
Endothelial Progenitor Cells Promote Osteosarcoma Progression and Invasiveness via AKT/PI3K Signaling
 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation, Expansion, and Characterization of EPCs
2.2. Osteosarcoma Cell Line Cultures
2.3. Preparation of EPC Conditioned Medium (CM)
2.4. Invasion Assay
2.5. Scratch Wound Healing Assay
2.6. VEGF-A and FGF2 Neutralization and PI3K Pathway Inhibition
2.7. RNA Isolation and Quantitative Real-Time PCR
2.8. Western Blot Analysis
2.9. Enzyme-Linked Immunosorbent Assay (ELISA)
2.10. Silencing VEGF Gene Expression
2.11. Proteomics Analysis of EPCs Conditioned Medium
2.12. Animal Model-Orthotopic Cell Injection to the Distal Femur
2.13. Histological Preparation
2.14. Immunohistochemistry
2.15. Statistical Analysis
3. Results
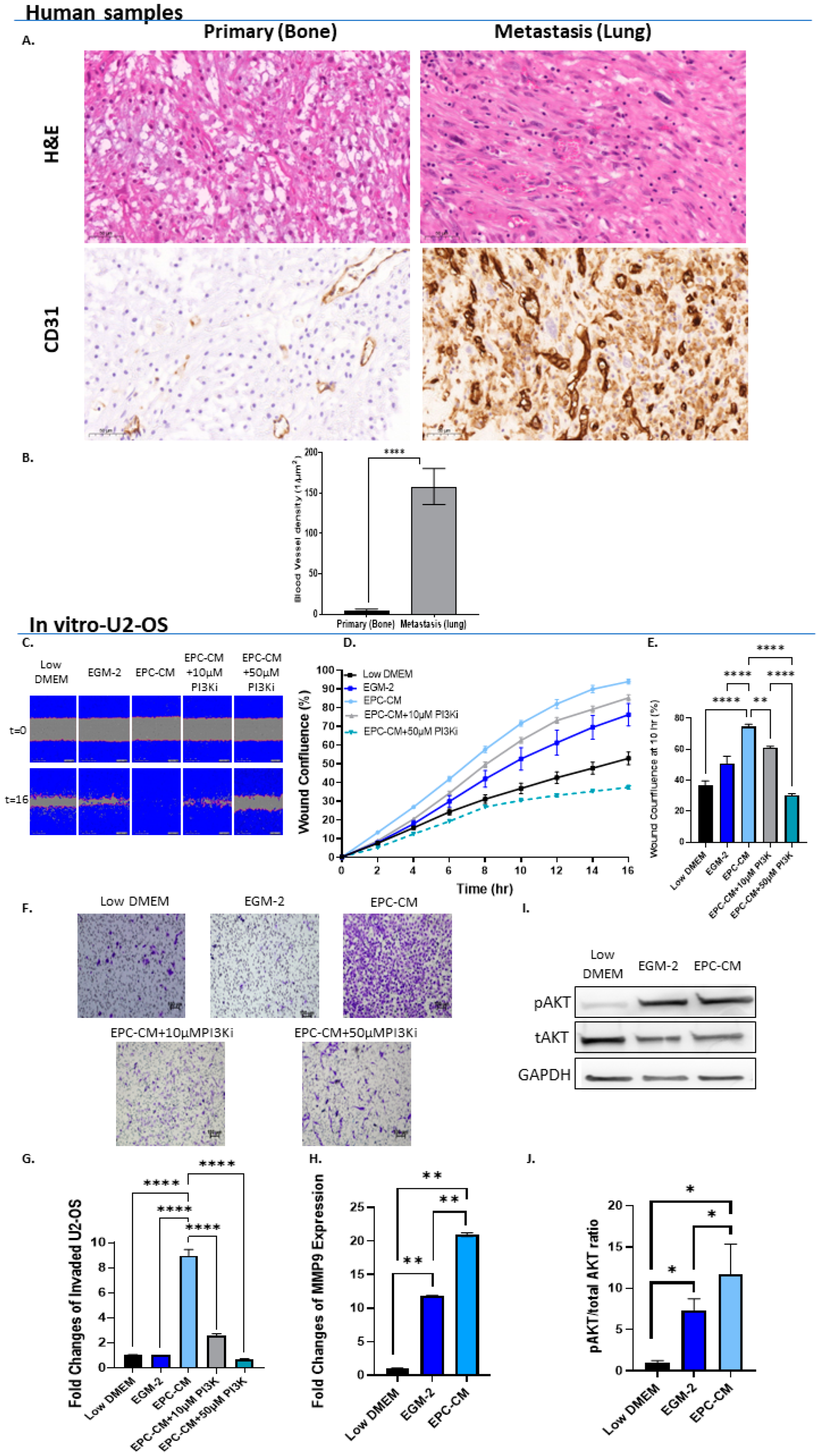
3.1. EPCs Promote OS In Vitro Migration and Invasion via PI3K/AKT Signaling
3.2. VEGF-A and FGF2 in the EPC-CM Influence OS Migration
3.3. OS Secretes VEGF Which Promotes Its Invasiveness in an Autocrine Manner
3.4. High VEGF-A and FGF2 Labeling Levels in Primary Mouse OS Tumors and Human Metastatic OS Specimens
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Durfee, R.A.; Mohammed, M.; Luu, H.H. Review of Osteosarcoma and Current Management. Rheumatol. Ther. 2016, 3, 221–243. [Google Scholar] [CrossRef] [PubMed]
- Mirabello, L.; Troisi, R.J.; Savage, S.A. Osteosarcoma Incidence and Survival Rates from 1973 to 2004: Data from the Surveillance, Epidemiology, and End Results Program. Cancer 2009, 115, 1531–1543. [Google Scholar] [CrossRef] [PubMed]
- Meazza, C.; Scanagatta, P. Metastatic Osteosarcoma: A Challenging Multidisciplinary Treatment. Expert. Rev. Anticancer Ther. 2016, 16, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Odri, G.A.; Tchicaya-Bouanga, J.; Yoon, D.J.Y.; Modrowski, D. Metastatic Progression of Osteosarcomas: A Review of Current Knowledge of Environmental versus Oncogenic Drivers. Cancers 2022, 14, 360. [Google Scholar] [CrossRef]
- Saha, D.; Saha, K.; Banerjee, A.; Jash, D. Osteosarcoma Relapse as Pleural Metastasis. South Asian J. Cancer 2013, 2, 56. [Google Scholar] [CrossRef]
- Carrle, D.; Bielack, S.S. Current Strategies of Chemotherapy in Osteosarcoma. Int. Orthop. 2006, 30, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Geller, D.S.; Gorlick, R. Osteosarcoma: A Review of Diagnosis, Management, and Treatment Strategies. Clin. Adv. Hematol. Oncol. 2010, 8, 705–718. [Google Scholar]
- Medina, R.J.; O’Neill, C.L.; Sweeney, M.; Guduric-Fuchs, J.; Gardiner, T.A.; Simpson, D.A.; Stitt, A.W. Molecular Analysis of Endothelial Progenitor Cell (EPC) Subtypes Reveals Two Distinct Cell Populations with Different Identities. BMC Med. Genom. 2010, 3, 18. [Google Scholar] [CrossRef]
- Salybekov, A.A.; Kobayashi, S.; Asahara, T. Characterization of Endothelial Progenitor Cell: Past, Present, and Future. Int. J. Mol. Sci. 2022, 23, 7697. [Google Scholar] [CrossRef]
- Tu, T.C.; Nagano, M.; Yamashita, T.; Hamada, H.; Ohneda, K.; Kimura, K.; Ohneda, O. A Chemokine Receptor, CXCR4, Which Is Regulated by Hypoxia-Inducible Factor 2α, Is Crucial for Functional Endothelial Progenitor Cells Migration to Ischemic Tissue and Wound Repair. Stem Cells Dev. 2016, 25, 266–276. [Google Scholar] [CrossRef]
- Hur, J.; Yoon, C.-H.; Kim, H.-S.; Choi, J.-H.; Kang, H.-J.; Hwang, K.-K.; Oh, B.-H.; Lee, M.-M.; Park, Y.-B. Characterization of Two Types of Endothelial Progenitor Cells and Their Different Contributions to Neovasculogenesis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Reale, A.; Melaccio, A.; Lamanuzzi, A.; Saltarella, I.; Dammacco, F.; Vacca, A.; Ria, R. Functional and Biological Role of Endothelial Precursor Cells in Tumour Progression: A New Potential Therapeutic Target in Haematological Malignancies. Stem Cells Int. 2016, 2016, 7954580. [Google Scholar] [CrossRef] [PubMed]
- Flamini, V.; Jiang, W.G.; Lane, J.; Cui, Y.-X. Significance and Therapeutic Implications of Endothelial Progenitor Cells in Angiogenic-Mediated Tumour Metastasis. Crit. Rev. Oncol./Hematol. 2016, 100, 177–189. [Google Scholar] [CrossRef]
- Yan, X.; Dai, X.; He, L.; Ling, X.; Shao, M.; Zhang, C.; Wang, Y.; Xiao, J.; Cai, L.; Li, X.; et al. A Novel CXCR4 Antagonist Enhances Angiogenesis via Modifying the Ischaemic Tissue Environment. J. Cell. Mol. Med. 2017, 21, 2298–2307. [Google Scholar] [CrossRef]
- Asahara, T.; Kawamoto, A. Endothelial Progenitor Cells for Postnatal Vasculogenesis. Am. J. Physiol.-Cell. Physiol. 2004, 287, C572–C579. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.; Willis, A.; Kornhauser, N.; Ward, M.M.; Lee, S.B.; Nackos, E.; Seo, B.R.; Chuang, E.; Cigler, T.; Moore, A.; et al. Influencing the Tumor Microenvironment: A Phase II Study of Copper Depletion Using Tetrathiomolybdate in Patients with Breast Cancer at High Risk for Recurrence and in Preclinical Models of Lung Metastases. Clin. Cancer Res. 2017, 23, 666–676. [Google Scholar] [CrossRef]
- Eveno, C.; Pocard, M. VEGF Levels and the Angiogenic Potential of the Microenvironment Can Affect Surgical Strategy for Colorectal Liver Metastasis. Cell. Adhes. Migr. 2012, 6, 569–573. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-Positive Haematopoietic Bone Marrow Progenitors Initiate the Pre-Metastatic Niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef]
- George, A.L.; Bangalore-Prakash, P.; Rajoria, S.; Suriano, R.; Shanmugam, A.; Mittelman, A.; Tiwari, R.K. Endothelial Progenitor Cell Biology in Disease and Tissue Regeneration. J. Hematol. Oncol. 2011, 4, 24. [Google Scholar] [CrossRef]
- Le Bourhis, X.; Romon, R.; Hondermarck, H. Role of Endothelial Progenitor Cells in Breast Cancer Angiogenesis: From Fundamental Research to Clinical Ramifications. Breast Cancer Res. Treat. 2010, 120, 17–24. [Google Scholar] [CrossRef]
- Tamari, T.; Kawar-Jaraisy, R.; Doppelt, O.; Giladi, B.; Sabbah, N.; Zigdon-Giladi, H. The Paracrine Role of Endothelial Cells in Bone Formation via CXCR4/SDF-1 Pathway. Cells 2020, 9, 1325. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, N.; Tamari, T.; Elimelech, R.; Doppelt, O.; Rudich, U.; Zigdon-Giladi, H. Predicting Angiogenesis by Endothelial Progenitor Cells Relying on In-Vitro Function Assays and VEGFR-2 Expression Levels. Biomolecules 2019, 9, 717. [Google Scholar] [CrossRef] [PubMed]
- Zigdon-Giladi, H.; Elimelech, R.; Michaeli-Geller, G.; Rudich, U.; Machtei, E.E. Safety Profile and Long-Term Engraftment of Human CD31+ Blood Progenitors in Bone Tissue Engineering. Cytotherapy 2017, 19, 895–908. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhu, L.B.; Peng, A.F.; Wang, T.F.; Long, X.H.; Gao, S.; Zhou, R.P.; Liu, Z.L. LY294002 Inhibits the Malignant Phenotype of Osteosarcoma Cells by Modulating the Phosphatidylinositol 3-kinase/Akt/Fatty Acid Synthase Signaling Pathway in Vitro. Mol. Med. Rep. 2015, 11, 1352–1357. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.; Gianferante, D.M.; Zhu, B.; Mirabello, L. Osteosarcoma: A Surveillance, Epidemiology, and End Results Program-Based Analysis from 1975 to 2017. Cancer 2022, 128, 2107–2118. [Google Scholar] [CrossRef] [PubMed]
- Ottaviani, G.; Jaffe, N. The Epidemiology of Osteosarcoma. Cancer Treat. Res. 2009, 152, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Cortini, M.; Massa, A.; Avnet, S.; Bonuccelli, G.; Baldini, N. Tumor-Activated Mesenchymal Stromal Cells Promote Osteosarcoma Stemness and Migratory Potential via IL-6 Secretion. PLoS ONE 2016, 11, e0166500. [Google Scholar] [CrossRef]
- Baglio, S.R.; Lagerweij, T.; Pérez-Lanzón, M.; Ho, X.D.; Léveillé, N.; Melo, S.A.; Cleton-Jansen, A.-M.; Jordanova, E.S.; Roncuzzi, L.; Greco, M.; et al. Blocking Tumor-Educated MSC Paracrine Activity Halts Osteosarcoma Progression. Clin. Cancer Res. 2017, 23, 3721–3733. [Google Scholar] [CrossRef]
- Li, A.; Cheng, X.J.; Moro, A.; Singh, R.K.; Hines, O.J.; Eibl, G. CXCR2-Dependent Endothelial Progenitor Cell Mobilization in Pancreatic Cancer Growth. Transl. Oncol. 2011, 4, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Nolan, D.J.; Ciarrocchi, A.; Mellick, A.S.; Jaggi, J.S.; Bambino, K.; Gupta, S.; Heikamp, E.; McDevitt, M.R.; Scheinberg, D.A.; Benezra, R.; et al. Bone Marrow-Derived Endothelial Progenitor Cells Are a Major Determinant of Nascent Tumor Neovascularization. Genes Dev. 2007, 21, 1546–1558. [Google Scholar] [CrossRef]
- Kuo, Y.-H.; Lin, C.-H.; Shau, W.-Y.; Chen, T.-J.; Yang, S.-H.; Huang, S.-M.; Hsu, C.; Lu, Y.-S.; Cheng, A.-L. Dynamics of Circulating Endothelial Cells and Endothelial Progenitor Cells in Breast Cancer Patients Receiving Cytotoxic Chemotherapy. BMC Cancer 2012, 12, 620. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zhou, F.; Zhou, H.; Huang, J.; Yu, D.; Wu, G. Endothelial Progenitor Cells Contribute to Neovascularization of Non-Small Cell Lung Cancer via Histone Deacetylase 7-Mediated Cytoskeleton Regulation and Angiogenic Genes Transcription. Int. J. Cancer 2018, 143, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Tamari, T.; Elimelech, R.; Cohen, G.; Cohen, T.; Doppelt, O.; Eskander-Hashoul, L.; Zigdon-Giladi, H. Endothelial Progenitor Cells Inhibit Jaw Osteonecrosis in a Rat Model: A Major Adverse Effect of Bisphosphonate Therapy. Sci. Rep. 2019, 9, 18896. [Google Scholar] [CrossRef] [PubMed]
- Huang, H. Matrix Metalloproteinase-9 (MMP-9) as a Cancer Biomarker and MMP-9 Biosensors: Recent Advances. Sensors 2018, 18, 3249. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, T.; Wang, W. Prognostic Significance of Matrix Metalloproteinase 9 Expression in Osteosarcoma. Medicine 2018, 97, e13051. [Google Scholar] [CrossRef]
- Li, H.; Zhang, K.; Liu, L.; Ouyang, Y.; Bu, J.; Guo, H.; Xiao, T. A Systematic Review of Matrix Metalloproteinase 9 as a Biomarker of Survival in Patients with Osteosarcoma. Tumour Biol. 2014, 35, 5487–5491. [Google Scholar] [CrossRef] [PubMed]
- Rundhaug, J.E. Matrix Metalloproteinases and Angiogenesis. J. Cell. Mol. Med. 2005, 9, 267–285. [Google Scholar] [CrossRef]
- Quintero-Fabián, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argáez, V.; Lara-Riegos, J.; Ramírez-Camacho, M.A.; Alvarez-Sánchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370. [Google Scholar] [CrossRef]
- Wang, Q.; Shi, Z.; Zhang, H.; Liu, W.; Yu, Z.; Zhang, Q. Upregulation of Exosomal Integrin Β4 Causes Osteosarcoma Cell Proliferation via the PI3K-Akt-MTOR Signaling Pathway. Transl. Cancer Res. 2018, 7, 1209–1220. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell. 2018, 173, 321–337. [Google Scholar] [CrossRef]
- Xu, C.-J.; Song, J.-F.; Su, Y.-X.; Liu, X.-L. Expression of B-FGF and Endostatin and Their Clinical Significance in Human Osteosarcoma. Orthop. Surg. 2010, 2, 291–298. [Google Scholar] [CrossRef]
- Sheen, M.R.; Marotti, J.D.; Allegrezza, M.J.; Rutkowski, M.; Conejo-Garcia, J.R.; Fiering, S. Constitutively Activated PI3K Accelerates Tumor Initiation and Modifies Histopathology of Breast Cancer. Oncogenesis 2016, 5, e267. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.; Kapur, R. Regulation of Hematopoietic Stem Cell Self-Renewal and Leukemia Maintenance by the PI3K-MTORC1 Pathway. Curr. Stem Cell. Rep. 2016, 2, 368–378. [Google Scholar] [CrossRef]
- Karar, J.; Maity, A. PI3K/AKT/MTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Pemigatinib: First Approval. Drugs 2020, 80, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Chen, T.; Ding, Z.; Wang, Y.; Wei, Y.; Wei, X. Inhibition of FGF-FGFR and VEGF-VEGFR Signalling in Cancer Treatment. Cell Prolif. 2021, 54, e13009. [Google Scholar] [CrossRef]
- Attar-Schneider, O.; Drucker, L.; Zismanov, V.; Tartakover-Matalon, S.; Rashid, G.; Lishner, M. Bevacizumab Attenuates Major Signaling Cascades and EIF4E Translation Initiation Factor in Multiple Myeloma Cells. Lab. Investig. 2012, 92, 178–190. [Google Scholar] [CrossRef]
- Okada, T.; Enkhjargal, B.; Travis, Z.D.; Ocak, U.; Tang, J.; Suzuki, H.; Zhang, J.H. FGF-2 Attenuates Neuronal Apoptosis via FGFR3/PI3k/Akt Signaling Pathway after Subarachnoid Hemorrhage. Mol. Neurobiol. 2019, 56, 8203–8219. [Google Scholar] [CrossRef]
- Assi, T.; Watson, S.; Samra, B.; Rassy, E.; Le Cesne, A.; Italiano, A.; Mir, O. Targeting the VEGF Pathway in Osteosarcoma. Cells 2021, 10, 1240. [Google Scholar] [CrossRef]
- Goel, H.L.; Mercurio, A.M. VEGF Targets the Tumour Cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef]
- Bachelder, R.E.; Crago, A.; Chung, J.; Wendt, M.A.; Shaw, L.M.; Robinson, G.; Mercurio, A.M. Vascular Endothelial Growth Factor Is an Autocrine Survival Factor for Neuropilin-Expressing Breast Carcinoma Cells. Cancer Res. 2001, 61, 5736–5740. [Google Scholar] [PubMed]
- Perrot-Applanat, M.; Di Benedetto, M. Autocrine Functions of VEGF in Breast Tumor Cells: Adhesion, Survival, Migration and Invasion. Cell. Adhes. Migr. 2012, 6, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.L.; Pursell, B.; Chang, C.; Shaw, L.M.; Mao, J.; Simin, K.; Kumar, P.; Vander Kooi, C.W.; Shultz, L.D.; Greiner, D.L.; et al. GLI1 Regulates a Novel Neuropilin-2/A6β1 Integrin Based Autocrine Pathway That Contributes to Breast Cancer Initiation. EMBO Mol. Med. 2013, 5, 488–508. [Google Scholar] [CrossRef]
- Duda, D.G.; Batchelor, T.T.; Willett, C.G.; Jain, R.K. VEGF-Targeted Cancer Therapy Strategies: Current Progress, Hurdles and Future Prospects. Trends Mol. Med. 2007, 13, 223–230. [Google Scholar] [CrossRef]
- Sitohy, B.; Nagy, J.A.; Dvorak, H.F. Anti-VEGF/VEGFR Therapy for Cancer: Reassessing the Target. Cancer Res. 2012, 72, 1909–1914. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Niu, X.; Yao, W. Receptor Tyrosine Kinases in Osteosarcoma Treatment: Which Is the Key Target? Front. Oncol. 2020, 10, 1642. [Google Scholar] [CrossRef]
- Pan, P.-J.; Liu, Y.-C.; Hsu, F.-T. Protein Kinase B and Extracellular Signal-Regulated Kinase Inactivation Is Associated with Regorafenib-Induced Inhibition of Osteosarcoma Progression In Vitro and In Vivo. J. Clin. Med. 2019, 8, 900. [Google Scholar] [CrossRef]
- Albarrán, V.; Villamayor, M.L.; Chamorro, J.; Rosero, D.I.; Pozas, J.; San Román, M.; Calvo, J.C.; Pérez de Aguado, P.; Moreno, J.; Guerrero, P.; et al. Receptor Tyrosine Kinase Inhibitors for the Treatment of Recurrent and Unresectable Bone Sarcomas. Int. J. Mol. Sci. 2022, 23, 13784. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doppelt-Flikshtain, O.; Younis, A.; Tamari, T.; Ginesin, O.; Shentzer-Kutiel, T.; Nikomarov, D.; Bar-Sela, G.; Coyac, B.R.; Assaraf, Y.G.; Zigdon-Giladi, H. Endothelial Progenitor Cells Promote Osteosarcoma Progression and Invasiveness via AKT/PI3K Signaling. Cancers 2023, 15, 1818. https://doi.org/10.3390/cancers15061818
Doppelt-Flikshtain O, Younis A, Tamari T, Ginesin O, Shentzer-Kutiel T, Nikomarov D, Bar-Sela G, Coyac BR, Assaraf YG, Zigdon-Giladi H. Endothelial Progenitor Cells Promote Osteosarcoma Progression and Invasiveness via AKT/PI3K Signaling. Cancers. 2023; 15(6):1818. https://doi.org/10.3390/cancers15061818
Chicago/Turabian StyleDoppelt-Flikshtain, Ofri, Amin Younis, Tal Tamari, Ofir Ginesin, Talia Shentzer-Kutiel, David Nikomarov, Gil Bar-Sela, Benjamin R. Coyac, Yehuda G. Assaraf, and Hadar Zigdon-Giladi. 2023. "Endothelial Progenitor Cells Promote Osteosarcoma Progression and Invasiveness via AKT/PI3K Signaling" Cancers 15, no. 6: 1818. https://doi.org/10.3390/cancers15061818
APA StyleDoppelt-Flikshtain, O., Younis, A., Tamari, T., Ginesin, O., Shentzer-Kutiel, T., Nikomarov, D., Bar-Sela, G., Coyac, B. R., Assaraf, Y. G., & Zigdon-Giladi, H. (2023). Endothelial Progenitor Cells Promote Osteosarcoma Progression and Invasiveness via AKT/PI3K Signaling. Cancers, 15(6), 1818. https://doi.org/10.3390/cancers15061818