, targeting therapies and therapeutic implications. Pharmacol. Res. 2021, 163, 105320. [Google Scholar] [CrossRef]
- Peiris-Pages, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef]
- Ciavardelli, D.; Rossi, C.; Barcaroli, D.; Volpe, S.; Consalvo, A.; Zucchelli, M.; De Cola, A.; Scavo, E.; Carollo, R.; D’Agostino, D.; et al. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death Dis. 2014, 5, e1336. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.C.; Kim, J.H. Cancer stem cell metabolism: Target for cancer therapy. BMB Rep. 2018, 51, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, M.; Sotgia, F.; Lisanti, M.P. “Energetic” Cancer Stem Cells (e-CSCs): A New Hyper-Metabolic and Proliferative Tumor Cell Phenotype, Driven by Mitochondrial Energy. Front. Oncol. 2018, 8, 677. [Google Scholar] [CrossRef]
- Farnie, G.; Sotgia, F.; Lisanti, M.P. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget 2015, 6, 30472–30486. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, L.R.; Varella-Garcia, M.; Liang, B.C. Diminished tumorigenic phenotype after depletion of mitochondrial DNA. Cell Growth Differ. 1997, 8, 1189–1198. [Google Scholar]
- Yu, M.; Shi, Y.; Wei, X.; Yang, Y.; Zhou, Y.; Hao, X.; Zhang, N.; Niu, R. Depletion of mitochondrial DNA by ethidium bromide treatment inhibits the proliferation and tumorigenesis of T47D human breast cancer cells. Toxicol. Lett. 2007, 170, 83–93. [Google Scholar] [CrossRef]
- Abad, E.; García-Mayea, Y.; Mir, C.; Sebastian, D.; Zorzano, A.; Potesil, D.; Zdrahal, Z.; Lyakhovich, A.; Lleonart, M.E. Common Metabolic Pathways Implicated in Resistance to Chemotherapy Point to a Key Mitochondrial Role in Breast Cancer. Mol. Cell. Proteomics 2019, 18, 231–244. [Google Scholar] [CrossRef]
- Greer, Y.E.; Hernandez, L.; Fennell, E.M.J.; Kundu, M.; Voeller, D.; Chari, R.; Gilbert, S.F.; Gilbert, T.S.K.; Ratnayake, S.; Tang, B.; et al. Mitochondrial Matrix Protease ClpP Agonists Inhibit Cancer Stem Cell Function in Breast Cancer Cells by Disrupting Mitochondrial Homeostasis. Cancer Res. Commun. 2022, 2, 1144–1161. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef]
- Evans, K.W.; Yuca, E.; Scott, S.S.; Zhao, M.; Paez Arango, N.; Cruz Pico, C.X.; Saridogan, T.; Shariati, M.; Class, C.A.; Bristow, C.A.; et al. Oxidative Phosphorylation Is a Metabolic Vulnerability in Chemotherapy-Resistant Triple-Negative Breast Cancer. Cancer Res. 2021, 81, 5572–5581. [Google Scholar] [CrossRef]
- Echeverria, G.V.; Ge, Z.; Seth, S.; Zhang, X.; Jeter-Jones, S.; Zhou, X.; Cai, S.; Tu, Y.; McCoy, A.; Peoples, M.; et al. Resistance to neoadjuvant chemotherapy in triple-negative breast cancer mediated by a reversible drug-tolerant state. Sci. Transl. Med. 2019, 11, eaav0936. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Fiorillo, M.; Peiris-Pagès, M.; Ozsvari, B.; Smith, D.L.; Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Cappello, A.R.; Pezzi, V.; Lisanti, M.P.; et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget 2015, 6, 14777–14795. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, J.; Guerrouahen, B.S.; Al Thawadi, H.; Ghiabi, P.; Maleki, M.; Abu-Kaoud, N.; Jacob, A.; Mirshahi, M.; Galas, L.; Rafii, S.; et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J. Transl. Med. 2013, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Wu, S.; Wang, Y.; Shi, D. Circulating tumor cell isolation for cancer diagnosis and prognosis. EBioMedicine 2022, 83, 104237. [Google Scholar] [CrossRef] [PubMed]
- Papadaki, M.A.; Stoupis, G.; Theodoropoulos, P.A.; Mavroudis, D.; Georgoulias, V.; Agelaki, S. Circulating Tumor Cells with Stemness and Epithelial-to-Mesenchymal Transition Features Are Chemoresistant and Predictive of Poor Outcome in Metastatic Breast Cancer. Mol. Cancer Ther. 2019, 18, 437–447. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003, 1001–1015. [Google Scholar] [CrossRef]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef]
- Zheng, Y.; Miyamoto, D.T.; Wittner, B.S.; Sullivan, J.P.; Aceto, N.; Jordan, N.V.; Yu, M.; Karabacak, N.M.; Comaills, V.; Morris, R.; et al. Expression of β-globin by cancer cells promotes cell survival during blood-borne dissemination. Nat. Commun. 2017, 8, 14344. [Google Scholar] [CrossRef]
- Marro, M.; Nieva, C.; de Juan, A.; Sierra, A. Unravelling the Metabolic Progression of Breast Cancer Cells to Bone Metastasis by Coupling Raman Spectroscopy and a Novel Use of Mcr-Als Algorithm. Anal. Chem. 2018, 90, 5594–5602. [Google Scholar] [CrossRef]
- Andrzejewski, S.; Klimcakova, E.; Johnson, R.M.; Tabariès, S.; Annis, M.G.; McGuirk, S.; Northey, J.J.; Chénard, V.; Sriram, U.; Papadopoli, D.J.; et al. PGC-1α Promotes Breast Cancer Metastasis and Confers Bioenergetic Flexibility against Metabolic Drugs. Cell Metab. 2017, 26, 778–787.e775. [Google Scholar] [CrossRef]
- Chen, E.I.; Hewel, J.; Krueger, J.S.; Tiraby, C.; Weber, M.R.; Kralli, A.; Becker, K.; Yates, J.R.; Felding-Habermann, B. Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res. 2007, 67, 1472–1486. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.; Willis, A.; Kornhauser, N.; Ward, M.M.; Lee, S.B.; Nackos, E.; Seo, B.R.; Chuang, E.; Cigler, T.; Moore, A.; et al. Influencing the Tumor Microenvironment: A Phase II Study of Copper Depletion Using Tetrathiomolybdate in Patients with Breast Cancer at High Risk for Recurrence and in Preclinical Models of Lung Metastases. Clin. Cancer Res. 2017, 23, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Ramchandani, D.; Berisa, M.; Tavarez, D.A.; Li, Z.; Miele, M.; Bai, Y.; Lee, S.B.; Ban, Y.; Dephoure, N.; Hendrickson, R.C.; et al. Copper depletion modulates mitochondrial oxidative phosphorylation to impair triple negative breast cancer metastasis. Nat. Commun. 2021, 12, 7311. [Google Scholar] [CrossRef]
- Sotgia, F.; Ozsvari, B.; Fiorillo, M.; De Francesco, E.M.; Bonuccelli, G.; Lisanti, M.P. A mitochondrial based oncology platform for targeting cancer stem cells (CSCs): MITO-ONC-RX. Cell Cycle 2018, 17, 2091–2100. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Uchiumi, T.; Yagi, M.; Matsumoto, S.; Amamoto, R.; Takazaki, S.; Yamaza, H.; Nonaka, K.; Kang, D. Dihydro-orotate dehydrogenase is physically associated with the respiratory complex and its loss leads to mitochondrial dysfunction. Biosci. Rep. 2013, 33, e00021. [Google Scholar] [CrossRef]
- Zhou, Y.; Tao, L.; Zhou, X.; Zuo, Z.; Gong, J.; Liu, X.; Liu, C.; Sang, N.; Liu, H.; Zou, J.; et al. DHODH and cancer: Promising prospects to be explored. Cancer Metab. 2021, 9, 22. [Google Scholar] [CrossRef]
- Han, Y.; Wu, P.; Wang, Z.; Zhang, Z.; Sun, S.; Liu, J.; Gong, S.; Gao, P.; Iwakuma, T.; Molina-Vila, M.A.; et al. Ubiquinol-cytochrome C reductase core protein II promotes tumorigenesis by facilitating p53 degradation. EBioMedicine 2019, 40, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Bott, A.J.; Maimouni, S.; Zong, W.X. The Pleiotropic Effects of Glutamine Metabolism in Cancer. Cancers 2019, 11, 770. [Google Scholar] [CrossRef]
- Jin, L.; Li, D.; Alesi, G.N.; Fan, J.; Kang, H.B.; Lu, Z.; Boggon, T.J.; Jin, P.; Yi, H.; Wright, E.R.; et al. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell 2015, 27, 257–270. [Google Scholar] [CrossRef]
- Jaggupilli, A.; Ly, S.; Nguyen, K.; Anand, V.; Yuan, B.; El-Dana, F.; Yan, Y.; Arvanitis, Z.; Piyarathna, D.W.B.; Putluri, N.; et al. Metabolic stress induces GD2. Br. J. Cancer 2021, 126, 615–627. [Google Scholar] [CrossRef]
- Jariyal, H.; Gupta, C.; Andhale, S.; Gadge, S.; Srivastava, A. Comparative stemness and differentiation of luminal and basal breast cancer stem cell type under glutamine-deprivation. J. Cell Commun. Signal. 2021, 15, 207–222. [Google Scholar] [CrossRef]
- Lukey, M.J.; Cluntun, A.A.; Katt, W.P.; Lin, M.J.; Druso, J.E.; Ramachandran, S.; Erickson, J.W.; Le, H.H.; Wang, Z.E.; Blank, B.; et al. Liver-Type Glutaminase GLS2 Is a Druggable Metabolic Node in Luminal-Subtype Breast Cancer. Cell Rep. 2019, 29, 76–88.e77. [Google Scholar] [CrossRef]
- McGuirk, S.; Gravel, S.P.; Deblois, G.; Papadopoli, D.J.; Faubert, B.; Wegner, A.; Hiller, K.; Avizonis, D.; Akavia, U.D.; Jones, R.G.; et al. PGC-1α supports glutamine metabolism in breast cancer. Cancer Metab. 2013, 1, 22. [Google Scholar] [CrossRef]
- Lampa, M.; Arlt, H.; He, T.; Ospina, B.; Reeves, J.; Zhang, B.; Murtie, J.; Deng, G.; Barberis, C.; Hoffmann, D.; et al. Glutaminase is essential for the growth of triple-negative breast cancer cells with a deregulated glutamine metabolism pathway and its suppression synergizes with mTOR inhibition. PLoS ONE 2017, 12, e0185092. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.; Dias, S.M.; Dang, C.V.; et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Kung, H.N.; Marks, J.R.; Chi, J.T. Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia. PLoS Genet. 2011, 7, e1002229. [Google Scholar] [CrossRef]
- Timmerman, L.A.; Holton, T.; Yuneva, M.; Louie, R.J.; Padró, M.; Daemen, A.; Hu, M.; Chan, D.A.; Ethier, S.P.; van’t Veer, L.J.; et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell 2013, 24, 450–465. [Google Scholar] [CrossRef] [PubMed]
- van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef]
- Quek, L.E.; van Geldermalsen, M.; Guan, Y.F.; Wahi, K.; Mayoh, C.; Balaban, S.; Pang, A.; Wang, Q.; Cowley, M.J.; Brown, K.K.; et al. Glutamine addiction promotes glucose oxidation in triple-negative breast cancer. Oncogene 2022, 41, 4066–4078. [Google Scholar] [CrossRef]
- Budczies, J.; Pfitzner, B.M.; Györffy, B.; Winzer, K.J.; Radke, C.; Dietel, M.; Fiehn, O.; Denkert, C. Glutamate enrichment as new diagnostic opportunity in breast cancer. Int. J. Cancer 2015, 136, 1619–1628. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, S.; Lu, J.; Zhang, Z.; Wu, D.; Wu, Z.; Zheng, Y. GLUL Promotes Cell Proliferation in Breast Cancer. J. Cell. Biochem. 2017, 118, 2018–2025. [Google Scholar] [CrossRef]
- Knott, S.R.V.; Wagenblast, E.; Khan, S.; Kim, S.Y.; Soto, M.; Wagner, M.; Turgeon, M.O.; Fish, L.; Erard, N.; Gable, A.L.; et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 2018, 554, 378–381. [Google Scholar] [CrossRef]
- Shen, N.; Korm, S.; Karantanos, T.; Li, D.; Zhang, X.; Ritou, E.; Xu, H.; Lam, A.; English, J.; Zong, W.X.; et al. DLST-dependence dictates metabolic heterogeneity in TCA-cycle usage among triple-negative breast cancer. Commun. Biol. 2021, 4, 1289. [Google Scholar] [CrossRef] [PubMed]
- You, D.; Du, D.; Zhao, X.; Li, X.; Ying, M.; Hu, X. Mitochondrial malic enzyme 2 promotes breast cancer metastasis via stabilizing HIF-1α under hypoxia. Chin. J. Cancer Res. 2021, 33, 308–322. [Google Scholar] [CrossRef] [PubMed]
- Geng, P.; Qin, W.; Xu, G. Proline metabolism in cancer. Amino. Acids 2021, 53, 1769–1777. [Google Scholar] [CrossRef]
- Liu, W.; Hancock, C.N.; Fischer, J.W.; Harman, M.; Phang, J.M. Proline biosynthesis augments tumor cell growth and aerobic glycolysis: Involvement of pyridine nucleotides. Sci. Rep. 2015, 5, 17206. [Google Scholar] [CrossRef]
- Ding, J.; Kuo, M.L.; Su, L.; Xue, L.; Luh, F.; Zhang, H.; Wang, J.; Lin, T.G.; Zhang, K.; Chu, P.; et al. Human mitochondrial pyrroline-5-carboxylate reductase 1 promotes invasiveness and impacts survival in breast cancers. Carcinogenesis 2017, 38, 519–531. [Google Scholar] [CrossRef]
- Loayza-Puch, F.; Rooijers, K.; Buil, L.C.; Zijlstra, J.; Oude Vrielink, J.F.; Lopes, R.; Ugalde, A.P.; van Breugel, P.; Hofland, I.; Wesseling, J.; et al. Tumour-specific proline vulnerability uncovered by differential ribosome codon reading. Nature 2016, 530, 490–494. [Google Scholar] [CrossRef]
- Shenoy, A.; Belugali Nataraj, N.; Perry, G.; Loayza Puch, F.; Nagel, R.; Marin, I.; Balint, N.; Bossel, N.; Pavlovsky, A.; Barshack, I.; et al. Proteomic patterns associated with response to breast cancer neoadjuvant treatment. Mol. Syst. Biol. 2020, 16, e9443. [Google Scholar] [CrossRef]
- Elia, I.; Broekaert, D.; Christen, S.; Boon, R.; Radaelli, E.; Orth, M.F.; Verfaillie, C.; Grünewald, T.G.P.; Fendt, S.M. Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat. Commun. 2017, 8, 15267. [Google Scholar] [CrossRef]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Pollari, S.; Käkönen, S.M.; Edgren, H.; Wolf, M.; Kohonen, P.; Sara, H.; Guise, T.; Nees, M.; Kallioniemi, O. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Res. Treat. 2011, 125, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Ngo, B.; Kim, E.; Osorio-Vasquez, V.; Doll, S.; Bustraan, S.; Liang, R.J.; Luengo, A.; Davidson, S.M.; Ali, A.; Ferraro, G.B.; et al. Limited Environmental Serine and Glycine Confer Brain Metastasis Sensitivity to PHGDH Inhibition. Cancer Discov. 2020, 10, 1352–1373. [Google Scholar] [CrossRef]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef]
- Bernhardt, S.; Bayerlová, M.; Vetter, M.; Wachter, A.; Mitra, D.; Hanf, V.; Lantzsch, T.; Uleer, C.; Peschel, S.; John, J.; et al. Proteomic profiling of breast cancer metabolism identifies SHMT2 and ASCT2 as prognostic factors. Breast Cancer Res. 2017, 19, 112. [Google Scholar] [CrossRef]
- Nilsson, R.; Jain, M.; Madhusudhan, N.; Sheppard, N.G.; Strittmatter, L.; Kampf, C.; Huang, J.; Asplund, A.; Mootha, V.K. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 2014, 5, 3128. [Google Scholar] [CrossRef]
- Lehtinen, L.; Ketola, K.; Mäkelä, R.; Mpindi, J.P.; Viitala, M.; Kallioniemi, O.; Iljin, K. High-throughput RNAi screening for novel modulators of vimentin expression identifies MTHFD2 as a regulator of breast cancer cell migration and invasion. Oncotarget 2013, 4, 48–63. [Google Scholar] [CrossRef]
- Showalter, S.L.; Showalter, T.N.; Witkiewicz, A.; Havens, R.; Kennedy, E.P.; Hucl, T.; Kern, S.E.; Yeo, C.J.; Brody, J.R. Evaluating the drug-target relationship between thymidylate synthase expression and tumor response to 5-fluorouracil. Is it time to move forward? Cancer Biol. Ther. 2008, 7, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Tian, B.; Zhang, M.; Gao, X.; Jie, L.; Liu, P.; Li, J. Diagnostic and prognostic value of thymidylate synthase expression in breast cancer. Clin. Exp. Pharmacol. Physiol. 2021, 48, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Cowan, K.H.; Goldsmith, M.E.; Levine, R.M.; Aitken, S.C.; Douglass, E.; Clendeninn, N.; Nienhuis, A.W.; Lippman, M.E. Dihydrofolate reductase gene amplification and possible rearrangement in estrogen-responsive methotrexate-resistant human breast cancer cells. J. Biol. Chem. 1982, 257, 15079–15086. [Google Scholar] [CrossRef] [PubMed]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Rios Garcia, M.; Steinbauer, B.; Srivastava, K.; Singhal, M.; Mattijssen, F.; Maida, A.; Christian, S.; Hess-Stumpp, H.; Augustin, H.G.; Müller-Decker, K.; et al. Acetyl-CoA Carboxylase 1-Dependent Protein Acetylation Controls Breast Cancer Metastasis and Recurrence. Cell Metab. 2017, 26, 842–855.e845. [Google Scholar] [CrossRef]
- Sardesai, S.D.; Thomas, A.; Gallagher, C.; Lynce, F.; Ottaviano, Y.L.; Ballinger, T.J.; Schneider, B.P.; Storniolo, A.M.; Bauchle, A.; Althouse, S.K.; et al. Inhibiting Fatty Acid Synthase with Omeprazole to Improve Efficacy of Neoadjuvant Chemotherapy in Patients with Operable TNBC. Clin. Cancer Res. 2021, 27, 5810–5817. [Google Scholar] [CrossRef]
- Giró-Perafita, A.; Sarrats, A.; Pérez-Bueno, F.; Oliveras, G.; Buxó, M.; Brunet, J.; Viñas, G.; Miquel, T.P. Fatty acid synthase expression and its association with clinico-histopathological features in triple-negative breast cancer. Oncotarget 2017, 8, 74391–74405. [Google Scholar] [CrossRef]
- Schroeder, B.; Vander Steen, T.; Espinoza, I.; Venkatapoorna, C.M.K.; Hu, Z.; Silva, F.M.; Regan, K.; Cuyàs, E.; Meng, X.W.; Verdura, S.; et al. Fatty acid synthase (FASN) regulates the mitochondrial priming of cancer cells. Cell Death Dis. 2021, 12, 977. [Google Scholar] [CrossRef] [PubMed]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Pniewski, K.; Perry, C.E.; Papp, S.B.; Shaffer, J.D.; Velasco-Silva, J.N.; Casciano, J.C.; Aramburu, T.M.; Srikanth, Y.V.V.; Cassel, J.; et al. Targeting ACSS2 with a Transition-State Mimetic Inhibits Triple-Negative Breast Cancer Growth. Cancer Res. 2021, 81, 1252–1264. [Google Scholar] [CrossRef]
- Wang, D.; Yin, L.; Wei, J.; Yang, Z.; Jiang, G. ATP citrate lyase is increased in human breast cancer, depletion of which promotes apoptosis. Tumour. Biol. 2017, 39, 1010428317698338. [Google Scholar] [CrossRef]
- Kumari, A. Chapter 4—Beta Oxidation of Fatty Acids; Academic Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Gatza, M.L.; Silva, G.O.; Parker, J.S.; Fan, C.; Perou, C.M. An integrated genomics approach identifies drivers of proliferation in luminal-subtype human breast cancer. Nat. Genet. 2014, 46, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Linher-Melville, K.; Zantinge, S.; Sanli, T.; Gerstein, H.; Tsakiridis, T.; Singh, G. Establishing a relationship between prolactin and altered fatty acid β-oxidation via carnitine palmitoyl transferase 1 in breast cancer cells. BMC Cancer 2011, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Pucci, S.; Zonetti, M.J.; Fisco, T.; Polidoro, C.; Bocchinfuso, G.; Palleschi, A.; Novelli, G.; Spagnoli, L.G.; Mazzarelli, P. Carnitine palmitoyl transferase-1A (CPT1A): A new tumor specific target in human breast cancer. Oncotarget 2016, 7, 19982–19996. [Google Scholar] [CrossRef]
- Loo, S.Y.; Toh, L.P.; Xie, W.H.; Pathak, E.; Tan, W.; Ma, S.; Lee, M.Y.; Shatishwaran, S.; Yeo, J.Z.Z.; Yuan, J.; et al. Fatty acid oxidation is a druggable gateway regulating cellular plasticity for driving metastasis in breast cancer. Sci. Adv. 2021, 7, eabh2443. [Google Scholar] [CrossRef]
- Park, J.H.; Vithayathil, S.; Kumar, S.; Sung, P.L.; Dobrolecki, L.E.; Putluri, V.; Bhat, V.B.; Bhowmik, S.K.; Gupta, V.; Arora, K.; et al. Fatty Acid Oxidation-Driven Src Links Mitochondrial Energy Reprogramming and Oncogenic Properties in Triple-Negative Breast Cancer. Cell Rep. 2016, 14, 2154–2165. [Google Scholar] [CrossRef]
- Li, Y.J.; Fahrmann, J.F.; Aftabizadeh, M.; Zhao, Q.; Tripathi, S.C.; Zhang, C.; Yuan, Y.; Ann, D.; Hanash, S.; Yu, H. Fatty acid oxidation protects cancer cells from apoptosis by increasing mitochondrial membrane lipids. Cell Rep. 2022, 39, 110870. [Google Scholar] [CrossRef]
- Wang, T.; Fahrmann, J.F.; Lee, H.; Li, Y.J.; Tripathi, S.C.; Yue, C.; Zhang, C.; Lifshitz, V.; Song, J.; Yuan, Y.; et al. JAK/STAT3-Regulated Fatty Acid β-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab. 2018, 27, 136–150.e135. [Google Scholar] [CrossRef]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef]
- Ehmsen, S.; Pedersen, M.H.; Wang, G.; Terp, M.G.; Arslanagic, A.; Hood, B.L.; Conrads, T.P.; Leth-Larsen, R.; Ditzel, H.J. Increased Cholesterol Biosynthesis Is a Key Characteristic of Breast Cancer Stem Cells Influencing Patient Outcome. Cell Rep. 2019, 27, 3927–3938.e3926. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.R.; Wardell, S.E.; Jasper, J.S.; Park, S.; Suchindran, S.; Howe, M.K.; Carver, N.J.; Pillai, R.V.; Sullivan, P.M.; Sondhi, V.; et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science 2013, 342, 1094–1098. [Google Scholar] [CrossRef] [PubMed]
- Baek, A.E.; Yu, Y.A.; He, S.; Wardell, S.E.; Chang, C.Y.; Kwon, S.; Pillai, R.V.; McDowell, H.B.; Thompson, J.W.; Dubois, L.G.; et al. The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat. Commun. 2017, 8, 864. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Liang, B.; Zhang, L.; Li, X.; Li, H.; Jing, W.; Jiang, Y.; Zhou, F.; Zhang, J.; Meng, Y.; et al. Enhanced CHOLESTEROL biosynthesis promotes breast cancer metastasis via modulating CCDC25 expression and neutrophil extracellular traps formation. Sci. Rep. 2022, 12, 17350. [Google Scholar] [CrossRef]
- Liu, B.; Yi, Z.; Guan, X.; Zeng, Y.X.; Ma, F. The relationship between statins and breast cancer prognosis varies by statin type and exposure time: A meta-analysis. Breast Cancer Res. Treat. 2017, 164, 1–11. [Google Scholar] [CrossRef]
- Zhou, C.; Wang, Z.; Cao, Y.; Zhao, L. Pan-cancer analysis reveals the oncogenic role of 3-hydroxy-3-methylglutaryl-CoA synthase 1. Cancer Rep. 2022, 5, e1562. [Google Scholar] [CrossRef]
- Walsh, C.A.; Akrap, N.; Garre, E.; Magnusson, Y.; Harrison, H.; Andersson, D.; Jonasson, E.; Rafnsdottir, S.; Choudhry, H.; Buffa, F.; et al. The mevalonate precursor enzyme HMGCS1 is a novel marker and key mediator of cancer stem cell enrichment in luminal and basal models of breast cancer. PLoS ONE 2020, 15, e0236187. [Google Scholar] [CrossRef]
- Clendening, J.W.; Pandyra, A.; Boutros, P.C.; El Ghamrasni, S.; Khosravi, F.; Trentin, G.A.; Martirosyan, A.; Hakem, A.; Hakem, R.; Jurisica, I.; et al. Dysregulation of the mevalonate pathway promotes transformation. Proc. Natl. Acad. Sci. USA 2010, 107, 15051–15056. [Google Scholar] [CrossRef]
- Chen, Y.; Olopade, O.I. MYC in breast tumor progression. Expert Rev. Anticancer Ther. 2008, 8, 1689–1698. [Google Scholar] [CrossRef]
- Hynes, N.E.; Stoelzle, T. Key signalling nodes in mammary gland development and cancer: Myc. Breast Cancer Res. 2009, 11, 210. [Google Scholar] [CrossRef]
- Efstratiadis, A.; Szabolcs, M.; Klinakis, A. Notch, Myc and breast cancer. Cell Cycle 2007, 6, 418–429. [Google Scholar] [CrossRef]
- Lee, K.M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sanchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647.e637. [Google Scholar] [CrossRef] [PubMed]
- Alles, M.C.; Gardiner-Garden, M.; Nott, D.J.; Wang, Y.; Foekens, J.A.; Sutherland, R.L.; Musgrove, E.A.; Ormandy, C.J. Meta-analysis and gene set enrichment relative to er status reveal elevated activity of MYC and E2F in the “basal” breast cancer subgroup. PLoS ONE 2009, 4, e4710. [Google Scholar] [CrossRef]
- Chandriani, S.; Frengen, E.; Cowling, V.H.; Pendergrass, S.A.; Perou, C.M.; Whitfield, M.L.; Cole, M.D. A core MYC gene expression signature is prominent in basal-like breast cancer but only partially overlaps the core serum response. PLoS ONE 2009, 4, e6693. [Google Scholar] [CrossRef]
- Network, C.G.A. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Deming, S.L.; Nass, S.J.; Dickson, R.B.; Trock, B.J. C-myc amplification in breast cancer: A meta-analysis of its occurrence and prognostic relevance. Br. J. Cancer 2000, 83, 1688–1695. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, Y.; Zeller, K.I.; Potter, J.J.; Wonsey, D.R.; O’Donnell, K.A.; Kim, J.W.; Yustein, J.T.; Lee, L.A.; Dang, C.V. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell. Biol. 2005, 25, 6225–6234. [Google Scholar] [CrossRef]
- Popay, T.M.; Wang, J.; Adams, C.M.; Howard, G.C.; Codreanu, S.G.; Sherrod, S.D.; McLean, J.A.; Thomas, L.R.; Lorey, S.L.; Machida, Y.J.; et al. MYC regulates ribosome biogenesis and mitochondrial gene expression programs through its interaction with host cell factor-1. Elife 2021, 10, e60191. [Google Scholar] [CrossRef]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef]
- Morrish, F.; Hockenbery, D. MYC and mitochondrial biogenesis. Cold Spring Harb. Perspect. Med. 2014, 4, a014225. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef]
- Psathas, J.N.; Thomas-Tikhonenko, A. MYC and the art of microRNA maintenance. Cold Spring Harb. Perspect. Med. 2014, 4, a014175. [Google Scholar] [CrossRef]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal Transduct. Target. Ther. 2020, 5, 124. [Google Scholar] [CrossRef]
- Bott, A.J.; Peng, I.C.; Fan, Y.; Faubert, B.; Zhao, L.; Li, J.; Neidler, S.; Sun, Y.; Jaber, N.; Krokowski, D.; et al. Oncogenic Myc Induces Expression of Glutamine Synthetase through Promoter Demethylation. Cell Metab. 2015, 22, 1068–1077. [Google Scholar] [CrossRef]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef]
- Liu, W.; Le, A.; Hancock, C.; Lane, A.N.; Dang, C.V.; Fan, T.W.; Phang, J.M. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc. Natl. Acad. Sci. USA 2012, 109, 8983–8988. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Song, L.; Wan, Q.; Wu, G.; Li, X.; Wang, Y.; Wang, J.; Liu, Z.; Zhong, X.; He, X.; et al. cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res. 2015, 25, 429–444. [Google Scholar] [CrossRef]
- Casciano, J.C.; Perry, C.; Cohen-Nowak, A.J.; Miller, K.D.; Vande Voorde, J.; Zhang, Q.; Chalmers, S.; Sandison, M.E.; Liu, Q.; Hedley, A.; et al. MYC regulates fatty acid metabolism through a multigenic program in claudin-low triple negative breast cancer. Br. J. Cancer 2020, 122, 868–884. [Google Scholar] [CrossRef]
- Camarda, R.; Zhou, A.Y.; Kohnz, R.A.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G.; et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med. 2016, 22, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, R.; Rein, J.; D’Arcy, M.; Herschkowitz, J.I.; Hoadley, K.A.; Troester, M.A. Overexpression of miR-146a in basal-like breast cancer cells confers enhanced tumorigenic potential in association with altered p53 status. Carcinogenesis 2014, 35, 2567–2575. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Ryan, K.M. p53 and metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumour-associated mutant p53 drives the Warburg effect. Nat. Commun. 2013, 4, 2935. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Ambroise, G.; Ouchida, A.T.; Lima Queiroz, A.; Smith, D.; Gimenez-Cassina, A.; Iwanicki, M.P.; Muller, P.A.; Norberg, E.; Vakifahmetoglu-Norberg, H. Effect of Mutant p53 Proteins on Glycolysis and Mitochondrial Metabolism. Mol. Cell Biol. 2017, 37, e00328-17. [Google Scholar] [CrossRef] [PubMed]
- Kamp, W.M.; Wang, P.Y.; Hwang, P.M. TP53 mutation, mitochondria and cancer. Curr. Opin. Genet. Dev. 2016, 38, 16–22. [Google Scholar] [CrossRef]
- Zhou, G.; Wang, J.; Zhao, M.; Xie, T.X.; Tanaka, N.; Sano, D.; Patel, A.A.; Ward, A.M.; Sandulache, V.C.; Jasser, S.A.; et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol. Cell 2014, 54, 960–974. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerød, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef]
- McCarthy, A.M.; Kumar, N.P.; He, W.; Regan, S.; Welch, M.; Moy, B.; Iafrate, A.J.; Chan, A.T.; Bardia, A.; Armstrong, K. Different associations of tumor PIK3CA mutations and clinical outcomes according to aspirin use among women with metastatic hormone receptor positive breast cancer. BMC Cancer 2020, 20, 347. [Google Scholar] [CrossRef]
- Lau, C.E.; Tredwell, G.D.; Ellis, J.K.; Lam, E.W.; Keun, H.C. Metabolomic characterisation of the effects of oncogenic PIK3CA transformation in a breast epithelial cell line. Sci. Rep. 2017, 7, 46079. [Google Scholar] [CrossRef]
- Yi, J.; Zhu, J.; Wu, J.; Thompson, C.B.; Jiang, X. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 31189–31197. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Carrington, E.M.; Zhan, Y.; Brady, J.L.; Zhang, J.G.; Sutherland, R.M.; Anstee, N.S.; Schenk, R.L.; Vikstrom, I.B.; Delconte, R.B.; Segal, D.; et al. Anti-apoptotic proteins BCL-2, MCL-1 and A1 summate collectively to maintain survival of immune cell populations both in vitro and in vivo. Cell Death Differ. 2017, 24, 878–888. [Google Scholar] [CrossRef]
- Merino, D.; Lok, S.W.; Visvader, J.E.; Lindeman, G.J. Targeting BCL-2 to enhance vulnerability to therapy in estrogen receptor-positive breast cancer. Oncogene 2016, 35, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Olopade, O.I.; Adeyanju, M.O.; Safa, A.R.; Hagos, F.; Mick, R.; Thompson, C.B.; Recant, W.M. Overexpression of BCL-x protein in primary breast cancer is associated with high tumor grade and nodal metastases. Cancer J. Sci. Am. 1997, 3, 230–237. [Google Scholar] [PubMed]
- Martin, S.S.; Ridgeway, A.G.; Pinkas, J.; Lu, Y.; Reginato, M.J.; Koh, E.Y.; Michelman, M.; Daley, G.Q.; Brugge, J.S.; Leder, P. A cytoskeleton-based functional genetic screen identifies Bcl-xL as an enhancer of metastasis, but not primary tumor growth. Oncogene 2004, 23, 4641–4645. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.J.; Dhayade, S.; Ferrari, N.; Sims, A.H.; Johnson, E.; Mason, S.M.; Dickson, A.; Ryan, K.M.; Kalna, G.; Edwards, J.; et al. MCL-1 is a prognostic indicator and drug target in breast cancer. Cell Death Dis. 2018, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.M.; Cook, R.S. Bcl-2 family proteins in breast development and cancer: Could Mcl-1 targeting overcome therapeutic resistance? Oncotarget 2015, 6, 3519–3530. [Google Scholar] [CrossRef] [PubMed]
- Lucantoni, F.; Salvucci, M.; Dussmann, H.; Prehn, J.H.M. BCL(X)L and BCL2 increase mitochondrial dynamics in breast cancer cell: Evidence from functional and genetic studies. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119095. [Google Scholar] [CrossRef] [PubMed]
- Lucantoni, F.; Salvucci, M.; Düssmann, H.; Lindner, A.U.; Lambrechts, D.; Prehn, J.H.M. BCL(X)L and BCL2 increase the metabolic fitness of breast cancer cells: A single-cell imaging study. Cell Death Differ. 2021, 28, 1512–1531. [Google Scholar] [CrossRef]
- Lucantoni, F.; Düssmann, H.; Llorente-Folch, I.; Prehn, J.H.M. BCL2 and BCL(X)L selective inhibitors decrease mitochondrial ATP production in breast cancer cells and are synthetically lethal when combined with 2-deoxy-D-glucose. Oncotarget 2018, 9, 26046–26063. [Google Scholar] [CrossRef]
- Ciccarese, F.; Ciminale, V. Escaping Death: Mitochondrial Redox Homeostasis in Cancer Cells. Front. Oncol. 2017, 7, 117. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Li, C.; Cheng, N.; Cui, X.; Xu, X.; Zhou, G. Redox Regulation in Cancer Stem Cells. Oxid. Med. Cell. Longev. 2015, 2015, 750798. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A.; Rogoff, H.A. Implications of reactive oxygen species on cancer formation and its treatment. Semin. Oncol. 2021, 48, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Havas, K.M.; Milchevskaya, V.; Radic, K.; Alladin, A.; Kafkia, E.; Garcia, M.; Stolte, J.; Klaus, B.; Rotmensz, N.; Gibson, T.J.; et al. Metabolic shifts in residual breast cancer drive tumor recurrence. J. Clin. Investig. 2017, 127, 2091–2105. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento-Salinas, F.L.; Delgado-Magallón, A.; Montes-Alvarado, J.B.; Ramírez-Ramírez, D.; Flores-Alonso, J.C.; Cortés-Hernández, P.; Reyes-Leyva, J.; Herrera-Camacho, I.; Anaya-Ruiz, M.; Pelayo, R.; et al. Breast Cancer Subtypes Present a Differential Production of Reactive Oxygen Species (ROS) and Susceptibility to Antioxidant Treatment. Front. Oncol. 2019, 9, 480. [Google Scholar] [CrossRef]
- Hecht, F.; Cazarin, J.M.; Lima, C.E.; Faria, C.C.; Leitão, A.A.; Ferreira, A.C.; Carvalho, D.P.; Fortunato, R.S. Redox homeostasis of breast cancer lineages contributes to differential cell death response to exogenous hydrogen peroxide. Life Sci. 2016, 158, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Sanz, A.; Caro, P.; Gómez, J.; Barja, G. Testing the vicious cycle theory of mitochondrial ROS production: Effects of H2O2 and cumene hydroperoxide treatment on heart mitochondria. J. Bioenerg. Biomembr. 2006, 38, 121–127. [Google Scholar] [CrossRef]
- Inigo, J.R.; Chandra, D. The mitochondrial unfolded protein response (UPR). J. Hematol. Oncol. 2022, 15, 98. [Google Scholar] [CrossRef]
- Lu, T.; Finkel, T. Free radicals and senescence. Exp. Cell Res. 2008, 314, 1918–1922. [Google Scholar] [CrossRef] [PubMed]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 3565127. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Missiroli, S.; Genovese, I.; Perrone, M.; Vezzani, B.; Vitto, V.A.M.; Giorgi, C. The Role of Mitochondria in Inflammation: From Cancer to Neurodegenerative Disorders. J. Clin. Med. 2020, 9, 740. [Google Scholar] [CrossRef]
- Zhou, D.; Shao, L.; Spitz, D.R. Reactive oxygen species in normal and tumor stem cells. Adv. Cancer Res. 2014, 122, 1–67. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.; Qin, S.; Townsend, D.; Schulte, B.A.; Tew, K.D.; Wang, G.Y. Oxidative stress induces senescence in breast cancer stem cells. Biochem. Biophys. Res. Commun. 2019, 514, 1204–1209. [Google Scholar] [CrossRef]
- Neophytou, C.M.; Panagi, M.; Stylianopoulos, T.; Papageorgis, P. The Role of Tumor Microenvironment in Cancer Metastasis: Molecular Mechanisms and Therapeutic Opportunities. Cancers 2021, 13, 53. [Google Scholar] [CrossRef]
- Saha, T.; Dash, C.; Jayabalan, R.; Khiste, S.; Kulkarni, A.; Kurmi, K.; Mondal, J.; Majumder, P.K.; Bardia, A.; Jang, H.L.; et al. Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat. Nanotechnol. 2022, 17, 98–106. [Google Scholar] [CrossRef]
- Kay, E.J.; Paterson, K.; Riera-Domingo, C.; Sumpton, D.; Däbritz, J.H.M.; Tardito, S.; Boldrini, C.; Hernandez-Fernaud, J.R.; Athineos, D.; Dhayade, S.; et al. Cancer-associated fibroblasts require proline synthesis by PYCR1 for the deposition of pro-tumorigenic extracellular matrix. Nat. Metab. 2022, 4, 693–710. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Attané, C.; Milhas, D.; Dirat, B.; Dauvillier, S.; Guerard, A.; Gilhodes, J.; Lazar, I.; Alet, N.; Laurent, V.; et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2017, 2, e87489. [Google Scholar] [CrossRef]
- Garcia-Martinez, L.; Zhang, Y.; Nakata, Y.; Chan, H.L.; Morey, L. Epigenetic mechanisms in breast cancer therapy and resistance. Nat. Commun. 2021, 12, 1786. [Google Scholar] [CrossRef]
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol. 2014, 12, 34. [Google Scholar] [CrossRef]
- Terunuma, A.; Putluri, N.; Mishra, P.; Mathé, E.A.; Dorsey, T.H.; Yi, M.; Wallace, T.A.; Issaq, H.J.; Zhou, M.; Killian, J.K.; et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J. Clin. Investig. 2014, 124, 398–412. [Google Scholar] [CrossRef]
- Fletcher, J.I.; Williams, R.T.; Henderson, M.J.; Norris, M.D.; Haber, M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist. Updat. 2016, 26, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Giddings, E.L.; Champagne, D.P.; Wu, M.H.; Laffin, J.M.; Thornton, T.M.; Valenca-Pereira, F.; Culp-Hill, R.; Fortner, K.A.; Romero, N.; East, J.; et al. Mitochondrial ATP fuels ABC transporter-mediated drug efflux in cancer chemoresistance. Nat. Commun. 2021, 12, 2804. [Google Scholar] [CrossRef] [PubMed]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef]
- Han, X.J.; Yang, Z.J.; Jiang, L.P.; Wei, Y.F.; Liao, M.F.; Qian, Y.; Li, Y.; Huang, X.; Wang, J.B.; Xin, H.B.; et al. Mitochondrial dynamics regulates hypoxia-induced migration and antineoplastic activity of cisplatin in breast cancer cells. Int. J. Oncol. 2015, 46, 691–700. [Google Scholar] [CrossRef]
- Humphries, B.A.; Cutter, A.C.; Buschhaus, J.M.; Chen, Y.C.; Qyli, T.; Palagama, D.S.W.; Eckley, S.; Robison, T.H.; Bevoor, A.; Chiang, B.; et al. Enhanced mitochondrial fission suppresses signaling and metastasis in triple-negative breast cancer. Breast Cancer Res. 2020, 22, 60. [Google Scholar] [CrossRef]
- Sánchez-Alvarez, R.; De Francesco, E.M.; Fiorillo, M.; Sotgia, F.; Lisanti, M.P. Mitochondrial Fission Factor (MFF) Inhibits Mitochondrial Metabolism and Reduces Breast Cancer Stem Cell (CSC) Activity. Front. Oncol. 2020, 10, 1776. [Google Scholar] [CrossRef]
- Li, Q.; Chu, Y.; Li, S.; Yu, L.; Deng, H.; Liao, C.; Liao, X.; Yang, C.; Qi, M.; Cheng, J.; et al. The oncoprotein MUC1 facilitates breast cancer progression by promoting Pink1-dependent mitophagy via ATAD3A destabilization. Cell Death Dis. 2022, 13, 899. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, G.; Zheng, Y.; Shen, H.M.; Hu, X.; Ming, Q.L.; Huang, C.; Li, P.; Gao, N. A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy 2015, 11, 1259–1279. [Google Scholar] [CrossRef] [PubMed]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous. liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Vidal, C.; Dey, S.; Zhang, L. Mitochondria Targeting as an Effective Strategy for Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 3363. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, M.; Stiller, S.B.; Lübbert, P.; Peikert, C.D.; Dannenmaier, S.; Drepper, F.; Weill, U.; Höß, P.; Feuerstein, R.; Gebert, M.; et al. Definition of a High-Confidence Mitochondrial Proteome at Quantitative Scale. Cell Rep. 2017, 19, 2836–2852. [Google Scholar] [CrossRef] [PubMed]
- Varabyova, A.; Stojanovski, D.; Chacinska, A. Mitochondrial protein homeostasis. IUBMB Life 2013, 65, 191–201. [Google Scholar] [CrossRef]
- Levytskyy, R.M.; Germany, E.M.; Khalimonchuk, O. Mitochondrial Quality Control Proteases in Neuronal Welfare. J. Neuroimmune Pharmacol. 2016, 11, 629–644. [Google Scholar] [CrossRef]
- Bulteau, A.L.; Bayot, A. Mitochondrial proteases and cancer. Biochim. Biophys. Acta 2011, 1807, 595–601. [Google Scholar] [CrossRef]
- Rugarli, E.I.; Langer, T. Mitochondrial quality control: A matter of life and death for neurons. EMBO J. 2012, 31, 1336–1349. [Google Scholar] [CrossRef]
- König, T.; Tröder, S.E.; Bakka, K.; Korwitz, A.; Richter-Dennerlein, R.; Lampe, P.A.; Patron, M.; Mühlmeister, M.; Guerrero-Castillo, S.; Brandt, U.; et al. The m-AAA Protease Associated with Neurodegeneration Limits MCU Activity in Mitochondria. Mol. Cell 2016, 64, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Nouri, K.; Feng, Y.; Schimmer, A.D. Mitochondrial ClpP serine protease-biological function and emerging target for cancer therapy. Cell Death Dis. 2020, 11, 841. [Google Scholar] [CrossRef] [PubMed]
- Deshwal, S.; Fiedler, K.U.; Langer, T. Mitochondrial Proteases: Multifaceted Regulators of Mitochondrial Plasticity. Annu. Rev. Biochem. 2020, 89, 501–528. [Google Scholar] [CrossRef]
- Song, J.; Herrmann, J.M.; Becker, T. Quality control of the mitochondrial proteome. Nat. Rev. Mol. Cell Biol. 2021, 22, 54–70. [Google Scholar] [CrossRef]
- Ng, M.Y.W.; Wai, T.; Simonsen, A. Quality control of the mitochondrion. Dev. Cell 2021, 56, 881–905. [Google Scholar] [CrossRef] [PubMed]
- Sauer, R.T.; Fei, X.; Bell, T.A.; Baker, T.A. Structure and function of ClpXP, a AAA+ proteolytic machine powered by probabilistic ATP hydrolysis. Crit. Rev. Biochem. Mol. Biol. 2022, 57, 188–204. [Google Scholar] [CrossRef]
- Glynn, S.E.; Martin, A.; Nager, A.R.; Baker, T.A.; Sauer, R.T. Structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+ protein-unfolding machine. Cell 2009, 139, 744–756. [Google Scholar] [CrossRef]
- Wang, J.; Hartling, J.A.; Flanagan, J.M. The structure of ClpP at 2.3 A resolution suggests a model for ATP-dependent proteolysis. Cell 1997, 91, 447–456. [Google Scholar] [CrossRef]
- Lee, M.E.; Baker, T.A.; Sauer, R.T. Control of substrate gating and translocation into ClpP by channel residues and ClpX binding. J. Mol. Biol. 2010, 399, 707–718. [Google Scholar] [CrossRef]
- Baker, T.A.; Sauer, R.T. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim. Biophys. Acta 2012, 1823, 15–28. [Google Scholar] [CrossRef]
- Malik, I.T.; Brötz-Oesterhelt, H. Conformational control of the bacterial Clp protease by natural product antibiotics. Nat. Prod. Rep. 2017, 34, 815–831. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Ologbenla, A.; Houry, W.A. Dynamics of the ClpP serine protease: A model for self-compartmentalized proteases. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 400–412. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Sieber, S.A. An amino acid domino effect orchestrates ClpP’s conformational states. Curr. Opin. Chem. Biol. 2017, 40, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Gribun, A.; Kimber, M.S.; Ching, R.; Sprangers, R.; Fiebig, K.M.; Houry, W.A. The ClpP double ring tetradecameric protease exhibits plastic ring-ring interactions, and the N termini of its subunits form flexible loops that are essential for ClpXP and ClpAP complex formation. J. Biol. Chem. 2005, 280, 16185–16196. [Google Scholar] [CrossRef]
- Lowth, B.R.; Kirstein-Miles, J.; Saiyed, T.; Brötz-Oesterhelt, H.; Morimoto, R.I.; Truscott, K.N.; Dougan, D.A. Substrate recognition and processing by a Walker B mutant of the human mitochondrial AAA+ protein CLPX. J. Struct. Biol. 2012, 179, 193–201. [Google Scholar] [CrossRef]
- Mabanglo, M.F.; Bhandari, V.; Houry, W.A. Substrates and interactors of the ClpP protease in the mitochondria. Curr. Opin. Chem. Biol. 2021, 66, 102078. [Google Scholar] [CrossRef]
- Fischer, F.; Langer, J.D.; Osiewacz, H.D. Identification of potential mitochondrial CLPXP protease interactors and substrates suggests its central role in energy metabolism. Sci. Rep. 2015, 5, 18375. [Google Scholar] [CrossRef]
- Nguyen, T.A.; Gronauer, T.F.; Nast-Kolb, T.; Sieber, S.A.; Lang, K. Substrate Profiling of Mitochondrial Caseinolytic Protease P via a Site-Specific Photocrosslinking Approach. Angew. Chem. Int. Ed. Engl. 2022, 61, e202111085. [Google Scholar] [CrossRef]
- Haynes, C.M.; Petrova, K.; Benedetti, C.; Yang, Y.; Ron, D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 2007, 13, 467–480. [Google Scholar] [CrossRef]
- Wu, G.; Xiong, Q.; Wei, X.; Wang, Y.; Hu, X.; He, G.; Liu, L.; Lai, Q.; Dai, Z.; Anushesh, D.; et al. Mitochondrial unfolded protein response gene CLPP changes mitochondrial dynamics and affects mitochondrial function. PeerJ 2019, 7, e7209. [Google Scholar] [CrossRef]
- Wang, G.; Fan, Y.; Cao, P.; Tan, K. Insight into the mitochondrial unfolded protein response and cancer: Opportunities and challenges. Cell Biosci. 2022, 12, 18. [Google Scholar] [CrossRef]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 2018, 19, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Inigo, J.R.; Kumar, R.; Chandra, D. Targeting the mitochondrial unfolded protein response in cancer: Opportunities and challenges. Trends Cancer 2021, 7, 1050–1053. [Google Scholar] [CrossRef] [PubMed]
- Kenny, T.C.; Craig, A.J.; Villanueva, A.; Germain, D. Mitohormesis Primes Tumor Invasion and Metastasis. Cell Rep. 2019, 27, 2292–2303.e2296. [Google Scholar] [CrossRef]
- Chen, F.M.; Huang, L.J.; Ou-Yang, F.; Kan, J.Y.; Kao, L.C.; Hou, M.F. Activation of mitochondrial unfolded protein response is associated with Her2-overexpression breast cancer. Breast Cancer Res. Treat. 2020, 183, 61–70. [Google Scholar] [CrossRef]
- Bross, P.; Andresen, B.S.; Knudsen, I.; Kruse, T.A.; Gregersen, N. Human ClpP protease: cDNA sequence, tissue-specific expression and chromosomal assignment of the gene. FEBS Lett. 1995, 377, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Gispert, S.; Parganlija, D.; Klinkenberg, M.; Dröse, S.; Wittig, I.; Mittelbronn, M.; Grzmil, P.; Koob, S.; Hamann, A.; Walter, M.; et al. Loss of mitochondrial peptidase Clpp leads to infertility, hearing loss plus growth retardation via accumulation of CLPX, mtDNA and inflammatory factors. Hum. Mol. Genet. 2013, 22, 4871–4887. [Google Scholar] [CrossRef] [PubMed]
- Jenkinson, E.M.; Rehman, A.U.; Walsh, T.; Clayton-Smith, J.; Lee, K.; Morell, R.J.; Drummond, M.C.; Khan, S.N.; Naeem, M.A.; Rauf, B.; et al. Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am. J. Hum. Genet. 2013, 92, 605–613. [Google Scholar] [CrossRef]
- Brodie, E.J.; Zhan, H.; Saiyed, T.; Truscott, K.N.; Dougan, D.A. Perrault syndrome type 3 caused by diverse molecular defects in CLPP. Sci. Rep. 2018, 8, 12862. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.H.; Rivadeneira, D.B.; Caino, M.C.; Chae, Y.C.; Speicher, D.W.; Tang, H.Y.; Vaira, V.; Bosari, S.; Palleschi, A.; Rampini, P.; et al. The Mitochondrial Unfoldase-Peptidase Complex ClpXP Controls Bioenergetics Stress and Metastasis. PLoS Biol. 2016, 14, e1002507. [Google Scholar] [CrossRef]
- Cole, A.; Wang, Z.; Coyaud, E.; Voisin, V.; Gronda, M.; Jitkova, Y.; Mattson, R.; Hurren, R.; Babovic, S.; Maclean, N.; et al. Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2015, 27, 864–876. [Google Scholar] [CrossRef]
- Cormio, A.; Musicco, C.; Gasparre, G.; Cormio, G.; Pesce, V.; Sardanelli, A.M.; Gadaleta, M.N. Increase in proteins involved in mitochondrial fission, mitophagy, proteolysis and antioxidant response in type I endometrial cancer as an adaptive response to respiratory complex I deficiency. Biochem. Biophys. Res. Commun. 2017, 491, 85–90. [Google Scholar] [CrossRef]
- Zhang, J.; Qiao, W.; Luo, Y. Mitochondrial quality control proteases and their modulation for cancer therapy. Med. Res. Rev. 2022, 43, 399–436. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Zeng, B.; Tao, C.; Lu, M.; Ren, G. ClpP regulates breast cancer cell proliferation, invasion and apoptosis by modulating the Src/PI3K/Akt signaling pathway. PeerJ 2020, 8, e8754. [Google Scholar] [CrossRef] [PubMed]
- Cormio, A.; Sanguedolce, F.; Pesce, V.; Musicco, C. Mitochondrial Caseinolytic Protease P: A Possible Novel Prognostic Marker and Therapeutic Target in Cancer. Int. J. Mol. Sci. 2021, 22, 6228. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.S.; Houry, W.A. Chemical Modulation of Human Mitochondrial ClpP: Potential Application in Cancer Therapeutics. ACS Chem. Biol. 2019, 14, 2349–2360. [Google Scholar] [CrossRef]
- Graves, P.R.; Aponte-Collazo, L.J.; Fennell, E.M.J.; Graves, A.C.; Hale, A.E.; Dicheva, N.; Herring, L.E.; Gilbert, T.S.K.; East, M.P.; McDonald, I.M.; et al. Mitochondrial Protease ClpP is a Target for the Anticancer Compounds ONC201 and Related Analogues. ACS Chem. Biol. 2019, 14, 1020–1029. [Google Scholar] [CrossRef]
- Greer, Y.E.; Porat-Shliom, N.; Nagashima, K.; Stuelten, C.; Crooks, D.; Koparde, V.N.; Gilbert, S.F.; Islam, C.; Ubaldini, A.; Ji, Y.; et al. ONC201 kills breast cancer cells in vitro by targeting mitochondria. Oncotarget 2018, 9, 18454–18479. [Google Scholar] [CrossRef]
- Fennell, E.M.J.; Aponte-Collazo, L.J.; Wynn, J.D.; Drizyte-Miller, K.; Leung, E.; Greer, Y.E.; Graves, P.R.; Iwanowicz, A.A.; Ashamalla, H.; Holmuhamedov, E.; et al. Characterization of TR-107, a novel chemical activator of the human mitochondrial protease ClpP. Pharmacol. Res. Perspect. 2022, 10, e00993. [Google Scholar] [CrossRef]
- Ishizawa, J.; Zarabi, S.F.; Davis, R.E.; Halgas, O.; Nii, T.; Jitkova, Y.; Zhao, R.; St-Germain, J.; Heese, L.E.; Egan, G.; et al. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell 2019, 35, 721–737.e729. [Google Scholar] [CrossRef]
- Nguyen, T.T.T.; Shang, E.; Schiffgens, S.; Torrini, C.; Shu, C.; Akman, H.O.; Prabhu, V.V.; Allen, J.E.; Westhoff, M.A.; Karpel-Massler, G.; et al. Induction of Synthetic Lethality by Activation of Mitochondrial ClpP and Inhibition of HDAC1/2 in Glioblastoma. Clin. Cancer Res. 2022, 28, 1881–1895. [Google Scholar] [CrossRef] [PubMed]
- Bhandary, L.; Bailey, P.C.; Chang, K.T.; Underwood, K.F.; Lee, C.J.; Whipple, R.A.; Jewell, C.M.; Ory, E.; Thompson, K.N.; Ju, J.A.; et al. Lipid tethering of breast tumor cells reduces cell aggregation during mammosphere formation. Sci. Rep. 2021, 11, 3214. [Google Scholar] [CrossRef] [PubMed]
- Brötz-Oesterhelt, H.; Beyer, D.; Kroll, H.P.; Endermann, R.; Ladel, C.; Schroeder, W.; Hinzen, B.; Raddatz, S.; Paulsen, H.; Henninger, K.; et al. Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat. Med. 2005, 11, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Cinos, C.; Goossens, K.; Salado, I.G.; Van Der Veken, P.; De Winter, H.; Augustyns, K. ClpP Protease, a Promising Antimicrobial Target. Int. J. Mol. Sci. 2019, 20, 2232. [Google Scholar] [CrossRef]
- Leung, E.; Datti, A.; Cossette, M.; Goodreid, J.; McCaw, S.E.; Mah, M.; Nakhamchik, A.; Ogata, K.; El Bakkouri, M.; Cheng, Y.Q.; et al. Activators of cylindrical proteases as antimicrobials: Identification and development of small molecule activators of ClpP protease. Chem. Biol. 2011, 18, 1167–1178. [Google Scholar] [CrossRef]
- Binepal, G.; Mabanglo, M.F.; Goodreid, J.D.; Leung, E.; Barghash, M.M.; Wong, K.S.; Lin, F.; Cossette, M.; Bansagi, J.; Song, B.; et al. Development of Antibiotics That Dysregulate the. ACS Infect. Dis. 2020, 6, 3224–3236. [Google Scholar] [CrossRef]
- Conlon, B.P.; Nakayasu, E.S.; Fleck, L.E.; LaFleur, M.D.; Isabella, V.M.; Coleman, K.; Leonard, S.N.; Smith, R.D.; Adkins, J.N.; Lewis, K. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 2013, 503, 365–370. [Google Scholar] [CrossRef]
- Socha, A.M.; Tan, N.Y.; LaPlante, K.L.; Sello, J.K. Diversity-oriented synthesis of cyclic acyldepsipeptides leads to the discovery of a potent antibacterial agent. Bioorg. Med. Chem. 2010, 18, 7193–7202. [Google Scholar] [CrossRef]
- Carney, D.W.; Schmitz, K.R.; Truong, J.V.; Sauer, R.T.; Sello, J.K. Restriction of the conformational dynamics of the cyclic acyldepsipeptide antibiotics improves their antibacterial activity. J. Am. Chem. Soc. 2014, 136, 1922–1929. [Google Scholar] [CrossRef]
- Goodreid, J.D.; Janetzko, J.; Santa Maria, J.P.; Wong, K.S.; Leung, E.; Eger, B.T.; Bryson, S.; Pai, E.F.; Gray-Owen, S.D.; Walker, S.; et al. Development and Characterization of Potent Cyclic Acyldepsipeptide Analogues with Increased Antimicrobial Activity. J. Med. Chem. 2016, 59, 624–646. [Google Scholar] [CrossRef]
- Hinzen, B.; Raddatz, S.; Paulsen, H.; Lampe, T.; Schumacher, A.; Häbich, D.; Hellwig, V.; Benet-Buchholz, J.; Endermann, R.; Labischinski, H.; et al. Medicinal chemistry optimization of acyldepsipeptides of the enopeptin class antibiotics. ChemMedChem 2006, 1, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.S.; Mabanglo, M.F.; Seraphim, T.V.; Mollica, A.; Mao, Y.Q.; Rizzolo, K.; Leung, E.; Moutaoufik, M.T.; Hoell, L.; Phanse, S.; et al. Acyldepsipeptide Analogs Dysregulate Human Mitochondrial ClpP Protease Activity and Cause Apoptotic Cell Death. Cell Chem. Biol. 2018, 25, 1017–1030.e1019. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.E.; Krigsfeld, G.; Mayes, P.A.; Patel, L.; Dicker, D.T.; Patel, A.S.; Dolloff, N.G.; Messaris, E.; Scata, K.A.; Wang, W.; et al. Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, TRAIL gene induction, and potent antitumor effects. Sci. Transl. Med. 2013, 5, 171ra117. [Google Scholar] [CrossRef]
- Stein, M.N.; Bertino, J.R.; Kaufman, H.L.; Mayer, T.; Moss, R.; Silk, A.; Chan, N.; Malhotra, J.; Rodriguez, L.; Aisner, J.; et al. First-in-Human Clinical Trial of Oral ONC201 in Patients with Refractory Solid Tumors. Clin. Cancer Res. 2017, 23, 4163–4169. [Google Scholar] [CrossRef]
- Kline, C.L.B.; Ralff, M.D.; Lulla, A.R.; Wagner, J.M.; Abbosh, P.H.; Dicker, D.T.; Allen, J.E.; El-Deiry, W.S. Role of Dopamine Receptors in the Anticancer Activity of ONC201. Neoplasia 2018, 20, 80–91. [Google Scholar] [CrossRef]
- Prabhu, V.V.; Morrow, S.; Rahman Kawakibi, A.; Zhou, L.; Ralff, M.; Ray, J.; Jhaveri, A.; Ferrarini, I.; Lee, Y.; Parker, C.; et al. ONC201 and imipridones: Anti-cancer compounds with clinical efficacy. Neoplasia 2020, 22, 725–744. [Google Scholar] [CrossRef]
- Chi, A.S.; Tarapore, R.S.; Hall, M.D.; Shonka, N.; Gardner, S.; Umemura, Y.; Sumrall, A.; Khatib, Z.; Mueller, S.; Kline, C.; et al. Pediatric and adult H3 K27M-mutant diffuse midline glioma treated with the selective DRD2 antagonist ONC201. J. Neurooncol. 2019, 145, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Odia, Y.; Allen, J.E.; Tarapore, R.; Khatib, Z.; Niazi, T.N.; Daghistani, D.; Schalop, L.; Chi, A.S.; Oster, W.; et al. First clinical experience with DRD2/3 antagonist ONC201 in H3 K27M-mutant pediatric diffuse intrinsic pontine glioma: A case report. J. Neurosurg. Pediatr. 2019, 23, 719–725. [Google Scholar] [CrossRef]
- Ni, X.; Zhang, X.; Hu, C.H.; Langridge, T.; Tarapore, R.S.; Allen, J.E.; Oster, W.; Duvic, M. ONC201 selectively induces apoptosis in cutaneous T-cell lymphoma cells via activating pro-apoptotic integrated stress response and inactivating JAK/STAT and NF-κB pathways. Oncotarget 2017, 8, 61761–61776. [Google Scholar] [CrossRef] [PubMed]
- Jacques, S.; van der Sloot, A.M.; C Huard, C.; Coulombe-Huntington, J.; Tsao, S.; Tollis, S.; Bertomeu, T.; Culp, E.J.; Pallant, D.; Cook, M.A.; et al. Imipridone Anticancer Compounds Ectopically Activate the ClpP Protease and Represent a New Scaffold for Antibiotic Development. Genetics 2020, 214, 1103–1120. [Google Scholar] [CrossRef] [PubMed]
- Przystal, J.M.; Cianciolo Cosentino, C.; Yadavilli, S.; Zhang, J.; Laternser, S.; Bonner, E.R.; Prasad, R.; Dawood, A.A.; Lobeto, N.; Chin Chong, W.; et al. Imipridones affect tumor bioenergetics and promote cell lineage differentiation in diffuse midline gliomas. Neuro. Oncol. 2022, 24, 1438–1451. [Google Scholar] [CrossRef] [PubMed]
- Mabanglo, M.F.; Houry, W.A. Recent structural insights into the mechanism of ClpP protease regulation by AAA+ chaperones and small molecules. J. Biol. Chem. 2022, 298, 101781. [Google Scholar] [CrossRef] [PubMed]
- Mabanglo, M.F.; Wong, K.S.; Barghash, M.M.; Leung, E.; Chuang, S.H.W.; Ardalan, A.; Majaesic, E.M.; Wong, C.J.; Zhang, S.; Lang, H.; et al. Potent ClpP agonists with anticancer properties bind with improved structural complementarity and alter the mitochondrial N-terminome. Structure 2023, 31, 185–200.e110. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Guo, P.; Gao, Y.; Shi, Q.; He, D.; Zhang, H. Acyldepsipeptides inhibit the growth of renal cancer cells through G1 phase cell cycle arrest. Biochem. Biophys. Res. Commun. 2013, 438, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.E.; Prabhu, V.V.; Talekar, M.; van den Heuvel, A.P.; Lim, B.; Dicker, D.T.; Fritz, J.L.; Beck, A.; El-Deiry, W.S. Genetic and Pharmacological Screens Converge in Identifying FLIP, BCL2, and IAP Proteins as Key Regulators of Sensitivity to the TRAIL-Inducing Anticancer Agent ONC201/TIC10. Cancer Res. 2015, 75, 1668–1674. [Google Scholar] [CrossRef]
- Kline, C.L.; Van den Heuvel, A.P.; Allen, J.E.; Prabhu, V.V.; Dicker, D.T.; El-Deiry, W.S. ONC201 kills solid tumor cells by triggering an integrated stress response dependent on ATF4 activation by specific eIF2α kinases. Sci. Signal. 2016, 9, ra18. [Google Scholar] [CrossRef]
- Yuan, X.; Kho, D.; Xu, J.; Gajan, A.; Wu, K.; Wu, G.S. ONC201 activates ER stress to inhibit the growth of triple-negative breast cancer cells. Oncotarget 2017, 8, 21626–21638. [Google Scholar] [CrossRef] [PubMed]
- Ralff, M.D.; Kline, C.L.B.; Küçükkase, O.C.; Wagner, J.; Lim, B.; Dicker, D.T.; Prabhu, V.V.; Oster, W.; El-Deiry, W.S. ONC201 Demonstrates Antitumor Effects in Both Triple-Negative and Non-Triple-Negative Breast Cancers through TRAIL-Dependent and TRAIL-Independent Mechanisms. Mol. Cancer Ther. 2017, 16, 1290–1298. [Google Scholar] [CrossRef]
- Wagner, J.; Kline, C.L.; Zhou, L.; Campbell, K.S.; MacFarlane, A.W.; Olszanski, A.J.; Cai, K.Q.; Hensley, H.H.; Ross, E.A.; Ralff, M.D.; et al. Dose intensification of TRAIL-inducing ONC201 inhibits metastasis and promotes intratumoral NK cell recruitment. J. Clin. Investig. 2018, 128, 2325–2338. [Google Scholar] [CrossRef]
- Ralff, M.D.; Jhaveri, A.; Ray, J.E.; Zhou, L.; Lev, A.; Campbell, K.S.; Dicker, D.T.; Ross, E.A.; El-Deiry, W.S. TRAIL receptor agonists convert the response of breast cancer cells to ONC201 from anti-proliferative to apoptotic. Oncotarget 2020, 11, 3753–3769. [Google Scholar] [CrossRef]
- Lim, B.; Peterson, C.B.; Davis, A.; Cho, E.; Pearson, T.; Liu, H.; Hwang, M.; Ueno, N.T.; Lee, J. ONC201 and an MEK Inhibitor Trametinib Synergistically Inhibit the Growth of Triple-Negative Breast Cancer Cells. Biomedicines 2021, 9, 1410. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, L.; Safran, H.; Borsuk, R.; Lulla, R.; Tapinos, N.; Seyhan, A.A.; El-Deiry, W.S. EZH2i EPZ-6438 and HDACi vorinostat synergize with ONC201/TIC10 to activate integrated stress response, DR5, reduce H3K27 methylation, ClpX and promote apoptosis of multiple tumor types including DIPG. Neoplasia 2021, 23, 792–810. [Google Scholar] [CrossRef] [PubMed]
- Mishukov, A.; Odinokova, I.; Mndlyan, E.; Kobyakova, M.; Abdullaev, S.; Zhalimov, V.; Glukhova, X.; Galat, V.; Galat, Y.; Senotov, A.; et al. ONC201-Induced Mitochondrial Dysfunction, Senescence-like Phenotype, and Sensitization of Cultured BT474 Human Breast Cancer Cells to TRAIL. Int. J. Mol. Sci. 2022, 23, 15551. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Kline, C.L.; Ralff, M.D.; Lev, A.; Lulla, A.; Zhou, L.; Olson, G.L.; Nallaganchu, B.R.; Benes, C.H.; Allen, J.E.; et al. Preclinical evaluation of the imipridone family, analogs of clinical stage anti-cancer small molecule ONC201, reveals potent anti-cancer effects of ONC212. Cell Cycle 2017, 16, 1790–1799. [Google Scholar] [CrossRef] [PubMed]
- Endo Greer, Y.; Lipkowitz, S. ONC201: Stressing tumors to death. Sci. Signal. 2016, 9, fs1. [Google Scholar] [CrossRef] [PubMed]
- Madka, V.; De La Cruz, A.; Pathuri, G.; Panneerselvam, J.; Zhang, Y.; Stratton, N.; Hacking, S.; Finnberg, N.K.; Safran, H.P.; Sei, S.; et al. Oral administration of TRAIL-inducing small molecule ONC201/TIC10 prevents intestinal polyposis in the. Am. J. Cancer Res. 2022, 12, 2118–2131. [Google Scholar] [PubMed]
- Ishizawa, J.; Kojima, K.; Chachad, D.; Ruvolo, P.; Ruvolo, V.; Jacamo, R.O.; Borthakur, G.; Mu, H.; Zeng, Z.; Tabe, Y.; et al. ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Sci. Signal. 2016, 9, ra17. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, V.V.; Talekar, M.K.; Lulla, A.R.; Kline, C.L.B.; Zhou, L.; Hall, J.; Van den Heuvel, A.P.J.; Dicker, D.T.; Babar, J.; Grupp, S.A.; et al. Single agent and synergistic combinatorial efficacy of first-in-class small molecule imipridone ONC201 in hematological malignancies. Cell Cycle 2018, 17, 468–478. [Google Scholar] [CrossRef]
- Aponte-Collazo, L.J.; Fennell, E.M.J.; East, M.P.; Gilbert, T.S.K.; Graves, P.R.; Ashamalla, H.; Iwanowicz, E.J.; Greer, Y.E.; Lipkowitz, S.; Graves, L.M. Small molecule clpp agonists induce senescence and alter trail-mediated apoptotic response of triple-negative breast cancer cells. bioRxiv 2022. [Google Scholar] [CrossRef]
- Liguori, N.R.; Sanchez Sevilla Uruchurtu, A.; Zhang, L.; Abbas, A.E.; Lee, Y.S.; Zhou, L.; Azzoli, C.G.; El-Deiry, W.S. Preclinical studies with ONC201/TIC10 and lurbinectedin as a novel combination therapy in small cell lung cancer (SCLC). Am. J. Cancer Res. 2022, 12, 729–743. [Google Scholar]
- Nii, T.; Prabhu, V.V.; Ruvolo, V.; Madhukar, N.; Zhao, R.; Mu, H.; Heese, L.; Nishida, Y.; Kojima, K.; Garnett, M.J.; et al. Imipridone ONC212 activates orphan G protein-coupled receptor GPR132 and integrated stress response in acute myeloid leukemia. Leukemia 2019, 33, 2805–2816. [Google Scholar] [CrossRef]
- Yeung CL, A.; Hu, W.; Ferri-Borgogno, S.; Tarapore, R.S.; Allen, J.E.; Lu, K.H.; Mok, S.C. Abstract 1440: Novel imipridone ONC206 suppresses ovarian cancer progression through modulating immune cell response 2021. Cancer Res. 2021, 81, 1440. [Google Scholar] [CrossRef]
- Stein, M.N.; Malhotra, J.; Tarapore, R.S.; Malhotra, U.; Silk, A.W.; Chan, N.; Rodriguez, L.; Aisner, J.; Aiken, R.D.; Mayer, T.; et al. Safety and enhanced immunostimulatory activity of the DRD2 antagonist ONC201 in advanced solid tumor patients with weekly oral administration. J. Immunother. Cancer 2019, 7, 136. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, V.V.; Lulla, A.R.; Madhukar, N.S.; Ralff, M.D.; Zhao, D.; Kline, C.L.B.; Van den Heuvel, A.P.J.; Lev, A.; Garnett, M.J.; McDermott, U.; et al. Cancer stem cell-related gene expression as a potential biomarker of response for first-in-class imipridone ONC201 in solid tumors. PLoS ONE 2017, 12, e0180541. [Google Scholar] [CrossRef]
- Ishida, C.T.; Zhang, Y.; Bianchetti, E.; Shu, C.; Nguyen, T.T.T.; Kleiner, G.; Sanchez-Quintero, M.J.; Quinzii, C.M.; Westhoff, M.A.; Karpel-Massler, G.; et al. Metabolic Reprogramming by Dual AKT/ERK Inhibition through Imipridones Elicits Unique Vulnerabilities in Glioblastoma. Clin. Cancer Res. 2018, 24, 5392–5406. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.H.; Trikalinos, N.A. Neuroendocrine and Rare Tumor Advances: A New and Promising TRAIL Emerges. Clin. Cancer Res. 2022, 28, 1748–1750. [Google Scholar] [CrossRef] [PubMed]
- Ferrarini, I.; Louie, A.; Zhou, L.; El-Deiry, W.S. ONC212 is a Novel Mitocan Acting Synergistically with Glycolysis Inhibition in Pancreatic Cancer. Mol. Cancer Ther. 2021, 20, 1572–1583. [Google Scholar] [CrossRef]
- Hubens, W.H.G.; Vallbona-Garcia, A.; de Coo, I.F.M.; van Tienen, F.H.J.; Webers, C.A.B.; Smeets, H.J.M.; Gorgels, T.G.M.F. Blood biomarkers for assessment of mitochondrial dysfunction: An expert review. Mitochondrion 2022, 62, 187–204. [Google Scholar] [CrossRef]
- Suomalainen, A.; Elo, J.M.; Pietiläinen, K.H.; Hakonen, A.H.; Sevastianova, K.; Korpela, M.; Isohanni, P.; Marjavaara, S.K.; Tyni, T.; Kiuru-Enari, S.; et al. FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: A diagnostic study. Lancet Neurol. 2011, 10, 806–818. [Google Scholar] [CrossRef]
- Ji, X.; Zhao, L.; Ji, K.; Zhao, Y.; Li, W.; Zhang, R.; Hou, Y.; Lu, J.; Yan, C. Growth Differentiation Factor 15 Is a Novel Diagnostic Biomarker of Mitochondrial Diseases. Mol. Neurobiol. 2017, 54, 8110–8116. [Google Scholar] [CrossRef]
- Greer, Y.E.; Gilbert, S.F.; Gril, B.; Narwal, R.; Peacock Brooks, D.L.; Tice, D.A.; Steeg, P.S.; Lipkowitz, S. MEDI3039, a novel highly potent tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) receptor 2 agonist, causes regression of orthotopic tumors and inhibits outgrowth of metastatic triple-negative breast cancer. Breast Cancer Res. 2019, 21, 27. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Davis, S.R.; Pumphrey, J.G.; Bao, J.; Nau, M.M.; Meltzer, P.S.; Lipkowitz, S. TRAIL induces apoptosis in triple-negative breast cancer cells with a mesenchymal phenotype. Breast Cancer Res. Treat. 2009, 113, 217–230. [Google Scholar] [CrossRef]
- Lev, A.; Lulla, A.R.; Wagner, J.; Ralff, M.D.; Kiehl, J.B.; Zhou, Y.; Benes, C.H.; Prabhu, V.V.; Oster, W.; Astsaturov, I.; et al. Anti-pancreatic cancer activity of ONC212 involves the unfolded protein response (UPR) and is reduced by IGF1-R and GRP78/BIP. Oncotarget 2017, 8, 81776–81793. [Google Scholar] [CrossRef] [PubMed]
- Gillissen, B.; Wendt, J.; Richter, A.; Müer, A.; Overkamp, T.; Gebhardt, N.; Preissner, R.; Belka, C.; Dörken, B.; Daniel, P.T. Endogenous Bak inhibitors Mcl-1 and Bcl-xL: Differential impact on TRAIL resistance in Bax-deficient carcinoma. J. Cell Biol. 2010, 188, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Finlay, D.; Vamos, M.; González-López, M.; Ardecky, R.J.; Ganji, S.R.; Yuan, H.; Su, Y.; Cooley, T.R.; Hauser, C.T.; Welsh, K.; et al. Small-molecule IAP antagonists sensitize cancer cells to TRAIL-induced apoptosis: Roles of XIAP and cIAPs. Mol. Cancer Ther. 2014, 13, 5–15. [Google Scholar] [CrossRef]
- Cortesi, L.; Rugo, H.S.; Jackisch, C. An Overview of PARP Inhibitors for the Treatment of Breast Cancer. Target Oncol. 2021, 16, 255–282. [Google Scholar] [CrossRef]
- Baumeister, M.D.; Küçükkase, O.C.; Prabhu, V.V.; Dicker, D.T.; Allen, J.E.; El-Deiry, W.S. Abstract 3212: ONC201 shows efficacy in BRCA-deficient cancer cells and synergy with PARP inhibitors in glioblastoma, breast, prostate, and ovarian cancers. Cancer Res. 2017, 77, 3212. [Google Scholar] [CrossRef]
- Yomtoubian, S.; Lee, S.B.; Verma, A.; Izzo, F.; Markowitz, G.; Choi, H.; Cerchietti, L.; Vahdat, L.; Brown, K.A.; Andreopoulou, E.; et al. Inhibition of EZH2 Catalytic Activity Selectively Targets a Metastatic Subpopulation in Triple-Negative Breast Cancer. Cell Rep. 2020, 30, 755–770.e756. [Google Scholar] [CrossRef]
- Wawruszak, A.; Borkiewicz, L.; Okon, E.; Kukula-Koch, W.; Afshan, S.; Halasa, M. Vorinostat (SAHA) and Breast Cancer: An Overview. Cancers 2021, 13, 4700. [Google Scholar] [CrossRef]
- Borsuk, R.; Zhou, L.; Chang, W.I.; Zhang, Y.; Sharma, A.; Prabhu, V.V.; Tapinos, N.; Lulla, R.R.; El-Deiry, W.S. Potent preclinical sensitivity to imipridone-based combination therapies in oncohistone H3K27M-mutant diffuse intrinsic pontine glioma is associated with induction of the integrated stress response, TRAIL death receptor DR5, reduced ClpX and apoptosis. Am. J. Cancer Res. 2021, 11, 4607–4623. [Google Scholar]
- Bosc, C.; Saland, E.; Bousard, A.; Gadaud, N.; Sabatier, M.; Cognet, G.; Farge, T.; Boet, E.; Gotanègre, M.; Aroua, N.; et al. Mitochondrial inhibitors circumvent adaptive resistance to venetoclax and cytarabine combination therapy in acute myeloid leukemia. Nat. Cancer 2021, 2, 1204–1223. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A. Breast Cancer Immunotherapy: Facts and Hopes. Clin. Cancer Res. 2018, 24, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Chiossone, L.; Dumas, P.Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef]
- Hodgins, J.J.; Khan, S.T.; Park, M.M.; Auer, R.C.; Ardolino, M. Killers 2.0: NK cell therapies at the forefront of cancer control. J. Clin. Investig. 2019, 129, 3499–3510. [Google Scholar] [CrossRef] [PubMed]
- Verma, C.; Kaewkangsadan, V.; Eremin, J.M.; Cowley, G.P.; Ilyas, M.; El-Sheemy, M.A.; Eremin, O. Natural killer (NK) cell profiles in blood and tumour in women with large and locally advanced breast cancer (LLABC) and their contribution to a pathological complete response (PCR) in the tumour following neoadjuvant chemotherapy (NAC): Differential restoration of blood profiles by NAC and surgery. J. Transl. Med. 2015, 13, 180. [Google Scholar] [CrossRef]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.H.; Jones, R.G. Fueling immunity: Insights into metabolism and lymphocyte function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef]
- van der Windt, G.J.; Pearce, E.L. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol. Rev. 2012, 249, 27–42. [Google Scholar] [CrossRef]
- Rangel Rivera, G.O.; Knochelmann, H.M.; Dwyer, C.J.; Smith, A.S.; Wyatt, M.M.; Rivera-Reyes, A.M.; Thaxton, J.E.; Paulos, C.M. Fundamentals of T Cell Metabolism and Strategies to Enhance Cancer Immunotherapy. Front. Immunol. 2021, 12, 645242. [Google Scholar] [CrossRef]
- Elia, I.; Haigis, M.C. Metabolites and the tumour microenvironment: From cellular mechanisms to systemic metabolism. Nat. Metab. 2021, 3, 21–32. [Google Scholar] [CrossRef]
- Surace, L.; Doisne, J.M.; Escoll, P.; Marie, S.; Dardalhon, V.; Croft, C.; Thaller, A.; Topazio, D.; Sparaneo, A.; Cama, A.; et al. Polarized mitochondria as guardians of NK cell fitness. Blood Adv. 2021, 5, 26–38. [Google Scholar] [CrossRef]
- Wu, S.Y.; Fu, T.; Jiang, Y.Z.; Shao, Z.M. Natural killer cells in cancer biology and therapy. Mol. Cancer 2020, 19, 120. [Google Scholar] [CrossRef]
- Jedlička, M.; Feglarová, T.; Janstová, L.; Hortová-Kohoutková, M.; Frič, J. Lactate from the tumor microenvironment—A key obstacle in NK cell-based immunotherapies. Front. Immunol. 2022, 13, 932055. [Google Scholar] [CrossRef] [PubMed]
- Hayes, C.; Donohoe, C.L.; Davern, M.; Donlon, N.E. The oncogenic and clinical implications of lactate induced immunosuppression in the tumour microenvironment. Cancer Lett. 2021, 500, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Myers, C.R.; Wang, Y.; You, M. Mitochondria as a Novel Target for Cancer Chemoprevention: Emergence of Mitochondrial-targeting Agents. Cancer Prev. Res. 2021, 14, 285–306. [Google Scholar] [CrossRef]
- Kafkova, A.; Trnka, J. Mitochondria-targeted compounds in the treatment of cancer. Neoplasma 2020, 67, 450–460. [Google Scholar] [CrossRef]
- Dong, L.; Gopalan, V.; Holland, O.; Neuzil, J. Mitocans Revisited: Mitochondrial Targeting as Efficient Anti-cancer Therapy. Int. J. Mol. Sci. 2020, 21, 7941. [Google Scholar] [CrossRef]
- Sandoval-Acuña, C.; Fuentes-Retamal, S.; Guzmán-Rivera, D.; Peredo-Silva, L.; Madrid-Rojas, M.; Rebolledo, S.; Castro-Castillo, V.; Pavani, M.; Catalán, M.; Maya, J.D.; et al. Destabilization of mitochondrial functions as a target against breast cancer progression: Role of TPP + -linked-polyhydroxybenzoates. Toxicol. Appl. Pharmacol. 2016, 309, 2–14. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Ózsvári, B.; Sotgia, F.; Lisanti, M.P. Dodecyl-TPP Targets Mitochondria and Potently Eradicates Cancer Stem Cells (CSCs): Synergy With FDA-Approved Drugs and Natural Compounds (Vitamin C and Berberine). Front. Oncol. 2019, 9, 615. [Google Scholar] [CrossRef]
- Fuentes-Retamal, S.; Sandoval-Acuña, C.; Peredo-Silva, L.; Guzmán-Rivera, D.; Pavani, M.; Torrealba, N.; Truksa, J.; Castro-Castillo, V.; Catalán, M.; Kemmerling, U.; et al. Complex Mitochondrial Dysfunction Induced by TPP+-Gentisic Acid and Mitochondrial Translation Inhibition by Doxycycline Evokes Synergistic Lethality in Breast Cancer Cells. Cells 2020, 9, 407. [Google Scholar] [CrossRef]
- Gazzano, E.; Lazzarato, L.; Rolando, B.; Kopecka, J.; Guglielmo, S.; Costamagna, C.; Chegaev, K.; Riganti, C. Mitochondrial Delivery of Phenol Substructure Triggers Mitochondrial Depolarization and Apoptosis of Cancer Cells. Front. Pharmacol. 2018, 9, 580. [Google Scholar] [CrossRef]
- Ózsvári, B.; Sotgia, F.; Lisanti, M.P. First-in-class candidate therapeutics that target mitochondria and effectively prevent cancer cell metastasis: Mitoriboscins and TPP compounds. Aging 2020, 12, 10162–10179. [Google Scholar] [CrossRef] [PubMed]
- Rohlenova, K.; Sachaphibulkij, K.; Stursa, J.; Bezawork-Geleta, A.; Blecha, J.; Endaya, B.; Werner, L.; Cerny, J.; Zobalova, R.; Goodwin, J.; et al. Selective Disruption of Respiratory Supercomplexes as a New Strategy to Suppress Her2high Breast Cancer. Antioxid. Redox Signal. 2017, 26, 84–103. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Daver, N.; Mahendra, M.; Zhang, J.; Kamiya-Matsuoka, C.; Meric-Bernstam, F.; Kantarjian, H.M.; Ravandi, F.; Collins, M.E.; Di Francesco, M.E.; et al. Complex I inhibitor of oxidative phosphorylation in advanced solid tumors and acute myeloid leukemia: Phase I trials. Nat. Med. 2023, 29, 115–126. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.; Ye, Z.; Townsend, D.M.; Tew, K.D. Pharmacology of ME-344, a novel cytotoxic isoflavone. Adv. Cancer Res. 2019, 142, 187–207. [Google Scholar] [CrossRef] [PubMed]
- Quintela-Fandino, M.; Morales, S.; Cortés-Salgado, A.; Manso, L.; Apala, J.V.; Muñoz, M.; Cudos, A.G.; Fortuny, J.S.; Gion, M.; Lopez-Alonso, A.; et al. Randomized Phase 0/I Trial of the Mitochondrial Inhibitor ME-344 or Placebo Added to Bevacizumab in Early HER2-Negative Breast Cancer. Clin. Cancer Res. 2020, 26, 35–45. [Google Scholar] [CrossRef]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348 Pt 3, 607–614. [Google Scholar] [PubMed]
- Degli Esposti, M. Inhibitors of NADH–ubiquinone reductase: An overview. Biochim. Biophys. Acta (BBA)-Bioenerg. 1998, 1364, 222–235. [Google Scholar] [CrossRef]
- Evans, J.M.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. Br. Med. J. 2005, 330, 1304–1305. [Google Scholar] [CrossRef]
- Shi, P.; Liu, W.; Tala; Wang, H.; Li, F.; Zhang, H.; Wu, Y.; Kong, Y.; Zhou, Z.; Wang, C.; et al. Metformin suppresses triple-negative breast cancer stem cells by targeting KLF5 for degradation. Cell Discov. 2017, 3, 17010. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin Selectively Targets Cancer Stem Cells, and Acts Together with Chemotherapy to Block Tumor Growth and Prolong Remission. Cancer Res 2009, 69, 7507–7511. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fan, Z.; Edgerton, S.M.; Deng, X.-S.; Alimova, I.N.; Lind, S.E.; Thor, A.D. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle 2009, 8, 2031–2040. [Google Scholar] [CrossRef]
- Wang, J.-C.; Li, G.-Y.; Wang, B.; Han, S.-X.; Sun, X.; Jiang, Y.-N.; Shen, Y.-W.; Zhou, C.; Feng, J.; Lu, S.-Y.; et al. Metformin inhibits metastatic breast cancer progression and improves chemosensitivity by inducing vessel normalization via PDGF-B downregulation. J. Exp. Clin. Cancer Res. 2019, 38, 235. [Google Scholar] [CrossRef] [PubMed]
- Barakat, H.E.; Hussein, R.R.S.; Elberry, A.A.; Zaki, M.A.; Ramadan, M.E. The impact of metformin use on the outcomes of locally advanced breast cancer patients receiving neoadjuvant chemotherapy: An open-labelled randomized controlled trial. Sci. Rep. 2022, 12, 1–16. [Google Scholar] [CrossRef]
- Pimentel, I.; Lohmann, A.E.; Ennis, M.; Dowling, R.J.O.; Cescon, D.; Elser, C.; Potvin, K.R.; Haq, R.; Hamm, C.; Chang, M.C.; et al. A phase II randomized clinical trial of the effect of metformin versus placebo on progression-free survival in women with metastatic breast cancer receiving standard chemotherapy. Breast 2019, 48, 17–23. [Google Scholar] [CrossRef]
- Goodwin, P.J.; Chen, B.E.; Gelmon, K.A.; Whelan, T.J.; Ennis, M.; Lemieux, J.; Ligibel, J.A.; Hershman, D.L.; Mayer, I.A.; Hobday, T.J.; et al. Effect of Metformin vs Placebo on Invasive Disease–Free Survival in Patients With Breast Cancer. JAMA 2022, 327, 1963–1973. [Google Scholar] [CrossRef]
- Cejuela, M.; Martin-Castillo, B.; Menendez, J.A.; Pernas, S. Metformin and Breast Cancer: Where Are We Now? Int. J. Mol. Sci. 2022, 23, 2705. [Google Scholar] [CrossRef]
- Fontaine, E. Metformin and respiratory chain complex I: The last piece of the puzzle? Biochem. J. 2014, 463, e3–e5. [Google Scholar] [CrossRef]
- Pernicova, I.; Korbonits, M. Metformin—mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2014, 10, 143–156. [Google Scholar] [CrossRef]
- Van Der Heijden, J.W.; Oerlemans, R.; Tak, P.P.; Assaraf, Y.G.; Kraan, M.C.; Scheffer, G.L.; Van Der Laken, C.J.; Lems, W.F.; Scheper, R.J.; Dijkmans, B.A.C.; et al. Involvement of breast cancer resistance protein expression on rheumatoid arthritis synovial tissue macrophages in resistance to methotrexate and leflunomide. Arthritis Rheum. 2009, 60, 669–677. [Google Scholar] [CrossRef]
- Córdova-Delgado, M.; Fuentes-Retamal, S.; Palominos, C.; López-Torres, C.; Guzmán-Rivera, D.; Ramírez-Rodríguez, O.; Araya-Maturana, R.; Urra, F.A. FRI-1 Is an Anti-Cancer Isoquinolinequinone That Inhibits the Mitochondrial Bioenergetics and Blocks Metabolic Shifts by Redox Disruption in Breast Cancer Cells. Antioxidants 2021, 10, 1618. [Google Scholar] [CrossRef]
- Alistar, A.; Morris, B.B.; Desnoyer, R.; Klepin, H.D.; Hosseinzadeh, K.; Clark, C.; Cameron, A.; Leyendecker, J.; D’Agostino, R.; Topaloglu, U.; et al. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: A single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2017, 18, 770–778. [Google Scholar] [CrossRef] [PubMed]
- A Philip, P.; E Buyse, M.; Alistar, A.T.; Lima, C.M.R.; Luther, S.; Pardee, T.S.; Van Cutsem, E. A Phase III open-label trial to evaluate efficacy and safety of CPI-613 plus modified FOLFIRINOX (mFFX) versus FOLFIRINOX (FFX) in patients with metastatic adenocarcinoma of the pancreas. Futur. Oncol. 2019, 15, 3189–3196. [Google Scholar] [CrossRef]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef]
- Falchook, G.; Infante, J.; Arkenau, H.-T.; Patel, M.R.; Dean, E.; Borazanci, E.; Brenner, A.; Cook, N.; Lopez, J.; Pant, S.; et al. First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors. Eclinicalmedicine 2021, 34, 100797. [Google Scholar] [CrossRef]
- Velez, B.C.; Petrella, C.P.; DiSalvo, K.H.; Cheng, K.; Kravtsov, R.; Krasniqi, D.; Krucher, N.A. Combined inhibition of ACLY and CDK4/6 reduces cancer cell growth and invasion. Oncol. Rep. 2022, 49, 1–11. [Google Scholar] [CrossRef]
- Wang, C.J.; Li, D.; Danielson, J.A.; Zhang, E.H.; Dong, Z.; Miller, K.D.; Li, L.; Zhang, J.-T.; Liu, J.-Y. Proton pump inhibitors suppress DNA damage repair and sensitize treatment resistance in breast cancer by targeting fatty acid synthase. Cancer Lett. 2021, 509, 1–12. [Google Scholar] [CrossRef]
- Lee, E.Y.; Gong, E.-Y.; Shin, J.-S.; Moon, J.-H.; Shim, H.J.; Kim, S.-M.; Lee, S.; Jeong, J.; Gong, J.H.; Kim, M.J.; et al. Human breast cancer cells display different sensitivities to ABT-263 based on the level of survivin. Toxicol. Vitr. 2018, 46, 229–236. [Google Scholar] [CrossRef]
- Lee, A.; Jin, H.-O.; Haque, M.; Kim, H.Y.; Jung, H.; Park, J.H.; Kim, I.; Song, J.Y.; Yoon, H.K.; Kim, H.K.; et al. Synergism of a novel MCL-1 downregulator, acriflavine, with navitoclax (ABT-263) in triple-negative breast cancer, lung adenocarcinoma and glioblastoma multiforme. Int. J. Oncol. 2021, 60, 1–12. [Google Scholar] [CrossRef]
- Zoeller, J.J.; Vagodny, A.; Daniels, V.W.; Taneja, K.; Tan, B.Y.; DeRose, Y.S.; Fujita, M.; Welm, A.L.; Letai, A.; Leverson, J.D.; et al. Navitoclax enhances the effectiveness of EGFR-targeted antibody-drug conjugates in PDX models of EGFR-expressing triple-negative breast cancer. Breast Cancer Res. 2020, 22, 1–13. [Google Scholar] [CrossRef]
- Tiwari, S.; Kaur, H.; Anees, M.; Gupta, P.; Dalela, M.; Kharbanda, S.; Singh, H. Co-encapsulation of PI3-Kδ/HDAC6 dual inhibitor and Navitoclax in Quatramer™ nanoparticles for synergistic effect in ER+ breast cancer. Int. J. Pharm. 2022, 628. [Google Scholar] [CrossRef]
- Vivo-Llorca, G.; Candela-Noguera, V.; Alfonso, M.; García-Fernández, A.; Orzáez, M.; Sancenón, F.; Martínez-Máñez, R. MUC1 Aptamer-Capped Mesoporous Silica Nanoparticles for Navitoclax Resistance Overcoming in Triple-Negative Breast Cancer. Chem.–A Eur. J. 2020, 26, 16318–16327. [Google Scholar] [CrossRef]
- Pesch, A.M.; Chandler, B.C.; Michmerhuizen, A.R.; Carter, H.M.; Hirsh, N.H.; Wilder-Romans, K.; Liu, M.; Ward, T.; Ritter, C.L.; Nino, C.A.; et al. Bcl-xL Inhibition Radiosensitizes PIK3CA/PTEN Wild-type Triple-negative Breast Cancers with Low Mcl-1 Expression. Cancer Res. Commun. 2022, 2, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Zoeller, J.J.; Vagodny, A.; Taneja, K.; Tan, B.Y.; O’Brien, N.; Slamon, D.J.; Sampath, D.; Leverson, J.D.; Bronson, R.T.; Dillon, D.A.; et al. Neutralization of BCL-2/XL Enhances the Cytotoxicity of T-DM1 In Vivo. Mol. Cancer Ther. 2019, 18, 1115–1126. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, L.; Camidge, D.R.; de Oliveira, M.R.; Bonomi, P.; Gandara, D.; Khaira, D.; Hann, C.L.; McKeegan, E.M.; Litvinovich, E.; Hemken, P.M.; et al. Phase I Study of Navitoclax (ABT-263), a Novel Bcl-2 Family Inhibitor, in Patients With Small-Cell Lung Cancer and Other Solid Tumors. J. Clin. Oncol. 2011, 29, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Lindeman, G.J.; Fernando, T.M.; Bowen, R.; Jerzak, K.J.; Song, X.; Decker, T.; Boyle, F.; McCune, S.; Armstrong, A.; Shannon, C.; et al. VERONICA: Randomized Phase II Study of Fulvestrant and Venetoclax in ER-Positive Metastatic Breast Cancer Post-CDK4/6 Inhibitors – Efficacy, Safety, and Biomarker Results. Clin. Cancer Res. 2022, 28, 3256–3267. [Google Scholar] [CrossRef]
- Muttiah, C.; Whittle, J.R.; Oakman, C.; Lindeman, G.J. PALVEN: Phase Ib trial of palbociclib, letrozole and venetoclax in estrogen receptor- and BCL2-positive advanced breast cancer. Futur. Oncol. 2022, 18, 1805–1816. [Google Scholar] [CrossRef]
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Le Toumelin-Braizat, G.; Chanrion, M.; Kelly, G.L.; Gong, J.-N.; Moujalled, D.M.; et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef]
- Merino, D.; Whittle, J.R.; Vaillant, F.; Serrano, A.; Gong, J.-N.; Giner, G.; Maragno, A.L.; Chanrion, M.; Schneider, E.; Pal, B.; et al. Synergistic action of the MCL-1 inhibitor S63845 with current therapies in preclinical models of triple-negative and HER2-amplified breast cancer. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Lee, T.; Christov, P.P.; Shaw, S.; Tarr, J.C.; Zhao, B.; Veerasamy, N.; Jeon, K.O.; Mills, J.J.; Bian, Z.; Sensintaffar, J.L.; et al. Discovery of Potent Myeloid Cell Leukemia-1 (Mcl-1) Inhibitors That Demonstrate in Vivo Activity in Mouse Xenograft Models of Human Cancer. J. Med. Chem. 2019, 62, 3971–3988. [Google Scholar] [CrossRef]
- Campbell, K.J.; Mason, S.M.; Winder, M.L.; Willemsen, R.B.E.; Cloix, C.; Lawson, H.; Rooney, N.; Dhayade, S.; Sims, A.H.; Blyth, K.; et al. Breast cancer dependence on MCL-1 is due to its canonical anti-apoptotic function. Cell Death Differ. 2021, 28, 2589–2600. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xu, L.; Zhang, F.; Vlashi, E. Doxycycline inhibits the cancer stem cell phenotype and epithelial-to-mesenchymal transition in breast cancer. Cell Cycle 2017, 16, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Ózsvári, B.; Magalhães, L.G.; Latimer, J.; Kangasmetsa, J.; Sotgia, F.; Lisanti, M.P. A Myristoyl Amide Derivative of Doxycycline Potently Targets Cancer Stem Cells (CSCs) and Prevents Spontaneous Metastasis, Without Retaining Antibiotic Activity. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Massó-Vallés, D.; Beaulieu, M.-E.; Jauset, T.; Giuntini, F.; Zacarías-Fluck, M.F.; Foradada, L.; Martínez-Martín, S.; Serrano, E.; Martín-Fernández, G.; Casacuberta-Serra, S.; et al. MYC Inhibition Halts Metastatic Breast Cancer Progression by Blocking Growth, Invasion, and Seeding. Cancer Res. Commun. 2022, 2, 110–130. [Google Scholar] [CrossRef]
- Sainero-Alcolado, L.; Liaño-Pons, J.; Ruiz-Pérez, M.V.; Arsenian-Henriksson, M. Targeting mitochondrial metabolism for precision medicine in cancer. Cell Death Differ. 2022, 29, 1304–1317. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2021, 21, 141–162. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).