
TWEAK/Fn14 Signalling Regulates the Tissue Microenvironment in Chronic Pancreatitis

 ,
,  , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Models
2.2. Histology, Immunohistochemistry, and Immunofluorescence Experiments
2.3. Triple Fluorescent Staining of Pancreatic Islet Cells
2.4. TNFRSF12A Gene Expression Analysis
2.5. Statistical Analysis
3. Results
3.1. The Fn14 Receptor Is Upregulated in Chronic Pancreatitis and Pancreatic Adenocarcinoma
3.2. Long-Term Choline-Deficient, Ethionine Diet Treatment Leads to Chronic Pancreatitis
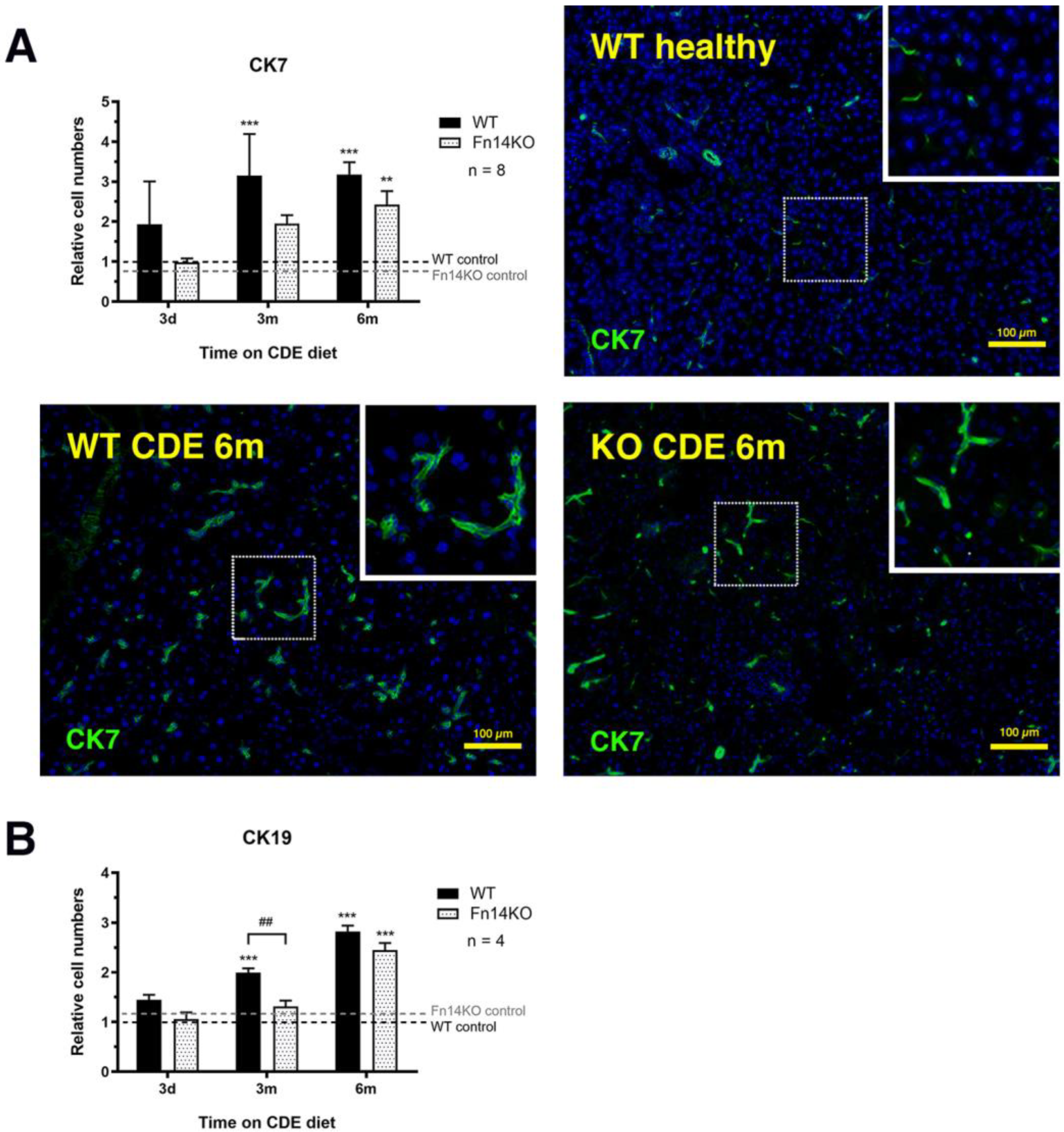
3.3. TWEAK/Fn14 Signalling Regulates the Pancreatic Ductular Reaction
3.4. Fibrosis Is Reduced in CDE-Treated Fn14 Knockout Mice
3.5. Pancreatic TWEAK/Fn14 Signalling Is Important for Inflammatory Cell Recruitment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Petrov, M.S.; Yadav, D. Global epidemiology and holistic prevention of pancreatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Kirkegard, J.; Mortensen, F.V.; Cronin-Fenton, D. Chronic Pancreatitis and Pancreatic Cancer Risk: A Systematic Review and Meta-analysis. Am. J. Gastroenterol. 2017, 112, 1366–1372. [Google Scholar] [CrossRef] [PubMed]
- Connor, A.A.; Gallinger, S. Pancreatic cancer evolution and heterogeneity: Integrating omics and clinical data. Nat. Rev. Cancer 2022, 22, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Silva-Vaz, P.; Abrantes, A.M.; Castelo-Branco, M.; Gouveia, A.; Botelho, M.F.; Tralhao, J.G. Murine Models of Acute Pancreatitis: A Critical Appraisal of Clinical Relevance. Int. J. Mol. Sci. 2019, 20, 2794. [Google Scholar] [CrossRef] [PubMed]
- Lerch, M.M.; Gorelick, F.S. Models of acute and chronic pancreatitis. Gastroenterology 2013, 144, 1180–1193. [Google Scholar] [CrossRef]
- Gogoi-Tiwari, J.; Kohn-Gaone, J.; Giles, C.; Schmidt-Arras, D.; Gratte, F.D.; Elsegood, C.L.; McCaughan, G.W.; Ramm, G.A.; Olynyk, J.K.; Tirnitz-Parker, J.E.E. The Murine Choline-Deficient, Ethionine-Supplemented (CDE) Diet Model of Chronic Liver Injury. J. Vis. Exp. 2017, 21, e56138. [Google Scholar] [CrossRef]
- Kohn-Gaone, J.; Dwyer, B.J.; Grzelak, C.A.; Miller, G.; Shackel, N.A.; Ramm, G.A.; McCaughan, G.W.; Elsegood, C.L.; Olynyk, J.K.; Tirnitz-Parker, J.E.E. Divergent Inflammatory, Fibrogenic, and Liver Progenitor Cell Dynamics in Two Common Mouse Models of Chronic Liver Injury. Am. J. Pathol. 2016, 186, 1762–1774. [Google Scholar] [CrossRef]
- Tirnitz-Parker, J.E.; Viebahn, C.S.; Jakubowski, A.; Klopcic, B.R.; Olynyk, J.K.; Yeoh, G.C.; Knight, B. Tumor necrosis factor-like weak inducer of apoptosis is a mitogen for liver progenitor cells. Hepatology 2010, 52, 291–302. [Google Scholar] [CrossRef]
- Tirnitz-Parker, J.E.; Olynyk, J.K.; Ramm, G.A. Role of TWEAK in coregulating liver progenitor cell and fibrogenic responses. Hepatology 2014, 59, 1198–1201. [Google Scholar] [CrossRef]
- Dwyer, B.J.; Jarman, E.J.; Gogoi-Tiwari, J.; Ferreira-Gonzalez, S.; Boulter, L.; Guest, R.V.; Kendall, T.J.; Kurian, D.; Kilpatrick, A.M.; Robson, A.J.; et al. TWEAK/Fn14 signalling promotes cholangiocarcinoma niche formation and progression. J. Hepatol. 2021, 74, 860–872. [Google Scholar] [CrossRef]
- Girgenrath, M.; Weng, S.; Kostek, C.A.; Browning, B.; Wang, M.; Brown, S.A.; Winkles, J.A.; Michaelson, J.S.; Allaire, N.; Schneider, P.; et al. TWEAK, via its receptor Fn14, is a novel regulator of mesenchymal progenitor cells and skeletal muscle regeneration. EMBO J. 2006, 25, 5826–5839. [Google Scholar] [CrossRef]
- Akhurst, B.; Croager, E.J.; Farley-Roche, C.A.; Ong, J.K.; Dumble, M.L.; Knight, B.; Yeoh, G.C. A modified choline-deficient, ethionine-supplemented diet protocol effectively induces oval cells in mouse liver. Hepatology 2001, 34, 519–522. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Enge, M.; Arda, H.E.; Mignardi, M.; Beausang, J.; Bottino, R.; Kim, S.K.; Quake, S.R. Single-Cell Analysis of Human Pancreas Reveals Transcriptional Signatures of Aging and Somatic Mutation Patterns. Cell 2017, 171, 321–330.e314. [Google Scholar] [CrossRef] [PubMed]
- Speir, M.L.; Bhaduri, A.; Markov, N.S.; Moreno, P.; Nowakowski, T.J.; Papatheodorou, I.; Pollen, A.A.; Raney, B.J.; Seninge, L.; Kent, W.J.; et al. UCSC Cell Browser: Visualize Your Single-Cell Data. Bioinformatics 2021, 37, 4578–4580. [Google Scholar] [CrossRef]
- La Rosa, S.; Sessa, F.; Capella, C. Acinar Cell Carcinoma of the Pancreas: Overview of Clinicopathologic Features and Insights into the Molecular Pathology. Front. Med. 2015, 2, 41. [Google Scholar] [CrossRef]
- Prakoso, E.; Tirnitz-Parker, J.E.; Clouston, A.D.; Kayali, Z.; Lee, A.; Gan, E.K.; Ramm, G.A.; Kench, J.G.; Bowen, D.G.; Olynyk, J.K.; et al. Analysis of the intrahepatic ductular reaction and progenitor cell responses in hepatitis C virus recurrence after liver transplantation. Liver Transpl. 2014, 20, 1508–1519. [Google Scholar] [CrossRef] [PubMed]
- Haber, P.S.; Keogh, G.W.; Apte, M.V.; Moran, C.S.; Stewart, N.L.; Crawford, D.H.; Pirola, R.C.; McCaughan, G.W.; Ramm, G.A.; Wilson, J.S. Activation of pancreatic stellate cells in human and experimental pancreatic fibrosis. Am. J. Pathol. 1999, 155, 1087–1095. [Google Scholar] [CrossRef]
- Chicheportiche, Y.; Bourdon, P.R.; Xu, H.D.; Hsu, Y.M.; Scott, H.; Hession, C.; Garcia, I.; Browning, J.L. TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J. Biol. Chem. 1997, 272, 32401–32410. [Google Scholar] [CrossRef]
- Winkles, J.A. The TWEAK-Fn14 cytokine-receptor axis: Discovery, biology and therapeutic targeting. Nat. Rev. Drug. Discov. 2008, 7, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Meighan-Mantha, R.L.; Hsu, D.K.; Guo, Y.; Brown, S.A.; Feng, S.L.; Peifley, K.A.; Alberts, G.F.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; et al. The mitogen-inducible Fn14 gene encodes a type I transmembrane protein that modulates fibroblast adhesion and migration. J. Biol. Chem. 1999, 274, 33166–33176. [Google Scholar] [CrossRef]
- Zaitseva, O.; Hoffmann, A.; Otto, C.; Wajant, H. Targeting fibroblast growth factor (FGF)-inducible 14 (Fn14) for tumor therapy. Front. Pharmacol. 2022, 13, 935086. [Google Scholar] [CrossRef]
- Zhang, Y.; Zeng, W.; Xia, Y. TWEAK/Fn14 axis is an important player in fibrosis. J. Cell. Physiol. 2021, 236, 3304–3316. [Google Scholar] [CrossRef] [PubMed]
- Karaca, G.; Swiderska-Syn, M.; Xie, G.; Syn, W.K.; Kruger, L.; Machado, M.V.; Garman, K.; Choi, S.S.; Michelotti, G.A.; Burkly, L.C.; et al. TWEAK/Fn14 signaling is required for liver regeneration after partial hepatectomy in mice. PLoS ONE 2014, 9, e83987. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, B.J.; Olynyk, J.K.; Ramm, G.A.; Tirnitz-Parker, J.E. TWEAK and LTbeta Signaling during Chronic Liver Disease. Front. Immunol. 2014, 5, 39. [Google Scholar] [CrossRef]
- Jakubowski, A.; Ambrose, C.; Parr, M.; Lincecum, J.M.; Wang, M.Z.; Zheng, T.S.; Browning, B.; Michaelson, J.S.; Baetscher, M.; Wang, B.; et al. TWEAK induces liver progenitor cell proliferation. J. Clin. Investig. 2005, 115, 2330–2340. [Google Scholar] [CrossRef]
- Thomas, J.A.; Pope, C.; Wojtacha, D.; Robson, A.J.; Gordon-Walker, T.T.; Hartland, S.; Ramachandran, P.; Van Deemter, M.; Hume, D.A.; Iredale, J.P.; et al. Macrophage therapy for murine liver fibrosis recruits host effector cells improving fibrosis, regeneration, and function. Hepatology 2011, 53, 2003–2015. [Google Scholar] [CrossRef]
- Wilhelm, A.; Shepherd, E.L.; Amatucci, A.; Munir, M.; Reynolds, G.; Humphreys, E.; Resheq, Y.; Adams, D.H.; Hubscher, S.; Burkly, L.C.; et al. Interaction of TWEAK with Fn14 leads to the progression of fibrotic liver disease by directly modulating hepatic stellate cell proliferation. J. Pathol. 2016, 239, 109–121. [Google Scholar] [CrossRef]
- Burke, Z.D.; Tosh, D. Ontogenesis of hepatic and pancreatic stem cells. Stem Cell Rev. Rep. 2012, 8, 586–596. [Google Scholar] [CrossRef]
- Ghurburrun, E.; Borbath, I.; Lemaigre, F.P.; Jacquemin, P. Liver and Pancreas: Do Similar Embryonic Development and Tissue Organization Lead to Similar Mechanisms of Tumorigenesis? Gene Expr. 2018, 18, 149–155. [Google Scholar] [CrossRef]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Cannon, A.; Thompson, C.M.; Bhatia, R.; Armstrong, K.A.; Solheim, J.C.; Kumar, S.; Batra, S.K. Molecular mechanisms of pancreatic myofibroblast activation in chronic pancreatitis and pancreatic ductal adenocarcinoma. J. Gastroenterol. 2021, 56, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Bridle, K.R.; Wells, L.D.; Marcu, M.; Ramm, G.A. Repetitive acute pancreatic injury in the mouse induces procollagen alpha1(I) expression colocalized to pancreatic stellate cells. Lab. Investig. 2000, 80, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xie, D.; Wei, D. Pancreatic Acinar-to-Ductal Metaplasia and Pancreatic Cancer. Methods Mol. Biol. 2019, 1882, 299–308. [Google Scholar] [CrossRef]
- Jain, R.; Fischer, S.; Serra, S.; Chetty, R. The use of Cytokeratin 19 (CK19) immunohistochemistry in lesions of the pancreas, gastrointestinal tract, and liver. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 9–15. [Google Scholar] [CrossRef]
- Matros, E.; Bailey, G.; Clancy, T.; Zinner, M.; Ashley, S.; Whang, E.; Redston, M. Cytokeratin 20 expression identifies a subtype of pancreatic adenocarcinoma with decreased overall survival. Cancer 2006, 106, 693–702. [Google Scholar] [CrossRef]
- Egberts, J.H.; Cloosters, V.; Noack, A.; Schniewind, B.; Thon, L.; Klose, S.; Kettler, B.; von Forstner, C.; Kneitz, C.; Tepel, J.; et al. Anti-tumor necrosis factor therapy inhibits pancreatic tumor growth and metastasis. Cancer Res. 2008, 68, 1443–1450. [Google Scholar] [CrossRef]
- Zhao, X.; Fan, W.; Xu, Z.; Chen, H.; He, Y.; Yang, G.; Yang, G.; Hu, H.; Tang, S.; Wang, P.; et al. Inhibiting tumor necrosis factor-alpha diminishes desmoplasia and inflammation to overcome chemoresistance in pancreatic ductal adenocarcinoma. Oncotarget 2016, 7, 81110–81122. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abu Bakar, N.D.B.; Carlessi, R.; Gogoi-Tiwari, J.; Köhn-Gaone, J.; Williams, V.; Falasca, M.; Olynyk, J.K.; Ramm, G.A.; Tirnitz-Parker, J.E.E. TWEAK/Fn14 Signalling Regulates the Tissue Microenvironment in Chronic Pancreatitis. Cancers 2023, 15, 1807. https://doi.org/10.3390/cancers15061807
Abu Bakar NDB, Carlessi R, Gogoi-Tiwari J, Köhn-Gaone J, Williams V, Falasca M, Olynyk JK, Ramm GA, Tirnitz-Parker JEE. TWEAK/Fn14 Signalling Regulates the Tissue Microenvironment in Chronic Pancreatitis. Cancers. 2023; 15(6):1807. https://doi.org/10.3390/cancers15061807
Chicago/Turabian StyleAbu Bakar, N. Dianah B., Rodrigo Carlessi, Jully Gogoi-Tiwari, Julia Köhn-Gaone, Vincent Williams, Marco Falasca, John K. Olynyk, Grant A. Ramm, and Janina E. E. Tirnitz-Parker. 2023. "TWEAK/Fn14 Signalling Regulates the Tissue Microenvironment in Chronic Pancreatitis" Cancers 15, no. 6: 1807. https://doi.org/10.3390/cancers15061807
APA StyleAbu Bakar, N. D. B., Carlessi, R., Gogoi-Tiwari, J., Köhn-Gaone, J., Williams, V., Falasca, M., Olynyk, J. K., Ramm, G. A., & Tirnitz-Parker, J. E. E. (2023). TWEAK/Fn14 Signalling Regulates the Tissue Microenvironment in Chronic Pancreatitis. Cancers, 15(6), 1807. https://doi.org/10.3390/cancers15061807