Simple Summary
This review reports the risk and management of the hepatotoxicity of all the approved protein kinase inhibitors (PKIs) for cancer. Hepatotoxicity is one of the major safety concerns of these drugs, as reflected by the discontinuation of the development of some of them due to liver injury, or by the significant number of warnings for hepatotoxicity reported in drug labeling. Although these side effects are usually reversible by dose adjustment or therapy suspension, or by switching to an alternative PKI, and fatality is uncommon, all patients undergoing these drugs should be carefully pre-evaluated and monitored during treatment.
Abstract
Small molecule protein kinase inhibitors (PKIs) have become an effective strategy for cancer patients. However, hepatotoxicity is a major safety concern of these drugs, since the majority are reported to increase transaminases, and few of them (Idelalisib, Lapatinib, Pazopanib, Pexidartinib, Ponatinib, Regorafenib, Sunitinib) have a boxed label warning. The exact rate of PKI-induced hepatoxicity is not well defined due to the fact that the majority of data arise from pre-registration or registration trials on fairly selected patients, and the post-marketing data are often based only on the most severe described cases, whereas most real practice studies do not include drug-related hepatotoxicity as an end point. Although these side effects are usually reversible by dose adjustment or therapy suspension, or by switching to an alternative PKI, and fatality is uncommon, all patients undergoing PKIs should be carefully pre-evaluated and monitored. The management of this complication requires an individually tailored reappraisal of the risk/benefit ratio, especially in patients who are responding to therapy. This review reports the currently available data on the risk and management of hepatotoxicity of all the approved PKIs.
1. Introduction
The phosphorylation of proteins by protein kinases is an important activating mechanism in the communication of signals within a cell and the regulation of both cellular activity and function [1,2]. These kinases play a major role in the cellular biochemical pathways involved in the transduction of extracellular signals regulating cellular responses including differentiation, proliferation, survival apoptosis, protein synthesis and other metabolic aspects of the cell cycle. Each kinase functions as an “on” switch, but they can also be overexpressed, become mutated or get blocked in the “on” position, resulting in the unregulated growth of the cell. Therefore, every overexpression, dysregulation or interference of these proteins’ activity may cause metabolic, autoimmune or inflammatory disorders, or lead to cancer [3]. For these reasons, the suppression of aberrantly expressed kinases has become an attractive strategy for patients with cancer. After Imatinib, the first U.S. Food and Drug Administration (FDA) approved small molecule tyrosine kinase inhibitor (TKI) in 2001, many others have been approved, and as of now there are 62 PKIs approved for cancer treatment. While these agents are generally well tolerated, their association with serious adverse events on a number of different organs, including the liver, have been reported. The liver injury secondary to PKIs involves:
- The production of several potentially toxic or immunogenic intermediates occurring within the CYP 3A4 pathway, that might alter both endogenous metabolism and cellular function leading to organ damage;
- The inhibition of cellular kinases or the “off-target effect” due to the production of toxic metabolites;
- The intrinsic hepatotoxicity by direct activity against essential intracellular kinases in hepatocytes;
- The immunologically mediated effect of the accumulation of immunogenic intermediates or B cell-driven autoimmunity (Figure 1).
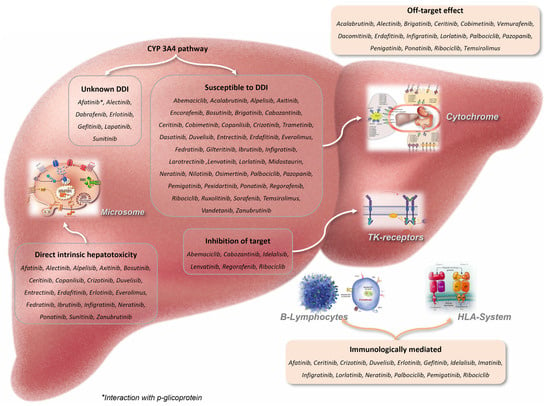 Figure 1. The different pathways of liver injury of the PKIs approved for cancer.
Figure 1. The different pathways of liver injury of the PKIs approved for cancer.
However, the rates of confirmed PKI-hepatotoxicity remain poorly defined because the majority of data comes from pre-registration or registration trials on highly selected patients, and the comprehensive safety information coming from clinical practice is often missing as a result of the lack of a routinely planned on-treatment determination of aminotransferase levels. Therefore, only the most striking adverse events, leading to liver injury, are eventually published as case reports. The real risk for PKI-related severe hepatotoxicity in cancer patients remains an open issue, particularly due to the fact that PKIs are frequently utilized either sequentially or in combination with other chemotherapies. These other chemotherapies can be hepatotoxic in individuals with advanced cancers whose immune system or liver regeneration capacity may be hampered by an already impaired liver function, either due to preexisting conditions or therapies, or by malignant involvement of the liver. It must be noted that causality assessment methods remain controversial because some PKIs were marketed for a relatively short period of time, exposing only a small number of patients and therefore preventing the identification of idiosyncratic hepatotoxicity. Despite the fact that the majority of PKIs is reported to cause an increase in transaminases, and some of them have warnings for liver injury in regulatory documents, they are not at the top of the list of drugs that most frequently can cause hepatotoxicity [4,5,6,7,8,9,10]. Moreover, some PKIs may induce a hepatitis B virus reactivation (HBVr) either directly, by acting as an immunosuppressant, or indirectly, after a tumor response, supporting the hypothesis of a relation with immune restoration. In vitro studies have indeed reported that such therapies are capable of inhibiting T cell activation and proliferation, thus restoring the function of plasmacytoid dendritic cells, the well-known crucial effectors of innate immunity [11,12]. The rate of HBVr in oncologic HBsAg positive patients treated with PKI without antiviral prophylaxis is nearly 10% [13,14,15,16,17,18,19,20]. For this reason, the patients who would be the best candidates to receive these drugs should be screened for serologic markers of HBV infection, including hepatitis B surface antigen (HBsAg) and anti-hepatitis B core antigen (anti-HBc). HBsAg-positive patients should receive prophylaxis against HBVr with an oral antiviral agent, whereas patients with resolved infection, i.e., HBsAg negative/anti-HBc positive carriers with or without antibodies against HBsAg (anti-HBs), should be monitored with serum HBV DNA and/or HBsAg every three months, with a prompt initiation of an antiviral agent before the clinical onset of HBVr-related liver injury [21].
A systematic search on the PubMed/MEDLINE and LiverTox database of the data on PKI-hepatotoxicity using the PKI names and ‘liver’, ‘hepatotoxicity’, ‘hepatitis’, ‘drug induced liver injury’, ‘liver failure’ and ‘hepatic failure’ was carried out, and the resulting records were reviewed to collect relevant information in combination with those available on drug labeling for all the approved PKIs for cancer.
2. Relevant Section
In Table 1, the primary target, indications and rate of any alanine aminotransferase (ALT) increase, the Grade 3/4 hepatotoxicity according to the Common Terminology Criteria for Adverse Events (CTCAE) is reported for each PKI: Grade 1 [ALT > 3 × upper limit of normal (ULN)]; Grade 2 (ALT > 3 − 5 × ULN) without increase in total bilirubin > 2 × ULN; Grade 3 (ALT > 5 − 20 × ULN) without increase in total bilirubin > 2 × ULN; Grade 4 (ALT > 20 × ULN), and the drug labeling warnings for liver injury or boxed warnings for liver injury are taken into account.

Table 1.
Main factors involved in liver injury secondary to PKIs approved for cancer.
2.1. Abemaciclib
ALT elevations were not reported during the prelicensure study [22], whereas phase II/III trials showed a moderate rate (30–48%) of any ALT elevation (G3/4: 4.6–7%) without reported cases of liver failure [23,24,25,26]. The median time to onset of G3/4 ALT increase ranged from 57 to 87 days, and the median time to resolution to <G3 was 14 days. Since the approval and more widescale use of Abemaciclib, there have been no published reports of its hepatotoxicity. Liver injury tests (LITs), i.e., ALT, AST and serum bilirubin, should be performed before starting treatment and every two weeks for the first two months, monthly for the next two months and then as clinically indicated. For G1/2 hepatotoxicity, no dose modification is required; with persistent or recurrent G2/3 hepatotoxicity, the dose should be suspended until toxicity resolves to baseline, or G1 and drug may be resumed at the next lower dose. The elevation of aminotransferase >3 × ULN with total bilirubin >2 × ULN, in the absence of cholestasis or G4 hepatotoxicity, requires Abemaciclib discontinuation. There is no information regarding cross-reactivity and the risk for hepatic injury between Abemaciclib and the other cyclin-dependent kinase (CDK) inhibitors, Ribociclib or Palbociclib, for breast cancers [27]. No dosage adjustments are necessary in patients with mild or moderate hepatic impairment (Child-Pugh A or B). The dosing frequency should be reduced to 150 mg once daily if Abemaciclib is given in combination with fulvestrant, tamoxifen or an aromatase inhibitor, or 200 mg once daily as monotherapy in patients with severe hepatic impairment (Child-Pugh C).
2.2. Acalabrutinib
There were no cases of ALT increase during prelicensure study [28], whereas phase II/III trials reported a mild rate (4.5–20%) of ALT elevations (G3/4: 1–1.9%) in monotherapy, which significantly increased when the drug was used in combination with Obinutuzumab (30% and 7%, respectively), however without cases of liver failure [29,30,31,32,33,34]. This drug has been associated with cases of HBVr. Acalabrutinib should be avoided in patients with severe hepatic impairment (Child-Pugh C), whereas no dosage adjustment is recommended in patients with mild to moderate hepatic impairment (Child-Pugh A and B).
2.3. Afatinib
Phase II/III trials reported a mild rate (10–20%) of any ALT elevations (G3/4: 1.7%), with a 0.2% risk of hepatic failure [35,36,37,38,39,40]. The periodic monitoring of LITs is recommended, and confirmed ALT elevations >5 × ULN should lead to temporary discontinuation, which should become permanent if laboratory values do not improve significantly or resolve within a few weeks, or if symptoms or jaundice arise. Restarting therapy is usually, but not always, followed by recurrence of the serum enzyme elevations. Afatinib hepatotoxicity appears to be less frequent and less severe than with Gefitinib [41,42], and due to the lack of cross-reactivity with other TKIs of EGFR, in some situations, Afatinib may represent an important treatment option when Gefitinib or Erlotinib-induced hepatotoxicity develops [43,44,45,46]. Afatinib has not been studied in patients with severe (Child Pugh C) hepatic impairment, whereas adjustments to the starting doses are not necessary in patients with mild/moderate (Child Pugh A and B) hepatic impairment.
2.4. Alectinib
Pre-registration studies reported a mild rate (13–26%) of any ALT elevations (G3/4: 0–5.1%) [47,48,49], similar (8.3–34%, G3/4: 5.7%) to that reported in phase III trials at a dose of 600 mg twice a day. Alectinib therapy was also associated with frequent elevations (47%) in alkaline phosphatase (ALP) and bilirubin (39%), but these abnormalities were usually mild-to-moderate in degree, as well as asymptomatic and transient in nature [50,51,52]. LITs should be performed every two weeks during the first three months of treatment, then once a month and as clinically indicated, with more frequent testing in patients who develop ALT and bilirubin elevations. For ≥G3 hepatotoxicity, the dose should be suspended until toxicity resolves and the value returns to baseline, or <G1 and drug may be resumed at a reduced dose (450 mg twice daily). In case of ALT increase ≥3 × ULN with bilirubin >2 × ULN, Alectinib should be permanently discontinued. There is no evidence of the cross-reactivity of ALK with other TKIs (such as Crizotinib or Ceritinib), and switching to another TKI may be appropriate [53,54]. Increased blood levels of Alectinib occurred in patients with severe hepatic impairment (Child Pugh C), and therefore, the recommended dose in such patients is 450 mg twice daily.
2.5. Alpelisib
In Alpelisib and Fulvestrant-treated patients, some degree of ALT elevation was shown in up to 44% (G3/4 in 3.5%). Confirmed ALT elevation >5 × ULN should lead to a dose reduction or temporary interruption. In patients with clinically evident liver injury and/or jaundice, restarting therapy should be done very cautiously, whereas in patients with ALT elevations without jaundice or symptoms, the reintroduction of therapy can be attempted with tight LITs controls. Cross-sensitivity to liver injury is uncommon among this class of TKIs, but there is no clear information or shared adverse event sensitivity of Alpelisib with other TKIs [55,56,57,58].
2.6. Avapritinib
Although the rate of all grades of ALT increase was not mentioned in prelicensure and registration studies, a 0.8% risk of liver failure was reported in a phase I dose escalating study [59,60,61]. No dose adjustment is recommended for patients with mild to moderate hepatic impairment, whereas the recommended dose of Avapritinib has not been established for patients with severe hepatic impairment.
2.7. Axitinib
Any elevation in serum ALT was reported in up to 22% of patients (G3/4 events occurring in 1%), with no cases of liver failure [62,63,64,65,66,67]. No starting dose adjustment is required in patients with mild hepatic impairment (Child Pugh A). The dose should be reduced by approximately half in patients with baseline moderate hepatic impairment (Child Pugh B), since the blood levels of the drug were shown to be higher in these subjects. Axitinib has not been studied in patients with severe hepatic impairment (Child Pugh C). Weekly monitoring of LITs is recommended, and confirmed serum ALT elevation >5 × ULN should lead to dose reduction or temporary discontinuation. Since a cross-reactivity risk for hepatic injury between Axitinib and other TKIs of this class is not reported, switching to other TKIs in the same class may be appropriate.
2.8. Binimetinib + Encorafenib
With this TKIs combination, any elevation in serum ALT was reported in up to 26% of patients (G3/4: 6%), although ALT elevation was generally transient and asymptomatic and there were no reports of liver failure [68,69]. LITs should be monitored before initiation, monthly during treatment and as clinically indicated. For G2 hepatotoxicity, doses should be maintained, but if there is no improvement within two weeks, doses should be suspended until toxicity resolves to baseline or <G1 and the treatment may be resumed at the same dose. With G3/4 hepatotoxicity, the permanent discontinuation of the drug is recommended. There is no evidence of cross-reactivity for hepatic injury between Binimetinib + Encorafenib and other TKIs, such as Dabrafenib + Vermurafenib, and switching to other BRAF/MEK inhibitors may be appropriate.
2.9. Bosutinib
In large clinical trials of Bosutinib, serum ALT elevations were common, occurring in up to 59% of recipients (G3/4 hepatotoxicity: 10% to 19%). Most cases of ALT elevations occurred early in treatment, with a median onset time of 30 days (more than 80% of patients experienced their first event within the first three months) and with a median duration of ALT flare of 21 days. These abnormalities were usually asymptomatic, leading to the discontinuation of therapy in approximately 2% of patients [70,71,72,73]. LITs should be monitored before initiation, monthly for the first three months and as clinically indicated. ALT levels >5 × ULN should lead to dose suspension until recovery to ≤2.5 × ULN and the restarting dose should be 400 mg once daily. However, if recovery lasts longer than four weeks, permanent drug discontinuation should be considered. Bilirubin ≥2 × ULN requires Bosutinib discontinuation. In patients with hepatic impairment (Child Pugh A to C), blood levels of Bosutinib are increased compared to healthy controls and a daily dose of 200 mg is recommended.
2.10. Brigatinib
This TKI is associated with a mild rate (11–22%) of ALT elevations (G3/4: up to 4%) without liver injury. Brigatinib therapy was also associated with frequent elevations in ALP (15% to 29%), but these elevations were usually asymptomatic, mild-to-moderate in degree and transient [74,75,76]. The product label does not recommend monitoring LITs, but hepatotoxicity ≥G3 should lead to temporary discontinuation, which should be permanent if laboratory values do not improve significantly or resolve within a few weeks, or if symptoms or jaundice arise. In patients with severe hepatic impairment (Child Pugh C), Brigatinib dose should be reduced by approximately 40%.
2.11. Cabozantinib
A moderate rate (12–73%) of serum ALT elevations (G3/4: up to 6%) has been reported during treatment with 60 mg once daily, without cases of liver failure. When used in combination with Nivolumab, a higher frequency of G3/4 hepatotoxicity has been reported. Serum ALP elevations were also common and were >3 × ULN in 3% of patients [77,78,79,80,81]. Monitoring LITs before initiation and throughout treatment is recommended in the product label, as well as tighter LITs monitoring when in combination with Nivolumab. Serum ALT elevations >5 × ULN should lead to dose reduction or temporary discontinuation. Cabozantinib dosage should be reduced in patients with moderate hepatic impairment, whereas it should be avoided in those with severe hepatic impairment. There is little information on cross-reactivity for hepatic injury between Cabozantinib and other TKIs of the same class.
2.12. Capmatinib
Increased ALT levels occurred in 13% of treated patients (G3/4: 4.4–7%). The median time to ALT increase was 1.4 months (range: 0.5 to 4.1 months) [82,83]. Monitoring LITs is recommended prior to the start and every two weeks during the first three months of treatment, then once a month or as clinically indicated, with more frequent testing in patients who develop increased LITs. G3 ALT increase should lead to temporary discontinuation until recovery to baseline. If ALT recovers to baseline within seven days, Capmatinib can be resumed at the same dose; otherwise, it should be resumed at a reduced dose. Any ALT increase combined with total bilirubin >2 × ULN in the absence of cholestasis or hemolysis required permanent Capmatinib discontinuation.
2.13. Ceritinib
ALT elevations occurred in up to 60% of patients (G3/4: 17–31%) [84,85,86,87], and approximately 1% of patients required permanent discontinuation due to hepatotoxicity. Patients should be monitored with LITs once a month and as clinically indicated, with weekly testing in patients who develop ALT elevations. Restarting therapy is usually, but not always, followed by recurrence of aminotransferase elevation. G3 hepatotoxicity should lead to temporary discontinuation until recovery to baseline or <G1. Ceritinib can be resumed with a 150 mg daily dose reduction. Any ALT elevation with total bilirubin elevation >2 × ULN requires permanent drug discontinuation. Despite the fact that hepatotoxicity seems to be a class effect of ALK inhibitors, liver injury appears to be less frequent and less severe with Ceritinib or Alectinib than Crizotinib, and a report of two patients with a successful treatment with Ceritinib after Crizotinib-induced hepatitis has been published [88].
2.14. Cobimetinib + Vemurafenib
This TKIs combination is commonly associated with serum ALT elevations occurring in 11% to 25% of patients (G3/4: 5% to 11%) [89,90,91,92,93]. The product label recommends monitoring LITs during treatment, and the first ALT G4 elevation should lead to four weeks’ discontinuation, which should become permanent if laboratory values do not improve to ≤G1. The drug should be resumed at the next lower dosing level. Recurrent G4 hepatotoxicity requires permanent drug discontinuation. Adjustment of the starting dose is not required in patients with mild to moderate hepatic impairment (Child Pugh A and B), whereas in severe hepatic impairment (Child Pugh C), the appropriate dose has not been established.
2.15. Copanlisib
The rates of ALT elevations ranged from 23% to 26% (G3/4: 2–4%) in patients without cases of liver injury [94,95]. Since its approval and more general use, no published reports of liver injury with jaundice are available. Although Idelalisib, a small molecule with a similar therapeutic target (PI3K), has been linked to acute liver failure, some of which have been fatal, for Copanlisib, there are no product label recommendations for routine LITs assessment or dosage modifications in case of baseline hepatic impairment or hepatotoxicity development. Moreover, there is no published information on cross-sensitivity to hepatic injury among the different PI3K TKIs.
2.16. Crizotinib
Elevations in serum ALT levels occurred in up to 38% of patients (G3/4: 1–3%), leading to early drug discontinuation in 2% to 4% of patients. Serum ALT elevation typically occurs 4 to 12 weeks into treatment, usually without jaundice or ALP elevation. Most cases of liver damage due to Crizotinib have been minimal or asymptomatic and the alteration resolved itself within 1–2 months after stopping the drug. Fatal liver failure has been reported in 0.1% of the cases [96,97]. Routine periodic monitoring of LITs every two weeks for the first two months and monthly thereafter and as clinically indicated, is recommended. ALT elevation >5 × ULN with total bilirubin ≤ 1.5 times ULN should lead to temporary drug discontinuation until recovery to baseline or ≤3 × ULN, and then the drug can be resumed at a reduced dose. ALT elevation >3 × ULN with concurrent total bilirubin elevation >1.5 × ULN requires permanent discontinuation. Patients with liver abnormalities during Crizotinib therapy may tolerate other TKIs, such as Erlotinib or Gefitinib. Crizotinib has not been studied in patients with hepatic impairment, however, since it is extensively metabolized within the liver, and plasma concentrations are expected to increase in case of hepatic impairment, the drug should be used with caution in such patients.
2.17. Dabrafenib + Trametinib
This TKIs combination is commonly associated with serum ALT elevations occurring in 35% to 42% of treated patients (G3/4: up to 4%), however, severe liver injury has not been reported [98,99,100,101,102]. Similarly, serum ALP elevations occurred in up to 67% of patients. These abnormalities were largely asymptomatic and fully reversible. Monitoring LITs before starting and during therapy is warranted. Serum ALT elevations >5 × ULN or elevations accompanied by jaundice or liver-related clinical events should lead to temporary cessation and not restart until the LITs abnormalities improve or resolve. Restarting requires careful weekly monitoring. A recommended dosage has not been established for patients with moderate to severe (Child Pugh B, C) hepatic impairment.
2.18. Dasatinib
Elevation in serum ALT levels occurred in up to 50% of patients, but was usually mild and self-limiting (G3/4: 1–9%), generally responding to dose adjustment and/or temporary discontinuation. The restarting should be at a lower dose. There have been no published reports of severe liver injury, although other TKIs for chronic myeloid leukemia, i.e., Imatinib, Nilotinib and Ponatinib with a similar therapeutic target (BCR-Abl), have been associated with cases of acute liver injury with jaundice. HBVr in a HBsAg positive patient has been reported with Dasatinib, as well as with Imatinib and Nilotinib [103,104,105,106,107,108,109,110,111,112,113]. Serum ALT elevation >5 × ULN should lead to dose reduction or temporary cessation. Therapy can be restarted, possibly with concurrent prednisone use. In patients with clinically evident liver injury and jaundice, restarting therapy should be done with extreme caution. There does not appear to be cross-reactivity with other TKIs and switching to another drug may be the most appropriate approach.
2.19. Dacomitinib
A moderate (23%) rate of transient serum ALT elevation (G3/4: 1%) has been reported without liver injury and, importantly, these rates are lower than those reported with other EGRF inhibitors such as Erlotinib and Gefitinib [114,115]. Serum ALT elevations >5 × ULN should lead to treatment interruption and if the alterations persist, are severe or associated with liver-related symptoms or jaundice, or reoccur on restarting treatment, the drug should be permanently discontinued. Patients with liver abnormalities during Dacomitinib may tolerate treatment with other EGFR inhibitors without recurrence of severe injury. No dose adjustment is recommended in patients with mild or moderate hepatic impairment (Child Pugh A, B) whereas the recommended dose has not been established for patients with severe hepatic impairment (Child Pugh C).
2.20. Duvelisib
ALT elevation developed in up to 40% of patients (Grade 3/4: 8%). The median time to onset of ALT elevation was two months, with a median duration of one month. Usually, the ALT elevations resolved spontaneously after drug withholding, and most patients were able to restart treatment without recurrence. While there were no reported cases of liver injury with jaundice, up to 35% of patients discontinued Duvelisib because of ALT elevations [116,117,118,119,120]. LITs should be monitored during treatment because Duvelisib affects B cell function, which may be capable of inducing HBVr, although in published trials, reactivation was not reported. For G2 ALT elevation, the same dose should be maintained while monitoring at least every week until ALT <3 × ULN. For G3 ALT elevation, drug should be suspended and weekly monitoring should be started until ALT <3 × ULN. Duvelisib can be restarted at the same dose (first occurrence) or at a reduced dose (15 mg twice daily) for subsequent occurrence. With G4 ALT elevation, Duvelisib needs to be permanently discontinued. Corticosteroids are often used if the liver injury does not resolve rapidly and maintenance of corticosteroids treatment may help to prevent recurrence of injury when restarting therapy. There is no known cross-sensitivity of hepatic injury among the different TKIs of this class.
2.21. Entrectinib
Serum ALT elevations are common during therapy (38%; G3/4: 2.9%), however, there are no reported cases of severe liver injury. The median time of ALT increase was two weeks (range: 1 day to 9.2 months) leading to dose interruptions or reductions in 0.8% of patients [121,122]. The product label recommends monitoring LITs every two weeks during the first month of treatment, then monthly thereafter and as clinically indicated. No dose adjustment is recommended for patients with mild hepatic impairment, whereas Entrectinib has not been studied in patients with moderate and severe hepatic impairment. Serum ALT elevations >5 × ULN should lead to dose interruption until recovery to baseline or <G1 and, if resolution occurs within four weeks, the drug could be resumed at the same dose, or otherwise should be permanently discontinued. For recurrent G3, doses should be resumed at 400 mg, a reduced dose if resolution occurs within four weeks. G4 ALT elevations should lead to dose interruption until recovery to baseline or <G1 and then the drug could be resumed at a reduced dose if resolution occurs within four weeks, or otherwise should be permanently discontinued in case of recurrent G4. In patients with clinically evident liver injury and jaundice, restarting therapy should be done with extreme caution. Cross-sensitivity to liver injury is uncommon among the TKIs, but there is no information or shared adverse event of Entrectinib with other antineoplastic TKIs.
2.22. Erdafitinib
In the prelicensure clinical trials, LITs abnormalities were frequent (41%) although usually mild (G3/4: up to 2%), without reports of liver injury or liver-related deaths. Since drug approval and a more widespread use, there have been no reports of liver injury. Regular monitoring of LITs is not specifically recommended during therapy, however G3 ALT elevations should lead to dose interruption until recovery to baseline or <G1, and then the drug can be resumed at a reduced dose, whereas G4 ALT elevation requires permanent drug discontinuation. There is no evidence to suggest cross-reactivity for adverse events between Erdafitinib and other FGFR TKIs [123].
2.23. Erlotinib
When combined with chemotherapy, an elevation in serum ALT levels is common (45%) (G3/4: 14%) [124,125,126]. Routine monitoring of LITs before starting and during therapy is recommended. Erlotinib is eliminated by hepatic metabolism and biliary excretion; therefore, patients with any hepatic impairment should be closely monitored during therapy. Serum ALT elevation >5 × ULN should lead to temporary discontinuation, which should be permanent if laboratory values do not improve significantly or resolve within three weeks. Erlotinib should be interrupted if total bilirubin is >3 × ULN. Restarting therapy is usually, but not always, followed by the recurrence of ALT elevations. Since a partial cross-sensitivity for liver injury among similar TKIs appears to exist, switching to another TKI of the same class requires careful monitoring. Patients with acute liver failure due to Erlotinib have been treated with corticosteroids, with uncertain results [44,45,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146].
2.24. Everolimus
Serum ALT elevation occurs in nearly a quarter of patients (G3/4: 1–3%), but the alteration is usually mild, asymptomatic and self-limiting, rarely requiring dose modification or discontinuation, and acute liver injury has not been reported despite its wide scale use. Since exposure to Everolimus was increased in patients with moderate hepatic impairment (Child Pugh B), dose reduction is recommended in such patients. Subsequent dosing should be individualized based on therapeutic drug monitoring. Everolimus has not been studied in patients with severe hepatic impairment (Child Pugh C) and therefore should not be used in this population. Importantly, Everolimus is an immunosuppressive agent and has been associated with HBVr (which can be severe and even fatal) in patients with cancer. Importantly enough, this drug has been associated also with reverse seroconversion, i.e., re-appearance of HBsAg in patients with resolved HBV infection, therefore routine screening for HBsAg, anti HBs and anti-HBc before starting therapy is mandatory [147,148,149,150,151,152,153,154,155,156,157].
2.25. Fedratinib
All grades of ALT elevations occurred in 43–53% (G3/4: 1–3%) of patients. The median time to onset of ALT elevation is one month, with 75% of cases occurring within three months. Monitoring LITs at baseline, during treatment and as clinically indicated is recommended. For G3/4 ALT elevation, the drug should be interrupted until resolved to baseline or ≤G1, at which point the drug can be restarted at a dose of 100 mg daily below the last given dose. In cases of the re-occurrence of ALT G3/4 elevation, Fedratinib should be permanently discontinued. The drug has not been evaluated in patients with severe hepatic impairment and (Child Pugh C), and therefore should be avoided in such patients. Cross-sensitivity for liver injury is uncommon among these TKIs class, but there is no information on the shared adverse event between other JAK inhibitors (such as Ruxolitinib) [158,159,160].
2.26. Gefitinib
In clinical trials, ALT elevation has been reported in 5 to 55% of the cases (G3/4: 2–27%). Despite the frequency of ALT elevation, severe liver injury was never reported. ALT elevation typically occurs 4 to 12 weeks into treatment, and restarting therapy is usually, but not always, followed by rapid recurrence of ALT elevation. Corticosteroid therapy did not appear to prevent the recurrence. Monitoring LITs during therapy is recommended and the drug should be discontinued in patients with worsening liver function or with severe hepatic impairment. G3/4 ALT elevation requires drug interruption until a reduction to baseline or <G1. The drug can be restarted at 100 mg daily dose below the last given dose. Re-occurrence of a G3/4 ALT elevation requires permanent treatment discontinuation. Patients with liver abnormalities during Gefitinib therapy generally tolerate other TKIs without the recurrence of severe injury [161,162,163,164,165,166,167,168,169,170]. The drug exposure was increased by 40% to 166% in patients with mild to severe hepatic impairment, and therefore, such patients should be monitored during treatment for adverse reactions.
2.27. Gilteritinib
Elevation in serum ALT levels is common during therapy, occurring in 42% of patients and rising above 5 × ULN in 14%, but without cases of acute liver injury. ALT elevations >5 × ULN should lead to temporary discontinuation, which should be permanent if laboratory values do not improve significantly or resolve within a few weeks, or if symptoms or jaundice arise [171].
2.28. Ibrutinib
In the prelicensure clinical trials, ALT elevation during therapy occurred in 20% to 30%, generally mild and self-limited. In controlled trials, there were no reports of liver injury or a need for early discontinuation because of hepatotoxicity [172,173,174]. Nevertheless, rare cases of acute liver injury, including acute liver failure and severe onset of HBVr, have been reported. Serum ALT elevations >5 × ULN should lead to dose reduction or temporary cessation. In patients with clinically apparent liver injury and jaundice, restarting therapy should be done with caution. Cross-sensitivity to liver injury is uncommon among other BKT TKIs and, in many situations, switching to another one may be appropriate. Importantly, patients who are to receive therapy with Ibrutinib should be screened for HBV markers and, if positive, managed according with HBV prophylaxis guidelines [17,19,20,175].
2.29. Idelalisib
The rates of ALT elevations ranged from 35% to 50%. Serum ALT elevations typically arise within 4 to 12 weeks of starting therapy and usually resolved rapidly with temporary discontinuation. Fatal and/or serious hepatotoxicity occurred in up to 14% of treated patients and for this reason, this drug has a warning box for hepatotoxicity, recommending LITs monitoring prior and during treatment. In some instances, when ALT remained high despite stopping therapy, corticosteroids appeared to have a beneficial effect. Due to the effects on B cell function, it may also be capable of inducing HBVr, although in published trials, this event has never been reported. Idelalisib should not be used with other agents with a potential hepatotoxic effect, and regular monitoring of LITs every 2 to 4 weeks is recommended during the first six months and every 1 to 3 months thereafter, with more frequent monitoring if serum ALT values rise. For ALT >3 − 5 × ULN, Idelalisib can be maintained at the same dose with weekly monitoring until ALT < 1 × ULN. The drug should be withheld with ALT >5 × ULN, continuing weekly LITs monitoring until the abnormality is resolved. The treatment may be resumed at a reduced dose (100 mg twice a day). Idelalisib should be permanently discontinued. Corticosteroids are often successfully used for treatment of liver injury that does not resolve rapidly after stopping the TKI. Moreover, continuing the corticosteroids may help to prevent the recurrence of injury when restarting therapy. There is no known cross-sensitivity to hepatic injury among the different TKIs of this class. Patients with any baseline hepatic impairment should be closely monitored for signs of toxicity [32,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190].
2.30. Imatinib
Elevation in ALT levels occurred in approximately 20% of treated patients (G3/4: 2–6.8%), requiring treatment discontinuation in <1% of patients. Assessment of LITs before and monthly thereafter, or as clinically indicated, is recommended. Bilirubin >3 × ULN or ALT >5 × ULN requires drug withholding until levels return to <1.5 and <2.5 × ULN, respectively, and then Imatinib can be resumed at a reduced daily dose (i.e., 400 mg to 300 mg, 600 mg to 400 mg or 800 mg to 600 mg daily), depending on the initial severity of the event [111]. Patients with severe hepatic impairment tend to have higher exposure to both Imatinib and its metabolites than patients with normal hepatic function. Therefore, those with mild and moderate hepatic impairment (Child Pugh A, B) do not require a dose adjustment and should be treated with the recommended doses, whereas a 25% dose reduction is recommended for patients with severe hepatic impairment (Child Pugh C).
2.31. Infigratinib
In pre-registration trials, ALT elevations arise in 51% (G3/4: 6%) of patients. ALT elevation was generally self-limited and rapidly resolving with or without dose adjustments, and none of the patients developed liver injury or jaundice. Indeed, since its approval, there have been no reports of severe liver injury. Regular monitoring of LITs is not specifically recommended, but in case of ALT elevations LITs monitoring is advisable, due to the risk of liver injury. The recommended dosage of Infigratinib for patients with mild or moderate hepatic impairment is 100 mg or 75 mg once daily for 21 consecutive days, followed by seven days off therapy in 28-day cycles, respectively. In patients with severe hepatic impairment, a recommended dose has not been established. ALT elevations > 5 × ULN or any elevations accompanied by jaundice or symptoms should lead to dose reduction or temporary cessation until the abnormalities resolve, or an alternative cause is identified. There is no evidence to suggest cross-reactivity for adverse events, hypersensitivity or hepatic injury between Infigratinib and other FGFR TKIs [191].
2.32. Lapatinib
Hepatotoxicity has been observed in clinical trials and post marketing experience (12–40%; G3/4 up to 13%), and may occur from days to several months after initiation of treatment. This adverse event may be severe and fatalities have been reported, although the causality of deaths is uncertain. For this reason, Lapatinib has a warning box for hepatotoxicity recommending LITs monitoring prior to and every 4 to 6 weeks during treatment, and as clinically indicated thereafter. The reduction or permanent discontinuation of the drug is required in case of severe changes in LITs [192,193]. Patients with severe hepatic impairment (Child Pugh C) should have their dose reduced from 1.250 mg/day to 750 mg/day (HER2 positive metastatic breast cancer indication) or from 1.500 mg/day to 1.000 mg/day (hormone receptor positive, HER2 positive breast cancer indication).
2.33. Larotrectinib
ALT increase occurred in up to 45% (G3/4: 11%) of patients. The median time to onset of the alteration was two months (range: 1 month to 13 months) with increased ALT levels leading to dose modifications in 6% and permanent drug discontinuation in 2% of patients, respectively. LITs should be monitored every two weeks during the first month, then monthly thereafter and as clinically indicated. Larotrectinib clearance is reduced in subjects with moderate to severe (Child Pugh B and C) hepatic impairment; therefore, the recommended starting dose should be reduced by 50% in such patients, whereas no dose adjustment is recommended for patients with mild hepatic impairment (Child Pugh A) [194,195,196].
2.34. Lenvatinib
Across clinical studies, enrolling patients with malignancies other than hepatocellular carcinoma (HCC), G3/4 ALT flare occurred in 4% of patients, and fatal events, including hepatic failure, acute hepatitis and hepatorenal syndrome, occurred in 0.5% of patients. Among patients treated for HCC, G3/4 ALT flare occurred in 3% of patients, and 2% percent of patients discontinued Lenvatinib. LITs should be monitored at baseline and then every two weeks for the first two months, and at least monthly thereafter during treatment, whereas patients with HCC should be closely monitored for signs of liver failure. G3/4 ALT elevations should lead to a dose interruption until improvement to baseline or <G1 ALT levels, and then the drug could be resumed at a reduced dose or discontinued, depending on the severity and persistence of hepatotoxicity. G4 ALT elevation and hepatic failure requires permanent drug discontinuation [197,198].
2.35. Lorlatinib
Increased ALT of any grade occurred in up to 28% of patients (G3/4: 1–2.1%), usually within three days, and returned to normal limits after a median of 15 days (7 to 34 days). This drug is contraindicated in patients treated with both moderate and strong CYP3A inducers, which should be discontinued prior to initiating Lorlatinib. If concomitant use of moderate CYP3A inducers cannot be avoided, LITs should be monitored 48 h after initiating Lorlatinib and at least thrice during the first week of treatment. Monitoring LITs is recommended at baseline and during treatment. ALT elevations >5 × ULN should lead to dose interruption and if the alterations persist, are severe or reoccur upon restarting, Lorlatinib should be permanently discontinued. Patients developing liver injury under treatment with a specific ALK inhibitor can often be treated with other ALK TKI without recurrence of liver injury, but careful monitoring is necessary during treatment after hepatotoxicity from a related TKI agent [199]. No dose adjustment is recommended for patients with mild hepatic impairment (Child Pugh A), whereas the recommended dose has not been established for patients with moderate or severe hepatic impairment.
2.36. Midostaurin
Elevation in serum ALT levels is common during therapy, occurring in up to 71% of patients (G3/4: 4–20). Hyperbilirubinemia was also reported to occur commonly, however liver injury with jaundice, severe hepatoxicity and deaths from hepatic failure have not been reported. It must be noted that, because of the limited clinical experience with the use of Midostaurin and other FLT3 TKIs, their potential for causing liver injury is not well defined as yet. Serum ALT elevations >5 × ULN should lead to temporary discontinuation, which should be permanent if laboratory values do not improve significantly or resolve within a few weeks, or if symptoms or jaundice arise [200].
2.37. Mobocertinib
Increased ALT of any grade occurred in up to 22% of patients (G3/4: 1–2.7%). No LITs monitoring is recommended. Intolerable or recurrent G2 or G3 hepatotoxicity should lead to temporary discontinuation until reaching levels ≤G1, when the drug can be resumed at either the same dose or the next lower dose. G4 toxicity should lead to temporary discontinuation until ≤G1, and the drug can be resumed at the next lower dose if recovery occurs within two weeks, or permanently discontinued if recovery does not occur within two weeks as well as in case of liver toxicity recurrence. No dose adjustment is recommended for patients with mild or moderate hepatic impairment (Child Pugh A, B) whereas the recommended dosage has not been established for patients with severe hepatic impairment (Child Pugh C) [201,202].
2.38. Neratinib
Elevation in serum ALT levels was uncommon during therapy, occurring in up to 13% of patients, (G3/4: 1–5%), and was typically mild, self-limited and not associated with symptoms or jaundice. Drug discontinuation due to hepatotoxicity was reported in 1.7% of patients and this side effect may be a class effect among TKI of HER2, although both the frequency and severity vary among different agents. Monitoring LITs is recommended before treatment, monthly for the first three months and every three months thereafter or as clinically indicated. Serum ALT elevations >5 × ULN should lead to temporary discontinuation, which should be permanent if laboratory values worsen or do not resolve or improve significantly within a few weeks, or if clinical symptoms or jaundice arise. Therapy can be restarted at the next lower dose if recovery to ≤G1 is achieved and it is usually, but not always, followed by the recurrence of ALT elevations. G4 ALT elevation requires permanent drug discontinuation [203,204].
2.39. Nilotinib
Elevation in serum ALT levels is reported in up to 24% of patients (G3/4: 3–9%), but these abnormalities are usually asymptomatic. There has been only a single published case report of liver injury attributed to Nilotinib. Most other TKIs have been linked to rare instances of clinically evident liver injury, usually arising 1 to 8 weeks into treatment. Monitoring of LITs is recommended at baseline and monthly or as clinically indicated during treatment. ALT levels >5 × ULN should lead to dose reduction or temporary discontinuation. Cross-reactivity of the hepatic injury with other TKI is uncommon, but can occur, therefore drug switching needs to be carefully evaluated [205,206].
2.40. Osimertinib
Elevation in serum ALT levels is uncommon (5%; G3/4: 1%). In pre-registration trials, one single evident case of liver injury was reported, however both the clinical features and the causality with Osimertinib were not clearly defined. Since its approval and more widespread use, there have been no reported cases of liver injury. Routine monitoring of LITs is not recommended, but ALT >5 × ULN should lead to discontinuation for up to three weeks. If the level drops to G0-2, the drug can be resumed at 80 mg or 40 mg daily whereas if no improvement is reached within three weeks, Osimertinib should be permanently discontinued. There does not appear to be cross-reactivity with other EGFR inhibitors and, when needed, switching to another TKI may be appropriate [207].
2.41. Palbociclib
In large clinical trials, serum ALT elevations occurred in up to 43% of patients (G3/4: 2%), and since its approval and widespread use, there have been several reports of ALT elevation arising after two or three cycles. Aminotransferase levels generally improved after discontinuation but recurred rapidly when Palbociclib was restarted. Serum ALT >5 × ULN or any elevations accompanied by jaundice or clinical symptoms should lead to dose reduction or temporary discontinuation. No dose adjustment is required in patients with mild or moderate hepatic impairment (Child Pugh A and B), whereas for patients with severe hepatic impairment (Child Pugh C), the recommended dose is 75 mg once daily for 21 consecutive days, followed by seven days off treatment to complete a full cycle of 28 days. There is no evidence to suggest cross-reactivity for adverse events, hypersensitivity or hepatic injury between Palbociclib and other CDK inhibitors [208].
2.42. Pazopanib
In large clinical trials, serum ALT elevation occurred in up to 60% of patients (G3/4: 9–17%), with total serum bilirubin elevation in approximately one-third. In preliminary trials in different solid tumors, there were rare reports of hepatitis with jaundice (<1%), but subsequent reports showed fatal instances of liver injury. This drug has a warning box for hepatotoxicity recommending to monitor LITs before initiation of treatment and at least once every four weeks for the first four months, or as clinically indicated. Patients with ALT elevation >3 − 8 × ULN may be maintained on treatment with the weekly monitoring of LITs until ALT levels return to G1 or baseline. In patients with isolated ALT elevations >8 × ULN, the drug should be withheld until they return to G1 or baseline; if the potential benefit for reinitiating treatment outweighs the risk of hepatotoxicity, the drug could be reintroduced at a reduced dose of no more than 400 mg once daily, maintaining LITs monitored weekly for eight weeks. Following the reintroduction of Pazopanib, a recurrent ALT increase >3 × ULN requires drug discontinuation. The dosage of Pazopanib in patients with moderate hepatic impairment (Child Pugh B) should be reduced to 200 mg per day, whereas there are no data for patients with severe hepatic impairment (Child Pugh C); therefore, the use of Pazopanib is not recommended in such patients [209,210,211,212,213].
2.43. Pemigatinib
In pre-registration clinical trials, ALT elevation was reported in 43% of patients (G3/4: 4.1%). The elevation was typically self-limited and resolved rapidly with or without dose adjustments. Since its approval, there have been no reports of clinically evident severe liver injury. Elevations in serum bilirubin were also common, but usually in the context of cholangiocarcinoma with partial or complete biliary obstruction. The recommended drug dose for patients with severe hepatic impairment (Child Pugh C) is 9 mg within the specific schedule (intermittent or continuous), designed for the specific indication [214].
2.44. Pexidartinib
Elevation in serum ALT levels is common, occurring in 50% to 90% of patients (G3/4: 12–20%). In addition, elevation in ALP levels occur in up to 20% of patients. In registration trials, liver injury with jaundice developed in 5% of patients. The time of onset of liver injury was usually between 2 and 6 weeks, and the pattern of liver enzyme elevations was either mixed or solely cholestatic. Liver biopsy documented both bile duct injury and loss, and three patients under treatment for conditions other than Tenosynovial giant cell tumors, developed bile duct paucity and features of vanishing bile duct syndrome that ultimately led to liver transplantation in one subject. For this reason, this drug has a warning box for hepatotoxicity, recommending monitoring of LITs before initiation of treatment, weekly for the first eight weeks, every two weeks for the next month and every three months thereafter. Patients with ALT elevations >3 − 5 × ULN should discontinue the treatment with a weekly monitoring of LITs until returning to G1 or baseline; if this occurs within four weeks from discontinuation, the drug can be resumed at a reduced dose, otherwise Pexidartinib needs to be permanently discontinued. For patients with ALT >5 − 10 × ULN, treatment should be interrupted and LITs should be checked twice weekly until they return to G1 or baseline; if this happens within four weeks, the drug could be resumed at a reduced dose, otherwise Pexidartinib needs to be permanently discontinued. In patients with ALT >10 × ULN, the drug should be discontinued permanently. Rechallenge with a reduced dose of Pexidartinib may result in a recurrence of increased ALT, bilirubin or ALP. Pexidartinib should be avoided in patients with pre-existing increased serum ALT, total bilirubin or direct bilirubin >ULN, or active liver or biliary tract disease, including increased ALP [215,216,217,218].
2.45. Ponatinib
In large clinical trials, elevation in ALT levels occurred in up to 56% of patients (G3/4: 8%). While these abnormalities were reversible in most patients, they were prolonged or severe in many others. Episodes of clinically evident progressive hepatic failure and death were reported in clinical trials. This drug has a warning box for hepatotoxicity, which recommends monitoring LITs before initiation of treatment and at least monthly or as clinically indicated. A ≥G2 ALT elevation occurring at 45 mg initial dose requires drug interruption until ≤G1, and a new start at 30 mg after recovery. When the starting dose is 30 mg, drug interruption is required until ≤G1 and a new start at 15 mg after recovery; when starting dose is 15 mg, permanent drug discontinuation is required. Hepatotoxicity ≥G2 with bilirubin >2 × ULN requires permanent treatment discontinuation. Ponatinib is metabolized in the liver largely through the CYP 3A4 pathway, and liver injury may be related to the production of a toxic intermediate. Because of this metabolic pathway, Ponatinib is susceptible to drug-drug interactions (DDI) when using agents that induce or inhibit CYP 3A4. As hepatic elimination is a major route of excretion for Ponatinib, hepatic impairment may result in increased drug exposure and increased risk for adverse reactions; therefore, in patients with moderate to severe (Child Pugh B or C) hepatic impairment, Ponatinib should be avoided unless the benefit outweighs the possible risk of overexposure [219,220].
2.46. Pralsetinib
ALT increase occurred in up to 46% of patients (G3/4: up to 6%). The median time to first onset was 22 days (range: 7 days to 1.7 years). LITs should be monitored prior to initiation, every two weeks during the first three months, then monthly thereafter and as clinically indicated. G3 or G4 toxicity requires the withholding of Pralsetinib with once-a-week ALT monitoring until resolution to baseline or G1; then, the drug can be resumed at a reduced dose. If hepatotoxicity recurs at G3 or greater, the drug needs to be permanently discontinued. Pralsetinib has not been studied in patients with moderate to severe hepatic impairment (Child Pugh B, C), whereas no dose adjustment is required for patients with mild hepatic impairment (Child Pugh A) [221,222].
2.47. Regorafenib
Severe and sometimes fatal hepatotoxicity events have been observed in clinical trials; therefore, this drug has a warning box for hepatotoxicity and recommends monitoring LITs before initiation of treatment and at least every two weeks during the first two months of treatment, or more frequently as clinically indicated. No clinically important differences in the mean exposure of Regorafenib or the active metabolites M-2 and M-5 were observed in patients with HCC and mild/moderate hepatic impairment (Child Pugh A and B), compared to patients with normal hepatic function, therefore no dose adjustment is recommended in such patients. Regorafenib is not recommended in patients with severe hepatic impairment (Child Pugh C) as it has not been studied in this population [223,224].
2.48. Ribociclib
In preregistration and large clinical trials, ALT elevation occurred in 46% of patients (G3/4: 9–14%). LITs should be evaluated initiating treatment and then every two weeks for the first two cycles, at the beginning of the subsequent four cycles and as clinically indicated. Ribociclib is extensively metabolized in the liver, largely through the CYP 3A4 pathway, and liver injury might be induced by the production of a toxic or immunogenic intermediate. On the other hand, the inhibition of CDK4/6 may also affect hepatocytes and cause direct hepatotoxicity. G3 ALT elevation requires dose interruption until recovery to baseline with a possible drug restart at the next lower dose level. G3 recurrence or G4 ALT elevation requires permanent treatment discontinuation. No dose adjustment is necessary in patients with mild hepatic impairment (Child Pugh A), whereas a reduced starting dose of 400 mg is recommended in patients with moderate and severe hepatic impairment (Child Pugh B or C) [225,226].
2.49. Ripretinib
ALT increase occurred in 12% of patients (G3/4: 1.2%). Monitoring LITs, as well as dose adjustment in patients with mild hepatic impairment (Child Pugh A), is not required. A recommended dosage and specific monitoring protocol have not been established for patients with moderate or severe hepatic impairment (Child Pugh B, C) [227,228].
2.50. Ruxolitinib
ALT elevation occurred in 25–38% of patients (G3/4: 1%) and were generally self-limited, asymptomatic and mild, with no cases of clinically evident liver injury. Importantly, there have been several published reports of HBVr, therefore patients undergoing Ruxolitinib should be screened for HBV markers and managed according to HBV reactivation guidelines (21). Ruxolitinib is metabolized in the liver largely through the CYP 3A4 pathway and therefore is susceptible to DDI agents that inhibit or induce this cytochrome activity. Blood levels of Ruxolitinib increase in mild to severe hepatic impairment (Child Pugh A to C), and therefore dose reduction is recommended in such patients [229,230].
2.51. Selpercatinib
Increased ALT occurred in 56% of patients (G3/4: 12%). The median time to onset of ALT elevation was 5.8 weeks (range: 1 day to 2.5 years). Monitoring LITs prior to initiation and every two weeks during the first three months, then monthly thereafter and as clinically indicated, is recommended. G3/4 ALT elevation requires dose interruption with LITs monitoring once weekly until resolution to baseline or G1; the drug can be restarted at two-dose levels reduction with LITs monitoring once weekly. Dosage can be increased by one dose after a minimum of two weeks without recurrence, and then further increased prior to the hepatotoxicity after a minimum of four weeks without recurrence. The Selpercatinib dose should be reduced when administered to patients with severe hepatic impairment (Child Pugh C), whereas no modification is recommended for patients with mild or moderate hepatic impairment [231,232].
2.52. Sorafenib
Sorafenib-induced liver dysfunction has been reported in 11% of patients (G3/4: 3%), although liver injury has been reported only in 0.06% of treated subjects. Monitoring LITs is recommended and in case of a significant increase of ALT without alternative explanation (such as viral hepatitis or progression of the underlying liver malignancy), treatment should be discontinued [233,234].
2.53. Sunitinib
In large clinical trials, ALT elevation was common, occurring in 39–61% of patients (G3/4: 2–5%), and in clinical trials and post-marketing experiences, the treatment has been associated with liver failure or death (0.3%). Therefore, this drug has a warning box for hepatotoxicity; monitoring LITs is recommended before initiation, during each cycle of treatment, and as clinically indicated. Sunitinib should be interrupted for G3/4 drug-related hepatic adverse events and discontinued if there is no resolution, a further increase of LITs or signs and symptoms of liver failure. Sunitinib has also been reported to cause hyperammonemia and encephalopathy with confusion and irritability, even with minimal elevation in serum enzymes and bilirubin and marked increases (4 − 10 × ULN) in serum ammonia levels. Recovery is rapid once Sunitinib is stopped and the syndrome can recur with re-exposure. Interestingly, there appears to be little cross-reactivity of this complication with other same class TKIs. The clinical features of the reported cases of severe acute liver injury due to Sunitinib have suggested a possible mechanism of ischemic damage related to hypotension and anoxia rather than direct hepatic injury.
No dose adjustment is required in subjects with mild or moderate hepatic impairment (Child Pugh A and B), whereas no data exist in subjects with severe hepatic impairment (Child Pugh C) since most studies have excluded patients with significant underlying liver disease at baseline [235,236,237,238].
2.54. Temsirolimus
Serum ALT elevation occurred in 30% to 40% of patients (G3/4: 1–3%), but the abnormalities are usually mild, asymptomatic and self-limited, rarely requiring dose modification or discontinuation. Since approval and widespread clinical use, there have been no case reports of severe liver injury. Although to date there have been no reports of HBVr, since Temsirolimus is an immunosuppressive agent, HBVr should be considered a possible complication of this drug. Concentrations of Temsirolimus as well as its metabolite Sirolimus are increased in patients with elevated ALT or bilirubin levels, and therefore, its use is contraindicated in patients with bilirubin >1.5 × ULN, and the dose should be reduced in patients with mild hepatic impairment (Child Pugh A) [239,240].
2.55. Tepotinib
Increased ALT occurred in 13% of patients (G3/4: 4.2%) with one reported fatal adverse reaction of hepatic failure (0.2%). The median time to onset of ≥G3 ALT increase was 30 days (range 1 to 178). LITs should be monitored prior to the start of treatment, every two weeks during the first three months and then once a month or as clinically indicated, with more frequent testing in patients who develop increased ALT or bilirubin. G3 hepatotoxicity requires dose interruption until recovery to baseline. If hepatotoxicity recovers to baseline within seven days, Tepotinib can be resumed at the same dose. Otherwise, it should be resumed at a reduced dose. G4 ALT increase or bilirubin >2 × ULN requires permanent drug discontinuation. No dose modification is recommended in patients with mild or moderate hepatic impairment (Child Pugh A and B), whereas the pharmacokinetics and safety of Tepotinib in patients with severe hepatic impairment (Child Pugh C) have not been studied [241].
2.56. Tivozanib
Increased ALT occurred in 30% of patients (G3/4: 4%). No dose modification is recommended for patients with mild hepatic impairment (Child Pugh A), but should be reduced in patients with moderate hepatic impairment (Child B), whereas the recommended dosage in patients with severe hepatic impairment (Child Pugh C) has not been established [242].
2.57. Tucatinib
Increased ALT occurred in 46% of patients (G3/4: 8%). LITs should be monitored prior to starting, every three weeks during treatment and as clinically indicated. G3 ALT increase or bilirubin >3 − 10 × ULN requires dose interruption until recovery to ≤G1, and then the drug could be resumed at the next lower dose level. G4 ALT increase or bilirubin >10 × ULN requires permanent drug discontinuation. Tucatinib exposure is increased in patients with severe hepatic impairment (Child Pugh C), therefore requiring a reduced dose. No dose adjustment is required for patients with mild or moderate hepatic impairment (Child Pugh A and B) [243].
2.58. Vandetanib
In large clinical trials, increased ALT occurred in 51% of patients (G3/4: 2%), without reports of severe liver injury or hepatic failure. Since approval and a wider scale use, there have been no published reports of hepatotoxicity, and the product label does not include discussion of hepatotoxicity. There are limited data in patients with liver dysfunction, however the use of Vandetanib is not recommended in patients with moderate and severe hepatic impairment (Child Pugh Band C), as safety and efficacy have not been established [244].
2.59. Zanobrutinib
Increased ALT occurred in 28% of patients (G3/4: 0.9%) without reports of liver injury or liver-related deaths. Nonetheless, other Bruton’s kinase inhibitors (Ibrutinib and Acalabrutinib) have been associated with rare cases of acute liver injury, including acute liver failure as well as cases of HBVr. No dosage modification is recommended in patients with mild to moderate hepatic impairment (Child Pugh A and B). Although Zanobrutinib has not been fully evaluated in patients with severe hepatic impairment (Child Pugh C), in such patients the recommended dosage should be reduced [245].
3. Discussion
The introduction of small-molecule PKIs in clinical oncology has dramatically improved the prognosis of certain types of cancers and in the last decades, the number of newly approved molecules has rapidly increased. Unfortunately, hepatotoxicity is a major safety concern of these drugs, with many of them having been implicated in cases of clinically evident liver injury and nearly 10% of the 62 approved compounds containing a drug labeling warning for hepatotoxicity. An interesting point, although not addressed by our review, is the issue of the combined use of a natural agent together with PKI. Data from the literature hypothesize that traditional Chinese medicinal herbs might increase efficacy while reducing toxicity if administered in combination with PKI, i.e., EGFR-TKI for Advanced Non-Small-Cell Lung Cancer [246]. However, only scattered evidence on this topic has been documented outside of China, and most of their reports do not specifically refer to liver toxicity. More importantly, all these evidences require further verification by well-designed studies. Above all, we should also highlight the fact that among the vast market of “natural products”, in certain cases, not all the components are clearly declared; this can prevent physicians from properly avoiding potentially major interferences with the CYP 3A4 pathway (with an inhibition or induction of this specific hepatic microsomal activity), leading to the production of several potentially toxic or immunogenic intermediates.
Moreover, several PKIs have also been implicated in causing HBV reactivation, both in patients with overt and resolved infection. Indeed, some PKIs act as immunosuppressants, lowering the normal immune surveillance pressure against HBV and enhancing HBV viral replication, which is responsible for the rapid spreading of the infection to most of the hepatocytes, followed by hepatitis flare when the host immunity recovers after PKI withdrawal. Some PKIs, even if not specifically identified as immunosuppressant agents, may induce HBVr after a tumor response, supporting the hypothesis of immune restoration leading to HBVr [11,12,247]. Overall, the severity and consequences of the side effects due to either HBVr or drug-related hepatotoxicity is driven by the pre-treatment stage of the liver function, extent of liver damage, patient’s age and performing status.
4. Future Directions
Despite the progress of PKIs for cancer treatment, the safety issues are often the main reason for their reduced effectiveness in the real practice since toxicity (and particularly hepatotoxicity) may cause their temporary or definitive discontinuation. For this reason, all the research efforts must aim to maximize the development of safer PKIs that can be defined not only in the pre-clinical and clinical phases, but also in the post-marketing phase. Research should focus on the identification of possible genetic predictors of hepatic metabolism and bio-transformation of the drugs, so that the therapeutic choice on the type and dose of the PKI should be tailored for each individual patient by identifying the one presenting both maximum efficacy and safety. Further efforts should be made in understanding the real rates of hepatotoxicity in cancer patients, overcoming all the confounding factors. Indeed, these patients may have already been treated with chemotherapies that could have impaired their liver function, or they may have metastatic liver involvement or may be on concomitant treatment with other drugs that could be either responsible for DDIs with PKIs or being primarily hepatotoxic themselves. Furthermore, as most of the data on hepatotoxicity comes from pre-clinical and clinical studies on fairly selected patients, and the post-marketing data are often based only on the most severe described cases, more information from the real practice with large-scale studies and possible long-term follow-ups are mostly needed. The aim should be to carefully assess the occurrence of liver injury or even liver failure. More data are needed in patients with pre-existing liver damage who need PKI for tumors arising in organs different from the liver, but above all, for those who need PKI for HCC or other tumors in the context of a pre-existing or even advanced liver disease. More information is also needed on the cross-reactivity of risk for hepatic injury and the possibility of safe therapeutic switches between different PKIs in order to provide each patient with the most effective treatment strategies.
The increased access of patients to these therapies and the risk of liver damage requires greater synergy and collaboration between oncologists and hepatologists, both in the correct definition of the patient at greater risk of adverse events before starting PKI, but also in the early identification of patients who develop liver damage for the most appropriate medical management of this complication. Although it is not easy to predict and treat the progress of most drug-related liver damages, it is essential, not only to ensure a prompt recovery, but mainly to prevent the evolution towards liver failure, as cancer patients currently do not have the opportunity for liver transplantation. Finally, the choice regarding the continuation of the PKI treatment could be shared between the oncologist and hepatologist on a case-by-case level and tailored to the risk/benefit ratio, especially for patients who are prone to respond to anticancer therapy.
5. Conclusions
The issue of potential drug hepatotoxicity and HBVr has always represented a major concern when treating cancer patients, although the current PKIs seem to be less dangerous than the previous standard chemotherapies [248]. Certainly, the occurrence of hepatotoxicity impacts both patient care and the survival rate since it deeply influences the management of a crucial oncological treatment by imposing either suspension or permanent discontinuation, or switching to a different drug. Therefore, any treatment choice after PKI-hepatotoxicity must take into account the potential beneficial risks while preventing any further liver deterioration. To minimize the consequences of this event, it is essential to:
Carefully select patients suitable for a specific PKI:
- Adapt the recommended PKI dose according to the pre-existing hepatic impairment;
- Avoid DDIs, also informing patients about the potential risks of concomitant use of over-the-counter drugs which could alter the metabolism of PKIs, thus favoring the formation of potentially hepatotoxic metabolites;
- Set and maintain the recommended monitoring protocol for LITs.
Finally, oncologists must be aware that is possible to avoid the use of a specific PKI when the risk of liver toxicity exceeds the possible benefits. In addition, they must be ready to promptly reduce or suspend the drug in the event of any LITs alteration, referring to hepatologists for an additional assessment of all the patients with a baseline hepatic dysfunction prior to starting PKI, or those developing liver damage during treatment for an appropriate medical management.
Author Contributions
Conceptualization, M.V. and S.F.; writing—original draft preparation, M.V., M.L.M., M.V.G., N.P. and M.D.G.; supervision, S.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Krause, D.S.; Van Etten, R.A. Tyrosine kinases as targets for cancer therapy. N. Engl. J. Med. 2005, 353, 172–187. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.H.; Kerkela, R.; Force, T. Mechanisms of cardiomyopathy associated with tyrosine kinase inhibitor cancer therapeutics. Circulation 2008, 118, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.J.; Linley, A.; Hammond, D.E.; Hood, F.E.; Coulson, J.M.; MacEwan, D.J.; Ross, S.J.; Slupsky, J.R.; Smith, P.D.; Eyers, P.A.; et al. New perspectives, opportunities, and challenges in exploring the human protein kinome. Cancer Res. 2018, 78, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Xu, J.J.; Bi, Y.A.; Mireles, R.; Davidson, R.; Duignan, D.B.; Campbell, S.; Kostrubsky, V.E.; Dunn, M.C.; Smith, A.R.; et al. Role of hepatic transporters in the disposition and hepatotoxicity of a HER2 tyrosine kinase inhibitor CP-724,714. Toxicol. Sci. 2009, 108, 492–500. [Google Scholar] [CrossRef]
- Shen, T.; Liu, Y.; Shang, J.; Xie, Q.; Li, J.; Yan, M.; Xu, J.; Niu, J.; Liu, J.; Watkins, P.B.; et al. Incidence and etiology of drug-induced liver injury in Mainland China. Gastroenterology 2019, 156, 2230–2241. [Google Scholar] [CrossRef]
- Bjornsson, E.S.; Bergmann, O.M.; Bjornsson, H.K.; Kvaran, R.B.; Olafsson, S. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology 2013, 144, 1419–1425. [Google Scholar] [CrossRef]
- Chalasani, N.; Bonkovsky, H.L.; Fontana, R.; Lee, W.; Stolz, A.; Talwalkar, J.; Reddy, K.R.; Watkins, P.B.; Navarro, V.; Barnhart, H.; et al. Features and outcomes of 899 patients with drug-induced liver injury: The DILIN prospective study. Gastroenterology 2015, 148, 1340–1352. [Google Scholar] [CrossRef]
- Bjornsson, E.S. Hepatotoxicity by drugs: The most common implicated agents. Int. J. Mol. Sci. 2016, 17, 224. [Google Scholar] [CrossRef]
- Bjornsson, E.S. Drug-induced liver injury: An overview over the most critical compounds. Arch. Toxicol. 2015, 89, 327–334. [Google Scholar] [CrossRef]
- Teschke, R. Top-ranking drugs out of 3312 drug-induced liver injury cases evaluated by the Roussel uclaf causality assessment method. Expert. Opin. Drug. Metab. Toxicol. 2018, 14, 1169–1187. [Google Scholar] [CrossRef]
- Seggewis, R.; Lore’, K.; Greiner, E.; Magnusson, M.K.; Price, D.A.; Douek, D.C.; Dunbar, C.E.; Wiestner, A. Imatininb inhibits T-cell-receptor mediated T-cell proliferation and activation in a dose-dependent manner. Blood 2005, 105, 2473–2479. [Google Scholar] [CrossRef]
- Cwynarski, K.; Laylor, R.; Macchiarulo, E.; Goldman, J.; Lombardi, G.; Melo, J.V.; Dazzi, F. Imatinib inhibits the activation and proliferation of normal T Lymphocytes in vitro. Leukemia 2004, 18, 1332–1339. [Google Scholar] [CrossRef]
- Orlandi, E.M.; Elena, C.; Bono, E. Risk of hepatitis B reactivation under treatment with tyrosine-kinase inhibitors for chronic myeloid leukemia. Leuk. Lymphoma 2017, 58, 1764–1766. [Google Scholar] [CrossRef]
- Sorà, F.; Ponziani, F.R.; Laurenti, L.; Chiusolo, P.; Autore, F.; Gasbarrini, A.; Sica, S.; Pompili, M. Low risk of hepatitis B virus reactivation in patients with resolved infection and chronic myeloid leukemia treated with tyrosine kinase inhibitors. Leuk. Lymphoma 2017, 58, 993–995. [Google Scholar] [CrossRef]
- Wang, Y.H.; Liang, J.D.; Sheng, W.H.; Tien, F.M.; Chen, C.Y.; Tien, H.F. Hepatitis B reactivation during treatment of tyrosine kinase inhibitors-Experience in 142 adult patients with chronic myeloid leukemia. Leuk. Res. 2019, 81, 95–97. [Google Scholar] [CrossRef]
- Yao, Z.H.; Liao, W.Y.; Ho, C.C.; Chen, K.Y.; Shih, J.Y.; Chen, J.S.; Lin, Z.Z.; Lin, C.C.; Yang, J.C.; Yu, C.J. Incidence of hepatitis B reactivation during epidermal growth factor receptor tyrosine kinase inhibitor treatment in non–small-cell lung cancer patients. Eur. J. Cancer 2019, 117, 107–115. [Google Scholar] [CrossRef]
- Hammond, S.P.; Chen, K.; Pandit, A.; Davids, M.S.; Issa, N.C.; Marty, F.M. Risk of hepatitis B virus reactivation in patients treated with ibrutinib. Blood 2018, 131, 1987–1989. [Google Scholar] [CrossRef]
- Lim, S.; Han, J.; Kim, G.M.; Han, K.H.; Choi, H.J. Hepatitis B viral load predicts survival in hepatocellular carcinoma patients treated with sorafenib. J. Gastroenterol. Hepatol. 2015, 30, 1024–1031. [Google Scholar] [CrossRef]
- Ni, Y.; Gao, L.; Lu, Y.; Ye, S.; Zhou, L.; Qian, W.; Liang, A.; Li, P. Risk of HBV reactivation in relapsed or refractory diffuse large B-cell lymphoma patients receiving Bruton tyrosine kinase inhibitors therapy. Front. Immunol. 2022, 13, 982346. [Google Scholar] [CrossRef]
- Yang, S.; Zhu, R.; Li, N.; Feng, Y.; Zuo, R.; Gale, R.P.; Huang, X. Ibrutinib in Advanced Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma: Lower Risk of Hepatitis B Virus Reactivation. Acta Haematol. 2022, 145, 54–62. [Google Scholar] [CrossRef]
- Viganò, M.; Serra, G.; Casella, G.; Grossi, G.; Lampertico, P. Reactivation of hepatitis B virus during targeted therapies for cancer and immune-mediated disorders. Expert Opin. Biol. Ther. 2016, 16, 917–926. [Google Scholar] [CrossRef]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non-small cell lung cancer, and other solid tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef]
- Dickler, M.N.; Tolaney, S.M.; Rugo, H.S.; Cortés, J.; Diéras, V.; Patt, D.; Wildiers, H.; Hudis, C.A.; O’Shaughnessy, J.; Zamora, E.; et al. MONARCH 1, A phase II study of abemaciclib, a CDK4 and CDK6 inhibitor, as a single agent, in patients with refractory HR(+)/HER2(-) metastatic breast cancer. Clin. Cancer Res. 2017, 23, 5218–5224. [Google Scholar] [CrossRef]
- Sledge, G.W., Jr.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in combination with fulvestrant in women with HR+/HER2- advanced breast cancer who had progressed while receiving endocrine therapy. J. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.C.; Manso, L.; et al. MONARCH 3: Abemaciclib as initial therapy for advanced breast cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Y.; Sun, T.; Mei, Y.Y.; Ping, L.H.; Yan, M.; Tong, Z.S.; Oppermann, C.P.; Liu, Y.P.; Costa, R.; Li, M.; et al. MONARCH plus: Abemaciclib plus endocrine therapy in women with HR+/HER2- advanced breast cancer: The multinational randomized phase III study. Ther. Adv. Med. Oncol. 2020, 12, 1758835920963925. [Google Scholar] [CrossRef] [PubMed]
- Spring, L.M.; Zangardi, M.L.; Moy, B.; Bardia, A. Clinical management of potential toxicities and drug interactions related to cyclin-dependent kinase 4/6 inhibitors in breast cancer: Practical considerations and recommendations. Oncologist 2017, 22, 1039–1048. [Google Scholar] [CrossRef]
- Byrd, J.C.; Harrington, B.; O’Brien, S.; Jones, J.A.; Schuh, A.; Devereux, S.; Chaves, J.; Wierda, W.G.; Awan, F.T.; Brown, J.R.; et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2016, 374, 323–332. [Google Scholar] [CrossRef]
- Wang, M.; Rule, S.; Zinzani, P.L.; Goy, A.; Casasnovas, O.; Smith, S.D.; Damaj, G.; Doorduijn, J.; Lamy, T.; Morschhauser, F.; et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): A single-arm, multicenter, phase 2 trial. Lancet 2018, 391, 659–667. [Google Scholar] [CrossRef]
- Awan, F.T.; Schuh, A.; Brown, J.R.; Furman, R.R.; Pagel, J.M.; Hillmen, P.; Stephens, D.M.; Woyach, J.; Bibikova, E.; Charuworn, P.; et al. Acalabrutinib monotherapy in patients with chronic lymphocytic leukemia who are intolerant to ibrutinib. Blood Adv. 2019, 3, 1553–1562. [Google Scholar] [CrossRef]
- Sharman, J.P.; Egyed, M.; Jurczak, W.; Skarbnik, A.; Pagel, J.M.; Flinn, I.W.; Kamdar, M.; Munir, T.; Walewska, R.; Corbett, G.; et al. Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzumab for treatment-naive chronic lymphocytic leukaemia (ELEVATE TN): A randomized, controlled, phase 3 trial. Lancet 2020, 395, 1278–1291. [Google Scholar] [CrossRef] [PubMed]
- Ghia, P.; Pluta, A.; Wach, M.; Lysak, D.; Kozak, T.; Simkovic, M.; Kaplan, P.; Kraychok, I.; Illes, A.; de la Serna, J.; et al. ASCEND: Phase III, randomized trial of acalabrutinib versus idelalisib plus rituximab or bendamustine plus rituximab in relapsed or refractory chronic lymphocytic leukemia. J. Clin. Oncol. 2020, 38, 2849–2861. [Google Scholar] [CrossRef]
- Davids, M.S.; Lampson, B.L.; Tyekucheva, S.; Wang, Z.; Lowney, J.C.; Pazienza, S.; Montegaard, J.; Patterson, V.; Weinstock, M.; Crombie, J.L.; et al. Acalabrutinib, venetoclax, and obinutuzumab as frontline treatment for chronic lymphocytic leukaemia: A single-arm, open-label, phase 2 study. Lancet Oncol. 2021, 22, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Cramer, P.; Fürstenau, M.; Robrecht, S.; Giza, A.; Zhang, C.; Fink, A.M.; Fischer, K.; Langerbeins, P.; Al-Sawaf, O.; Tausch, E.; et al. Obinutuzumab, acalabrutinib, and venetoclax, after an optional debulking with bendamustine in relapsed or refractory chronic lymphocytic leukaemia (CLL2-BAAG): A multicentre, open-label, phase 2 trial. Lancet Haematol. 2022, 9, e745–e755. [Google Scholar] [CrossRef]
- Miller, V.A.; Hirsh, V.; Cadranel, J.; Chen, Y.M.; Park, K.; Kim, S.W.; Zhou, C.; Wu Chou, S.; Mengzhao, W.; Yan, S.; et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): A phase 2b/3 randomized trial. Lancet Oncol. 2012, 13, 528–538. [Google Scholar] [CrossRef]
- Sequist, L.V.; Yang, J.C.; Yamamoto, N.; O’Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.M.; Boyer, M.; et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 2013, 31, 3327–3334. [Google Scholar] [CrossRef]
- Wu, Y.L.; Zhou, C.; Hu, C.P.; Feng, J.; Lu, S.; Huang, Y.; Li, W.; Hou, M.; Shi, J.H.; Lee, K.Y.; et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harboring EGFR mutations (LUX-Lung 6): An open-label, randomized phase 3 trial. Lancet Oncol. 2014, 15, 213–222. [Google Scholar] [CrossRef]
- Soria, J.C.; Felip, E.; Cobo, M.; Lu, S.; Syrigos, K.; Lee, K.H.; Göker, E.; Georgoulias, V.; Li, W.; Isla, D.; et al. LUX-Lung 8 Investigators. Afatinib versus erlotinib as second-line treatment of patients with advanced squamous cell carcinoma of the lung (LUX-Lung 8): An open-label randomized controlled phase 3 trial. Lancet Oncol. 2015, 16, 897–907. [Google Scholar] [CrossRef]
- Park, K.; Tan, E.H.; O’Byrne, K.; Zhang, L.; Boyer, M.; Mok, T.; Hirsh, V.; Yang, J.C.; Lee, K.H.; Lu, S.; et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): A phase 2B, open-label, randomized controlled trial. Lancet Oncol. 2016, 17, 577–589. [Google Scholar] [CrossRef]
- Yang, J.C.; Wu, Y.L.; Schuler, M.; Sebastian, M.; Popat, S.; Yamamoto, N.; Zhou, C.; Hu, C.P.; O’Byrne, K.; Feng, J.; et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomized, phase 3 trials. Lancet Oncol. 2015, 16, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Okamoto, I.; Nakagawa, K. Pooled safety analysis of EGFR-TKI treatment for EGFR mutation-positive non-small cell lung cancer. Lung Cancer 2015, 88, 74–79. [Google Scholar] [CrossRef]
- Ding, P.N.; Lord, S.J.; Gebski, V.; Links, M.; Bray, V.; Gralla, R.J.; Yang, J.C.; Lee, C.K. Risk of treatment-related toxicities from EGFR tyrosine kinase inhibitors: A meta-analysis of clinical trials of gefitinib, erlotinib, and afatinib in advanced EGFR-mutated non-small cell lung cancer. J. Thorac. Oncol. 2017, 12, 633–643. [Google Scholar] [CrossRef]
- Ueda, H.; Hayashi, H.; Kudo, K.; Takeda, M.; Nakagawa, K. Successful treatment with afatinib after gefitinib- and erlotinib-induced hepatotoxicity. Investig. New Drugs 2016, 34, 797–799. [Google Scholar] [CrossRef]
- Zenke, Y.; Umemura, S.; Sugiyama, E.; Kirita, K.; Matsumoto, S.; Yoh, K.; Niho, S.; Ohmatsu, H.; Goto, K. Successful treatment with afatinib after grade 3 hepatotoxicity induced by both gefitinib and erlotinib in EGFR mutation-positive non-small cell lung cancer. Lung Cancer 2016, 99, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Toba, H.; Sakiyama, S.; Takizawa, H.; Tangoku, A. Safe and successful treatment with afatinib in three postoperative non-small cell lung cancer patients with recurrences following gefitinib/erlotinib-induced hepatotoxicity. J. Med. Investig. 2016, 63, 149–151. [Google Scholar] [CrossRef]
- Yamanaka, Y.; Sekine, A.; Kato, T.; Yamakawa, H.; Ikeda, S.; Baba, T.; Iwasawa, T.; Okudela, K.; Ogura, T. Afatinib therapy for brain metastases aggravated by a reduction in the dose of erlotinib due to the development of hepatotoxicity. Intern. Med. 2017, 56, 2895–2898. [Google Scholar] [CrossRef] [PubMed]
- Seto, T.; Kiura, K.; Nishio, M.; Nakagawa, K.; Maemondo, M.; Inoue, A.; Hida, T.; Yamamoto, N.; Yoshioka, H.; Harada, M. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): A single-arm, open-label, phase 1–2 study. Lancet Oncol. 2013, 14, 590–598. [Google Scholar] [CrossRef]
- Tamura, T.; Kiura, K.; Seto, T.; Nakagawa, K.; Maemondo, M.; Inoue, A.; Hida, T.; Yoshioka, H.; Harada, M.; Ohe, Y. Three-year follow-up of an alectinib phase I/II study in ALK-positive non-small-cell lung cancer: AF-001JP. J. Clin. Oncol. 2017, 35, 1515–1521. [Google Scholar] [CrossRef]
- Gadgeel, S.M.; Gandhi, L.; Riely, G.J.; Chiappori, A.A.; West, H.L.; Azada, M.C.; Morcos, P.N.; Azada, M.C.; Morcos, P.N.; Lee, R.M.; et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): Results from the dose-finding portion of a phase 1/2 study. Lancet Oncol. 2014, 15, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Larkins, E.; Blumenthal, G.M.; Chen, H.; He, K.; Agarwal, R.; Gieser, G.; Stephens, O.; Zahalka, E.; Ringgold, K.; Helms, W.; et al. FDA approval: Alectinib for the treatment of metastatic, ALK-positive non-small cell lung cancer following crizotinib. Clin. Cancer. Res. 2016, 22, 5171–5176. [Google Scholar] [CrossRef]
- Shaw, A.T.; Gandhi, L.; Gadgeel, S.; Riely, G.J.; Cetnar, J.; West, H.; Camidge, D.R.; Socinski, M.A.; Chiappori, A.; Mekhail, T.; et al. Alectinib in ALK-positive, crizotinib-resistant, non-small-cell lung cancer: A single-group, multicenter, phase 2 trial. Lancet Oncol. 2016, 17, 234–242. [Google Scholar] [CrossRef]
- Hida, T.; Nokihara, H.; Kondo, M.; Kim, Y.H.; Azuma, K.; Seto, T.; Takiguchi, Y.; Nishio, M.; Yoshioka, H.; Imamura, F.; et al. Alectinib versus crizotinib in patients with ALK-positive non-small-cell lung cancer (J-ALEX): An open-label, randomized phase 3 trial. Lancet 2017, 390, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Makimoto, G.; Kawakado, K.; Nakanishi, M.; Tamura, T.; Kuyama, S. Successful Treatment with Lorlatinib after the Development of Alectinib-Induced Liver Damage in ALK-Positive Non-Small-Cell Lung Cancer: A Case Report. Case Rep. Oncol. 2021, 14, 197–201. [Google Scholar] [CrossRef]
- Duarte, F.A.; Rodrigues, L.B.; Paes, F.R.; Diniz, P.H.C.; Lima, H.F.C.A. Successful treatment with alectinib after crizotinib-induced hepatitis in ALK-rearranged advanced lung cancer patient: A case report. BMC Pulm. Med. 2021, 21, 43. [Google Scholar] [CrossRef]
- Jain, S.; Shah, A.N.; Santa-Maria, C.A.; Siziopikou, K.; Rademaker, A.; Helenowski, I.; Cristofanilli, M.; Gradishar, W.J. Phase I study of alpelisib (BYL-719) and trastuzumab emtansine (T-DM1) in HER2-positive metastatic breast cancer (MBC) after trastuzumab and taxane therapy. Breast Cancer Res. Treat. 2018, 171, 371–381. [Google Scholar] [CrossRef]
- Juric, D.; Janku, F.; Rodón, J.; Burris, H.A.; Mayer, I.A.; Schuler, M.; Seggewiss-Bernhardt, R.; Gil-Martin, M.; Middleton, M.R.; Baselga, J.; et al. Alpelisib plus fulvestrant in PIK3CA-altered and PIK3CA-wild-type estrogen receptor-positive advanced breast cancer: A phase 1b clinical trial. JAMA Oncol. 2019, 5, e184475. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I.A.; Prat, A.; Egle, D.; Blau, S.; Fidalgo, J.A.P.; Gnant, M.; Fasching, P.A.; Colleoni, M.; Wolff, A.C.; Winer, E.P.; et al. A phase II randomized study of neoadjuvant letrozole plus alpelisib for hormone receptor-positive, human epidermal growth factor receptor 2-negative breast cancer (NEO-ORB). Clin. Cancer Res. 2019, 25, 2975–2987. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; SOLAR-1 Study Group; et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Jones, R.L.; Serrano, C.; von Mehren, M.; George, S.; Heinrich, M.C.; Kang, Y.K.; Schöffski, P.; Cassier, P.A.; Mir, O.; Chawla, S.P.; et al. Avapritinib in unresectable or metastatic PDGFRA D842V-mutant gastrointestinal stromal tumours: Long-term efficacy and safety data from the NAVIGATOR phase I trial. Eur. J. Cancer 2021, 145, 132–142. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Jones, R.L.; von Mehren, M.; Schöffski, P.; Serrano, C.; Kang, Y.K.; Cassier, P.A.; Mir, O.; Eskens, F.; Tap, W.D.; et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): A multicentre, open-label, phase 1 trial. Lancet Oncol. 2020, 21, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.K.; George, S.; Jones, R.L.; Rutkowski, P.; Shen, L.; Mir, O.; Patel, S.; Zhou, Y.; von Mehren, M.; Hohenberger, P.; et al. Avapritinib Versus Regorafenib in Locally Advanced Unresectable or Metastatic GI Stromal Tumor: A Randomized, Open-Label Phase III Study. J. Clin. Oncol. 2021, 39, 3128–3139. [Google Scholar] [CrossRef]
- Rixe, O.; Bukowski, R.M.; Michaelson, M.D.; Wilding, G.; Hudes, G.R.; Bolte, O.; Motzer, R.J.; Bycott, P.; Liau, K.F.; Freddo, J.; et al. Axitinib treatment in patients with cytokine-refractory metastatic renal-cell cancer: A phase II study. Lancet Oncol. 2007, 8, 975–984. [Google Scholar] [CrossRef]
- Rini, B.I.; Escudier, B.; Tomczak, P.; Kaprin, A.; Szczylik, C.; Hutson, T.E.; Michaelson, M.D.; Gorbunova, V.A.; Gore, M.E.; Rusakov, I.G.; et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): A randomized phase 3 trial. Lancet 2011, 378, 1931–1939. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Tomczak, P.; Hutson, T.E.; Michaelson, M.D.; Negrier, S.; Oudard, S.; Gore, M.E.; Tarazi, J.; Hariharan, S.; et al. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: Overall survival analysis and updated results from a randomized phase 3 trial. Lancet Oncol. 2013, 14, 552–562. [Google Scholar] [CrossRef]
- Hutson, T.E.; Lesovoy, V.; Al-Shukri, S.; Stus, V.P.; Lipatov, O.N.; Bair, A.H.; Rosbrook, B.; Chen, C.; Kim, S.; Vogelzang, N.J. Axitinib versus sorafenib as first-line therapy in patients with metastatic renal-cell carcinoma: A randomized open-label phase 3 trial. Lancet Oncol. 2013, 14, 1287–1294. [Google Scholar] [CrossRef]
- Rini, B.I.; Melichar, B.; Ueda, T.; Grünwald, V.; Fishman, M.N.; Arranz, J.A.; Bair, A.H.; Pithavala, Y.K.; Andrews, G.I.; Pavlov, D.; et al. Axitinib with or without dose titration for first-line metastatic renal-cell carcinoma: A randomized double-blind phase 2 trial. Lancet Oncol. 2013, 14, 1233–1242. [Google Scholar] [CrossRef]
- Miyake, H.; Harada, K.I.; Ozono, S.; Fujisawa, M. Assessment of efficacy, safety, and quality of life of 124 patients treated with axitinib as second-line therapy for metastatic renal-cell carcinoma: Experience in real-world clinical practice in Japan. Clin. Genitourin. Cancer 2017, 15, 122–128. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicenter, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef]
- Cortes, J.E.; Kantarjian, H.M.; Brümmendorf, T.H.; Kim, D.W.; Turkina, A.G.; Shen, Z.X.; Pasquini, R.; Khoury, H.J.; Arkin, S.; Volkert, A.; et al. Safety and efficacy of bosutinib (SKI-606) in chronic phase Philadelphia chromosome-positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood 2011, 118, 4567–4576. [Google Scholar] [CrossRef]
- Gambacorti-Passerini, C.; Cortes, J.E.; Lipton, J.H.; Dmoszynska, A.; Wong, R.S.; Rossiev, V.; Pavlov, D.; Gogat Marchant, K.; Duvillié, L.; Khattry, N.; et al. Safety of bosutinib versus imatinib in the phase 3 BELA trial in newly diagnosed chronic phase chronic myeloid leukemia. Am. J. Hematol. 2014, 89, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Gambacorti-Passerini, C.; Brümmendorf, T.H.; Kim, D.W.; Turkina, A.G.; Masszi, T.; Assouline, S.; Durrant, S.; Kantarjian, H.M.; Khoury, H.J.; Zaritskey, A.; et al. Bosutinib efficacy and safety in chronic phase chronic myeloid leukemia after imatinib resistance or intolerance: Minimum 24-month follow-up. Am. J. Hematol. 2014, 89, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Brümmendorf, T.H.; Cortes, J.E.; de Souza, C.A.; Guilhot, F.; Duvillié, L.; Pavlov, D.; Gogat, K.; Countouriotis, A.M.; Gambacorti-Passerini, C. Bosutinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukaemia: Results from the 24-month follow-up of the BELA trial. Br. J. Haematol. 2015, 168, 69–81. [Google Scholar] [CrossRef]
- Gettinger, S.N.; Bazhenova, L.A.; Langer, C.J.; Salgia, R.; Gold, K.A.; Rosell, R.; Shaw, A.T.; Weiss, G.J.; Tugnait, M.; Narasimhan, N.I.; et al. Activity and safety of brigatinib in ALK-rearranged non-small-cell lung cancer and other malignancies: A single-arm, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 1683–1696. [Google Scholar] [CrossRef]
- Kim, D.W.; Tiseo, M.; Ahn, M.J.; Reckamp, K.L.; Hansen, K.H.; Kim, S.W.; Huber, R.M.; West, H.L.; Groen, H.J.M.; Hochmair, M.J.; et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase-positive non-small-cell lung cancer: A randomized, multicenter phase II trial. J. Clin. Oncol. 2017, 35, 2490–2498. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Kim, H.R.; Ahn, M.J.; Yang, J.C.H.; Han, J.Y.; Hochmair, M.J.; Lee, K.H.; Delmonte, A.; García Campelo, M.R.; Kim, D.-W.; et al. Brigatinib Versus Crizotinib in Advanced ALK Inhibitor-Naive ALK-Positive Non-Small Cell Lung Cancer: Second Interim Analysis of the Phase III ALTA-1L Trial. Clin. Oncol. 2020, 38, 3592–3603. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Escudier, B.; Powles, T.; Mainwaring, P.N.; Rini, B.I.; Donskov, H.; Hammers, T.E.; Hutson, T.E.; Lee, J.L.; Peltola, K.; et al. METEOR Investigators. Cabozantinib versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1814–1823. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Escudier, B.; Powles, T.; Tannir, N.M.; Mainwaring, P.N.; Rini, B.I.; Hammers, H.J.; Donskov, F.; Roth, B.J.; Peltola, K.; et al. METEOR investigators. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): Final results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2016, 17, 917–927. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Tovoli, F.; Dadduzio, V.; De Lorenzo, S.; Rimassa, L.; Masi, G.; Iavarone, M.; Marra, F.; Garajova, I.; Brizzi, M.P.; Daniele, B.; et al. Real-Life Clinical Data of Cabozantinib for Unresectable Hepatocellular Carcinoma. Liver Cancer 2021, 10, 370–379. [Google Scholar] [CrossRef]
- Kudo, M.; Tsuchiya, K.; Kato, N.; Hagihara, A.; Numata, K.; Aikata, H.; Inaba, Y.; Kondo, S.; Motomura, K.; Furuse, J.; et al. Cabozantinib in japanese patients with advanced hepatocellular carcinoma: A Phase 2 Multicenter Study. J. Gastroenterol. 2021, 56, 181–190. [Google Scholar] [CrossRef]
- Wolf, J.; Seto, T.; Han, J.Y.; Reguart, N.; Garon, E.B.; Groen, H.J.M.; Tan, D.S.W.; Hida, T.; de Jonge, M.; Orlov, S.V.; et al. Capmatinib in MET Exon 14-Mutated or MET-Amplified Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 944–957. [Google Scholar] [CrossRef]
- Takashi, S.; Kadoaki, O.; Shunichi, S.; Makoto, N.; Masayuki, T.; Keisuke, A.; Sanae, M.; Satoshi, N.; Takeshi, T.; Toyoaki, H. Capmatinib in Japanese patients with MET exon 14 skipping-mutated or MET-amplified advanced NSCLC: GEOMETRY mono-1 study. Cancer Sci. 2021, 112, 1556–1566. [Google Scholar]
- Shaw, A.T.; Kim, D.W.; Mehra, R.; Tan, D.S.; Felip, E.; Chow, L.Q.; Camidge, D.R.; Vansteenkiste, J.; Sharma, S.; De Pas, T.; et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 370, 1189–1197. [Google Scholar] [CrossRef]
- Crinò, L.; Ahn, M.J.; De Marinis, F.; Groen, H.J.; Wakelee, H.; Hida, T.; Mok, T.; Spigel, D.; Felip, E.; Nishio, M.; et al. Multicenter phase II study of whole-body and intracranial activity with ceritinib in patients with ALK-rearranged non-small-cell lung cancer previously treated with chemotherapy and crizotinib: Results from ASCEND-2. J. Clin. Oncol. 2016, 34, 2866–2873. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Mehra, R.; Tan, D.S.; Felip, E.; Chow, L.Q.; Camidge, D.R.; Vansteenkiste, J.; Sharma, S.; De Pas, T.; Riely, G.J.; et al. Activity and safety of ceritinib in patients with ALK-rearranged non-small-cell lung cancer (ASCEND-1): Updated results from the multicentre, open-label, phase 1 trial. Lancet Oncol. 2016, 17, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Tan, D.S.W.; Chiari, R.; Wu, Y.L.; Paz-Ares, L.; Wolf, J.; Geater, S.L.; Orlov, S.; Cortinovis, D.; Yu, C.J.; et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): A randomised, open-label, phase 3 study. Lancet 2017, 389, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Sassier, M.; Mennecier, B.; Gschwend, A.; Rein, M.; Coquerel, A.; Humbert, X.; Alexandre, J.; Fedrizzi, S.; Gervais, R. Successful treatment with ceritinib after crizotinib induced hepatitis. Lung Cancer 2016, 95, 15–16. [Google Scholar] [CrossRef]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef]
- McArthur, G.A.; Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Dummer, R.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): Extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014, 15, 323–332. [Google Scholar] [CrossRef]
- Dreyling, M.; Santoro, A.; Mollica, L.; Leppä, S.; Follows, G.A.; Lenz, G.; Kim, W.S.; Nagler, A.; Panayiotidis, P.; Demeter, J.; et al. Phosphatidylinositol 3-Kinase Inhibition by Copanlisib in Relapsed or Refractory Indolent Lymphoma. J. Clin. Oncol. 2017, 35, 3898–3905. [Google Scholar] [CrossRef]
- Dreyling, M.; Morschhauser, F.; Bouabdallah, K.; Bron, D.; Cunningham, D.; Assouline, S.E.; Verhoef, G.; Linton, K.; Thieblemont, C.; Vitolo, U.; et al. Phase II study of copanlisib, a PI3K inhibitor, in relapsed or refractory, indolent or aggressive lymphoma. Ann. Oncol. 2017, 28, 2169–2178. [Google Scholar] [CrossRef]
- Camidge, D.R.; Bang, Y.J.; Kwak, E.L.; Iafrate, A.J.; Varella-Garcia, M.; Fox, S.B.; Riely, G.J.; Solomon, B.; Ou, S.H.; Kim, D.W.; et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: Updated results from a phase 1 study. Lancet Oncol. 2012, 13, 1011–1019. [Google Scholar] [CrossRef]
- Shaw, A.T.; Kim, D.W.; Nakagawa, K.; Seto, T.; Crinó, L.; Ahn, M.J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A. Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef]
- Davies, M.A.; Saiag, P.; Robert, C.; Grob, J.J.; Flaherty, K.T.; Arance, A.; Chiarion-Sileni, V.; Thomas, L.; Lesimple, T.; Mortier, L.; et al. Dabrafenib plus trametinib in patients with BRAF (V600)-mutant melanoma brain metastases (COMBI-MB): A multicentre, multicohort, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 863–873. [Google Scholar] [CrossRef]
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 2017, 28, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Amaria, R.N.; Prieto, P.A.; Tetzlaff, M.T.; Reuben, A.; Andrews, M.C.; Ross, M.I.; Glitza, I.C.; Cormier, J.; Hwu, W.J.; Tawbi, H.A.; et al. Neoadjuvant plus adjuvant dabrafenib and trametinib versus standard of care in patients with high-risk, surgically resectable melanoma: A single-centre, open-label, randomised, phase 2 trial. Lancet Oncol. 2018, 19, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Hjorth-Hansen, H.; Stenke, L.; Söderlund, S.; Dreimane, A.; Ehrencrona, H.; Gedde-Dahl, T.; Gjertsen, B.T.; Höglund, M.; Koskenvesa, P.; Lotfi, K.; et al. Nordic CML Study Group. Dasatinib induces fast and deep responses in newly diagnosed chronic myeloid leukaemia patients in chronic phase: Clinical results from a randomised phase-2 study (NordCML006). Eur. J. Haematol. 2015, 94, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Radich, J.P.; Kopecky, K.J.; Appelbaum, F.R.; Kamel-Reid, S.; Stock, W.; Malnassy, G.; Paietta, E.; Wadleigh, M.; Larson, R.A.; Emanuel, P.; et al. A randomized trial of dasatinib 100 mg versus imatinib 400 mg in newly diagnosed chronic-phase chronic myeloid leukemia. Blood 2012, 120, 3898–3905. [Google Scholar] [CrossRef]
- Guilhot, F.; Apperley, J.; Kim, D.W.; Bullorsky, E.O.; Baccarani, M.; Roboz, G.J.; Amadori, S.; de Souza, C.A.; Lipton, J.H.; Hochhaus, A.; et al. Dasatinib induces significant hematologic and cytogenetic responses in patients with imatinib-resistant or -intolerant chronic myeloid leukemia in accelerated phase. Blood 2007, 109, 4143–4150. [Google Scholar] [CrossRef]
- Apperley, J.F.; Cortes, J.E.; Kim, D.W.; Roy, L.; Roboz, G.J.; Rosti, G.; Bullorsky, E.O.; Abruzzese, E.; Hochhaus, A.; Heim, D.; et al. Dasatinib in the treatment of chronic myeloid leukemia in accelerated phase after imatinib failure: The START a trial. J. Clin. Oncol. 2009, 27, 3472–3479. [Google Scholar] [CrossRef]
- Kantarjian, H.; Shah, N.P.; Hochhaus, A.; Cortes, J.; Shah, S.; Ayala, M.; Moiraghi, B.; Shen, Z.; Mayer, J.; Pasquini, R.; et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 2010, 362, 2260–2270. [Google Scholar] [CrossRef]
- Sasaki, K.; Lahoti, A.; Jabbour, E.; Jain, P.; Pierce, S.; Borthakur, G.; Daver, N.; Kadia, T.; Pemmaraju, N.; Ferrajoli, A.; et al. Clinical safety and efficacy of nilotinib or dasatinib in patients with newly diagnosed chronic-phase chronic myelogenous leukemia and pre-existing liver and/or renal dysfunction. Clin. Lymphoma Myeloma Leuk. 2016, 16, 152–162. [Google Scholar] [CrossRef]
- Harbaum, L.; Marx, A.; Goekkurt, E.; Schafhausen, P.; Atanackovic, D. Treatment with dasatinib for chronic myeloid leukemia following imatinib-induced hepatotoxicity. Int. J. Hematol. 2014, 99, 91–94. [Google Scholar] [CrossRef]
- Hughes, T.P.; Lipton, J.H.; Spector, N.; Cervantes, F.; Pasquini, R.; Clementino, N.C.; Dorlhiac Llacer, P.E.; Schwarer, A.P.; Mahon, F.X.; Rea, D.; et al. Deep molecular responses achieved in patients with CML-CP who are switched to nilotinib after long-term imatinib. Blood 2014, 124, 729–736. [Google Scholar] [CrossRef]
- Saglio, G.; Kim, D.W.; Issaragrisil, S.; le Coutre, P.; Etienne, G.; Lobo, C.; Pasquini, R.; Clark, R.E.; Hochhaus, A.; Hughes, T.P.; et al. ENESTnd Investigators. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N. Engl. J. Med. 2010, 362, 2251–2259. [Google Scholar] [CrossRef]
- Wang, J.; Shen, Z.X.; Saglio, G.; Jin, J.; Huang, H.; Hu, Y.; Du, X.; Li, J.; Meng, F.; Zhu, H.; et al. Phase 3 study of nilotinib vs imatinib in Chinese patients with newly diagnosed chronic myeloid leukemia in chronic phase: ENESTchina. Blood 2015, 125, 2771–2778. [Google Scholar] [CrossRef]
- Lipton, J.H.; Chuah, C.; Guerci-Bresler, A.; Rosti, G.; Simpson, D.; Assouline, S.; Etienne, G.; Nicolini, F.E.; le Coutre, P.; Clark, R.E.; et al. Ponatinib versus imatinib for newly diagnosed chronic myeloid leukaemia: An international, randomised, open-label, phase 3 trial. Lancet Oncol. 2016, 17, 612–621. [Google Scholar] [CrossRef]
- Wu, Y.L.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Tsuji, F.; Tsuji, F.; Linke, R.; Rosell, R.; et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 1454–1466. [Google Scholar] [CrossRef]
- Mok, T.S.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Chawla, A.; Rosell, R.; Corral, J.; Migliorino, M.R.; et al. Updated overall survival in a randomized study comparing dacomitinib with gefitinib as first-line treatment in patients with advanced non-small-cell lung cancer and EGFR-activating mutations. Drugs 2021, 81, 257–266. [Google Scholar] [CrossRef]
- Flinn, I.W.; Hillmen, P.; Montillo, M.; Nagy, Z.; Illés, Á.; Etienne, G.; Delgado, J.; Kuss, B.J.; Tam, C.S.; Gasztonyi, Z.; et al. The phase 3 DUO trial: Duvelisib vs. ofatumumab in relapsed and refractory CLL/SLL. Blood 2018, 132, 2446–2455. [Google Scholar] [CrossRef]
- Davids, M.S.; Kuss, B.J.; Hillmen, P.; Montillo, M.; Moreno, C.; Essell, J.; Lamanna, N.; Nagy, Z.; Tam, C.S.; Stilgenbauer, S.; et al. Efficacy and safety of duvelisib following disease progression on ofatumumab in patients with relapsed/refractory CLL or SLL in the DUO crossover extension study. Clin. Cancer Res. 2020, 26, 2096–2103. [Google Scholar] [CrossRef]
- O’Brien, S.; Patel, M.; Kahl, B.S.; Horwitz, S.M.; Foss, F.M.; Porcu, P.; Jones, J.; Burger, J.; Jain, N.; Allen, K.; et al. Duvelisib, an oral dual PI3K-δ,γ inhibitor, shows clinical and pharmacodynamic activity in chronic lymphocytic leukemia and small lymphocytic lymphoma in a phase 1 study. Am. J. Hematol. 2018, 93, 1318–1326. [Google Scholar] [CrossRef]
- Flinn, I.W.; Cherry, M.A.; Maris, M.B.; Matous, J.V.; Berdeja, J.G.; Patel, M. Combination trial of duvelisib (IPI-145) with rituximab or bendamustine/rituximab in patients with non-Hodgkin lymphoma or chronic lymphocytic leukemia. Am. J. Hematol. 2019, 94, 1325–1334. [Google Scholar] [CrossRef]
- Davids, M.S.; Fisher, D.C.; Tyekucheva, S.; McDonough, M.; Hanna, J.; Lee, B.; Francoeur, K.; Montegaard, J.; Odejide, O.; Armand, P.; et al. A phase 1b/2 study of duvelisib in combination with FCR (DFCR) for frontline therapy for younger CLL patients. Leukemia 2021, 35, 1064–1072. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Blakely, C.M.; Seto, T.; Cho, B.C.; et al. trial investigators. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Siena, S.; Dziadziuszko, R.; Barlesi, F.; Krebs, M.G.; Shaw, A.T.; de Braud, F.; Rolfo, C.; Ahn, M.J.; Wolf, J.; et al. Trial investigators. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. BLC2001 Study Group. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. National Cancer Institute of Canada Clinical Trials Group. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Kulke, M.H.; Blaszkowsky, L.S.; Ryan, D.P.; Clark, J.W.; Meyerhardt, J.A.; Zhu, A.X.; Enzinger, P.C.; Kwak, E.L.; Muzikansky, A.; Lawrence, C.; et al. Capecitabine plus erlotinib in gemcitabine-refractory advanced pancreatic cancer. J. Clin. Oncol. 2007, 25, 4787–4792. [Google Scholar] [CrossRef]
- Yoshida, T.; Yamada, K.; Azuma, K.; Kawahara, A.; Abe, H.; Hattori, S.; Yamashita, F.; Zaizen, Y.; Kage, M.; Hoshino, T.; et al. Comparison of adverse events and efficacy between gefitinib and erlotinib in patients with non-small-cell lung cancer: A retrospective analysis. Med. Oncol. 2013, 30, 349. [Google Scholar] [CrossRef]
- Yonesaka, K.; Suzumura, T.; Tsukuda, H.; Hasegawa, Y.; Ozaki, T.; Sugiura, T.; Fukuoka, M. Erlotinib is a well-tolerated alternate treatment for non-small cell lung cancer in cases of gefitinib-induced hepatotoxicity. Anticancer Res. 2014, 34, 5211–5215. [Google Scholar]
- Saif, M.W. Erlotinib-induced acute hepatitis in a patient with pancreatic cancer. Serum enzyme elevations due to erlotinib therapy. Acute liver failure and death due to erlotinib therapy. Clin. Adv. Hematol. Oncol. 2008, 6, 191–199. [Google Scholar]
- Liu, W.; Makrauer, F.L.; Qamar, A.A.; Jänne, P.A.; Odze, R.D. Fulminant hepatic failure secondary to erlotinib. Clin. Gastroenterol. Hepatol. 2007, 5, 917–920. [Google Scholar] [CrossRef]
- Ramanarayanan, J.; Scarpace, S.L. Acute drug induced hepatitis due to erlotinib. JOP 2007, 8, 39–43. [Google Scholar]
- Ramanarayanan, J.; Krishnan, G.S. Review: Hepatotoxicity and EGFR inhibition. Rev. Clin. Adv. Hematol. Oncol. 2008, 6, 200–201. [Google Scholar]
- Schacher-Kaufmann, S.; Pless, M. Acute Fatal Liver Toxicity under Erlotinib. Case Rep. Oncol. 2010, 3, 182–188. [Google Scholar] [CrossRef]
- Saif, M.W. Hepatic failure and hepatorenal syndrome secondary to erlotinib. Saf. Remind. JOP 2008, 9, 748–752. [Google Scholar]
- Pellegrinotti, M.; Fimognari, F.L.; Franco, A.; Repetto, L.; Pastorelli, R. Erlotinib-induced hepatitis complicated by fatal lactic acidosis in an elderly man with lung cancer. Ann. Pharmacother. 2009, 43, 542–545. [Google Scholar] [CrossRef]
- Huang, Y.S.; An, S.J.; Chen, Z.H.; Wu, Y.L. Three cases of severe hepatic impairment caused by erlotinib. Br. J. Clin. Pharmacol. 2009, 68, 464–467. [Google Scholar] [CrossRef]
- Takeda, M.; Okamoto, I.; Fukuoka, M.; Nakagawa, K. Successful treatment with erlotinib after gefitinib-related severe hepatotoxicity. J. Clin. Oncol. 2010, 28, e273–e274. [Google Scholar] [CrossRef]
- Ku, G.Y.; Chopra, A.; de Lima Lopes, G., Jr. Successful treatment of two lung cancer patients with erlotinib following gefitinib-induced hepatotoxicity. Lung Cancer 2010, 70, 223–225. [Google Scholar] [CrossRef]
- Gunturu, K.S.; Abu-Khalaf, M.; Saif, M.W. Hepatic failure and hepatorenal syndrome secondary to erlotinib: A possible etiology of complications in a patient with pancreatic cancer. JOP 2010, 11, 484–485. [Google Scholar]
- Nakatomi, K.; Nakamura, Y.; Tetsuya, I.; Kohno, S. Treatment with gefitinib after erlotinib-induced liver injury: A case report. J. Med. Case Rep. 2011, 5, 593. [Google Scholar] [CrossRef]
- Lai, Y.C.; Lin, P.C.; Lai, J.I.; Hsu, S.Y.; Kuo, L.C.; Chang, S.C.; Wang, W.S. Successful treatment of erlotinib-induced acute hepatitis and acute interstitial pneumonitis with high-dose corticosteroid: A case report and literature review. Int. J. Clin. Pharmacol. Ther. 2011, 49, 461–466. [Google Scholar] [CrossRef]
- Kijima, T.; Shimizu, T.; Nonen, S.; Furukawa, M.; Otani, Y.; Minami, T.; Takahashi, R.; Hirata, H.; Nagatomo, I.; Takeda, Y.; et al. Safe and successful treatment with erlotinib after gefitinib-induced hepatotoxicity: Difference in metabolism as a possible mechanism. J. Clin. Oncol. 2011, 29, e588–e590. [Google Scholar] [CrossRef] [PubMed]
- Kunimasa, K.; Yoshioka, H.; Iwasaku, M.; Nishiyama, A.; Korogi, Y.; Masuda, G.; Takaiwa, T.; Ishida, T. Successful treatment of non-small cell lung cancer with gefitinib after severe erlotinib-related hepatotoxicity. Intern. Med. 2012, 51, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Kitade, H.; Yamada, T.; Igarashi, S.; Hokkoku, K.; Mori, M.; Shintaku, K.; Sagawa, M.; Nakai, M.; Yano, S. Efficacy of low-dose erlotinib against gefitinib-induced hepatotoxicity in a patient with lung adenocarcinoma harboring EGFR mutations. Gan Kagaku Ryoho 2013, 40, 79–81. [Google Scholar]
- Takimoto, T.; Kijima, T.; Otani, Y.; Nonen, S.; Namba, Y.; Mori, M.; Yokota, S.; Minami, S.; Komuta, K.; Uchida, J.; et al. Polymorphisms of CYP2D6 gene and gefitinib-induced hepatotoxicity. Clin. Lung Cancer 2013, 14, 502–507. [Google Scholar] [CrossRef]
- Durand, M.; Logerot, S.; Fonrose, X.; Schir, E. Treatment with erlotinib after gefitinib induced hepatotoxicity: Literature review and case report. Therapie 2014, 69, 163–168. [Google Scholar] [CrossRef]
- Bui, N.; Wong-Sefidan, I. Reactivation of hepatitis B virus after withdrawal of erlotinib. Curr. Oncol. 2015, 22, 430–432. [Google Scholar] [CrossRef]
- Baselga, J.; Semiglazov, V.; van Dam, P.; Manikhas, A.; Bellet, M.; Mayordomo, J.; Campone, M.; Kubista, E.; Greil, R.; Bianchi, G.; et al. Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plus letrozole in patients with estrogen receptor-positive breast cancer. J. Clin. Oncol. 2009, 27, 2630–2637. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grünwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. RECORD.1 Study Group. Phase 3 trial of everolimus for metastatic renal cell carcinoma: Final results and analysis of prognostic factors. Cancer 2010, 116, 4256–4265. [Google Scholar] [CrossRef]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A.; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef]
- Moscetti, L.; Vici, P.; Gamucci, T.; Natoli, C.; Cortesi, E.; Marchetti, P.; Santini, D.; Giuliani, R.; Sperduti, I.; Mauri, M.; et al. Safety analysis, association with response and previous treatments of everolimus and exemestane in 181 metastatic breast cancer patients: A multicenter Italian experience. Breast 2016, 29, 96–101. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Halabi, S.; Eisen, T.; Broderick, S.; Stadler, W.M.; Jones, R.J.; Garcia, J.A.; Vaishampayan, U.N.; Picus, J.; Hawkins, R.E.; et al. Everolimus versus sunitinib for patients with metastatic non-clear cell renal cell carcinoma (ASPEN): A multicentre, open-label, randomised phase 2 trial. Lancet Oncol. 2016, 17, 378–388. [Google Scholar] [CrossRef]
- Mizuno, S.; Yamagishi, Y.; Ebinuma, H.; Nakamoto, N.; Katahira, M.; Sasaki, A.; Sakamoto, M.; Suzuki, H.; Kanai, T.; Hibi, T. Progressive liver failure induced by everolimus for renal cell carcinoma in a 58-year-old male hepatitis B virus carrier. Clin. J. Gastroenterol. 2013, 6, 188–192. [Google Scholar] [CrossRef]
- Sezgin Göksu, S.; Bilal, S.; Coşkun, H.Ş. Hepatitis B reactivation related to everolimus. World J. Hepatol. 2013, 5, 43–45. [Google Scholar] [CrossRef]
- Teplinsky, E.; Cheung, D.; Weisberg, I.; Jacobs, R.E.; Wolff, M.; Park, J.; Friedman, K.; Muggia, F.; Jhaveri, K. Fatal hepatitis B reactivation due to everolimus in metastatic breast cancer: Case report and review of literature. Breast Cancer Res. Treat. 2013, 141, 167–172. [Google Scholar] [CrossRef]
- Schieren, G.; Bölke, E.; Scherer, A.; Raffel, A.; Gerber, P.A.; Kröpil, P.; Schott, M.; Hamilton, J.; Hayman, A.; Knoefel, W.T.; et al. Severe everolimus-induced steatohepatis: A case report. Eur. J. Med. Res. 2013, 18, 22. [Google Scholar] [CrossRef]
- Mir, O.; Toulmonde, M.; Coriat, R.; Ropert, S.; Loulergue, P. Hepatitis B reactivation during everolimus treatment. Acta Oncol. 2016, 55, 1505–1506. [Google Scholar] [CrossRef]
- D’Aniello, C.; Maruzzo, M.; Basso, U. Prevention of hepatitis B virus reactivation with lamivudine in a patient with advanced renal cell carcinoma treated with everolimus. Am. J. Ther. 2016, 23, e300–e303. [Google Scholar] [CrossRef]
- Pardanani, A.; Harrison, C.; Cortes, J.E.; Cervantes, F.; Mesa, R.A.; Milligan, D.; Masszi, T.; Mishchenko, E.; Jourdan, E.; Vannucchi, A.M.; et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: A randomized clinical trial. JAMA Oncol. 2015, 1, 643–651. [Google Scholar] [CrossRef]
- Harrison, C.N.; Schaap, N.; Vannucchi, A.M.; Kiladjian, J.J.; Tiu, R.V.; Zachee, P.; Jourdan, E.; Winton, E.; Silver, R.T.; Schouten, H.C.; et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): A single-arm, open-label, non-randomised, phase 2, multicenter study. Lancet Haematol. 2017, 4, e317–e324. [Google Scholar] [CrossRef]
- Pardanani, A.; Tefferi, A.; Masszi, T.; Mishchenko, E.; Drummond, M.; Jourdan, E.; Vannucchi, A.; Jurgutis, M.; Ribrag, V.; Rambaldi, A.; et al. Updated results of the placebo-controlled, phase III JAKARTA trial of fedratinib in patients with intermediate-2 or high-risk myelofibrosis. Br. J. Haematol. 2021, 195, 244–248. [Google Scholar] [CrossRef]
- Goss, G.D.; O’Callaghan, C.; Lorimer, I.; Tsao, M.S.; Masters, G.A.; Jett, J.; Edelman, M.J.; Lilenbaum, R.; Choy, H.; Khuri, F.; et al. Gefitinib versus placebo in completely resected non-small-cell lung cancer: Results of the NCIC CTG BR19 study. J. Clin. Oncol. 2013, 31, 3320–3326. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, L.; Liu, X.; Zhou, C.; Zhang, L.; Zhang, S.; Wang, D.; Li, Q.; Qin, S.; Hu, C.; et al. Icotinib versus gefitinib in previously treated advanced non-small-cell lung cancer (ICOGEN): A randomised, double-blind phase 3 non-inferiority trial. Lancet Oncol. 2013, 14, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Chu, D.T.; Han, B.; Liu, X.; Zhang, L.; Zhou, C.; Liao, M.; Mok, T.; Jiang, H.; Duffield, E.; et al. Phase III, randomized, open-label, first-line study in Asia of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer: Evaluation of patients recruited from mainland China. Asia Pac. J. Clin. Oncol. 2012, 8, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ma, S.; Song, X.; Han, B.; Cheng, Y.; Huang, C.; Yang, S.; Liu, X.; Liu, Y.; Lu, S.; et al. INFORM investigators. Gefitinib versus placebo as maintenance therapy in patients with locally advanced or metastatic non-small-cell lung cancer (INFORM.; C-TONG 0804): A multicenter, double-blind randomised phase 3 trial. Lancet Oncol. 2012, 13, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Gaafar, R.M.; Surmont, V.F.; Scagliotti, G.V.; Van Klaveren, R.J.; Papamichael, D.; Welch, J.J.; Hasan, B.; Torri, V.; Van Meerbeeck, J.P. EORTC Lung Cancer Group and the Italian Lung Cancer Project. A double-blind, randomised, placebo-controlled phase III intergroup study of gefitinib in patients with advanced NSCLC, non-progressing after first line platinum-based chemotherapy (EORTC 08021/ILCP 01/03). Eur. J. Cancer 2011, 47, 2331–2340. [Google Scholar]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. North-East Japan Study Group. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef]
- Lee, Y.J.; Kim, H.T.; Han, J.Y.; Yun, T.; Lee, G.K.; Kim, H.Y.; Sung, J.H.; Lee, J.S. First-line gefitinib treatment for patients with advanced non-small cell lung cancer with poor performance status. J. Thorac. Oncol. 2010, 5, 361–368. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. West Japan Oncology Group. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harboring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Natale, R.B.; Bodkin, D.; Govindan, R.; Sleckman, B.G.; Rizvi, N.A.; Capó, A.; Germonpré, P.; Eberhardt, W.E.; Stockman, P.K.; Kennedy, S.J.; et al. Vandetanib versus gefitinib in patients with advanced non-small-cell lung cancer: Results from a two-part, double-blind, randomized phase II study. J. Clin. Oncol. 2009, 27, 2523–2529. [Google Scholar] [CrossRef]
- Maruyama, R.; Nishiwaki, Y.; Tamura, T.; Yamamoto, N.; Tsuboi, M.; Nakagawa, K.; Shinkai, T.; Negoro, S.; Imamura, F.; Eguchi, K.; et al. Phase III study, V-15-32, of gefitinib versus docetaxel in previously treated Japanese patients with non-small-cell lung cancer. J. Clin. Oncol. 2008, 26, 4244–4252. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- De Jésus Ngoma, P.; Kabamba, B.; Dahlqvist, G.; Sempoux, C.; Lanthier, N.; Shindano, T.; Van Den Neste, E.; Horsmans, Y. Occult HBV reactivation induced by ibrutinib treatment: A case report. Acta Gastroenterol. Belg. 2015, 78, 424–426. [Google Scholar]
- Nandikolla, A.G.; Derman, O.; Nautsch, D.; Liu, Q.; Massoumi, H.; Venugopal, S.; Braunschweig, I.; Janakiram, M. Ibrutinib-induced severe liver injury. Clin. Case Rep. 2017, 5, 735–738. [Google Scholar] [CrossRef]
- Kahn, A.; Horsley-Silva, J.L.; Lam-Himlin, D.M.; Reeder, C.B.; Douglas, D.D.; Carey, E.J. Ibrutinib-induced acute liver failure. Leuk. Lymphoma 2018, 59, 512–514. [Google Scholar] [CrossRef]
- Herishanu, Y.; Katchman, H.; Polliack, A. Severe hepatitis B virus reactivation related to ibrutinib monotherapy. Ann. Hematol. 2017, 96, 689–690. [Google Scholar] [CrossRef]
- Furman, R.R.; Sharman, J.P.; Coutre, S.E.; Cheson, B.D.; Pagel, J.M.; Hillmen, P.; Barrientos, J.C.; Zelenetz, A.D.; Kipps, T.J.; Flinn, I.; et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2014, 370, 997–1007. [Google Scholar] [CrossRef]
- Gopal, A.K.; Kahl, B.S.; De Vos, S.; Wagner-Johnston, N.D.; Schuster, S.J.; Jurczak, W.J.; Flinn, I.W.; Flowers, C.R.; Martin, P.; Viardot, A.; et al. PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N. Engl. J. Med. 2014, 370, 1008–1018. [Google Scholar] [CrossRef]
- Kahl, B.S.; Spurgeon, S.E.; Furman, R.R.; Flinn, I.W.; Coutre, S.E.; Brown, J.R.; Benson, D.M.; Byrd, J.C.; Peterman, S.; Cho, Y.; et al. A phase 1 study of the PI3Kδ inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood 2014, 123, 3398–3405. [Google Scholar] [CrossRef]
- De Vos, S.; Wagner-Johnston, N.D.; Coutre, S.E.; Flinn, I.W.; Schreeder, M.T.; Fowler, N.H.; Sharman, J.P.; Boccia, R.V.; Barrientos, J.C.; Rai, K.R.; et al. Combinations of idelalisib with rituximab and/or bendamustine in patients with recurrent indolent non-Hodgkin lymphoma. Blood Adv. 2016, 1, 12231. [Google Scholar] [CrossRef]
- Lampson, B.L.; Kasar, S.N.; Matos, T.R.; Morgan, E.A.; Rassenti, L.; Davids, M.S.; Fisher, D.C.; Freedman, A.S.; Jacobson, C.A.; Armand, P.; et al. Idelalisib given front-line for treatment of chronic lymphocytic leukemia causes frequent immune-mediated hepatotoxicity. Blood 2016, 128, 195–203. [Google Scholar] [CrossRef]
- Smith, S.M.; Pitcher, B.N.; Jung, S.H.; Bartlett, N.L.; Wagner-Johnston, N.; Park, S.I.; Richards, K.L.; Cashen, A.F.; Jaslowski, A.; Smith, S.E.; et al. Safety and tolerability of idelalisib, lenalidomide, and rituximab in relapsed and refractory lymphoma: The Alliance for Clinical Trials in Oncology A051201 and A051202 phase 1 trials. Lancet Haematol. 2017, 4, e176–e182. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.A.; Robak, T.; Brown, J.R.; Awan, F.T.; Badoux, X.; Coutre, S.; Loscertales, J.; Taylor, K.; Vandenberghe, E.; Wach, M.; et al. Efficacy and safety of idelalisib in combination with ofatumumab for previously treated chronic lymphocytic leukaemia: An open-label, randomised phase 3 trial. Lancet Haematol. 2017, 4, e114–e126. [Google Scholar] [CrossRef] [PubMed]
- Zelenetz, A.D.; Barrientos, J.C.; Brown, J.R.; Coiffier, B.; Delgado, J.; Egyed, M.; Ghia, P.; Illés, Á.; Jurczak, W.; Marlton, P.; et al. Idelalisib or placebo in combination with bendamustine and rituximab in patients with relapsed or refractory chronic lymphocytic leukaemia: Interim results from a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2017, 18, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Coutre, S.E.; Flinn, I.W.; De Vos, S.; Barrientos, J.C.; Schreeder, M.T.; Wagner-Johnson, N.D.; Sharman, J.P.; Boyd, T.E.; Fowler, N.; Dreiling, L.; et al. Idelalisib in combination with rituximab or bendamustine or both in patients with relapsed/refractory chronic lymphocytic leukemia. Hemasphere 2018, 2, e39. [Google Scholar] [CrossRef] [PubMed]
- Sharman, J.P.; Coutre, S.E.; Furman, R.R.; Cheson, B.D.; Pagel, J.M.; Hillmen, P.; Barrientos, J.C.; Zelenetz, A.D.; Kipps, T.J.; Flinn, I.W.; et al. Final results of a randomized, phase III study of rituximab with or without idelalisib followed by open-label idelalisib in patients with relapsed chronic lymphocytic leukemia. J. Clin. Oncol. 2019, 37, 1391–1402. [Google Scholar] [CrossRef]
- Eyre, T.A.; Preston, G.; Kagdi, H.; Islam, A.; Nicholson, T.; Smith, H.W.; Cursley, A.P.; Ramroth, H.; Xing, G.; Gu, L.; et al. A retrospective observational study to evaluate the clinical outcomes and routine management of patients with chronic lymphocytic leukaemia treated with idelalisib and rituximab in the UK and Ireland (RETRO-idel). Br. J. Haematol. 2021, 194, 69–77. [Google Scholar] [CrossRef]
- Wagner-Johnston, N.D.; Schuster, S.J.; deVos, S.; Salles, G.; Jurczak, W.J.; Flowers, C.R.; Viardot, A.; Flinn, I.W.; Martin, P.; Xing, G.; et al. Outcomes of patients with up to 6 years of follow-up from a phase 2 study of idelalisib for relapsed indolent lymphomas. Leuk. Lymphoma. 2021, 62, 1077–1087. [Google Scholar] [CrossRef]
- Hoechstetter, M.A.; Knauf, W.; Dambacher, S.; Hucke, N.; Höhne, K.; van Troostenburg, A.; Ramroth, H.; Abenhardt, W.; Rummel, M. Results of a prospective non-Interventional post-authorization safety study of idelalisib in Germany. Clin. Lymphoma Myeloma Leuk. 2022, 22, e777–e787. [Google Scholar] [CrossRef]
- Casadei, B.; Argnani, L.; Broccoli, A.; Patti, C.; Stefani, P.M.; Cuneo, A.; Margiotta Casaluci, G.; Visco, C.; Gini, G.; Pane, F.; et al. Treatment with idelalisib in patients with relapsed or refractory follicular lymphoma: The observational Italian multicenter FolIdela Study. Cancers 2022, 14, 654. [Google Scholar] [CrossRef]
- Cuneo, A.; Barosi, G.; Danesi, R.; Fagiuoli, S.; Ghia, P.; Marzano, A.; Montillo, M.; Poletti, V.; Viale, P.; Zinzani, P.L. Management of adverse events associated with idelalisib treatment in chronic lymphocytic leukemia and follicular lymphoma: A multidisciplinary position paper. Hematol. Oncol. 2019, 37, 3–14. [Google Scholar] [CrossRef]
- Javle, M.; Roychowdhury, S.; Kelley, R.K.; Sadeghi, S.; Macarulla, T.; Weiss, K.H.; Waldschmidt, D.T.; Goyal, L.; Borbath, I.; El-Khoueiry, A.; et al. Infigratinib (BGJ398) in previously treated patients with advanced or metastatic cholangiocarcinoma with FGFR2 fusions or rearrangements: Mature results from a multicenter, open-label, single-arm, phase 2 study. Lancet Gastroenterol. Hepatol. 2021, 6, 803–815. [Google Scholar] [CrossRef]
- Ryan, Q.; Ibrahim, A.; Cohen, M.H.; Johnson, J.; Ko, C.W.; Sridhara, R.; Justice, R.; Pazdur, R. FDA drug approval summary: Lapatinib in combination with capecitabine for previously treated metastatic breast cancer that overexpresses HER-2. Oncologist 2008, 13, 1114–1119. [Google Scholar] [CrossRef]
- Gómez, H.L.; Neciosup, S.; Tosello, C.; Mano, M.; Bines, J.; Ismael, G.; Santi, P.X.; Pinczowski, H.; Nerón, Y.; Fanelli, M.; et al. A Phase II randomized study of lapatinib combined with capecitabine, vinorelbine, or gemcitabine in patients with HER2-positive metastatic breast cancer with progression after a taxane (Latin American Cooperative Oncology Group 0801 Study). Clin. Breast Cancer 2016, 16, 38–44. [Google Scholar] [CrossRef]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in patients with TRK fusion-positive solid tumors: A pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- Waguespack, S.G.; Drilon, A.; Lin, J.J.; Brose, M.S.; McDermott, R.; Almubarak, M.; Bauman, J.; Casanova, M.; Krishnamurthy, A.; Kummar, S.; et al. Efficacy and safety of larotrectinib in patients with TRK fusion-positive thyroid carcinoma. Eur. J. Endocrinol. 2022, 186, 631–643. [Google Scholar] [CrossRef]
- Laetsch, T.W.; DuBois, S.G.; Mascarenhas, L.; Turpin, B.; Federman, N.; Albert, C.M.; Nagasubramanian, R.; Davis, J.L.; Rudzinski, E.; Feraco, A.M.; et al. Larotrectinib for pediatric solid tumors harboring NTRK gene fusions: Phase 1 results from a multicenter, open-label, phase 1/2 study. Lancet Oncol. 2018, 19, 705–714. [Google Scholar] [CrossRef]
- Lee, C.H.; Shah, A.Y.; Rasco, D.; Rao, A.; Taylor, M.H.; Di Simone, C.; Hsieh, J.J.; Pinto, A.; Shaffer, D.R.; Girones Sarrio, R.; et al. Lenvatinib plus pembrolizumab in patients with either treatment-naive or previously treated metastatic renal cell carcinoma (Study 111/KEYNOTE-146): A phase 1b/2 study. Lancet Oncol. 2021, 22, 946–958. [Google Scholar] [CrossRef]
- Makker, V.; Rasco, D.; Vogelzang, N.J.; Brose, M.S.; Cohn, A.L.; Mier, J.; Di Simone, C.; Hyman, D.M.; Stepan, D.E.; Dutcus, C.E.; et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: An interim analysis of a multicenter, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 711–718. [Google Scholar] [CrossRef]
- Solomon, B.J.; Besse, B.; Bauer, T.M.; Felip, E.; Soo, R.A.; Camidge, D.R.; Chiari, R.; Bearz, A.; Lin, C.C.; Gadgeel, S.M.; et al. Lorlatinib in patients with ALK-positive non-small-cell lung cancer: Results from a global phase 2 study. Lancet Oncol. 2018, 19, 1654–1667. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Duke, E.S.; Stapleford, L.; Drezner, N.; Amatya, A.K.; Mishra-Kalyani, P.S.; Shen, Y.L.; Maxfield, K.; Fourie Zirkelbach, J.; Bi, Y.; Liu, J.; et al. FDA Approval Summary: Mobocertinib for Metastatic Non-Small Cell Lung Cancer with EGFR Exon 20 Insertion Mutations. Clin. Cancer Res. 2023, 29, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Ramalingam, S.S.; Kim, T.M.; Kim, S.W.; Yang, J.C.; Riely, G.J.; Mekhail, T.; Nguyen, D.; Garcia Campelo, M.R.; Felip, E.; et al. Treatment outcomes and safety of mobocertinib in platinum-pretreated patients with EGFR exon 20 insertion-positive metastatic non-small cell lung cancer: A Phase 1/2 open-label nonrandomized clinical trial. JAMA Oncol. 2021, 7, e214761. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Moy, B.; Mansi, J.; Ejlertsen, B.; Holmes, F.A.; Chia, S.; Iwata, H.; Gnant, M.; Loibl, S.; Barrios, C.H.; et al. ExteNET Study Group. Final efficacy results of neratinib in HER2-positive hormone receptor-positive early-stage breast cancer from the Phase III ExteNET Trial. Clin. Breast Cancer 2021, 21, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Awada, A.; Colomer, R.; Inoue, K.; Bondarenko, I.; Badwe, R.A.; Demetriou, G.; Lee, S.C.; Mehta, A.O.; Kim, S.B.; Bachelot, T.; et al. Neratinib plus paclitaxel vs trastuzumab plus paclitaxel in previously untreated metastatic ERBB2-positive breast cancer: The NEfERT-T Randomized Clinical Trial. JAMA Oncol. 2016, 2, 1557–1564. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Hochhaus, A.; Saglio, G.; De Souza, C.; Flinn, I.W.; Stenke, L.; Goh, Y.T.; Rosti, G.; Nakamae, H.; Gallagher, N.J.; et al. Nilotinib versus imatinib for the treatment of patients with newly diagnosed chronic phase, Philadelphia chromosome-positive, chronic myeloid leukaemia: 24-month minimum follow-up of the phase 3 randomised ENESTnd trial. Lancet Oncol. 2011, 12, 841–851. [Google Scholar] [CrossRef]
- Usuki, K.; Tojo, A.; Maeda, Y.; Kobayashi, Y.; Matsuda, A.; Ohyashiki, K.; Nakaseko, C.; Kawaguchi, T.; Tanaka, H.; Miyamura, K.; et al. Efficacy and safety of nilotinib in Japanese patients with imatinib-resistant or -intolerant Ph+ CML or relapsed/refractory Ph+ ALL: A 36-month analysis of a phase I and II study. Int. J. Hematol. 2012, 95, 409–419. [Google Scholar] [CrossRef]
- Goss, G.; Tsai, C.M.; Shepherd, F.A.; Bazhenova, L.; Lee, J.S.; Chang, G.C.; Crino, L.; Satouchi, M.; Chu, Q.; Hida, T.; et al. Osimertinib for pretreated EGFR Thr790Met-positive advanced non-small-cell lung cancer (AURA2): A multicenter, open-label, single-arm, phase 2 study. Lancet Oncol. 2016, 17, 1643–1652. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicenter, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar]
- Hutson, T.E.; Davis, I.D.; Machiels, J.P.; De Souza, P.L.; Rottey, S.; Hong, B.F.; Epstein, R.J.; Baker, K.L.; McCann, L.; Crofts, T.; et al. Efficacy and safety of pazopanib in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2010, 28, 475–480. [Google Scholar] [CrossRef]
- Sternberg, C.N.; Davis, I.D.; Mardiak, J.; Szczylik, C.; Lee, E.; Wagstaff, J.; Barrios, C.H.; Salman, P.; Gladkov, O.A.; Kavina, A.; et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: Results of a randomized phase III trial. J. Clin. Oncol. 2010, 28, 1061–1068. [Google Scholar] [CrossRef]
- Van der Graaf, W.T.; Blay, J.Y.; Chawla, S.P.; Kim, D.W.; Bui-Nguyen, B.; Casali, P.G.; Schöffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. PALETTE study group. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Cella, D.; Reeves, J.; Hawkins, R.; Guo, J.; Nathan, P.; Staehler, M.; de Souza, P.; Merchan, J.R.; et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N. Engl. J. Med. 2013, 369, 722–731. [Google Scholar] [CrossRef]
- Klempner, S.J.; Choueiri, T.K.; Yee, E.; Doyle, L.A.; Schuppan, D.; Atkins, M.B. Severe pazopanib-induced hepatotoxicity: Clinical and histologic course in two patients. J. Clin. Oncol. 2012, 30, e264–e268. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicenter, open-label, phase 2 study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Tap, W.D.; Wainberg, Z.A.; Anthony, S.P.; Ibrahim, P.N.; Zhang, C.; Healey, J.H.; Chmielowski, B.; Staddon, A.P.; Cohn, A.L.; Shapiro, G.I.; et al. Structure-Guided Blockade of CSF1R Kinase in Tenosynovial Giant-Cell Tumor. N. Engl. J. Med. 2015, 373, 428–437. [Google Scholar] [CrossRef]
- Tap, W.D.; Gelderblom, H.; Palmerini, E.; Desai, J.; Bauer, S.; Blay, J.Y.; Alcindor, T.; Ganjoo, K.; Martín-Broto, J.; Ryan, C.W.; et al. ENLIVEN investigators. Pexidartinib versus placebo for advanced tenosynovial giant cell tumor (ENLIVEN): A randomised phase 3 trial. Lancet 2019, 394, 478–487. [Google Scholar] [CrossRef]
- Piawah, S.; Hyland, C.; Umetsu, S.E.; Esserman, L.J.; Rugo, H.S.; Chien, A.J. A case report of vanishing bile duct syndrome after exposure to pexidartinib (PLX3397) and paclitaxel. NPJ Breast Cancer 2019, 5, 17. [Google Scholar] [CrossRef]
- Lee, J.H.; Chen, T.W.; Hsu, C.H.; Yen, Y.H.; Yang, J.C.; Cheng, A.L.; Ni, S.I.; Chiu, L.L.; Sugihara, M.; Ishizuka, T.; et al. A phase I study of pexidartinib, a colony-stimulating factor 1 receptor inhibitor, in Asian patients with advanced solid tumors. Investig. New Drugs 2020, 38, 99–110. [Google Scholar] [CrossRef]
- Cortes, J.E.; Kim, D.W.; Pinilla-Ibarz, J.; le Coutre, P.; Paquette, R.; Chuah, C.; Nicolini, F.E.; Apperley, J.F.; Khoury, H.J.; Talpaz, M.; et al. PACE Investigators. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2013, 369, 1783–1796. [Google Scholar] [CrossRef]
- Jain, P.; Kantarjian, H.; Jabbour, E.; Gonzalez, G.N.; Borthakur, G.; Pemmaraju, N.; Daver, N.; Gachimova, E.; Ferrajoli, A.; Kornblau, S.; et al. Ponatinib as first-line treatment for patients with chronic myeloid leukaemia in chronic phase: A phase 2 study. Lancet Haematol. 2015, 2, e376–e383. [Google Scholar] [CrossRef]
- Gainor, J.F.; Curigliano, G.; Kim, D.W.; Lee, D.H.; Besse, B.; Baik, C.S.; Doebele, R.C.; Cassier, P.A.; Lopes, G.; Tan, D.S.W.; et al. Pralsetinib for RET fusion-positive non-small-cell lung cancer (ARROW): A multi-cohort, open-label, phase 1/2 study. Lancet Oncol. 2021, 22, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Hu, M.I.; Wirth, L.J.; Schuler, M.; Mansfield, A.S.; Curigliano, G.; Brose, M.S.; Zhu, V.W.; Leboulleux, S.; Bowles, D.W.; et al. Pralsetinib for patients with advanced or metastatic RET-altered thyroid cancer (ARROW): A multi-cohort, open-label, registrational, phase 1/2 study. Lancet Diabetes Endocrinol. 2021, 9, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C.; et al. CORRECT Study Group. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicenter, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. RESORCE Investigators. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; André, F.; Winer, E.P.; et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1738–1748. [Google Scholar] [CrossRef]
- Slamon, D.J.; Neven, P.; Chia, S.; Jerusalem, G.; De Laurentiis, M.; Im, S.; Petrakova, K.; Bianchi, G.V.; Martín, M.; Nusch, A.; et al. Ribociclib plus fulvestrant for postmenopausal women with hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer in the phase III randomized MONALEESA-3 trial: Updated overall survival. Ann. Oncol. 2021, 32, 1015–1024. [Google Scholar] [CrossRef]
- Blay, J.Y.; Serrano, C.; Heinrich, M.C.; Zalcberg, J.; Bauer, S.; Gelderblom, H.; Schöffski, P.; Jones, R.L.; Attia, S.; D’Amato, G.; et al. Ripretinib in patients with advanced gastrointestinal stromal tumors (INVICTUS): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 923–934. [Google Scholar] [CrossRef]
- Bauer, S.; Jones, R.L.; Blay, J.Y.; Gelderblom, H.; George, S.; Schöffski, P.; Von Mehren, M.; Zalcberg, J.R.; Kang, Y.K.; Razak, A.A.; et al. Ripretinib versus sunitinib in patients with advanced gastrointestinal stromal tumor after treatment with imatinib (INTRIGUE): A randomized, open-label, phase III trial. J. Clin. Oncol. 2022, 40, 3918–3928. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Kiladjian, J.J.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Mesa, R.; et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N. Engl. J. Med. 2015, 372, 426–435. [Google Scholar] [CrossRef]
- Subbiah, V.; Gainor, J.F.; Oxnard, G.R.; Tan, D.S.W.; Owen, D.H.; Cho, B.C.; Loong, H.H.; McCoach, C.E.; Weiss, J.; Kim, Y.J.; et al. Intracranial efficacy of selpercatinib in RET fusion-positive non-small cell lung cancers on the LIBRETTO-001 Trial. Clin. Cancer Res. 2021, 27, 4160–4167. [Google Scholar] [CrossRef]
- Wirth, L.J.; Sherman, E.; Robinson, B.; Solomon, B.; Kang, H.; Lorch, J.; Worden, F.; Brose, M.; Patel, J.; Leboulleux, S.; et al. Efficacy of selpercatinib in RET-altered thyroid cancers. N. Engl. J. Med. 2020, 383, 825–835. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A.A.; et al. TARGET Study Group. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef]
- Demetri, G.D.; Van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumor after failure of imatinib: A randomised controlled trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- Raymond, E.; Dahan, L.; Raoul, J.L.; Bang, Y.J.; Borbath, I.; Lombard-Bohas, C.; Valle, J.; Metrakos, P.; Smith, D.; Vinik, A.; et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011, 364, 501–513. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Rixe, O.; Oudard, S.; Negrier, S.; Szczylik, C.; Kim, S.T.; et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 115–124. [Google Scholar] [CrossRef]
- Reichardt, P.; Kang, Y.K.; Rutkowski, P.; Schuette, J.; Rosen, L.S.; Seddon, B.; Yalcin, S.; Gelderblom, H.; Williams, C.C., Jr.; Fumagalli, E.; et al. Clinical outcomes of patients with advanced gastrointestinal stromal tumors: Safety and efficacy in a worldwide treatment-use trial of sunitinib. Cancer 2015, 121, 1405–1413. [Google Scholar] [CrossRef]
- Hutson, T.E.; Escudier, B.; Esteban, E.; Bjarnason, G.A.; Lim, H.Y.; Pittman, K.B.; Senico, P.; Niethammer, A.; Lu, D.R.; Hariharan, S.; et al. Randomized phase III trial of temsirolimus versus sorafenib as second-line therapy after sunitinib in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2014, 32, 760–767. [Google Scholar] [CrossRef]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I.; et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar] [CrossRef]
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in Non-Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. N. Engl. J. Med. 2020, 383, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Pal, S.K.; Escudier, B.J.; Atkins, M.B.; Hutson, T.E.; Porta, C.; Verzoni, E.; Needle, M.N.; McDermott, D.F. Tivozanib versus sorafenib in patients with advanced renal cell carcinoma (TIVO-3): A phase 3, multicenter, randomised, controlled, open-label study. Lancet Oncol. 2020, 21, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Murthy, R.K.; Loi, S.; Okines, A.; Paplomata, E.; Hamilton, E.; Hurvitz, S.A.; Lin, N.U.; Borges, V.; Abramson, V.; Anders, C.; et al. Tucatinib, Trastuzumab, and Capecitabine for HER2-Positive Metastatic Breast Cancer. N. Engl. J. Med. 2020, 382, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.A., Jr.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R.; et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized, double-blind phase III trial. J. Clin. Oncol. 2012, 30, 134–141. [Google Scholar] [CrossRef]
- Zhou, K.; Zou, D.; Zhou, J.; Hu, J.; Yang, H.; Zhang, H.; Ji, J.; Xu, W.; Jin, J.; Lv, F.; et al. Zanubrutinib monotherapy in relapsed/refractory mantle cell lymphoma: A pooled analysis of two clinical trials. J. Hematol. Oncol. 2021, 14, 167. [Google Scholar] [CrossRef]
- Zhang, X.-W.; Liu, W.; Jiang, H.-L.; Mao, B. Chinese Herbal Medicine for Advanced Non-Small-Cell Lung Cancer: A Systematic Review and Meta-Analysis. Am. J. Chin. Med. 2018, 46, 923–952. [Google Scholar] [CrossRef]
- Raimondo, G.; Pollicino, T.; Cacciola, I.; Squadrito, G. Occult hepatitis B infection. J. Hepatol. 2007, 46, 160–170. [Google Scholar] [CrossRef]
- Béchade, D.; Chakiba, C.; Desjardin, M.; Bécouarn, Y.; Fonck, M. Hepatotoxicity of tyrosine kinase inhibitors: Mechanisms involved and practical implications. Bull. Cancer 2018, 105, 290–298. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).