
Empowering the Potential of CAR-T Cell Immunotherapies by Epigenetic Reprogramming
 and
and
Abstract
Simple Summary
Abstract
1. Introduction
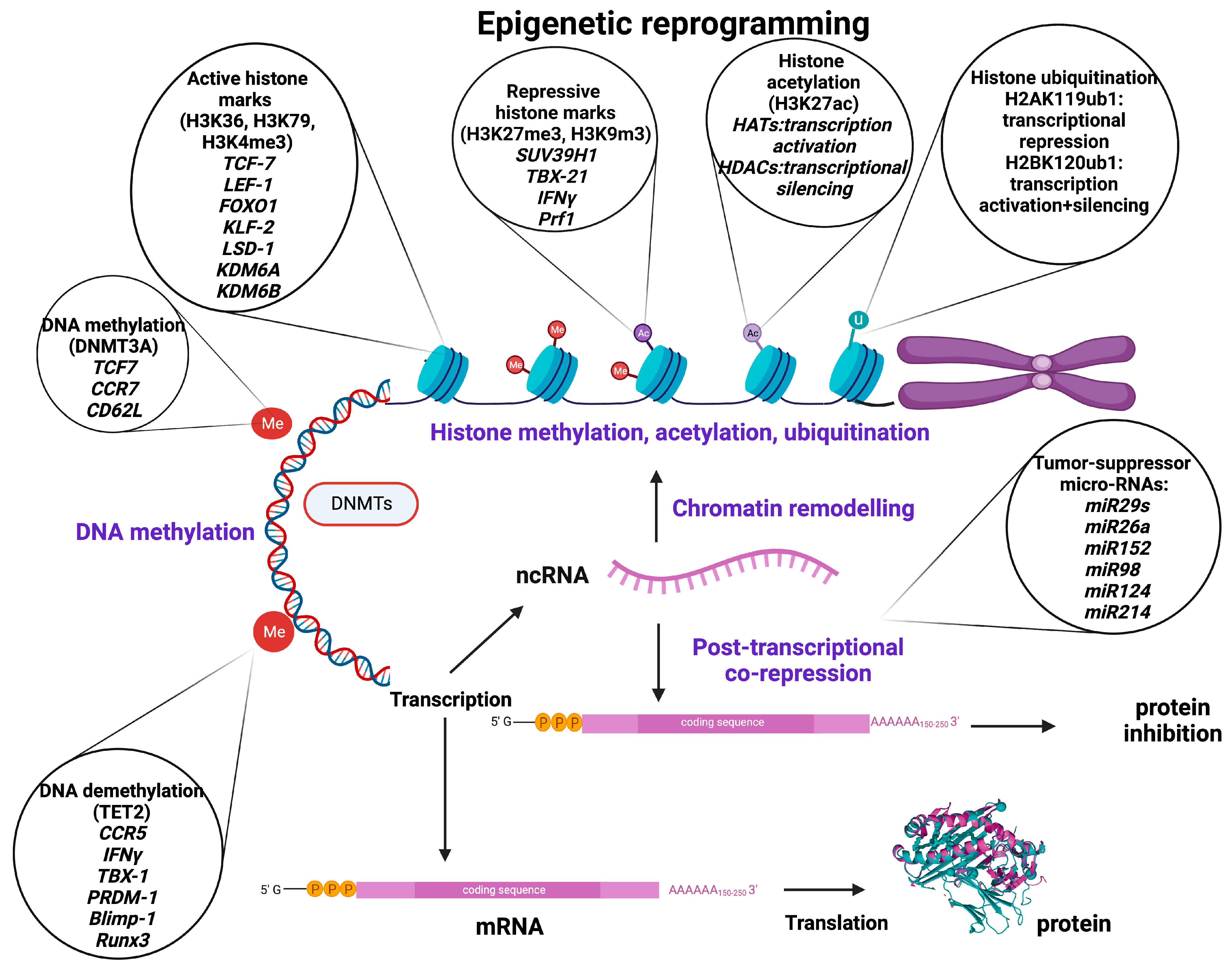
2. Epigenetic Modifications
2.1. DNA Modifications
2.2. Histone Modifications
2.3. Non-Coding RNA-Associated Modifications
2.4. Epigenome Editing
3. Epigenetic Reprogramming to Circumvent Challenges of CAR-T Cell In Vivo Performance
3.1. T-Cell Stemness and Memory
3.1.1. Epigenetic Regulation of T Cell Differentiation
3.1.2. Targeting Epigenetic Programs to Enhance CAR-Ts’ Long-Term Fate
3.2. Exhaustion
3.2.1. Epigenetic Regulation of T-Cell Exhaustion
3.2.2. Targeting Epigenetic Programs to Overcome CAR-Ts’ Exhaustion
3.3. The Tumor Microenvironment
3.3.1. Epigenetic Regulation of T-Cell Infiltration
3.3.2. Cell Metabolism and Epigenetics
3.3.3. Targeting Epigenetic Programs to Alter the TME and Enhance CAR-Ts’ Infiltration
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Maus, M.V.; Grupp, S.A.; Porter, D.L.; June, C.H. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood 2014, 123, 2625–2635. [Google Scholar] [CrossRef]
- Halim, L.; Maher, J. CAR T-cell immunotherapy of B-cell malignancy : The story so far. Ther. Adv. Vaccines Immunother. 2020, 8, 2515135520927164. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. FDA approves axicabtagene ciloleucel for large B-cell lymphoma. Case Med. Res. 2017, 1–2. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome. Case Med. Res. 2017, 1–2. [Google Scholar] [CrossRef]
- European Medicines Agency. First Two CAR-T Cell Medicines Recommended for Approval in the European Union; European Medicines Agency: Amsterdam, The Netherlands, 2018; Volume 44, pp. 1–4. [Google Scholar]
- Turtle, C.J.; Hay, K.A.; Hanafi, L.A.; Li, D.; Cherian, S.; Chen, X.; Wood, B.; Lozanski, A.; Byrd, J.C.; Heimfeld, S.; et al. Durable molecular remissions in chronic lymphocytic leukemia treated with CD19-Specific chimeric antigen Receptor-modified T cells after failure of ibrutinib. J. Clin. Oncol. 2017, 35, 3010–3020. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef]
- D’Aloia, M.M.; Zizzari, I.G.; Sacchetti, B.; Pierelli, L.; Alimandi, M. CAR-T cells: The long and winding road to solid tumors review-article. Cell. Death Dis. 2018, 9, 282. [Google Scholar] [CrossRef]
- Long, K.B.; Young, R.M.; Boesteanu, A.C.; Davis, M.M.; Melenhorst, J.J.; Lacey, S.F.; DeGaramo, D.A.; Levine, B.L.; Fraietta, J.A. CAR T Cell Therapy of Non-hematopoietic Malignancies: Detours on the Road to Clinical Success. Front. Immunol. 2018, 9, 2740. [Google Scholar] [CrossRef]
- Schmidts, A.; Maus, M.V. Making CAR T cells a solid option for solid tumors. Front. Immunol. 2018, 9, 2593. [Google Scholar] [CrossRef]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Kasakovski, D.; Xu, L.; Li, Y. T cell senescence and CAR-T cell exhaustion in hematological malignancies. J. Hematol. Oncol. 2018, 11, 91. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; An, L.; Huang, R.; Xiong, J.; Yang, H.; Wang, X.; Zhang, X. Strategies to enhance CAR-T persistence. Biomark. Res. 2022, 10, 86. [Google Scholar] [CrossRef] [PubMed]
- Pietrobon, V.; Todd, L.A.; Goswami, A.; Stefanson, O.; Yang, Z.; Marincola, F. Improving car t-cell persistence. Int. J. Mol. Sci. 2021, 22, 10828. [Google Scholar] [CrossRef]
- López-Cantillo, G.; Urueña, C.; Camacho, B.A.; Ramírez-Segura, C. CAR-T Cell. Performance: How to Improve Their Persistence? Front. Immunol. 2022, 13, 878209. [Google Scholar] [CrossRef]
- Koukoulias, K.; Papayanni, P.G.; Georgakopoulou, A.; Alvanou, M.; Laidou, S.; Kouimtzidis, A.; Pantazi, C.; Gkoliou, G.; Vyzantiadis, T.A.; Spyridonidis, A.; et al. “Cerberus” T Cells: A Glucocorticoid-Resistant, Multi-Pathogen Specific T Cell Product to Fight Infections in Severely Immunocompromised Patients. Front. Immunol. 2021, 11, 608701. [Google Scholar] [CrossRef]
- Linnér, A.; Almgren, M. Epigenetic programming—The important first 1000 days. Acta Paediatr. Int. J. Paediatr. 2020, 109, 443–452. [Google Scholar] [CrossRef]
- Jurkowska, R.Z.; Jurkowski, T.P.; Jeltsch, A. Structure and Function of Mammalian DNA Methyltransferases. ChemBioChem 2011, 12, 206–222. [Google Scholar] [CrossRef]
- Akbari, B.; Ghahri-Saremi, N.; Soltantoyeh, T.; Hadjati, J.; Ghassemi, S.; Mirzaei, H.R. Epigenetic strategies to boost CAR T cell therapy. Mol. Ther. 2021, 29, 2640–2659. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. XCRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442. [Google Scholar] [CrossRef]
- Holtzman, L.; Gersbach, C.A. Editing the epigenome: Reshaping the genomic landscape. Annu. Rev. Genomics Hum. Genet. 2018, 19, 43–71. [Google Scholar] [CrossRef] [PubMed]
- Jambhekar, A.; Dhall, A.; Shi, Y. Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell. Biol. 2019, 20, 625–641. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Whetstine, J.R. Dynamic Regulation of Histone Lysine Methylation by Demethylases. Mol. Cell. 2007, 25, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Li, Y.; Deng, C.; Hu, X.; Patel, B.; Fu, X.; Qiu, Y.; Brand, M.; Zhao, K.; Huang, S. Dynamic interaction between TAL1 oncoprotein and LSD1 regulates TAL1 function in hematopoiesis and leukemogenesis. Oncogene 2012, 31, 5007–5018. [Google Scholar] [CrossRef]
- Dimitrova, E.; Turberfield, A.H.; Klose, R.J. Histone demethylases in chromatin biology and beyond. EMBO Rep. 2015, 16, 1620–1639. [Google Scholar] [CrossRef]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, J.S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef]
- Kimura, H. Histone modifications for human epigenome analysis. J. Hum. Genet. 2013, 58, 439–445. [Google Scholar] [CrossRef]
- Oss-Ronen, L.; Sarusi, T.; Cohen, I. Histone Mono-Ubiquitination in Transcriptional Regulation and Its Mark on Life: Emerging Roles in Tissue Development and Disease. Cells 2022, 11, 2404. [Google Scholar] [CrossRef]
- Sana, J.; Faltejskova, P.; Svoboda, M.; Slaby, O. Novel classes of non-coding RNAs and cancer. J. Transl. Med. 2012, 10, 103. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Villeneuve, L.M.; Morris, K.V.; Rossi, J.J. Argonaute-1 directs siRNA-mediated transcriptional gene silencing in human cells. Nat. Struct. Mol. Biol. 2006, 13, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.G.; Santoso, S.; Adams, C.; Anest, V.; Morris, K.V. Promoter targeted small RNAs induce long-term transcriptional gene silencing in human cells. Nucleic Acids Res. 2009, 37, 2984–2995. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Gonzalez, E.A.; Rameshwar, P.; Etchegaray, J.P. Non-coding RNAs as mediators of epigenetic changes in malignancies. Cancers 2020, 12, 3657. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef]
- Hwang, H.W.; Mendell, J.T. MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br. J. Cancer 2006, 94, 776–780. [Google Scholar] [CrossRef]
- Bianchi, M.; Renzini, A.; Adamo, S.; Moresi, V. Coordinated actions of microRNAs with other epigenetic factors regulate skeletal muscle development and adaptation. Int. J. Mol. Sci. 2017, 18, 840. [Google Scholar] [CrossRef]
- Yao, Q.; Chen, Y.; Zhou, X. The roles of microRNAs in epigenetic regulation. Curr. Opin. Chem. Biol. 2019, 51, 11–17. [Google Scholar] [CrossRef]
- Duursma, A.M.; Kedde, M.; Schrier, M.; Le Sage, C.; Agami, R. miR-148 targets human DNMT3b protein coding region. RNA 2008, 14, 872–877. [Google Scholar] [CrossRef]
- Yong-Ming, H.; Ai-Jun, J.; Xiao-Yue, X.; Jian-Wei, L.; Chen, Y.; Ye, C. MiR-449a: A potential therapeutic agent for cancer. Anticancer Drugs 2017, 28, 1067–1078. [Google Scholar] [CrossRef]
- Noonan, E.J.; Place, R.F.; Pookot, D.; Basak, S.; Whitson, J.M.; Hirata, H.; Giardina, C.; Dahiya, R. MiR-449a targets HDAC-1 and induces growth arrest in prostate cancer. Oncogene 2009, 28, 1714–1724. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Maruyama, R.; Yamamoto, E.; Kai, M. Epigenetic alteration and microRNA dysregulation in cancer. Front. Genet. 2013, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Croce, C.M. 27 Causes and consequences of microRNA dysregulation in cancer. Eur. J. Cancer Suppl. 2010, 8, 8. [Google Scholar] [CrossRef]
- Psatha, N.; Paschoudi, K.; Papadopoulou, A.; Yannaki, E. In Vivo Hematopoietic Stem Cell Genome Editing: Perspectives and Limitations. Genes 2022, 13, 2222. [Google Scholar] [CrossRef] [PubMed]
- Pichon, X.; Bastide, A.; Safieddine, A.; Chouaib, R.; Samacoits, A.; Basyuk, E.; Peter, M.; Mueller, F.; Bertrand, E.; Halstead, J.M.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 135, 437–445. [Google Scholar]
- Brocken, D.J.W.; Tark-Dame, M.; Dame, R.T. dCas9: A versatile tool for epigenome editing. Curr. Issues Mol. Biol. 2018, 26, 15–32. [Google Scholar] [CrossRef]
- Amabile, A.; Migliara, A.; Capasso, P.; Biffi, M.; Cittaro, D.; Naldini, L.; Lombardo, A. Inheritable Silencing of Endogenous Genes by Hit-and-Run Targeted Epigenetic Editing. Cell 2016, 167, 219–232.e14. [Google Scholar] [CrossRef]
- Liu, Q.; Sun, Z.; Chen, L. Memory T cells: Strategies for optimizing tumor immunotherapy. Protein Cell. 2020, 11, 549–564. [Google Scholar] [CrossRef]
- Sprent, J.; Surh, C.D. T cell memory. Annu. Rev. Immunol. 2002, 20, 551–579. [Google Scholar] [CrossRef]
- Sprent, J.; Tough, D.F. T cell death and memory. Science 2001, 293, 245–248. [Google Scholar] [CrossRef]
- Sallusto, F.; Lenig, D.; Förster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Joe, G.; Hexner, E.; Zhu, J.; Emerson, S.G. Host-reactive CD8+ memory stem cells in graft-versus-host disease. Nat. Med. 2005, 11, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, D.; Yang, X.; Zhou, S. Immunotherapeutic Potential of T Memory Stem Cells. Front. Oncol. 2021, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.H.; Fiorenza, S.; Koldej, R.M.; Jaworowski, A.; Ritchie, D.S.; Quinn, K.M. T Cell Fitness and Autologous CAR T Cell Therapy in Haematologic Malignancy. Front. Immunol. 2021, 12, 780442. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, M.; Ramos, C.A.; Durett, A.; Liu, E.; Dakhova, O.; Liu, H.; Creighton, C.J.; Gee, A.P.; Heslop, H.E.; et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood 2014, 123, 3750–3759. [Google Scholar] [CrossRef] [PubMed]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef]
- Alvarez-Fernández, C.; Escribà-Garcia, L.; Caballero, A.C.; Escudero-López, E.; Ujaldón-Miró, C.; Montserrat-Torres, R.; Pujol-Fernández, P.; Sierra, J.; Briones, J. Memory stem T cells modified with a redesigned CD30-chimeric antigen receptor show an enhanced antitumor effect in Hodgkin lymphoma. Clin. Transl. Immunol. 2021, 10, e1268. [Google Scholar] [CrossRef]
- Lécuroux, C.; Girault, I.; Urrutia, A.; Doisne, J.M.; Deveau, C.; Goujard, C.; Meyer, L.; Sinet, M.; Venet, A. Identification of a particular HIV-specific CD8+ T-cell subset with a CD27+ CD45RO/RA+ phenotype and memory characteristics after initiation of HAART during acute primary HIV infection. Blood 2009, 113, 3209–3217. [Google Scholar] [CrossRef]
- Garfall, A.L.; Dancy, E.K.; Cohen, A.D.; Hwang, W.T.; Fraietta, J.A.; Davis, M.M.; Levine, B.L.; Siegel, D.L.; Stadtmauer, E.A.; Vogl, D.T.; et al. T-cell phenotypes associated with effective CAR T-cell therapy in postinduction vs relapsed multiple myeloma. Blood Adv. 2019, 3, 2812–2815. [Google Scholar] [CrossRef]
- Zebley, C.C.; Abdelsamed, H.A.; Ghoneim, H.E.; Alli, S.; Brown, C.; Haydar, D.; Mi, T.; Harris, T.; McGargill, M.A.; Krenciute, G.; et al. Proinflammatory cytokines promote TET2-mediated DNA demethylation during CD8 T cell effector differentiation. Cell. Rep. 2021, 37, 109796. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, M.; Hu, J.; Sommariva, M.; Gautam, S.; Fellowes, V.; Hocker, J.D.; Dougherty, S.; Qin, H.; Klebanoff, C.A.; Fry, T.J.; et al. Generation of clinical-grade CD19-specific CAR-modified CD81 memory stem cells for the treatment of human B-cell malignancies. Blood 2016, 128, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Blaeschke, F.; Stenger, D.; Kaeuferle, T.; Willier, S.; Lotfi, R.; Kaiser, A.D.; Assenmacher, M.; Döring, M.; Feucht, J.; Feuchtinger, T. Induction of a central memory and stem cell memory phenotype in functionally active CD4+ and CD8+ CAR T cells produced in an automated good manufacturing practice system for the treatment of CD19+ acute lymphoblastic leukemia. Cancer Immunol. Immunother. 2018, 67, 1053–1066. [Google Scholar] [CrossRef]
- Youngblood, B.; Hale, J.S.; Kissick, H.T.; Ahn, E.; Xu, X.; Wieland, A.; Araki, K.; West, E.E.; Ghoneim, H.E.; Fan, Y.; et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature 2017, 552, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Carty, S.A.; Gohil, M.; Banks, L.B.; Cotton, R.M.; Johnson, M.E.; Stelekati, E.; Wells, A.D.; Wherry, E.J.; Koretzky, G.A.; Jordan, M.S. The Loss of TET2 Promotes CD8 + T Cell Memory Differentiation. J. Immunol. 2018, 200, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xue, H.-H. Generation of memory precursors and functional memory CD8+ T cells depends on TCF-1 and LEF-1. J. Immunol. 2012, 189, 2722. [Google Scholar] [CrossRef]
- Crompton, J.G.; Narayanan, M.; Cuddapah, S.; Roychoudhuri, R.; Ji, Y.; Yang, W.; Patel, S.J.; Sukumar, M.; Palmer, D.C.; Peng, W.; et al. Lineage relationship of CD8+ T cell subsets is revealed by progressive changes in the epigenetic landscape. Cell. Mol. Immunol. 2016, 13, 502–513. [Google Scholar] [CrossRef]
- Shin, H.M.; Kapoor, V.N.; Guan, T.; Kaech, S.M.; Welsh, R.M.; Berg, L.J. Epigenetic modifications induced by blimp-1 regulate CD8+ T cell memory progression during acute virus infection. Immunity 2013, 39, 661–675. [Google Scholar] [CrossRef]
- Gyory, I.; Wu, J.; Fejér, G.; Seto, E.; Wright, K.L. PRDI-BF1 recruits the histone H3 methyltransferase G9a in transcriptional silencing. Nat. Immunol. 2004, 5, 299–308. [Google Scholar] [CrossRef]
- Pace, L.; Goudot, C.; Zueva, E.; Gueguen, P.; Burgdorf, N.; Waterfall, J.J.; Quivy, J.P.; Almouzni, G.; Amigorena, S. The epigenetic control of stemness in CD8+ T cell fate commitment. Science 2018, 359, 177–186. [Google Scholar] [CrossRef]
- Durek, P.; Nordström, K.; Gasparoni, G.; Salhab, A.; Kressler, C.; de Almeida, M.; Bassler, K.; Ulas, T.; Schmidt, F.; Xiong, J.; et al. Epigenomic Profiling of Human CD4+ T Cells Supports a Linear Differentiation Model and Highlights Molecular Regulators of Memory Development. Immunity 2016, 45, 1148–1161. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Prieto, C.A.; Villanueva, L.; Bueno-Costa, A.; Davalos, V.; González-Navarro, E.A.; Juan, M.; Urbano-Ispizua, Á.; Delgado, J.; Ortiz-Maldonado, V.; Del Bufalo, F.; et al. Epigenetic Profiling and Response to CD19 Chimeric Antigen Receptor T-Cell Therapy in B-Cell Malignancies. J. Natl. Cancer Inst. 2022, 114, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Sheih, A.; Voillet, V.; Hanafi, L.A.; DeBerg, H.A.; Yajima, M.; Hawkins, R.; Gersuk, V.; Riddell, S.R.; Maloney, D.G.; Wohlfahrt, M.E.; et al. Clonal kinetics and single-cell transcriptional profiling of CAR-T cells in patients undergoing CD19 CAR-T immunotherapy. Nat. Commun. 2020, 11, 2020. [Google Scholar] [CrossRef] [PubMed]
- Zebley, C.C.; Brown, C.; Mi, T.; Fan, Y.; Alli, S.; Boi, S.; Galletti, G.; Lugli, E.; Langfitt, D.; Metais, J.Y.; et al. CD19-CAR T cells undergo exhaustion DNA methylation programming in patients with acute lymphoblastic leukemia. Cell Rep. 2021, 37, 110079. [Google Scholar] [CrossRef]
- Chen, G.M.; Chen, C.; Das, R.K.; Gao, P.; Chen, C.H.; Bandyopadhyay, S.; Ding, Y.Y.; Uzun, Y.; Yu, W.; Zhu, Q.; et al. Integrative bulk and single-cell profiling of premanufacture t-cell populations reveals factors mediating long-term persistence of car t-cell therapy. Cancer Discov. 2021, 11, 2186–2199. [Google Scholar] [CrossRef]
- Prinzing, B.; Zebley, C.C.; Petersen, C.T.; Fan, Y.; Anido, A.A.; Yi, Z.; Nguyen, P.; Houke, H.; Bell, M.; Haydar, D.; et al. Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Sci. Transl. Med. 2021, 13, 6234. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Wu, Z.; Inoue, S.; Kasuya, H.; Matsushita, H.; Takahashi, Y.; Kuroda, H.; Hosoda, W.; Suzuki, S.; Kagoya, Y. Genetic ablation of PRDM1 in antitumor T cells enhances therapeutic efficacy of adoptive immunotherapy. Blood 2022, 139, 2156–2172. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.D.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018, 558, 307–312. [Google Scholar] [CrossRef]
- Chen, Z.; Stelekati, E.; Kurachi, M.; Yu, S.; Cai, Z.; Manne, S.; Khan, O.; Yang, X.; Wherry, E.J. miR-150 Regulates Memory CD8 T Cell Differentiation via c-Myb. Cell. Rep. 2017, 20, 2584–2597. [Google Scholar] [CrossRef]
- Ban, Y.H.; Oh, S.C.; Seo, S.H.; Kim, S.M.; Choi, I.P.; Greenberg, P.D.; Chang, J.; Kim, T.D.; Ha, S.J. miR-150-Mediated Foxo1 Regulation Programs CD8+ T Cell Differentiation. Cell. Rep. 2017, 20, 2598–2611. [Google Scholar] [CrossRef] [PubMed]
- Trifari, S.; Pipkin, M.E.; Rao, A. MicroRNA-directed program of cytotoxic CD8 + T- cell differentiation. Proc. Natl. Acad. Sci. USA 2014, 110, 18613. [Google Scholar] [CrossRef] [PubMed]
- Sang, W.; Sun, C.; Zhang, C.; Zhang, D.; Wang, Y.; Xu, L.; Zhang, Z.; Wei, X.; Pan, B.; Yan, D.; et al. MicroRNA-150 negatively regulates the function of CD4+ T cells through AKT3/Bim signaling pathway. Cell. Immunol. 2016, 306–307, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, L.; Massett, M.E.; Bunschoten, R.P.; Hoose, A.; Pirvan, P.A.; Liskamp, R.M.J.; Jørgensen, H.G.; Huang, X. The Emerging Role of H3K9me3 as a Potential Therapeutic Target in Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 705. [Google Scholar] [CrossRef]
- Youngblood, B.; Oestreich, K.J.; Ha, S.J.; Duraiswamy, J.; Akondy, R.S.; West, E.E.; Wei, Z.; Lu, P.; Austin, J.W.; Riley, J.L.; et al. Chronic Virus Infection Enforces Demethylation of the Locus that Encodes PD-1 in Antigen-Specific CD8+ T Cells. Immunity 2011, 35, 400–412. [Google Scholar] [CrossRef]
- Jindra, P.T.; Bagley, J.; Godwin, J.G.; Iacomini, J. Costimulation-Dependent Expression of MicroRNA-214 Increases the Ability of T Cells to Proliferate by Targeting Pten. J. Immunol. 2010, 185, 990–997. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, Z.; Li, F.; Ping, Y.; Qin, G.; Zhang, C.; Zhang, Y. miR-143 Regulates Memory T Cell Differentiation by Reprogramming T Cell Metabolism. J. Immunol. 2018, 201, 2165–2175. [Google Scholar] [CrossRef]
- Li, H.; Chiappinelli, K.B.; Guzzetta, A.A.; Easwaran, H.; Yen, R.W.C.; Vatapalli, R.; Topper, M.J.; Luo, J.; Connolly, R.M.; Azad, N.S.; et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget 2014, 5, 587–598. [Google Scholar] [CrossRef]
- Boldin, M.P.; Taganov, K.D.; Rao, D.S.; Yang, L.; Zhao, J.L.; Kalwani, M.; Garcia-Flores, Y.; Luong, M.; Devrekanli, A.; Xu, J.; et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med. 2011, 208, 1189–1201. [Google Scholar] [CrossRef]
- Cycle, C.; Gagnon, J.D.; Kageyama, R.; Shehata, H.M.; Matloubian, M.; Sanjabi, S.; Ansel, K.M. miR-15 / 16 Restrain Memory T Cell Differentiation, Article miR-15/16 Restrain Memory T Cell Differentiation, Cell Cycle, and Survival. Cell Rep. 2019, 28, 2169–2181.e4. [Google Scholar]
- Gao, L.; Guo, Q.; Li, X.; Yang, X.; Ni, H.; Wang, T.; Zhao, Q.; Liu, H.; Xing, Y.; Xi, T.; et al. MiR-873/PD-L1 axis regulates the stemness of breast cancer cells. EBioMedicine 2019, 41, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Li, C.; Olive, V.; Lykken, E.; Feng, F.; Sevilla, J.; Wan, Y.; He, L.; Li, Q.J. Molecular dissection of the miR-17-92 cluster’s critical dual roles in promoting Th1 responses and preventing inducible Treg differentiation. Blood 2011, 118, 5487–5497. [Google Scholar] [CrossRef] [PubMed]
- Tay, R.E.; Olawoyin, O.; Cejas, P.; Xie, Y.; Meyer, C.A.; Weng, Q.Y.; Fisher, D.E.; Long, H.W.; Brown, M.; Kim, H.J.; et al. Hdac3 is an epigenetic inhibitor of the cytotoxicity program in CD8 T cells. J. Exp. Med. 2020, 217, 20191453. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Zhang, Y.; Yu, L.; Wu, D. Cytotoxic effect of CLL-1 CAR-T cell immunotherapy with PD-1 silencing on relapsed/refractory acute myeloid leukemia. Mol. Med. Rep. 2021, 23, 2021. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, Q.; Li, D.; Zhang, L.; Gu, Z.; Liu, J.; Liu, G.; Yang, M.; Gu, J.; Cui, X.; et al. PD-1 silencing improves anti-tumor activities of human mesothelin-targeted CAR T cells. Hum. Immunol. 2021, 82, 130–138. [Google Scholar] [CrossRef]
- Zhu, H.; You, Y.; Shen, Z.; Shi, L. EGFRvIII-CAR-T Cells with PD-1 Knockout Have Improved Anti-Glioma Activity. Pathol. Oncol. Res. 2020, 26, 2135–2141. [Google Scholar] [CrossRef]
- Guo, X.; Jiang, H.; Shi, B.; Zhou, M.; Zhang, H.; Shi, Z.; Du, G.; Luo, H.; Wu, X.; Wang, Y.; et al. Disruption of PD-1 enhanced the anti-tumor activity of chimeric antigen receptor T cells against hepatocellular carcinoma. Front. Pharmacol. 2018, 9, 2018. [Google Scholar] [CrossRef]
- Si, J.; Shi, X.; Sun, S.; Zou, B.; Li, Y.; An, D.; Lin, X.; Gao, Y.; Long, F.; Pang, B.; et al. Hematopoietic Progenitor Kinase1 (HPK1) Mediates T Cell Dysfunction and Is a Druggable Target for T Cell-Based Immunotherapies. Cancer Cell. 2020, 38, 551–566.e11. [Google Scholar] [CrossRef]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Zou, F.; Lu, L.; Liu, J.; Xia, B.; Zhang, W.; Hu, Q.; Liu, W.; Zhang, Y.; Lin, Y.; Jing, S.; et al. Engineered triple inhibitory receptor resistance improves anti-tumor CAR-T cell performance via CD56. Nat. Commun. 2019, 10, 4109. [Google Scholar] [CrossRef]
- Li, Q.; Johnston, N.; Zheng, X.; Wang, H.; Zhang, X.; Gao, D.; Min, W. miR-28 modulates exhaustive differentiation of T cells through silencing programmed cell death-1 and regulating cytokine secretion. Oncotarget 2016, 7, 53735–53750. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Angelou, C.C.; Wells, A.C.; Vijayaraghavan, J.; Dougan, C.E.; Lawlor, R.; Iverson, E.; Lazarevic, V.; Kimura, M.Y.; Peyton, S.R.; Minter, L.M.; et al. Differentiation of Pathogenic Th17 Cells Is Negatively Regulated by Let-7 MicroRNAs in a Mouse Model of Multiple Sclerosis. Front. Immunol. 2020, 10, 3125. [Google Scholar] [CrossRef] [PubMed]
- Lind, E.F.; Elford, A.R.; Ohashi, P.S. Micro-RNA 155 Is Required for Optimal CD8 + T Cell Responses to Acute Viral and Intracellular Bacterial Challenges. J. Immunol. 2013, 190, 1210–1216. [Google Scholar] [CrossRef]
- Dudda, J.C.; Salaun, B.; Ji, Y.; Palmer, D.C.; Monnot, G.C.; Merck, E.; Boudousquie, C.; Utzschneider, D.T.; Escobar, T.M.; Perret, R.; et al. MicroRNA-155 is required for effector CD8+ T cell responses to virus infection and cancer. Immunity 2013, 38, 742–753. [Google Scholar] [CrossRef]
- Monnot, G.C.; Martinez-Usatorre, A.; Lanitis, E.; Lopes, S.F.; Cheng, W.C.; Ho, P.C.; Irving, M.; Coukos, G.; Donda, A.; Romero, P. miR-155 Overexpression in OT-1 CD8+ T Cells Improves Anti-Tumor Activity against Low-Affinity Tumor Antigen. Mol. Ther. Oncolytics 2020, 16, 111–123. [Google Scholar] [CrossRef]
- Ohno, M.; Ohkuri, T.; Kosaka, A.; Tanahashi, K.; June, C.H.; Natsume, A.; Okada, H. Expression of miR-17-92 enhances anti-tumor activity of T-cells transduced with the anti-EGFRvIII chimeric antigen receptor in mice bearing human GBM xenografts. J. Immunother. Cancer 2013, 1, 2051. [Google Scholar] [CrossRef]
- Li, Q.J.; Chau, J.; Ebert, P.J.R.; Sylvester, G.; Min, H.; Liu, G.; Braich, R.; Manoharan, M.; Soutschek, J.; Skare, P.; et al. miR-181a Is an Intrinsic Modulator of T Cell Sensitivity and Selection. Cell 2007, 129, 147–161. [Google Scholar] [CrossRef]
- Vignard, V.; Labbe, M.; Marec, N.; Andre-Gregoire, G.; Jouand, N.; Fonteneau, J.F.; Labarriere, N.; Fradin, D. MicroRNAs in tumor exosomes drive immune escape in melanoma. Cancer Immunol. Res. 2020, 8, 255–267. [Google Scholar] [CrossRef]
- Lou, Q.; Liu, R.; Yang, X.; Li, W.; Huang, L.; Wei, L.; Tan, H.; Xiang, N.; Chan, K.; Chen, J.; et al. MiR-448 targets IDO1 and regulates CD8+ T cell response in human colon cancer. J. Immunother. Cancer 2019, 7, 2019. [Google Scholar] [CrossRef]
- Huang, Q.; Xia, J.; Wang, L.; Wang, X.; Ma, X.; Deng, Q.; Lu, Y.; Kumar, M.; Zhou, Z.; Li, L.; et al. MiR-153 suppresses IDO1 expression and enhances CAR T cell immunotherapy. J. Hematol. Oncol. 2018, 11, 2018. [Google Scholar] [CrossRef]
- Yang, J.; Lan, H.; Huang, X.; Liu, B.; Tong, Y. MicroRNA-126 inhibits tumor cell growth and its expression level correlates with poor survival in non-small cell lung cancer patients. PLoS ONE 2012, 7, 42978. [Google Scholar] [CrossRef] [PubMed]
- Kong, R.; Ma, Y.; Feng, J.; Li, S.; Zhang, W.; Jiang, J.; Zhang, J.; Qiao, Z.; Yang, X.; Zhou, B. The crucial role of miR-126 on suppressing progression of esophageal cancer by targeting VEGF-A. Cell. Mol. Biol. Lett. 2016, 21, 2016. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Peng, X.C.; Zheng, X.L.; Wang, J.; Qin, Y.W. MiR-126 restoration down-regulate VEGF and inhibit the growth of lung cancer cell lines in vitro and in vivo. Lung Cancer 2009, 66, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; He, J.; Zhang, L. The down-regulation of microRNA-137 contributes to the up-regulation of retinoblastoma cell proliferation and invasion by regulating COX-2/PGE2 signaling. Biomed. Pharmacother. 2018, 106, 35–42. [Google Scholar] [CrossRef]
- Wei, L.; Sun, C.; Zhang, Y.; Han, N.; Sun, S. miR-503-5p inhibits colon cancer tumorigenesis, angiogenesis, and lymphangiogenesis by directly downregulating VEGF-A. Gene Ther. 2022, 29, 28–40. [Google Scholar] [CrossRef]
- Lv, M.; Xu, Y.; Tang, R.; Ren, J.; Shen, S.; Chen, Y.; Liu, B.; Hou, Y.; Wang, T. MiR141-CXCL1-CXCR2 signaling-induced treg recruitment regulates metastases and survival of non-small cell lung cancer. Mol. Cancer Ther. 2014, 13, 3152–3162. [Google Scholar] [CrossRef]
- Chen, S.; Pu, J.; Bai, J.; Yin, Y.; Wu, K.; Wang, J.; Shuai, X.; Gao, J.; Tao, K.; Wang, G.; et al. EZH2 promotes hepatocellular carcinoma progression through modulating miR-22/galectin-9 axis. J. Exp. Clin. Cancer Res. 2018, 37, 2018. [Google Scholar] [CrossRef]
- Zhu, J.; Zeng, Y.; Li, W.; Qin, H.; Lei, Z.; Shen, D.; Gu, D.; Huang, J.; Liu, Z. CD73/NT5E is a target of miR-30a-5p and plays an important role in the pathogenesis of non-small cell lung cancer. Mol. Cancer 2017, 16, 2017. [Google Scholar] [CrossRef]
- Bonnin, N.; Armandy, E.; Carras, J.; Ferrandon, S.; Battiston-Montagne, P.; Aubry, M.; Guihard, S.; Meyronet, D.; Foy, J.P.; Saintigny, P.; et al. MiR-422a promotes loco-regional recurrence by targeting NT5E/CD73 in head and neck squamous cell carcinoma. Oncotarget 2016, 7, 44023–44038. [Google Scholar] [CrossRef]
- Monteleone, N.J.; Lutz, C.S. miR-708-5p targets oncogenic prostaglandin E2 production to suppress a pro-tumorigenic phenotype in lung cancer cells. Oncotarget 2020, 11, 2464–2483. [Google Scholar] [CrossRef] [PubMed]
- McKinney, E.F.; Lee, J.C.; Jayne, D.R.W.; Lyons, P.A.; Smith, K.G.C. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature 2015, 523, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, A.; Tey, S.-K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Kagoya, Y. Epigenetic engineering for optimal chimeric antigen receptor T cell therapy. Cancer Sci. 2022, 113, 3664–3671. [Google Scholar] [CrossRef]
- Alfei, F.; Zehn, D. T Cell Exhaustion: An Epigenetically Imprinted Phenotypic and Functional Makeover. Trends Mol. Med. 2017, 23, 769–771. [Google Scholar] [CrossRef]
- Baitsch, L.; Baumgaertner, P.; Devêvre, E.; Raghav, S.K.; Legat, A.; Barba, L.; Wieckowski, S.; Bouzourene, H.; Deplancke, B.; Romero, P.; et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Investig. 2011, 121, 2350–2360. [Google Scholar] [CrossRef]
- Odorizzi, P.M.; Pauken, K.E.; Paley, M.A.; Sharpe, A.; John Wherry, E. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J. Exp. Med. 2015, 212, 1125–1137. [Google Scholar] [CrossRef]
- Deng, Q.; Han, G.; Puebla-osorio, N.; Chun, M.; Ma, J.; Strati, P.; Chasen, B.; Dai, E.; Dang, M.; Jain, N.; et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat. Med. 2020, 26, 1878–1887. [Google Scholar] [CrossRef]
- Tang, L.; Zhang, Y.; Hu, Y.; Mei, H. T Cell Exhaustion and CAR-T Immunotherapy in Hematological Malignancies. BioMed Res. Int. 2021, 2021, 6616391. [Google Scholar] [CrossRef]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef]
- Frigault, M.J.; Lee, J.; Basil, M.C.; Carpenito, C.; Motohashi, S.; Scholler, J.; Kawalekar, O.U.; Guedan, S.; McGettigan, S.E.; Posey, A.D.; et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol. Res. 2015, 3, 356–367. [Google Scholar] [CrossRef]
- Gomes-Silva, D.; Mukherjee, M.; Srinivasan, M.; Krenciute, G.; Dakhova, O.; Zheng, Y.; Cabral, J.M.S.; Rooney, C.M.; Orange, J.S.; Brenner, M.K.; et al. Tonic 4-1BB Costimulation in Chimeric Antigen Receptors Impedes T Cell Survival and Is Vector-Dependent. Cell. Rep. 2017, 21, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kohler, M.E.; Chien, C.D.; Sauter, C.T.; Jacoby, E.; Yan, C.; Hu, Y.; Wanhainen, K.; Qin, H.; Fry, T.J. TCR engagement negatively affects CD8 but not CD4 CAR T cell expansion and leukemic clearance. Sci. Transl. Med. 2017, 9, eaag1209. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Frey, N.V.; Engels, B.; Barrett, D.M.; Shestova, O.; Ravikumar, P.; Cummins, K.D.; Lee, Y.G.; Pajarillo, R.; Chun, I.; et al. Antigen-independent activation enhances the efficacy of 4-1BB-costimulated CD22 CAR T cells. Nat. Med. 2021, 27, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef]
- Abdel-hakeem, M.S.; Manne, S.; Beltra, J.C.; Stelekati, E.; Chen, Z.; Nzingha, K.; Ali, M.; Johnson, J.L.; Giles, J.R.; Mathew, D.; et al. Epigenetic scarring of exhausted T cells hinders memory differentiation upon eliminating chronic antigenic stimulation. Nat. Immunol. 2021, 22, 1008–1019. [Google Scholar] [CrossRef]
- Utzschneider, D.T.; Legat, A.; Fuertes Marraco, S.A.; Carrié, L.; Luescher, I.; Speiser, D.E.; Zehn, D. T cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nat. Immunol. 2013, 14, 603–610. [Google Scholar] [CrossRef]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; Barnitz, R.A.; et al. HHS Public Access reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Neale, G.; Thomas, P.G.; Youngblood, B.; Rejuvenation, P.-B.T.C.; Ghoneim, H.E.; Fan, Y.; Moustaki, A.; et al. Article De Novo Epigenetic Programs Inhibit. Cell 2017, 170, 142–157.e19. [Google Scholar] [CrossRef]
- Scott, A.C.; Dündar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S.; et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571, 270–274. [Google Scholar] [CrossRef]
- Alfei, F.; Kanev, K.; Hofmann, M.; Wu, M.; Ghoneim, H.E.; Roelli, P.; Utzschneider, D.T.; Von Hoesslin, M.; Cullen, J.G.; Fan, Y.; et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 2019, 571, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; López-Moyado, I.F.; Seo, H.; Lio, C.W.J.; Hempleman, L.J.; Sekiya, T.; Yoshimura, A.; Scott-Browne, J.P.; Rao, A. NR4A transcription factors limit CAR T cell function in solid tumours. Nature 2019, 567, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Belk, J.A.; Yao, W.; Ly, N.; Freitas, K.A.; Chen, Y.T.; Shi, Q.; Valencia, A.M.; Shifrut, E.; Kale, N.; Yost, K.E.; et al. Genome-wide CRISPR screens of T cell exhaustion identify chromatin remodeling factors that limit T cell persistence. Cancer Cell 2022, 40, 768–786.e7. [Google Scholar] [CrossRef] [PubMed]
- Yates, K.B.; Tonnerre, P.; Martin, G.E.; Gerdemann, U.; Al Abosy, R.; Comstock, D.E.; Weiss, S.A.; Wolski, D.; Tully, D.C.; Chung, R.T.; et al. Epigenetic scars of CD8+ T cell exhaustion persist after cure of chronic infection in humans. Nat. Immunol. 2021, 22, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Weber, E.W.; Parker, K.R.; Sotillo, E.; Lynn, R.C.; Anbunathan, H.; Lattin, J.; Daniel, B.; Klysz, D.; Malipatlolla, M.; Xu, P.; et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 2021, 372, 6537. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Han, Q.; Zhu, L.; Zhu, Y.; Bao, C.; Yang, C.; Lei, W.; Qian, W. Decitabine-Mediated Epigenetic Reprograming Enhances Anti-leukemia Efficacy of CD123-Targeted Chimeric Antigen Receptor T-Cells. Front. Immunol. 2020, 11, 787. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tong, C.; Dai, H.; Wu, Z.; Han, X.; Guo, Y.; Chen, D.; Wei, J.; Ti, D.; Liu, Z.; et al. Low-dose decitabine priming endows CAR T cells with enhanced and persistent antitumour potential via epigenetic reprogramming. Nat. Commun. 2021, 12, 409. [Google Scholar] [CrossRef]
- Zhang, F.; Zhou, X.; Dispirito, J.R.; Wang, C.; Wang, Y.; Shen, H. Epigenetic manipulation restores functions of defective CD8+ T Cells from chronic viral infection. Mol. Ther. 2014, 22, 1698–1706. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 2020. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell. 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Jitschin, R.; Braun, M.; Büttner, M.; Dettmer-Wilde, K.; Bricks, J.; Berger, J.; Eckart, M.J.; Krause, S.W.; Oefner, P.J.; Le Blanc, K.; et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood 2014, 124, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Dong, Y.; Yang, Q.; Xu, W.; Jiang, S.; Yu, Z.; Yu, K.; Zhang, S. Acute Myeloid Leukemia Cells Express ICOS Ligand to Promote the Expansion of Regulatory T Cells. Front. Immunol. 2018, 9, 2227. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.X.; Li, L.; Wang, W.; OuYang, B.S.; Cheng, S.; Wang, L.; Wu, W.; Xu, P.P.; Muftuoglu, M.; Hao, M.; et al. Clinical efficacy and tumor microenvironment influence in a dose-escalation study of anti-CD19 chimeric antigen receptor T cells in refractory B-cell non-Hodgkin’s lymphoma. Clin. Cancer Res. 2019, 25, 6995–7003. [Google Scholar] [CrossRef] [PubMed]
- White, L.G.; Goy, H.E.; Rose, A.J.; McLellan, A.D. Controlling Cell Trafficking: Addressing Failures in CAR T and NK Cell Therapy of Solid Tumours. Cancers 2022, 14, 978. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.T.; Chen, Q.; Appenheimer, M.M.; Skitzki, J.; Wang, W.C.; Odunsi, K.; Evans, S.S. Hurdles to lymphocyte trafficking in the tumor microenvironment: Implications for effective immunotherapy. Immunol. Investig. 2006, 35, 251–277. [Google Scholar] [CrossRef]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef]
- Sackstein, R.; Schatton, T.; Barthel, S.R. T-lymphocyte homing: An underappreciated yet critical hurdle for successful cancer immunotherapy. Lab. Investig. 2017, 97, 669–697. [Google Scholar] [CrossRef]
- Bam, M.; Chintala, S.; Fetcko, K.; Williamsen, B.C.; Siraj, S.; Liu, S.; Wan, J.; Xuei, X.; Liu, Y.; Leibold, A.T.; et al. Genome wide DNA methylation landscape reveals glioblastoma’s influence on epigenetic changes in tumor infiltrating CD4+ T cells. Oncotarget 2021, 12, 967–981. [Google Scholar] [CrossRef]
- Borgoni, S.; Iannello, A.; Cutrupi, S.; Allavena, P.; D’Incalci, M.; Novelli, F.; Cappello, P. Depletion of tumor-associated macrophages switches the epigenetic profile of pancreatic cancer infiltrating T cells and restores their anti-tumor phenotype. Oncoimmunology 2018, 7, 1393596. [Google Scholar] [CrossRef]
- Stoffel, N.U.; Zeder, C.; Brittenham, G.M.; Moretti, D.; Zimmermann, M.B. Iron absorption from supplements is greater with alternate day than with consecutive day dosing in iron-deficient anemic women. Haematologica 2020, 105, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Lyu, H.; Wang, J.; Liu, B. MicroRNA regulation and therapeutic targeting of survivin in cancer. Am. J. Cancer Res. 2015, 5, 20–31. [Google Scholar] [PubMed]
- Nabilsi, N.H.; Broaddus, R.R.; Loose, D.S. DNA methylation inhibits p53-mediated survivin repression. Oncogene 2009, 28, 2046–2050. [Google Scholar] [CrossRef]
- Acquati, S.; Greco, A.; Licastro, D.; Bhagat, H.; Ceric, D.; Rossini, Z.; Grieve, J.; Shaked-Rabi, M.; Henriquez, N.V.; Brandner, S.; et al. Epigenetic regulation of survivin by Bmi1 is cell type specific during corticogenesis and in gliomas. Stem Cells 2013, 31, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Abdi, J.; Rastgoo, N.; Chen, Y.; Chen, G.A.; Chang, H. Ectopic expression of BIRC5-targeting miR-101-3p overcomes bone marrow stroma-mediated drug resistance in multiple myeloma cells. BMC Cancer 2019, 19, 2019. [Google Scholar] [CrossRef] [PubMed]
- Tessema, M.; Klinge, D.M.; Yingling, C.M.; Do, K.; Van Neste, L.; Belinsky, S.A. Re-expression of CXCL14, a common target for epigenetic silencing in lung cancer, induces tumor necrosis. Oncogene 2010, 29, 5159–5170. [Google Scholar] [CrossRef] [PubMed]
- Nolz, J.C.; Harty, J.T. IL-15 regulates memory CD8+ T cell O-glycan synthesis and affects trafficking. J. Clin. Investig. 2014, 124, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhang, H.; Gao, P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 2022, 13, 877–919. [Google Scholar] [CrossRef] [PubMed]
- Yerinde, C.; Siegmund, B.; Glauben, R.; Weidinger, C. Metabolic Control of Epigenetics and Its Role in CD8+ T Cell Differentiation and Function. Front. Immunol. 2019, 10, 2718. [Google Scholar] [CrossRef] [PubMed]
- Thakur, C.; Chen, F. Connections between metabolism and epigenetics in cancers. Semin. Cancer Biol. 2019, 57, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.D.D.; O’Sullivan, D.; Klein Geltink, R.I.I.; Curtis, J.D.D.; Chang, C.H.; Sanin, D.E.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.J.W.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Jeng, M.Y.; Hull, P.A.; Fei, M.; Kwon, H.S.; Tsou, C.L.; Kasler, H.; Ng, C.P.; Gordon, D.E.; Johnson, J.; Krogan, N.; et al. Metabolic reprogramming of human CD8+ memory T cells through loss of SIRT1. J. Exp. Med. 2018, 215, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Hamaidi, I.; Zhang, L.; Kim, N.; Wang, M.H.; Iclozan, C.; Fang, B.; Liu, M.; Koomen, J.M.; Berglund, A.E.; Yoder, S.J.; et al. Sirt2 Inhibition Enhances Metabolic Fitness and Effector Functions of Tumor-Reactive T Cells. Cell. Metab. 2020, 32, 420–436.e12. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Kennemer, A.; Patrick, K.; Tsichlis, P.; Guerau-de-Arellano, M. Protein Arginine Methyltransferase 5 in T Lymphocyte Biology. Trends Immunol. 2020, 41, 918–931. [Google Scholar] [CrossRef]
- Webb, L.M.; Sengupta, S.; Edell, C.; Piedra-Quintero, Z.L.; Amici, S.A.; Miranda, J.N.; Bevins, M.; Kennemer, A.; Laliotis, G.; Tsichlis, P.N.; et al. Protein arginine methyltransferase 5 promotes cholesterol biosynthesis-mediated Th17 responses and autoimmunity. J. Clin. Investig. 2020, 130, 1683–1698. [Google Scholar] [CrossRef]
- Koss, B.; Shields, B.D.; Taylor, E.M.; Storey, A.J.; Byrum, S.D.; Gies, A.J.; Washam, C.L.; Choudhury, S.R.; Ahn, J.H.; Uryu, H.; et al. Epigenetic control of Cdkn2a.Arf protects tumor-infiltrating lymphocytes from metabolic exhaustion. Cancer Res. 2021, 80, 4707–4719. [Google Scholar] [CrossRef]
- Fitness, M. The Epigenetic Regulator EZH2 Instructs CD4 T Cell Response. J. Virol. 2020, 94, e01627-20. [Google Scholar]
- Verma, V.; Jafarzadeh, N.; Boi, S.; Kundu, S.; Jiang, Z.; Fan, Y.; Lopez, J.; Nandre, R.; Zeng, P.; Alolaqi, F.; et al. MEK inhibition reprograms CD8+ T lymphocytes into memory stem cells with potent antitumor effects. Nat. Immunol. 2021, 22, 53–66. [Google Scholar] [CrossRef]
- Dushyanthen, S.; Teo, Z.L.; Caramia, F.; Savas, P.; Mintoff, C.P.; Virassamy, B.; Henderson, M.A.; Luen, S.J.; Mansour, M.; Kershaw, M.H.; et al. Agonist immunotherapy restores T cell function following MEK inhibition improving efficacy in breast cancer. Nat. Commun. 2017, 8, 606. [Google Scholar] [CrossRef]
- Ji, Y.; Fioravanti, J.; Zhu, W.; Wang, H.; Wu, T.; Hu, J.; Lacey, N.E.; Gautam, S.; Le Gall, J.B.; Yang, X.; et al. miR-155 harnesses Phf19 to potentiate cancer immunotherapy through epigenetic reprogramming of CD8+ T cell fate. Nat. Commun. 2019, 10, 2157. [Google Scholar] [CrossRef]
- Ji, Y.; Wrzesinski, C.; Yu, Z.; Hu, J.; Gautam, S.; Hawk, N.V.; Telford, W.G.; Palmer, D.C.; Franco, Z.; Sukumar, M.; et al. MiR-155 augments CD8+ T-cell antitumor activity in lymphoreplete hosts by enhancing responsiveness to homeostatic γc cytokines. Proc. Natl. Acad. Sci. USA 2015, 112, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Ping, W.; Senyan, H.; Li, G.; Yan, C.; Long, L. Increased Lactate in Gastric Cancer Tumor-Infiltrating Lymphocytes Is Related to Impaired T Cell Function Due to miR-34a Deregulated Lactate Dehydrogenase A. Cell. Physiol. Biochem. 2018, 49, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.; Nattress, C.; Navarrete, A.S.; Barisa, M.; Fisher, J. Payload delivery: Engineering immune cells to disrupt the tumour microenvironment. Cancers 2021, 13, 6000. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Moon, E.K. CAR T cells for solid tumors: New strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Yeku, O.O.; Brentjens, R.J. Armored CAR T-cells: Utilizing cytokines and pro-inflammatory ligands to enhance CAR T-cell anti-tumour efficacy. Biochem. Soc. Trans. 2016, 44, 412–418. [Google Scholar] [CrossRef]
- Ma, R.; Rei, M.; Woodhouse, I.; Ferris, K.; Kirschner, S.; Chandran, A.; Gileadi, U.; Chen, J.-L.; Pereira Pinho, M.; Ariosa-Morejon, Y.; et al. Decitabine increases neoantigen and cancer testis antigen expression to enhance T-cell–mediated toxicity against glioblastoma. Neuro Oncol. 2022, 24, 2106. [Google Scholar] [CrossRef]
- Sheng, W.; LaFleur, M.W.; Nguyen, T.H.; Chen, S.; Chakravarthy, A.; Conway, J.R.; Li, Y.; Chen, H.; Yang, H.; Hsu, P.H.; et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 2018, 174, 549–563.e19. [Google Scholar] [CrossRef]
- De Haart, S.J.; Van De Donk, N.W.C.J.; Minnema, M.C.; Huang, J.H.; Aarts-Riemens, T.; Bovenschen, N.; Yuan, H.; Groen, R.W.J.; McMillin, D.W.; Jakubikova, J.; et al. Accessory cells of the microenvironment protect multiple myeloma from T-cell cytotoxicity through cell adhesion-mediated immune resistance. Clin. Cancer Res. 2013, 19, 5591–5601. [Google Scholar] [CrossRef]
- Holthof, L.C.; van der Schans, J.J.; Katsarou, A.; Poels, R.; Gelderloos, A.T.; Drent, E.; van Hal-Van Veen, S.E.; Li, F.; Zweegman, S.; van de Donk, N.W.C.J.; et al. Bone marrow mesenchymal stromal cells can render multiple myeloma cells resistant to cytotoxic machinery of CAR T cells through inhibition of apoptosis. Clin. Cancer Res. 2021, 27, 3793–3803. [Google Scholar] [CrossRef]
- Xia, L.; Liu, J.; Zheng, Z.; Chen, Y.; Ding, J.; Hu, Y.; Hu, G.; Xia, N.; Liu, W. BRD4 inhibition boosts the therapeutic effects of epidermal growth factor receptor-targeted chimeric antigen receptor T cells in glioblastoma. Mol. Ther. 2021, 29, 3011–3026. [Google Scholar] [CrossRef]
- Hogg, S.J.; Vervoort, S.J.; Deswal, S.; Ott, C.J.; Li, J.; Cluse, L.A.; Beavis, P.A.; Darcy, P.K.; Martin, B.P.; Spencer, A.; et al. BET-Bromodomain Inhibitors Engage the Host Immune System and Regulate Expression of the Immune Checkpoint Ligand PD-L1. Cell Rep. 2017, 18, 2162–2174. [Google Scholar] [CrossRef] [PubMed]
- Zhong, M.; Gao, R.; Zhao, R.; Huang, Y.; Chen, C.; Li, K.; Yu, X.; Nie, D.; Chen, Z.; Liu, X.; et al. BET bromodomain inhibition rescues PD-1-mediated T-cell exhaustion in acute myeloid leukemia. Cell. Death Dis. 2022, 13, 671. [Google Scholar] [CrossRef]
- Zhu, H.; Bengsch, F.; Svoronos, N.; Rutkowski, M.R.; Bitler, B.G.; Allegrezza, M.J.; Yokoyama, Y.; Kossenkov, A.V.; Bradner, J.E.; Conejo-Garcia, J.R.; et al. BET Bromodomain Inhibition Promotes Anti-tumor Immunity by Suppressing PD-L1 Expression. Cell Rep. 2016, 16, 2829–2837. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, Y.; Jing, Y.; Green, M.R.; Han, L. Advancing CAR T cell therapy through the use of multidimensional omics data. Nat. Rev. Clin. Oncol. 2023, 20, 211–228. [Google Scholar] [CrossRef] [PubMed]
- Philip, M.; Fairchild, L.; Sun, L.; Horste, E.L.; Camara, S.; Shakiba, M.; Scott, A.C.; Viale, A.; Lauer, P.; Merghoub, T.; et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017, 545, 452–456. [Google Scholar] [CrossRef]
- Raihani, N.J.; Bell, V. An evolutionary perspective on paranoia. Nat. Hum. Behav. 2019, 3, 114–121. [Google Scholar] [CrossRef]
- Thibault Flesher, D.L.; Sun, X.; Behrens, T.W.; Graham, R.R.; Criswell, L.A. Recent advances in the genetics of systemic lupus erythematosus. Expert Rev. Clin. Immunol. 2010, 6, 461–479. [Google Scholar] [CrossRef]
- Nardini, C.; Moreau, J.F.; Gensous, N.; Ravaioli, F.; Garagnani, P.; Bacalini, M.G. The epigenetics of inflammaging: The contribution of age-related heterochromatin loss and locus-specific remodelling and the modulation by environmental stimuli. Semin. Immunol. 2018, 40, 49–60. [Google Scholar] [CrossRef]
- Gabandé-Rodríguez, E.; Pfeiffer, M.; Mittelbrunn, M. Immuno(T)herapy for age-related diseases. EMBO Mol. Med. 2023, 15, e16301. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Epigenetic Reprogramming Strategies to Overcome CAR-Ts Roadblocks | Targets | Modification | Function on T Cells | Reference |
|---|---|---|---|---|
| Promoting stemness | CCR7 | Demethylation | Promoting the dedifferentiation of effector into memory cells | [65] |
| TCF7 | ||||
| TET2 | DNA Methylation—KO | Promoting memory T-cell differentiation | [66] | |
| Il2ra | Histone H3-acetylation and reduced histone H3K9-trimethylation | Improving memory cell formation and anti-tumor activity | [69] | |
| miR150 | Reduces effector function and proliferation | [83,84] | ||
| SUV39H1 | H3K9-trimethylation | Increasing long-term memory reprogramming capacity | [71,85] | |
| PDCD1 | DNA methylation | Inhibits naïve to effector CD8 T-cell differentiation | [86] | |
| TCF7 | DNA methylation | Maintaining of naïve and memory T-cell states | [76] | |
| LEF1 | ||||
| PRDM1 | DNA methylation—KO | Avoiding the maintenance of TEFF cell states | [77] | |
| TBX21 | ||||
| c-Myb | Non-coding RNA-mediated mechanisms by miR-150 (absence of miR-150) | Enhancing CD8+ T-cell memory differentiation | [81] | |
| PTEN | miR-214 | Enhances proliferation | [87] | |
| Glut-1 | miR-143 | Promotes memory development | [88] | |
| PRDM1 | miR-23a | Reduces T-cell differentiation and effector function | [89] | |
| NF-κB | miR-146a | Regulates and reduces effector function | [90] | |
| ARRB2 | miR-150 | Reduces effector function and proliferation | [83,84] | |
| Bcl-2 | miR-15/16 | Inhibits memory T-cell formation and differentiation | [91] | |
| Pim-1 | ||||
| Il7r | ||||
| CD28 | ||||
| EOMES | miR139–342 | Reduce effector function and differentiation | [83] | |
| Perforin | ||||
| PDL-1 | miR-873 | Attenuates stemness and resistance of tumor cells | [92] | |
| Promoting stemness | CREB-1 | miR-17 | Restrains i-Treg differentiation | [93] |
| Runx3 | Histone deacetylation | Inhibits differentiation into cytotoxic effector cells | [94] | |
| PRDM1 | ||||
| Overcoming exhaustion | PDCD1 | shRNA-mediated knockdown | Enhancing the anti-tumor efficacy of CLL-1-, mesothelin-, EGFR-, CD19-, and GPC3- CAR-Ts | [95,96,97,98] |
| DNA methylation | Reverses exhaustion | [86] | ||
| HPK1 | Genetic depletion or pharmacological inhibition | Improving the exhaustion of CAR-Ts and the immune responses | [99] | |
| TOX-bound HBO1 complex | Histone H3 and H4 deacetylation | Reversing exhaustion | [100] | |
| PD-1 | shRNA-mediated knockdown | Enhance the secretion of IFN-γ and the resistance to apoptosis | [101] | |
| TIM-3 | ||||
| LAG-3 | ||||
| CTLA-4 | miR-28 | Reduces exhausted T cells and regulates the cytokine secretion in the tumor microenvironment | [102] | |
| PD-1 | ||||
| Promoting infiltration | ΕΖH-2 | H3K27 trimethylation and DNA methylation | Preventing CXCR3 + Th1 cell infiltration in the TME | [103] |
| CCR2 | Let-7 miRNA | Impairs trafficking | [104] | |
| CCR5 | ||||
| SHIP-1 | miR-155 | Enhances trafficking and function of CD8+T cells | [105,106,107] | |
| SOCS-1 | ||||
| PHLPP2 | miR-19–92 | Enhances IFN-γ release and reduces inhibition of proliferation | [108] | |
| PTEN | ||||
| DUSP5 | miR-181a | Augments the sensitivity to peptide antigens and induces tolerance | [109] | |
| DUSP6 | ||||
| PTPN11 | ||||
| PTPN22 | ||||
| TNFα | miR-181a/b | Reduces cytotoxicity | [110] | |
| IDO1 | miR-448 | Enhance proliferation and antitumor function of CD8+ T cells | [111] | |
| miR-153 | [112] | |||
| VEGF-A | miR-126 | Reduces cell proliferation, inhibits angiogenesis | [113,114,115] | |
| COX-2 | miR-137 | Contributes to the upregulation of retinoblastoma cell proliferation and invasion | [116] | |
| VEGF-A | miR-503-5p | Inhibits angiogenesis | [117] | |
| CXCL-1 | miR-141 | Inhibits Tregs recruitment | [118] | |
| Galectin-9 | miR-22 | Suppresses cell growth, invasion, and metastasis | [119] | |
| Promoting infiltration | CD73 | miR-30a-5p | Inhibits cell proliferation, cell migration and invasion | [120] |
| CD73 | miR-422a | Inhibits adenosine production | [121] | |
| COX-2 | miR-708 | Decreases proliferation, survival, and migration of lung cancer cells | [122] | |
| mPGES-1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alvanou, M.; Lysandrou, M.; Christophi, P.; Psatha, N.; Spyridonidis, A.; Papadopoulou, A.; Yannaki, E. Empowering the Potential of CAR-T Cell Immunotherapies by Epigenetic Reprogramming. Cancers 2023, 15, 1935. https://doi.org/10.3390/cancers15071935
Alvanou M, Lysandrou M, Christophi P, Psatha N, Spyridonidis A, Papadopoulou A, Yannaki E. Empowering the Potential of CAR-T Cell Immunotherapies by Epigenetic Reprogramming. Cancers. 2023; 15(7):1935. https://doi.org/10.3390/cancers15071935
Chicago/Turabian StyleAlvanou, Maria, Memnon Lysandrou, Panayota Christophi, Nikoleta Psatha, Alexandros Spyridonidis, Anastasia Papadopoulou, and Evangelia Yannaki. 2023. "Empowering the Potential of CAR-T Cell Immunotherapies by Epigenetic Reprogramming" Cancers 15, no. 7: 1935. https://doi.org/10.3390/cancers15071935
APA StyleAlvanou, M., Lysandrou, M., Christophi, P., Psatha, N., Spyridonidis, A., Papadopoulou, A., & Yannaki, E. (2023). Empowering the Potential of CAR-T Cell Immunotherapies by Epigenetic Reprogramming. Cancers, 15(7), 1935. https://doi.org/10.3390/cancers15071935