
Novel Insights into RAD52’s Structure, Function, and Druggability for Synthetic Lethality and Innovative Anticancer Therapies

 , and
, and 
Abstract
Simple Summary
Abstract
1. Introduction
2. RAD52 Functions
2.1. Homologous Recombination (HR)
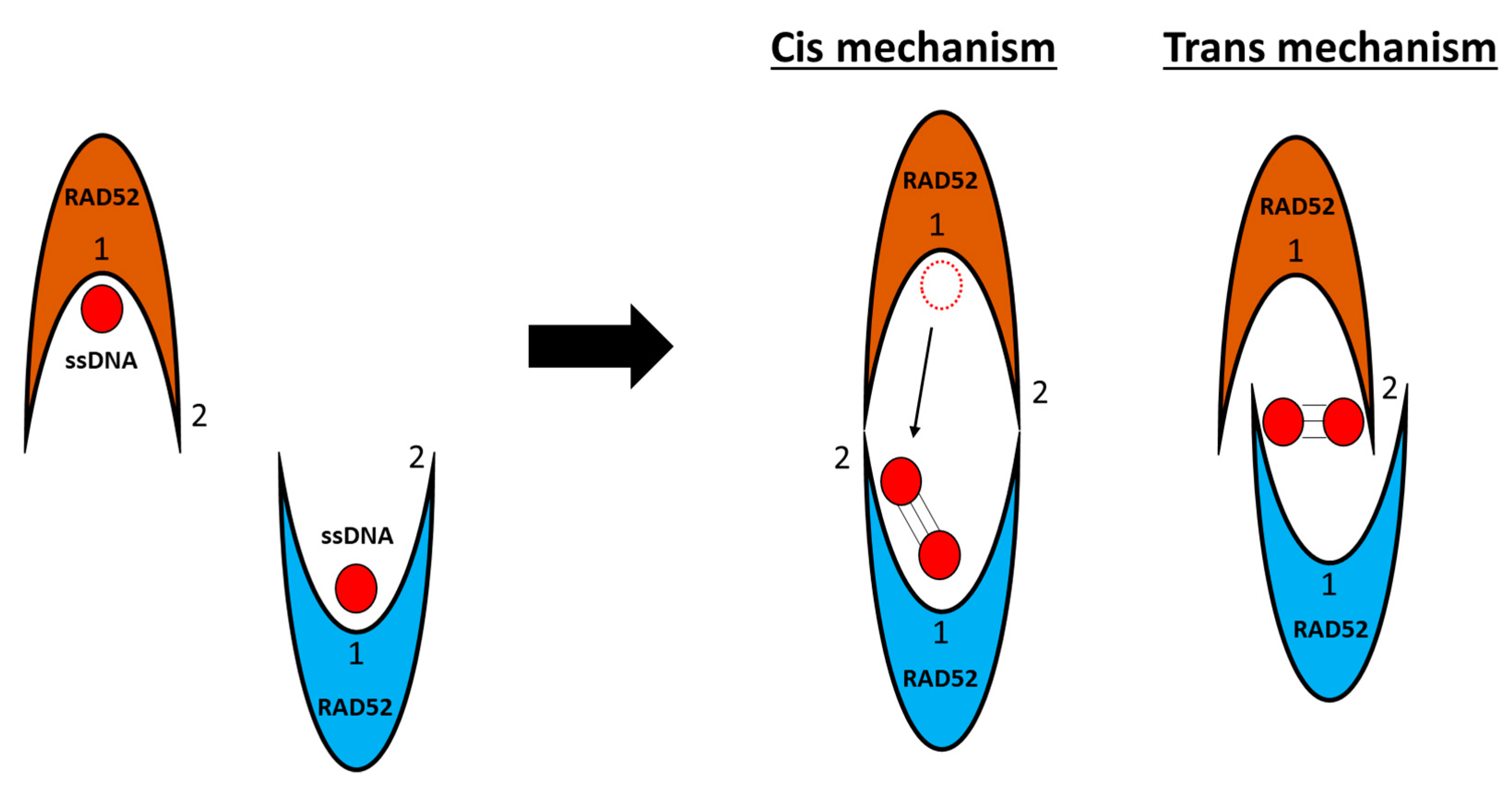
2.2. Single-Strand Annealing (SSA)
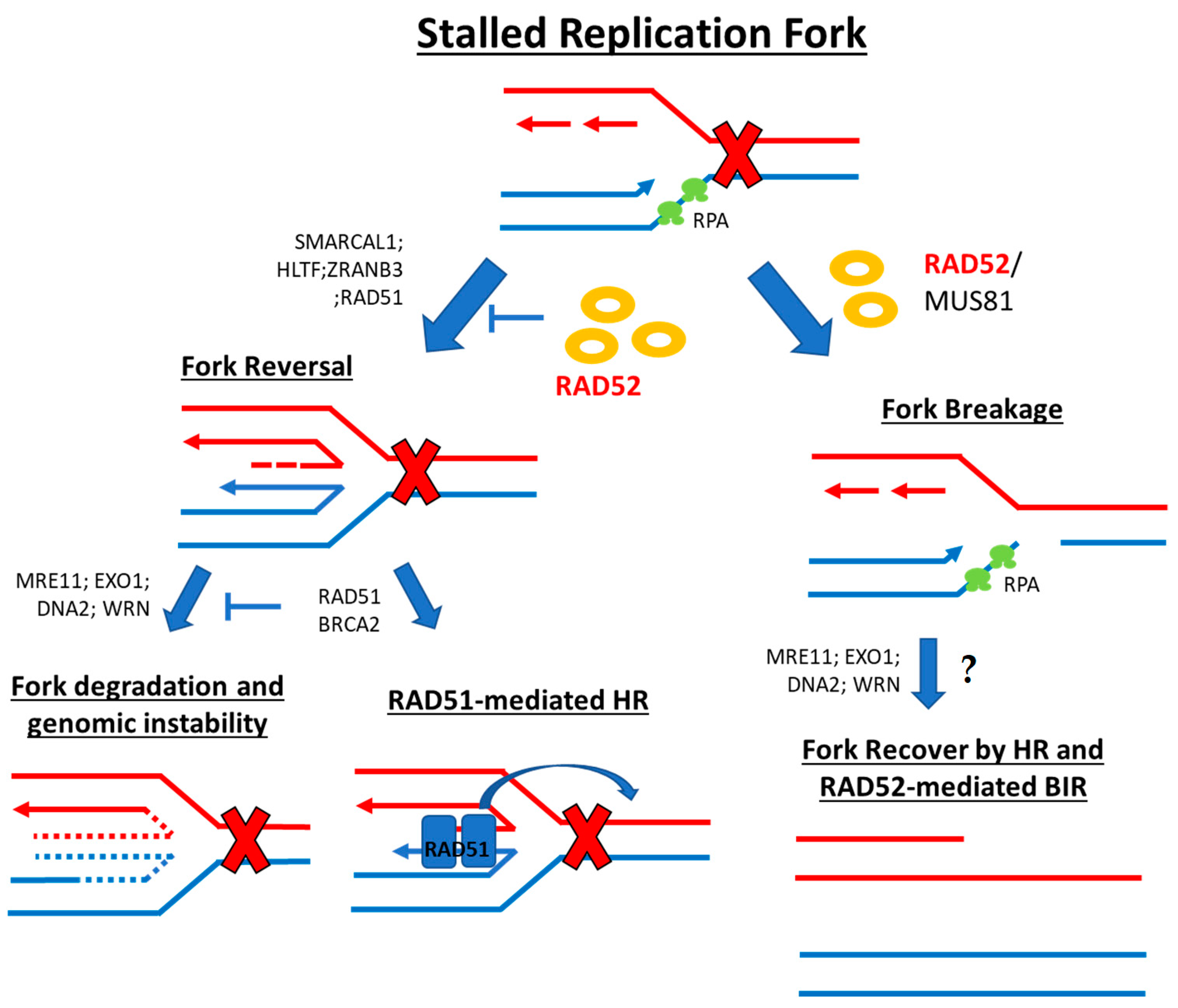
2.3. Stalled Replication Fork
2.4. RNA-Dependent DNA Repair
2.5. Regulatory Role
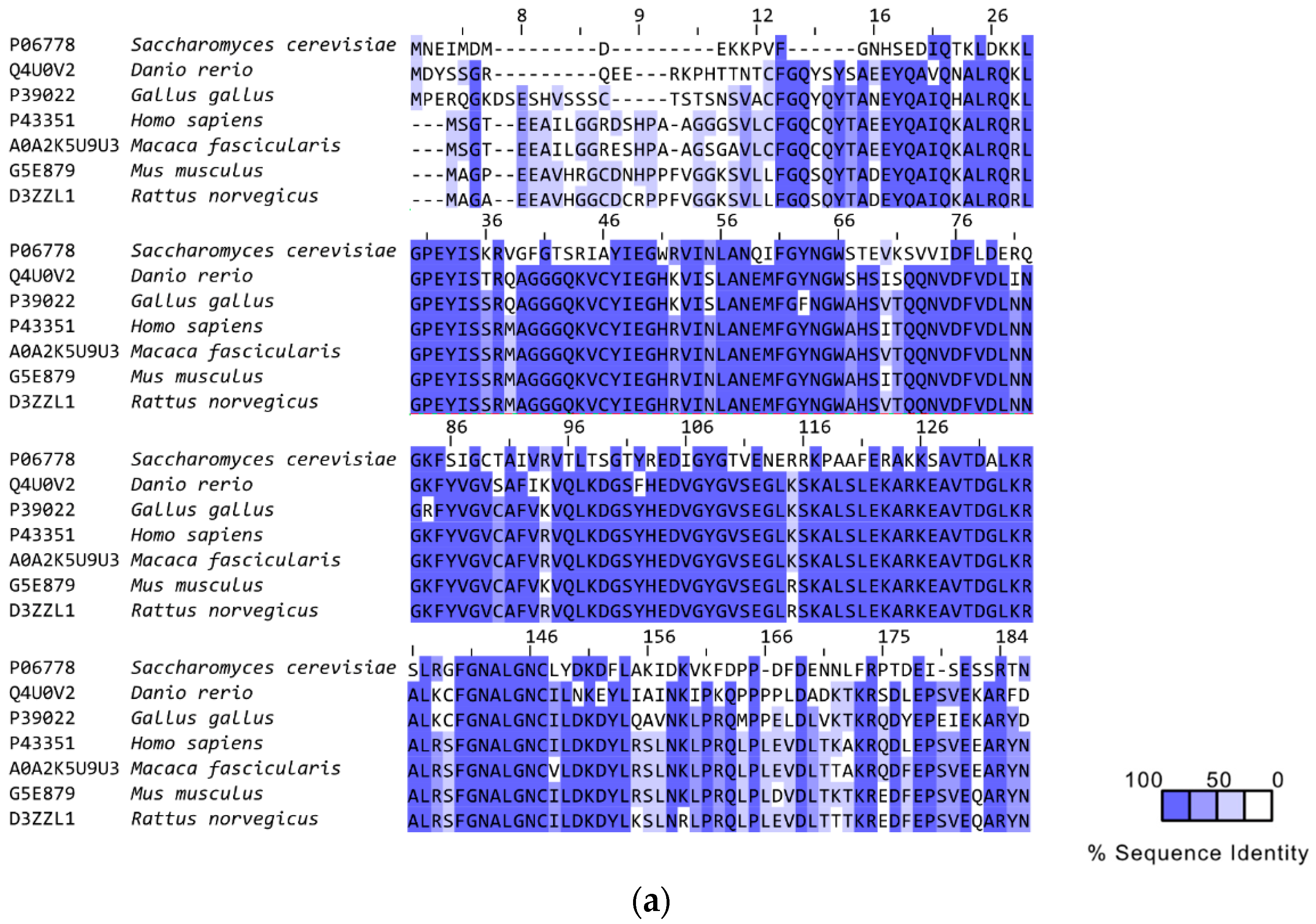
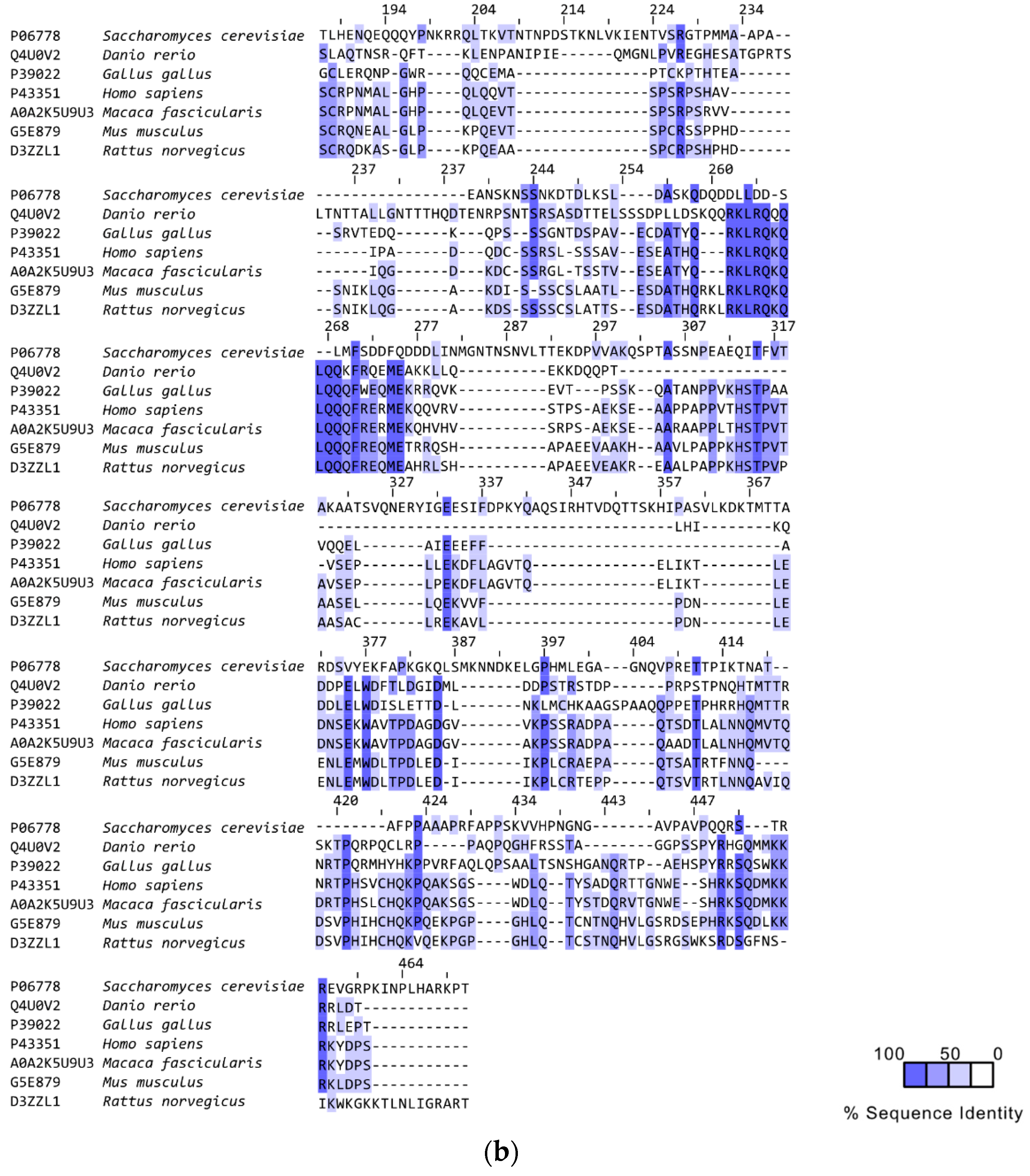
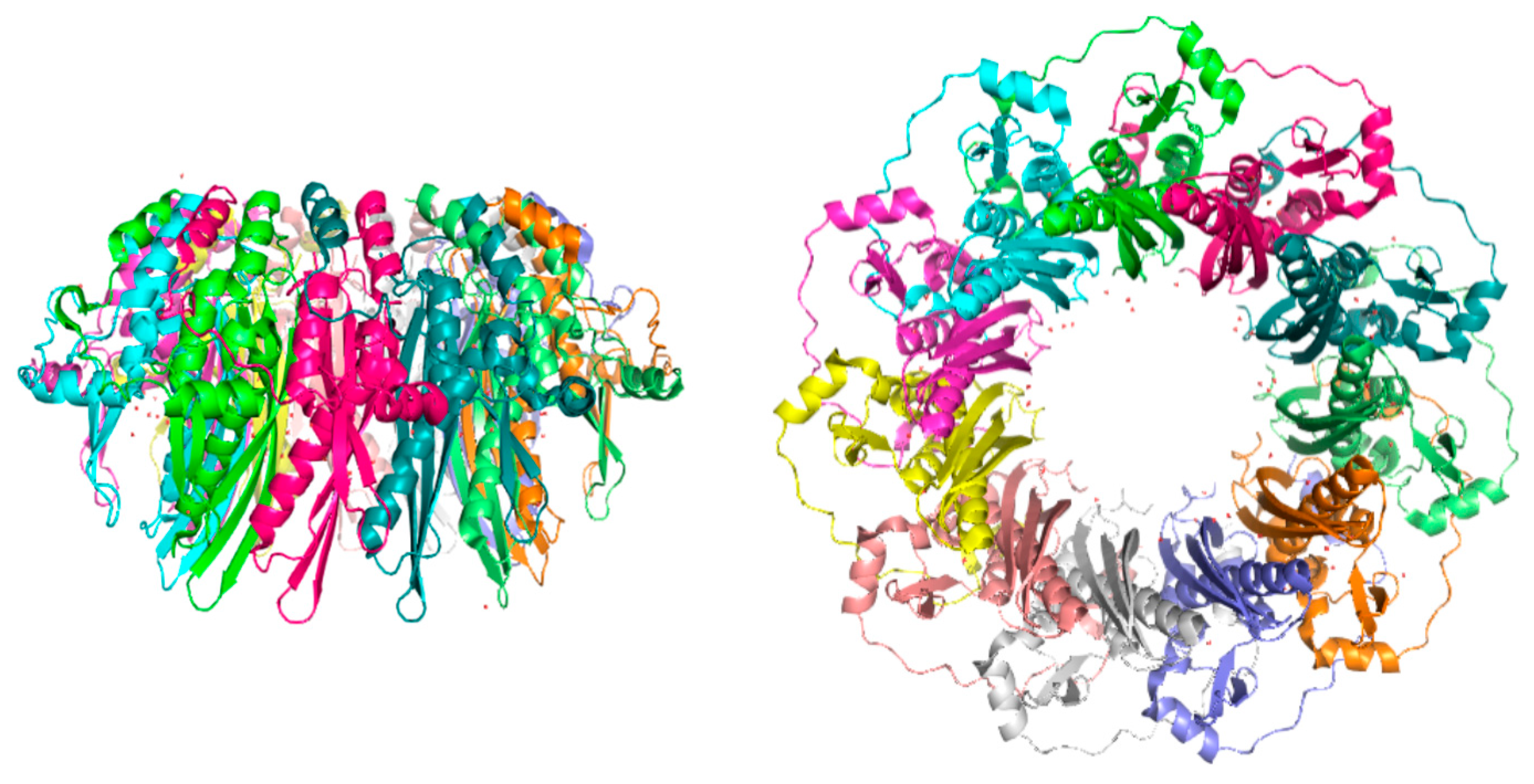
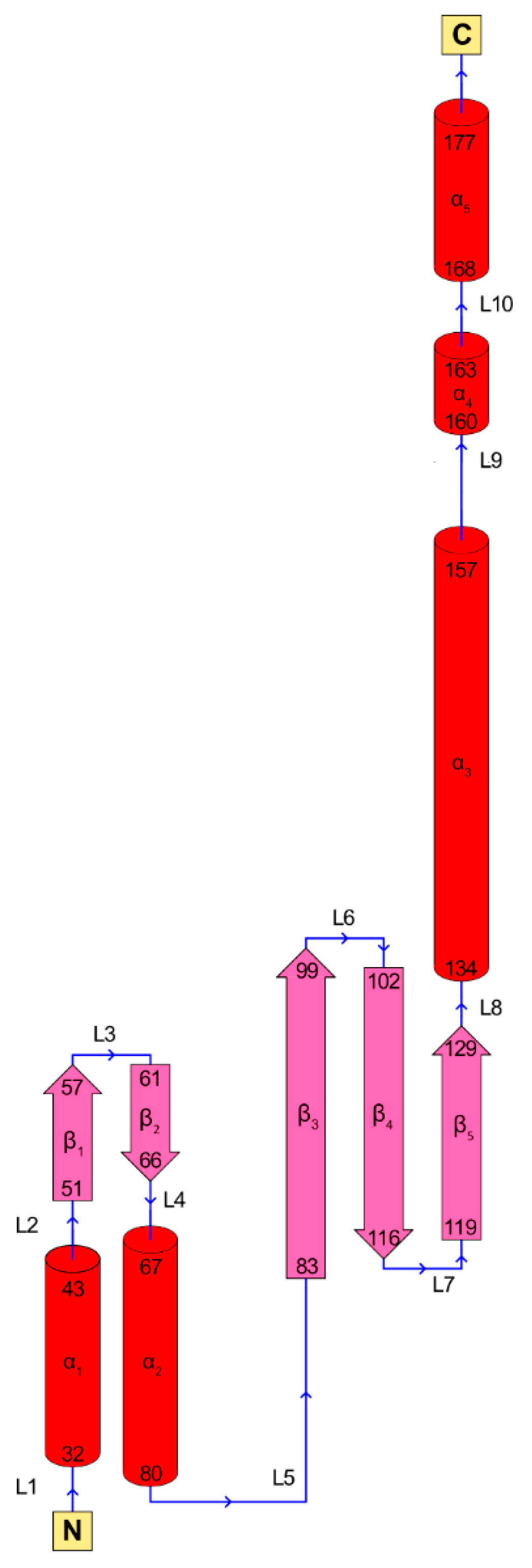
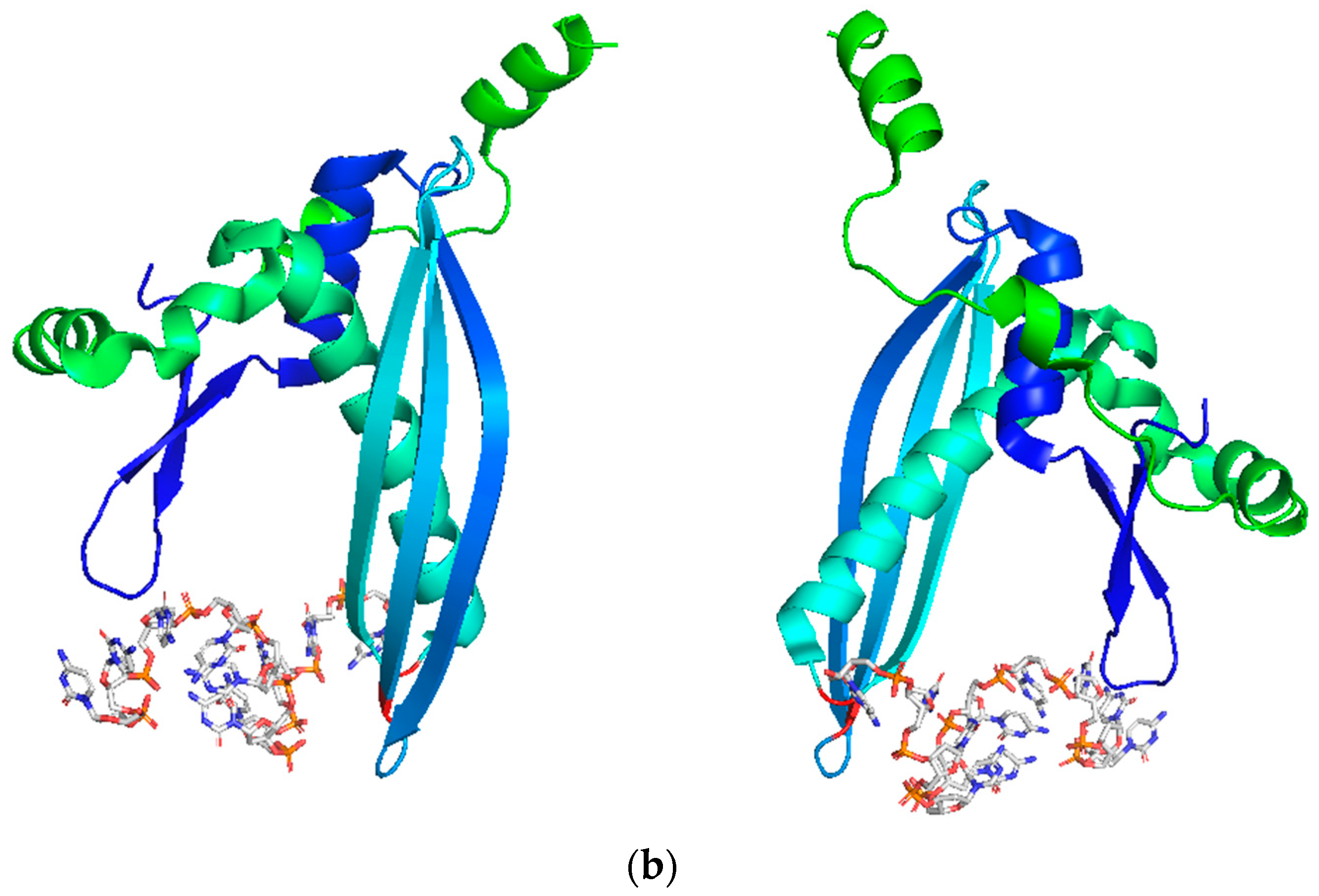
3. RAD52’s Structure
3.1. DNA/RNA Binding
3.2. Post-Translational Modification
4. RAD52 in Cancer
5. RAD52 Inhibition in Synthetic Lethality Therapies
5.1. F79 Peptide Aptamer
5.2. 6-OH-DOPA
5.3. A5MP/ZMP
5.4. D-I03
5.5. Epigallocatechin; Epigallocatechin-3-Monogallate; NP-004255
5.6. F779-0434
5.7. Curcumin
5.8. C791-0064
5.9. Quinacrine, Mitoxantrone, Doxorubicine
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Rossi, M.J.; DiDomenico, S.F.; Patel, M.; Mazin, A.V. RAD52: Paradigm of Synthetic Lethality and New Developments. Front. Genet. 2021, 12, 780293. [Google Scholar] [CrossRef]
- Toma, M.; Sullivan-Reed, K.; Śliwiński, T.; Skorski, T. RAD52 as a Potential Target for Synthetic Lethality-Based Anticancer Therapies. Cancers 2019, 11, 1561. [Google Scholar] [CrossRef]
- Rijkers, T.; Van Den Ouweland, J.; Morolli, B.; Rolink, A.G.; Baarends, W.M.; Van Sloun, P.P.; Lohman, P.H.; Pastink, A. Targeted inactivation of mouse RAD52 reduces homologous recombination but not resistance to ionizing radiation. Mol. Cell. Biol. 1998, 18, 6423–6429. [Google Scholar] [CrossRef]
- Yamaguchi-Iwai, Y.; Sonoda, E.; Buerstedde, J.M.; Bezzubova, O.; Morrison, C.; Takata, M.; Shinohara, A.; Takeda, S. Homologous recombination, but not DNA repair, is reduced in vertebrate cells deficient in RAD52. Mol. Cell. Biol. 1998, 18, 6430–6435. [Google Scholar] [CrossRef]
- Jalan, M.; Olsen, K.S.; Powell, S.N. Emerging Roles of RAD52 in Genome Maintenance. Cancers 2019, 11, 1038. [Google Scholar] [CrossRef]
- New, J.H.; Sugiyama, T.; Zaitseva, E.; Kowalczykowski, S.C. Rad52 protein stimulates DNA strand exchange by Rad51 and replication protein A. Nature 1998, 391, 407–410. [Google Scholar] [CrossRef]
- Sugiyama, T.; Kantake, N.; Wu, Y.; Kowalczykowski, S.C. Rad52-mediated DNA annealing after Rad51-mediated DNA strand exchange promotes second ssDNA capture. EMBO J. 2006, 25, 5539–5548. [Google Scholar] [CrossRef]
- Hanamshet, K.; Mazina, O.M.; Mazin, A.V. Reappearance from Obscurity: Mammalian Rad52 in Homologous Recombination. Genes 2016, 7, 63. [Google Scholar] [CrossRef]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691. [Google Scholar] [CrossRef]
- Lok, B.H.; Carley, A.C.; Tchang, B.; Powell, S.N. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene 2013, 32, 3552–3558. [Google Scholar] [CrossRef]
- Mahajan, S.; Raina, K.; Verma, S.; Rao, B.J. Human RAD52 protein regulates homologous recombination and checkpoint function in BRCA2 deficient cells. Int. J. Biochem. Cell Biol. 2019, 107, 128–139. [Google Scholar] [CrossRef]
- McIlwraith, M.J.; West, S.C. DNA repair synthesis facilitates RAD52-mediated second-end capture during DSB repair. Mol. Cell 2008, 29, 510–516. [Google Scholar] [CrossRef]
- Sugiyama, T.; Kantake, N. Dynamic regulatory interactions of rad51, rad52, and replication protein-a in recombination intermediates. J. Mol. Biol. 2009, 390, 45–55. [Google Scholar] [CrossRef]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef]
- Blasiak, J. Single-Strand Annealing in Cancer. Int. J. Mol. Sci. 2021, 22, 2167. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef]
- Chen, L.; Nievera, C.J.; Lee, A.Y.; Wu, X. Cell cycle-dependent complex formation of BRCA1-CtIP-MRN is important for DNA double-strand break repair. J. Biol. Chem. 2008, 283, 7713–7720. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Motycka, T.A.; Bessho, T.; Post, S.M.; Sung, P.; Tomkinson, A.E. Physical and functional interaction between the XPF/ERCC1 endonuclease and hRad52. J. Biol. Chem. 2004, 279, 13634–13639. [Google Scholar] [CrossRef]
- Al-Minawi, A.Z.; Saleh-Gohari, N.; Helleday, T. The ERCC1/XPF endonuclease is required for efficient single-strand annealing and gene conversion in mammalian cells. Nucleic Acids Res. 2008, 36, 1–9. [Google Scholar] [CrossRef]
- Stefanovie, B.; Hengel, S.R.; Mlcouskova, J.; Prochazkova, J.; Spirek, M.; Nikulenkov, F.; Nemecek, D.; Koch, B.G.; Bain, F.E.; Yu, L.; et al. DSS1 interacts with and stimulates RAD52 to promote the repair of DSBs. Nucleic Acids Res. 2020, 48, 694–708. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Kondratick, C.M.; Washington, M.T.; Spies, M. Making Choices: DNA Replication Fork Recovery Mechanisms. Semin. Cell Dev. Biol. 2021, 113, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Malacaria, E.; Pugliese, G.M.; Honda, M.; Marabitti, V.; Aiello, F.A.; Spies, M.; Franchitto, A.; Pichierri, P. Rad52 prevents excessive replication fork reversal and protects from nascent strand degradation. Nat. Commun. 2019, 10, 1412. [Google Scholar] [CrossRef] [PubMed]
- Mijic, S.; Zellweger, R.; Chappidi, N.; Berti, M.; Jacobs, K.; Mutreja, K.; Ursich, S.; Ray Chaudhuri, A.; Nussenzweig, A.; Janscak, P.; et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 2017, 8, 859. [Google Scholar] [CrossRef]
- Murfuni, I.; Basile, G.; Subramanyam, S.; Malacaria, E.; Bignami, M.; Spies, M.; Franchitto, A.; Pichierri, P. Survival of the replication checkpoint deficient cells requires MUS81-RAD52 function. PLoS Genet. 2013, 9, e1003910. [Google Scholar] [CrossRef]
- Malkova, A.; Ira, G. Break-induced replication: Functions and molecular mechanism. Curr. Opin. Genet. Dev. 2013, 23, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Costantino, L.; Sotiriou, S.K.; Rantala, J.K.; Magin, S.; Mladenov, E.; Helleday, T.; Haber, J.E.; Iliakis, G.; Kallioniemi, O.P.; Halazonetis, T.D. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 2014, 343, 88–91. [Google Scholar] [CrossRef]
- Sotiriou, S.K.; Kamileri, I.; Lugli, N.; Evangelou, K.; Da-Ré, C.; Huber, F.; Padayachy, L.; Tardy, S.; Nicati, N.L.; Barriot, S.; et al. Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks. Mol. Cell 2016, 64, 1127–1134. [Google Scholar] [CrossRef]
- Sharma, N.; Speed, M.C.; Allen, C.P.; Maranon, D.G.; Williamson, E.; Singh, S.; Hromas, R.; Nickoloff, J.A. Distinct roles of structure-specific endonucleases EEPD1 and Metnase in replication stress responses. NAR Cancer 2020, 2, zcaa008. [Google Scholar] [CrossRef]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Lukas, C.; Savic, V.; Bekker-Jensen, S.; Doil, C.; Neumann, B.; Pedersen, R.S.; Grøfte, M.; Chan, K.L.; Hickson, I.D.; Bartek, J.; et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 2011, 13, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Spies, J.; Lukas, C.; Somyajit, K.; Rask, M.B.; Lukas, J.; Neelsen, K.J. 53BP1 nuclear bodies enforce replication timing at under-replicated DNA to limit heritable DNA damage. Nat. Cell Biol. 2019, 21, 487–497. [Google Scholar] [CrossRef]
- Zhang, J.M.; Yadav, T.; Ouyang, J.; Lan, L.; Zou, L. Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cell Rep. 2019, 26, 955–968.e3. [Google Scholar] [CrossRef]
- Welty, S.; Teng, Y.; Liang, Z.; Zhao, W.; Sanders, L.H.; Greenamyre, J.T.; Rubio, M.E.; Thathiah, A.; Kodali, R.; Wetzel, R.; et al. RAD52 is required for RNA-templated recombination repair in post-mitotic neurons. J. Biol. Chem. 2018, 293, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Nandi, P.; Taylor, M.B.; Stuckey, S.; Bhadsavle, H.P.; Weiss, B.; Storici, F. RNA-driven genetic changes in bacteria and in human cells. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2011, 717, 91–98. [Google Scholar] [CrossRef]
- Keskin, H.; Shen, Y.; Huang, F.; Patel, M.; Yang, T.; Ashley, K.; Mazin, A.V.; Storici, F. Transcript-RNA-templated DNA recombination and repair. Nature 2014, 515, 436–439. [Google Scholar] [CrossRef]
- Mazina, O.M.; Keskin, H.; Hanamshet, K.; Storici, F.; Mazin, A.V. Rad52 Inverse Strand Exchange Drives RNA-Templated DNA Double-Strand Break Repair. Mol. Cell. 2017, 67, 19–29.e3. [Google Scholar] [CrossRef]
- McDevitt, S.; Rusanov, T.; Kent, T.; Chandramouly, G.; Pomerantz, R.T. How RNA transcripts coordinate DNA recombination and repair. Nat. Commun. 2018, 9, 1091. [Google Scholar] [CrossRef]
- Chandramouly, G.; Zhao, J.; McDevitt, S.; Rusanov, T.; Hoang, T.; Borisonnik, N.; Treddinick, T.; Lopezcolorado, F.W.; Kent, T.; Siddique, L.A.; et al. Polθ reverse transcribes RNA and promotes RNA-templated DNA repair. Sci. Adv. 2021, 7, eabf1771. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Ghodke, P.P.; Egli, M.; Li, L.; Wang, Y.; Guengerich, F.P. Human DNA polymerase η has reverse transcriptase activity in cellular environments. J. Biol. Chem. 2019, 294, 6073–6081. [Google Scholar] [CrossRef] [PubMed]
- Meers, C.; Keskin, H.; Banyai, G.; Mazina, O.; Yang, T.; Gombolay, A.L.; Mukherjee, K.; Kaparos, E.I.; Newnam, G.; Mazin, A.; et al. Genetic Characterization of Three Distinct Mechanisms Supporting RNA-Driven DNA Repair and Modification Reveals Major Role of DNA Polymerase ζ. Mol. Cell 2020, 79, 1037–1050.e5. [Google Scholar] [CrossRef]
- Aymard, F.; Bugler, B.; Schmidt, C.K.; Guillou, E.; Caron, P.; Briois, S.; Iacovoni, J.S.; Daburon, V.; Miller, K.M.; Jackson, S.P.; et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21, 366–374. [Google Scholar] [CrossRef]
- Teng, Y.; Yadav, T.; Duan, M.; Tan, J.; Xiang, Y.; Gao, B.; Xu, J.; Liang, Z.; Liu, Y.; Nakajima, S.; et al. ROS-induced R loops trigger a transcription-coupled but BRCA1/2-independent homologous recombination pathway through CSB. Nat. Commun. 2018, 9, 4115. [Google Scholar] [CrossRef]
- Yasuhara, T.; Kato, R.; Hagiwara, Y.; Shiotani, B.; Yamauchi, M.; Nakada, S.; Shibata, A.; Miyagawa, K. Human Rad52 Promotes XPG-Mediated R-loop Processing to Initiate Transcription-Associated Homologous Recombination Repair. Cell 2018, 175, 558–570.e11. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yang, H.; Zhu, X.; Yadav, T.; Ouyang, J.; Truesdell, S.S.; Tan, J.; Wang, Y.; Duan, M.; Wei, L.; et al. m5C modification of mRNA serves a DNA damage code to promote homologous recombination. Nat. Commun. 2020, 11, 2834. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Oh, Y.T.; Li, Z.; Dou, J.; Tang, S.; Wang, X.; Wang, H.; Takeda, S.; Wang, Y. RAD52 Adjusts Repair of Single-Strand Breaks via Reducing DNA-Damage-Promoted XRCC1/LIG3α Co-localization. Cell Rep. 2021, 34, 108625. [Google Scholar] [CrossRef]
- Kagawa, W.; Kurumizaka, H.; Ishitani, R.; Fukai, S.; Nureki, O.; Shibata, T.; Yokoyama, S. Crystal structure of the homologous-pairing domain from the human Rad52 recombinase in the undecameric form. Mol. Cell 2002, 10, 359–371. [Google Scholar] [CrossRef]
- Shen, Z.; Cloud, K.G.; Chen, D.J.; Park, M.S. Specific interactions between the human RAD51 and RAD52 proteins. J. Biol. Chem. 1996, 271, 148–152. [Google Scholar] [CrossRef]
- Ranatunga, W.; Jackson, D.; Lloyd, J.A.; Forget, A.L.; Knight, K.L.; Borgstahl, G.E. Human RAD52 exhibits two modes of self-association. J. Biol. Chem. 2001, 276, 15876–15880. [Google Scholar] [CrossRef] [PubMed]
- Stasiak, A.Z.; Larquet, E.; Stasiak, A.; Müller, S.; Engel, A.; Van Dyck, E.; West, S.C.; Egelman, E.H. The human Rad52 protein exists as a heptameric ring. Curr. Biol. 2000, 10, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Van Dyck, E.; Hajibagheri, N.M.; Stasiak, A.; West, S.C. Visualisation of human rad52 protein and its complexes with hRad51 and DNA. J. Mol. Biol. 1998, 284, 1027–1038. [Google Scholar] [CrossRef]
- Van Dyck, E.; Stasiak, A.Z.; Stasiak, A.; West, S.C. Visualization of recombination intermediates produced by RAD52-mediated single-strand annealing. EMBO Rep. 2001, 2, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, C.; Takizawa, Y.; Saotome, M.; Ogino, S.; Kurumizaka, H.; Kagawa, W. The cryo-EM structure of full-length RAD52 protein contains an undecameric ring. FEBS Open Bio 2023, 13, 408–418. [Google Scholar] [CrossRef]
- Ranatunga, W.; Jackson, D. Flowers II RA 2nd, Borgstahl GE. Human RAD52 protein has extreme thermal stability. Biochemistry 2001, 40, 8557–8562. [Google Scholar] [CrossRef]
- Kito, K.; Wada, H.; Yeh, E.T.; Kamitani, T. Identification of novel isoforms of human RAD52. Biochim. Biophys. Acta 1999, 1489, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol. Mol. Biol. Rev. 2002, 66, 630–670. [Google Scholar] [CrossRef]
- Saotome, M.; Saito, K.; Yasuda, T.; Ohtomo, H.; Sugiyama, S.; Nishimura, Y.; Kurumizaka, H.; Kagawa, W. Structural Basis of Homology-Directed DNA Repair Mediated by RAD52. iScience 2018, 3, 50–62. [Google Scholar] [CrossRef]
- Singleton, M.R.; Wentzell, L.M.; Liu, Y.; West, S.C.; Wigley, D.B. Structure of the single-strand annealing domain of human RAD52 protein. Proc. Natl. Acad. Sci. USA 2002, 99, 13492–13497. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Ferrin, T.E. Visualizing density maps with UCSF Chimera. J. Struct. Biol. 2007, 157, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Kagawa, W.; Kurumizaka, H.; Ikawa, S.; Yokoyama, S.; Shibata, T. Homologous pairing promoted by the human Rad52 protein. J. Biol. Chem. 2001, 276, 35201–35208. [Google Scholar] [CrossRef] [PubMed]
- Kagawa, W.; Kagawa, A.; Saito, K.; Ikawa, S.; Shibata, T.; Kurumizaka, H.; Yokoyama, S. Identification of a second DNA binding site in the human Rad52 protein. J. Biol. Chem. 2008, 283, 24264–24273. [Google Scholar] [CrossRef]
- Van Dyck, E.; Stasiak, A.Z.; Stasiak, A.; West, S.C. Binding of double-strand breaks in DNA by human Rad52 protein. Nature 1999, 398, 728–731, Erratum in Nature 1999, 401, 403. [Google Scholar] [CrossRef] [PubMed]
- Chandramouly, G.; McDevitt, S.; Sullivan, K.; Kent, T.; Luz, A.; Glickman, J.F.; Andrake, M.; Skorski, T.; Pomerantz, R.T. Small-Molecule Disruption of RAD52 Rings as a Mechanism for Precision Medicine in BRCA-Deficient Cancers. Chem. Biol. 2015, 22, 1491–1504. [Google Scholar] [CrossRef] [PubMed]
- Grimme, J.M.; Spies, M. FRET-based assays to monitor DNA binding and annealing by Rad52 recombination mediator protein. Methods Mol. Biol. 2011, 745, 463–483. [Google Scholar] [PubMed]
- Rothenberg, E.; Grimme, J.M.; Spies, M.; Ha, T. Human Rad52-mediated homology search and annealing occurs by continuous interactions between overlapping nucleoprotein complexes. Proc. Natl. Acad. Sci. USA 2008, 105, 20274–20279. [Google Scholar] [CrossRef]
- Sugiyama, T.; New, J.H.; Kowalczykowski, S.C. DNA annealing by Rad52 Protein is stimulated by specific interaction with the complex of replication protein A and single-stranded DNA. Proc. Natl. Acad. Sci. USA 1998, 95, 6049–6054. [Google Scholar] [CrossRef]
- Mortensen, U.H.; Bendixen, C.; Sunjevaric, I.; Rothstein, R. DNA strand annealing is promoted by the yeast Rad52 protein. Proc. Natl. Acad. Sci. USA 1996, 93, 10729–10734. [Google Scholar] [CrossRef]
- Lloyd, J.A.; McGrew, D.A.; Knight, K.L. Identification of residues important for DNA binding in the full-length human Rad52 protein. J. Mol. Biol. 2005, 345, 239–249. [Google Scholar] [CrossRef]
- Hanamshet, K.; Mazin, A.V. The function of RAD52 N-terminal domain is essential for viability of BRCA-deficient cells. Nucleic Acids Res. 2020, 48, 12778–12791. [Google Scholar] [CrossRef] [PubMed]
- Grimme, J.M.; Honda, M.; Wright, R.; Okuno, Y.; Rothenberg, E.; Mazin, A.V.; Ha, T.; Spies, M. Human Rad52 binds and wraps single-stranded DNA and mediates annealing via two hRad52-ssDNA complexes. Nucleic Acids Res. 2010, 38, 2917–2930. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yang, H.; Pavletich, N.P. Mechanism of homologous recombination from the RecA–ssDNA/dsDNA structures. Nature 2008, 453, 489–494. [Google Scholar] [CrossRef]
- Sugiman-Marangos, S.N.; Weiss, Y.M.; Junop, M.S. Mechanism for accurate, protein-assisted DNA annealing by Deinococcus radiodurans DdrB. Proc. Natl. Acad. Sci. USA 2016, 113, 4308–4313. [Google Scholar] [CrossRef]
- Ma, C.J.; Kwon, Y.; Sung, P.; Greene, E.C. Human RAD52 interactions with replication protein A and the RAD51 presynaptic complex. J. Biol. Chem. 2017, 292, 11702–11713. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, I.; Zhang, H.; Candelli, A.; Normanno, D.; Peterman, E.J.G.; Wuite, G.J.L.; Modesti, M. Human RAD52 Captures and Holds DNA Strands, Increases DNA Flexibility, and Prevents Melting of Duplex DNA: Implications for DNA Recombination. Cell Rep. 2017, 18, 2845–2853. [Google Scholar] [CrossRef]
- Yasuda, T.; Kagawa, W.; Ogi, T.; Kato, T.A.; Suzuki, T.; Dohmae, N.; Takizawa, K.; Nakazawa, Y.; Genet, M.D.; Saotome, M.; et al. Novel function of HATs and HDACs in homologous recombination through acetylation of human RAD52 at double-strand break sites. PLoS Genet. 2018, 14, e1007277. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Takizawa, K.; Ui, A.; Hama, M.; Kagawa, W.; Sugasawa, K.; Tajima, K. Human SIRT2 and SIRT3 deacetylases function in DNA homologous recombinational repair. Genes Cells 2021, 26, 328–335. [Google Scholar] [CrossRef]
- Charifi, F.; Churikov, D.; Eckert-Boulet, N.; Minguet, C.; Jourquin, F.; Hardy, J.; Lisby, M.; Simon, M.N.; Géli, V. Rad52 SUMOylation functions as a molecular switch that determines a balance between the Rad51- and Rad59-dependent survivors. iScience 2021, 24, 102231. [Google Scholar] [CrossRef]
- Saito, K.; Kagawa, W.; Suzuki, T.; Suzuki, H.; Yokoyama, S.; Saitoh, H.; Tashiro, S.; Dohmae, N.; Kurumizaka, H. The putative nuclear localization signal of the human RAD52 protein is a potential sumoylation site. J. Biochem. 2010, 147, 833–842. [Google Scholar] [CrossRef]
- Cramer, K.; Nieborowska-Skorska, M.; Koptyra, M.; Slupianek, A.; Penserga, E.T.; Eaves, C.J.; Aulitzky, W.; Skorski, T. BCR/ABL and other kinases from chronic myeloproliferative disorders stimulate single-strand annealing, an unfaithful DNA double-strand break repair. Cancer Res. 2008, 68, 6884–6888. [Google Scholar] [CrossRef]
- Fernandes, M.S.; Reddy, M.M.; Gonneville, J.R.; DeRoo, S.C.; Podar, K.; Griffin, J.D.; Weinstock, D.M.; Sattler, M. BCR-ABL promotes the frequency of mutagenic single-strand annealing DNA repair. Blood 2009, 114, 1813–1819. [Google Scholar] [CrossRef] [PubMed]
- Honda, M.; Okuno, Y.; Yoo, J.; Ha, T.; Spies, M. Tyrosine phosphorylation enhances RAD52-mediated annealing by modulating its DNA binding. EMBO J. 2011, 30, 3368–3382. [Google Scholar] [CrossRef]
- Cramer-Morales, K.; Nieborowska-Skorska, M.; Scheibner, K.; Padget, M.; Irvine, D.A.; Sliwinski, T.; Haas, K.; Lee, J.; Geng, H.; Roy, D.; et al. Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood 2013, 122, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Guo, Y.; Zhou, L.; Ge, Y.; Wei, L.; Li, L.; Zhou, C.; Wei, J.; Yuan, Q.; Li, J.; et al. Association of a functional RAD52 genetic variant locating in a miRNA binding site with risk of HBV-related hepatocellular carcinoma. Mol. Carcinog. 2015, 54, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Naccarati, A.; Rosa, F.; Vymetalkova, V.; Barone, E.; Jiraskova, K.; Di Gaetano, C.; Novotny, J.; Levy, M.; Vodickova, L.; Gemignani, F.; et al. Double-strand break repair and colorectal cancer: Gene variants within 3’ UTRs and microRNAs binding as modulators of cancer risk and clinical outcome. Oncotarget 2016, 7, 23156–23169. [Google Scholar] [CrossRef] [PubMed]
- Adamson, A.W.; Ding, Y.C.; Mendez-Dorantes, C.; Bailis, A.M.; Stark, J.M.; Neuhausen, S.L. The RAD52 S346X variant reduces risk of developing breast cancer in carriers of pathogenic germline BRCA2 mutations. Mol. Oncol. 2020, 14, 1124–1133. [Google Scholar] [CrossRef]
- Shi, T.Y.; Yang, G.; Tu, X.Y.; Yang, J.M.; Qian, J.; Wu, X.H.; Zhou, X.Y.; Cheng, X.; Wei, Q. RAD52 variants predict platinum resistance and prognosis of cervical cancer. PLoS ONE 2012, 7, e50461. [Google Scholar] [CrossRef]
- Ho, V.; Chung, L.; Singh, A.; Lea, V.; Abubakar, A.; Lim, S.H.; Chua, W.; Ng, W.; Lee, M.; Roberts, T.L.; et al. Aberrant Expression of RAD52, Its Prognostic Impact in Rectal Cancer and Association with Poor Survival of Patients. Int. J. Mol. Sci. 2020, 21, 1768. [Google Scholar] [CrossRef]
- Kan, Y.; Batada, N.N.; Hendrickson, E.A. Human somatic cells deficient for RAD52 are impaired for viral integration and compromised for most aspects of homology-directed repair. DNA Repair 2017, 55, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, R.; You, M. Corrupting the DNA damage response: A critical role for Rad52 in tumor cell survival. Aging 2017, 9, 1647–1659. [Google Scholar] [CrossRef]
- Hironaka, K.; Factor, V.M.; Calvisi, D.F.; Conner, E.A.; Thorgeirsson, S.S. Dysregulation of DNA repair pathways in a transforming growth factor alpha/c-myc transgenic mouse model of accelerated hepatocarcinogenesis. Lab. Investig. 2003, 83, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Treuner, K.; Helton, R.; Barlow, C. Loss of Rad52 partially rescues tumorigenesis and T-cell maturation in Atm-deficient mice. Oncogene 2004, 23, 4655–4661. [Google Scholar] [CrossRef] [PubMed]
- Kohzaki, M.; Ootsuyama, A.; Sun, L.; Moritake, T.; Okazaki, R. Human RECQL4 represses the RAD52-mediated single-strand annealing pathway after ionizing radiation or cisplatin treatment. Int. J. Cancer 2020, 146, 3098–3113. [Google Scholar] [CrossRef]
- Lieberman, R.; Pan, J.; Zhang, Q.; You, M. Rad52 deficiency decreases development of lung squamous cell carcinomas by enhancing immuno-surveillance. Oncotarget 2017, 8, 34032–34044. [Google Scholar] [CrossRef]
- Sullivan-Reed, K.; Bolton-Gillespie, E.; Dasgupta, Y.; Langer, S.; Siciliano, M.; Nieborowska-Skorska, M.; Hanamshet, K.; Belyaeva, E.A.; Bernhardy, A.J.; Lee, J.; et al. Simultaneous Targeting of PARP1 and RAD52 Triggers Dual Synthetic Lethality in BRCA-Deficient Tumor Cells. Cell Rep. 2018, 23, 3127–3136. [Google Scholar] [CrossRef]
- Bhat, D.S.; Spies, M.A.; Spies, M. A moving target for drug discovery: Structure activity relationship and many genome (de)stabilizing functions of the RAD52 protein. DNA Repair 2022, 120, 103421. [Google Scholar] [CrossRef]
- Myers, S.H.; Ortega, J.A.; Cavalli, A. Synthetic Lethality through the Lens of Medicinal Chemistry. J. Med. Chem. 2020, 63, 14151–14183. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Buechelmaier, E.S.; Powell, S.N. Rad51 paralog complexes BCDX2 and CX3 act at different stages in the BRCA1-BRCA2-dependent homologous recombination pathway. Mol. Cell. Biol. 2013, 33, 387–395. [Google Scholar] [CrossRef]
- Sharma, A.B.; Erasimus, H.; Pinto, L.; Caron, M.C.; Gopaul, D.; Peterlini, T.; Neumann, K.; Nazarov, P.V.; Fritah, S.; Klink, B.; et al. XAB2 promotes Ku eviction from single-ended DNA double-strand breaks independently of the ATM kinase. Nucleic Acids Res. 2021, 49, 9906–9925. [Google Scholar] [CrossRef]
- Hromas, R.; Kim, H.S.; Sidhu, G.; Williamson, E.; Jaiswal, A.; Totterdale, T.A.; Nole, J.; Lee, S.H.; Nickoloff, J.A.; Kong, K.Y. The endonuclease EEPD1 mediates synthetic lethality in RAD52-depleted BRCA1 mutant breast cancer cells. Breast Cancer Res. 2017, 19, 122. [Google Scholar] [CrossRef] [PubMed]
- Crosby, M.E.; Kulshreshtha, R.; Ivan, M.; Glazer, P.M. MicroRNA regulation of DNA repair gene expression in hypoxic stress. Cancer Res. 2009, 69, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Heng, C.; Li, Y.; Yu, L. MiR-302a sensitizes leukemia cells to etoposide by targeting Rad52. Oncotarget 2017, 8, 73884–73891. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.; Cramer-Morales, K.; McElroy, D.L.; Ostrov, D.A.; Haas, K.; Childers, W.; Hromas, R.; Skorski, T. Identification of a Small Molecule Inhibitor of RAD52 by Structure-Based Selection. PLoS ONE 2016, 11, e0147230. [Google Scholar] [CrossRef]
- Yang, Q.; Li, Y.; Sun, R.; Li, J. Identification of a RAD52 Inhibitor Inducing Synthetic Lethality in BRCA2-Deficient Cancer Cells. Front. Pharmacol. 2021, 12, 637825. [Google Scholar] [CrossRef]
- Huang, F.; Goyal, N.; Sullivan, K.; Hanamshet, K.; Patel, M.; Mazina, O.M.; Wang, C.X.; An, W.F.; Spoonamore, J.; Metkar, S.; et al. Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Res. 2016, 44, 4189–4199. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, Q.; Zhang, Y.; Huang, K.; Sun, R.; Zhao, Q. Compound F779-0434 causes synthetic lethality in BRCA2-deficient cancer cells by disrupting RAD52-ssDNA association. RSC Adv. 2018, 8, 18859–18869. [Google Scholar] [CrossRef] [PubMed]
- Hengel, S.R.; Malacaria, E.; Folly da Silva Constantino, L.; Bain, F.E.; Diaz, A.; Koch, B.G.; Yu, L.; Wu, M.; Pichierri, P.; Spies, M.A.; et al. Small-molecule inhibitors identify the RAD52-ssDNA interaction as critical for recovery from replication stress and for survival of BRCA2 deficient cells. eLife 2016, 5, e14740. [Google Scholar] [CrossRef]
- Al-Mugotir, M.; Lovelace, J.J.; George, J.; Bessho, M.; Pal, D.; Struble, L.; Kolar, C.; Rana, S.; Natarajan, A.; Bessho, T.; et al. Selective killing of homologous recombination-deficient cancer cell lines by inhibitors of the RPA:RAD52 protein-protein interaction. PLoS ONE 2021, 16, e0248941. [Google Scholar] [CrossRef]
- Xu, Y.; Lin, Y.; Luo, Y.; Yang, Y.; Long, B.; Fang, Z.; Liu, L.; Zhang, J.; Zhang, X. RAD52 aptamer regulates DNA damage repair and STAT3 in BRCA1/BRCA2-deficient human acute myeloid leukemia. Oncol. Rep. 2020, 44, 1455–1466. [Google Scholar] [CrossRef]
- Al Zaid Siddiquee, K.; Turkson, J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. 2008, 18, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, Ø.; Nepstad, I.; Hauge, M.; Hatfield, K.J.; Reikvam, H. STAT3 as a possible therapeutic target in human malignancies: Lessons from acute myeloid leukemia. Expert Rev. Hematol. 2015, 8, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Gunn, A.; Stark, J.M. I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol. Biol. 2012, 920, 379–391. [Google Scholar] [PubMed]
- Baell, J.B.; Nissink, J.W.M. Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017—Utility and Limitations. ACS Chem. Biol. 2018, 13, 36–44. [Google Scholar] [CrossRef]
- Olney, J.W.; Zorumski, C.F.; Stewart, G.R.; Price, M.T.; Wang, G.J.; Labruyere, J. Excitotoxicity of L-dopa and 6-OH-dopa: Implications for Parkinson’s and Huntington’s diseases. Exp. Neurol. 1990, 108, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Corton, J.M.; Gillespie, J.G.; Hawley, S.A.; Hardie, D.G. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 1995, 229, 558–565. [Google Scholar] [CrossRef]
- Elliott, T.S.; Slowey, A.; Ye, Y.; Conway, S.J. The use of phosphate bioisosteres in medicinal chemistry and chemical biology. Med. Chem. Comm. 2012, 3, 735–751. [Google Scholar] [CrossRef]
- Ronchetti, R.; Moroni, G.; Carotti, A.; Gioiello, A.; Camaioni, E. Recent advances in urea- And thiourea-containing compounds: Focus on innovative approaches in medicinal chemistry and organic synthesis. RSC Med. Chem. 2021, 12, 1046–1064. [Google Scholar] [CrossRef]
- Dalvit, C.; Flocco, M.; Knapp, S.; Mostardini, M.; Perego, R.; Stockman, B.J.; Veronesi, M.; Varasi, M. High-throughput NMR-based screening with competition binding experiments. J. Am. Chem. Soc. 2002, 124, 7702–7709. [Google Scholar] [CrossRef]
- Tseng, W.C.; Chen, C.Y.; Chern, C.Y.; Wang, C.A.; Lee, W.C.; Chi, Y.C.; Cheng, S.F.; Kuo, Y.T.; Chiu, Y.C.; Tseng, S.T.; et al. Targeting HR Repair as a Synthetic Lethal Approach to Increase DNA Damage Sensitivity by a RAD52 Inhibitor in BRCA2-Deficient Cancer Cells. Int. J. Mol. Sci. 2021, 22, 4422. [Google Scholar] [CrossRef]
- Wang, S.H.; Lin, P.Y.; Chiu, Y.C.; Huang, J.S.; Kuo, Y.T.; Wu, J.C.; Chen, C.C. Curcumin-Mediated HDAC Inhibition Suppresses the DNA Damage Response and Contributes to Increased DNA Damage Sensitivity. PLoS ONE 2015, 10, e0134110. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The Essential Medicinal Chemistry of Curcumin. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Initial Screening (HTS) | In Vitro Assays | In Cellulo Assays | In Vivo Assays | Group [Ref] |
|---|---|---|---|---|---|
| F79 aptamer | Rational design after mutagenic assay (ssDNA groove and protomer-protomer interaction) | RAD52-ssDNA (EMSA) | BRCA1/2± cell survival (viability and clonogenic: BRCA1/2− cells over BRCA1/2+ and combination with PARPi and leukemia standard treatments); foci (γH2AX: increase at 5 μM); apoptosis (flow cytometry: increase at 5 μM) | PARPi + F79 (2.5 mg/kg/day) synergic against BRCA1-deficient primary AML xenograft in NSG Mice | T. Skorski [84,96] |
| 6-OH-DOPA | RAD52-ssDNA (FP: 18,304 cpds – Sigma Lopac) | RAD52-ssDNA (FP: IC50 = 1.6 μM, EMSA); DSB repair (ssDNA annealing); RAD52 binding (ITC: Kd = 17.8 μM); oligomer dissociation (DLS; native gel analysis) | BRCA1/2± cell survival (viability and clonogenic: BRCA1/2− selective over BRCA1/2+); DSB repair (SSA selective over HR, NHEJ); foci (RAD52: decrease at 10 μM, γH2AX: increase at 10 μM); apoptosis (flow cytometry: increase at 20–40 μM); siRNA-RAD52 (western blot) | none | R. T. Pomerantz [65] |
| ZMP/A5MP | Virtual screening (docking – PDB: 1KN0 ssDNA groove: 1217 FDA and 139,735 NCI drug-like cpds – ZINC library, DOCK6.6 software) | RAD52-ssDNA (EMSA) | BRCA1/2± cell survival (growth rate: BRCA1/2− selective over BRCA1/2+); DSB repair (SSA: decrease at 20 μM); foci (RAD52: decrease at 20 μM) | none | T. Skorski [104] |
| D-I03 | DSB repair (ssDNA: 93,672 cpds from Diversity Oriented Synthesis (DOS) library and 279,231 cpds from Molecular Libraries Probe Center Network (MPLCN) - Broad Institute) | DSB repair (ssDNA annealing: IC50 = 5 μM, D-loop: IC50 = 8 μM); RAD52 binding (SPR: Kd = 25.8 μM) | BRCA1/2± cell survival (viability and clonogenic: IC50 = 14.5 μM for BRCA1/2− cells over BRCA1/2+ and combination with PARPi); foci (2.5 μM ≈ 2.0-fold reduction of RAD52 and not of RAD51); DSB repair (SSA, 30 μM ≈ 3.4-fold reduction and selective over HR) | PARPi (talazoparib)+D-I03 (50 mg/kg/day) synergic against BRCA1-deficient solid tumor growth in nude mice | A. V. Mazin [96,106] |
| Epigallocatechines (NP-004255) | RAD52-ssDNA (FRET: 2320 cpds from MicroSource SPECTRUM collection); Virtual screening (docking – PDB: 1KN0 ssDNA groove: AnalytiCon Discovery MEGx Natural Products Screen Library) | RAD52-ssDNA (FRET: IC50 = 1.5 μM); DSB repair (ssDNA annealing: IC50 = 7 μM); RAD52 binding (WaterLOGSY); | BRCA2± cell survival (viability); DSB repair (comet assay); siRNA-RAD52 (western blot) | none | M. A. Spies; M. Spies [108] |
| F779-0434 | Virtual screening (docking – PDB: 5JRB ssDNA groove: 47,737 cpds Targeted Diversity Library (TDL) – Chemdiv database, DOCK6.5 software) | RAD52-ssDNA (pull-down assay: 50% disruption at 5 μM); | BRCA2± cell survival (viability: BRCA2− selective over BRCA2+, 50% growth inhibition at 10 μM) | none | R. Sun; Q. Zhao [107] |
| C791-0064 | Virtual screening (docking – PDB: 5JRB self-association pocket: 66,608 cpds - Chemdiv database, DOCK6.5 software) | RAD52-ssDNA (EMSA); DSB repair (ssDNA annealing); RAD52 binding (MST); | BRCA2± cell survival (viability and clonogenic: IC50 = 29 μM for BRCA2− cells over BRCA+); siRNA-RAD52 and shRNA-BRCA (western blot); foci (γH2AX: increase at 40 μM); apoptosis (flow cytometry: increase at 40 μM, western blot) | none | J. Li [105] |
| mitoxantrone | RPA:RAD51 PPIs (FluorIA: 335 cpds – SellekChem, 1200 cpds - Prestwick, 100,000 cpds – Chembridge: EC50 = 30 μM) | - | BRCA1/2± cell survival (viability: EC50 = 0.1–2.5 μM for BRCA2− cells over BRCA+); DSB repair (SSA reduction and selective over HR up to 6 nM); apoptosis (western blot: increase at 0.2 μM); foci (RAD52: decrease at 3 nM over RAD51); RPA:RAD51 PPIs (co-immunoprecipitation) | none | T. Bessho; G. E. O. Borgstahl [109] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balboni, B.; Rinaldi, F.; Previtali, V.; Ciamarone, A.; Girotto, S.; Cavalli, A. Novel Insights into RAD52’s Structure, Function, and Druggability for Synthetic Lethality and Innovative Anticancer Therapies. Cancers 2023, 15, 1817. https://doi.org/10.3390/cancers15061817
Balboni B, Rinaldi F, Previtali V, Ciamarone A, Girotto S, Cavalli A. Novel Insights into RAD52’s Structure, Function, and Druggability for Synthetic Lethality and Innovative Anticancer Therapies. Cancers. 2023; 15(6):1817. https://doi.org/10.3390/cancers15061817
Chicago/Turabian StyleBalboni, Beatrice, Francesco Rinaldi, Viola Previtali, Andrea Ciamarone, Stefania Girotto, and Andrea Cavalli. 2023. "Novel Insights into RAD52’s Structure, Function, and Druggability for Synthetic Lethality and Innovative Anticancer Therapies" Cancers 15, no. 6: 1817. https://doi.org/10.3390/cancers15061817
APA StyleBalboni, B., Rinaldi, F., Previtali, V., Ciamarone, A., Girotto, S., & Cavalli, A. (2023). Novel Insights into RAD52’s Structure, Function, and Druggability for Synthetic Lethality and Innovative Anticancer Therapies. Cancers, 15(6), 1817. https://doi.org/10.3390/cancers15061817