
Biological Role and Clinical Implications of MYOD1L122R Mutation in Rhabdomyosarcoma

 , , , ,
, , , , 
Simple Summary
Abstract
1. Introduction
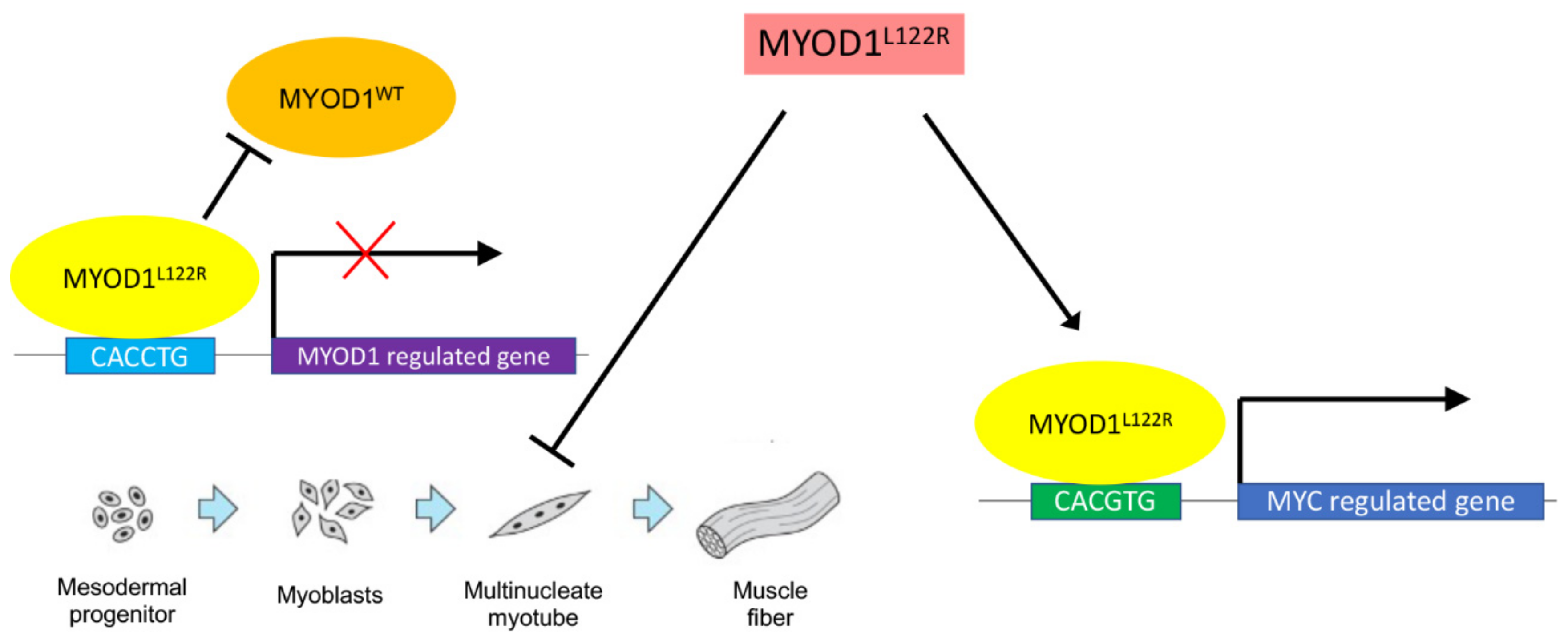
2. Normal Role for MYOD1 in Myogenesis
3. MYOD1L122R Mutation Blocks Differentiation of the Cells and Has an MYC-like Activity
4. MYOD1L122R Affects Tumorigenesis and Causes an Aggressive Biological Phenotype
5. MYOD1L122R Mutation Frequently Occurs with Other Mutations
6. MYOD1L122R Mutation Can Occur Both in Adults and Children
7. MYOD1L122R Is Frequent in Sc/Sp Cell Histology but also Present in a Subset of Embryonal RMS
8. MYOD1L122R Mutated Tumors Most Frequently Arise at Head and Neck and Extremity Sites
9. MYOD1L122R Mutation Is Associated with a Poor Outcome
10. Discussion
11. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Miller, R.W.; Young, J.L.; Novakovic, B. Childhood cancer. Cancer 1995, 75 (Suppl. 1), 395–405. [Google Scholar] [CrossRef]
- Bisogno, G.; De Salvo, G.L.; Bergeron, C.; Gallego Melcón, S.; Merks, J.H.; Kelsey, A.; Martelli, H.; Minard-Colin, V.; Orbach, D.; Glosli, H.; et al. Vinorelbine and continuous low-dose cyclophosphamide as maintenance chemotherapy in patients with high-risk rhabdomyosarcoma (RMS 2005): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2019, 20, 1566–1575. [Google Scholar] [CrossRef]
- Affinita, M.C.; Ferrari, A.; Chiaravalli, S.; Melchionda, F.; Quaglietta, L.; Casanova, M.; Zanetti, I.; Scarzello, G.; Di Pasquale, L.; Di Cataldo, A.; et al. Defining the impact of prognostic factors at the time of relapse for nonmetastatic rhabdomyosarcoma. Pediatr. Blood Cancer 2020, 67, e28674. [Google Scholar] [CrossRef]
- Chisholm, J.C.; Marandet, J.; Rey, A.; Scopinaro, M.; de Toledo, J.S.; Merks, J.H.M.; O’Meara, A.; Stevens, M.C.G.; Oberlin, O. Prognostic factors after relapse in nonmetastatic rhabdomyosarcoma: A nomogram to better define patients who can be salvaged with further therapy. J. Clin. Oncol. 2011, 29, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Pappo, A.S.; Anderson, J.R.; Crist, W.M.; Wharam, M.D.; Breitfeld, P.P.; Hawkins, D.; Raney, R.B.; Womer, R.B.; Parham, D.M.; Qualman, S.J.; et al. Survival after Relapse in Children and Adolescents with Rhabdomyosarcoma: A Report from the Intergroup Rhabdomyosarcoma Study Group. J. Clin. Oncol. 1999, 17, 3487–3493. [Google Scholar] [CrossRef]
- Mazzoleni, S.; Bisogno, G.; Garaventa, A.; Cecchetto, G.; Ferrari, A.; Sotti, G.; Donfrancesco, A.; Madon, E.; Casula, L.; Carli, M.; et al. Outcomes and prognostic factors after recurrence in children and adolescents with nonmetastatic rhabdomyosarcoma. Cancer 2005, 104, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Minn, A.Y.; Lyden, E.R.; Anderson, J.R.; Million, L.; Arndt, C.A.; Brown, K.; Hawkins, D.S.; Donaldson, S.S. Early treatment failure in intermediate-risk rhabdomyosarcoma: Results from IRS-IV and D9803--a report from the Children’s Oncology Group. J. Clin. Oncol. 2010, 28, 4228–4232. [Google Scholar] [CrossRef] [PubMed]
- Vaarwerk, B.; Hol, M.L.F.; Schoot, R.A.; Breunis, W.B.; de Win, M.M.L.; Westerveld, H.; Fajardo, R.D.; Saeed, P.; van den Brekel, M.W.; Pieters, B.R.; et al. AMORE treatment as salvage treatment in children and young adults with relapsed head-neck rhabdomyosarcoma. Radiother. Oncol. 2019, 131, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Koscielniak, E.; Harms, D.; Henze, G.; Jürgens, H.; Gadner, H.; Herbst, M.; Klingebiel, T.; Schmidt, B.F.; Morgan, M.; Knietig, R.; et al. Results of treatment for soft tissue sarcoma in childhood and adolescence: A final report of the German Cooperative Soft Tissue Sarcoma Study CWS-86. J. Clin. Oncol. 1999, 17, 3706–3719. [Google Scholar] [CrossRef] [PubMed]
- Newton, W.A.; Gehan, E.A.; Webber, B.L.; Marsden, H.B.; van Unnik, A.J.; Hamoudi, A.B.; Tsokos, M.G.; Shimada, H.; Harms, D.; Schmidt, D. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification--an Intergroup Rhabdomyosarcoma Study. Cancer 1995, 76, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Skapek, S.X.; Anderson, J.; Barr, F.G.; Bridge, J.A.; Gastier-Foster, J.M.; Parham, D.M.; Rudzinski, E.R.; Triche, T.; Hawkins, D.S. PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: A children’s oncology group report. Pediatr. Blood Cancer 2013, 60, 1411–1417. [Google Scholar] [CrossRef] [PubMed]
- Missiaglia, E.; Williamson, D.; Chisholm, J.; Wirapati, P.; Pierron, G.; Petel, F.; Concordet, J.-P.; Thway, K.; Oberlin, O.; Pritchard-Jones, K.; et al. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J. Clin. Oncol. 2012, 30, 1670–1677. [Google Scholar] [CrossRef]
- Alaggio, R.; Zhang, L.; Sung, Y.-S.; Huang, S.-C.; Chen, C.-L.; Bisogno, G.; Zin, A.; Agaram, N.P.; LaQuaglia, M.P.; Wexler, L.H.; et al. A Molecular Study of Pediatric Spindle and Sclerosing Rhabdomyosarcoma: Identification of Novel and Recurrent VGLL2-related Fusions in Infantile Cases. Am. J. Surg. Pathol. 2016, 40, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, D.; Chargari, C.; Scoazec, J.-Y.; Cotteret, S.; Felix, A.; Moalla, S.; Temam, S.; Minard-Colin, V. PAX3-NCOA1 alveolar rhabdomyosarcoma of the tongue: A rare entity with challenging diagnosis and management. Pediatr. Blood Cancer 2021, 68, e29288. [Google Scholar] [CrossRef]
- Mosquera, J.M.; Sboner, A.; Zhang, L.; Kitabayashi, N.; Chen, C.-L.; Sung, Y.S.; Wexler, L.H.; LaQuaglia, M.P.; Edelman, M.; Sreekantaiah, C.; et al. Recurrent NCOA2 gene rearrangements in congenital/infantile spindle cell rhabdomyosarcoma. Genes Chromosomes Cancer 2013, 52, 538–550. [Google Scholar] [CrossRef]
- Chrisinger, J.S.A.; Wehrli, B.; Dickson, B.C.; Fasih, S.; Hirbe, A.C.; Shultz, D.B.; Zadeh, G.; Gupta, A.A.; Demicco, E.G. Epithelioid and spindle cell rhabdomyosarcoma with FUS-TFCP2 or EWSR1-TFCP2 fusion: Report of two cases. Virchows Arch. 2020, 477, 725–732. [Google Scholar] [CrossRef]
- Szuhai, K.; de Jong, D.; Leung, W.Y.; Fletcher, C.D.; Hogendoorn, P.C. Transactivating mutation of the MYOD1 gene is a frequent event in adult spindle cell rhabdomyosarcoma: MYOD1 mutation in adult spindle cell rhabdomyosarcoma. J. Pathol. 2014, 232, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Shern, J.F.; Selfe, J.; Izquierdo, E.; Patidar, R.; Chou, H.-C.; Song, Y.K.; Yohe, M.E.; Sindiri, S.; Wei, J.; Wen, X.; et al. Genomic Classification and Clinical Outcome in Rhabdomyosarcoma: A Report From an International Consortium. JCO 2021, 39, 2859–2871. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana, A.O.; Schmidt, D.; Ninfo, V.; Harms, D.; Tollot, M.; Carli, M.; Treuner, J.; Betto, R.; Salviati, G. Spindle cell rhabdomyosarcoma. A prognostically favorable variant of rhabdomyosarcoma. Am. J. Surg. Pathol. 1992, 16, 229–235. [Google Scholar] [CrossRef]
- Folpe, A.L.; McKenney, J.K.; Bridge, J.A.; Weiss, S.W. Sclerosing rhabdomyosarcoma in adults: Report of four cases of a hyalinizing, matrix-rich variant of rhabdomyosarcoma that may be confused with osteosarcoma, chondrosarcoma, or angiosarcoma. Am. J. Surg. Pathol. 2002, 26, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Mentzel, T.; Katenkamp, D. Sclerosing, pseudovascular rhabdomyosarcoma in adults. Clinicopathological and immunohistochemical analysis of three cases. Virchows Arch. 2000, 436, 305–311. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Tian, Z.; Zhu, Y. Clinicopathologic features and molecular spectrum of spindle cell and sclerosing rhabdomyosarcomas in the head and neck region. Int. J. Clin. Exp. Pathol. 2018, 11, 3436. [Google Scholar]
- Jo, V.Y.; Fletcher, C.D.M. WHO classification of soft tissue tumours: An update based on the 2013 (4th) edition. Pathology 2014, 46, 95–104. [Google Scholar] [CrossRef]
- Choi, J.H.; Ro, J.Y. The 2020 WHO Classification of Tumors of Soft Tissue: Selected Changes and New Entities. Adv. Anat. Pathol. 2021, 28, 44–58. [Google Scholar] [CrossRef]
- Kohsaka, S.; Shukla, N.; Ameur, N.; Ito, T.; Ng, C.K.Y.; Wang, L.; Lim, D.; Marchetti, A.; Viale, A.; Pirun, M.; et al. A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K-AKT pathway mutations. Nat. Genet. 2014, 46, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.L.; Weintraub, H.; Lassar, A.B. Expression of a Single Transfected cDNA Converts Fibmblasts to Myoblasts. Cell 1987, 51, 987–1000. [Google Scholar] [CrossRef]
- Zammit, P.S. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin. Cell Dev. Biol. 2017, 72, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Emerson, C.P. Skeletal myogenesis: Genetics and embryology to the fore. Curr. Opin. Genet. Dev. 1993, 3, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Hasty, P.; Bradley, A.; Morris, J.H.; Edmondson, D.G.; Venuti, J.M.; Olson, E.N.; Klein, W.H. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 1993, 364, 501–506. [Google Scholar] [CrossRef]
- Tajbakhsh, S.; Rocancourt, D.; Cossu, G.; Buckingham, M. Redefining the Genetic Hierarchies Controlling Skeletal Myogenesis: Pax-3 and Myf-5 Act Upstream of MyoD. Cell 1997, 89, 127–138. [Google Scholar] [CrossRef]
- Hausburg, M.A.; Doles, J.D.; Clement, S.L.; Cadwallader, A.B.; Hall, M.N.; Blackshear, P.J.; Lykke-Andersen, J.; Olwin, B.B. Post-transcriptional regulation of satellite cell quiescence by TTP-mediated mRNA decay. eLife 2015, 4, e03390. [Google Scholar] [CrossRef] [PubMed]
- Crist, C.G.; Montarras, D.; Buckingham, M. Muscle Satellite Cells Are Primed for Myogenesis but Maintain Quiescence with Sequestration of Myf5 mRNA Targeted by microRNA-31 in mRNP Granules. Cell Stem Cell 2012, 11, 118–126. [Google Scholar] [CrossRef]
- Rudnicki, M.A.; Braun, T.; Hinuma, S.; Jaenisch, R. Inactivation of MyoD in Mice Leads to Up-Regulation of the Myogenic HLH Gene Myf-5 and Results in Apparently Normal Muscle Development. Cell 1992, 71, 383–390. [Google Scholar] [CrossRef]
- Braun, T.; Rudnicki, M.A.; Arnold, H.-H.; Jaenisch, R. Targeted inactivation of the muscle regulatory gene Myf-5 results in abnormal rib development and perinatal death. Cell 1992, 71, 369–382. [Google Scholar] [CrossRef]
- Haldar, M.; Karan, G.; Watanabe, S.; Guenther, S.; Braun, T.; Capecchi, M.R. Response: Contributions of the Myf5-Independent Lineage to Myogenesis. Dev. Cell 2014, 31, 539–541. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Megeney, L.A.; Kablar, B.; Garrett, K.; Anderson, J.E.; Rudnicki, M.A. MyoD is required for myogenic stem cell function in adult skeletal muscle. Genes Dev. 1996, 10, 1173–1183. [Google Scholar] [CrossRef]
- Lassar, A.B.; Davis, R.L.; Wright, W.E.; Kadesch, T.; Murre, C.; Voronova, A.; Baltimore, D.; Weintraub, H. Functional activity of myogenic HLH proteins requires hetero-oligomerization with E12/E47-like proteins in vivo. Cell 1991, 66, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yao, Z.; Sarkar, D.; Lawrence, M.; Sanchez, G.J.; Parker, M.H.; MacQuarrie, K.L.; Davison, J.; Morgan, M.T.; Ruzzo, W.L.; et al. Genome-wide MyoD Binding in Skeletal Muscle Cells: A Potential for Broad Cellular Reprogramming. Dev. Cell 2010, 18, 662–674. [Google Scholar] [CrossRef]
- Bergstrom, D.A.; Penn, B.H.; Strand, A.; Perry, R.L.S.; Rudnicki, M.A.; Tapscott, S.J. Promoter-Specific Regulation of MyoD Binding and Signal Transduction Cooperate to Pattern Gene Expression. Mol. Cell 2002, 9, 587–600. [Google Scholar] [CrossRef]
- Siles, L.; Sánchez-Tilló, E.; Lim, J.-W.; Darling, D.S.; Kroll, K.L.; Postigo, A. ZEB1 Imposes a Temporary Stage-Dependent Inhibition of Muscle Gene Expression and Differentiation via CtBP-Mediated Transcriptional Repression. Mol. Cell. Biol. 2013, 33, 1368–1382. [Google Scholar] [CrossRef]
- Van Antwerp, M.E.; Chen, D.G.; Chang, C.; Prochownik, E.V. A point mutation in the MyoD basic domain imparts c-Myc-like properties. Proc. Natl. Acad. Sci. USA 1992, 89, 9010–9014. [Google Scholar] [CrossRef]
- MacQuarrie, K.L.; Yao, Z.; Fong, A.P.; Diede, S.J.; Rudzinski, E.R.; Hawkins, D.S.; Tapscott, S.J. Comparison of Genome-Wide Binding of MyoD in Normal Human Myogenic Cells and Rhabdomyosarcomas Identifies Regional and Local Suppression of Promyogenic Transcription Factors. Mol. Cell. Biol. 2013, 33, 773–784. [Google Scholar] [CrossRef]
- Agaram, N.P.; Chen, C.-L.; Zhang, L.; LaQuaglia, M.P.; Wexler, L.; Antonescu, C.R. Recurrent MYOD1 mutations in pediatric and adult sclerosing and spindle cell rhabdomyosarcomas: Evidence for a common pathogenesis: MYOD1 Mutation in Rhabdomyosarcoma. Genes Chromosomes Cancer 2014, 53, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Rekhi, B.; Upadhyay, P.; Ramteke, M.P.; Dutt, A. MYOD1 (L122R) mutations are associated with spindle cell and sclerosing rhabdomyosarcomas with aggressive clinical outcomes. Mod. Pathol. 2016, 29, 1532–1540. [Google Scholar] [CrossRef]
- Owosho, A.A.; Chen, S.; Kashikar, S.; Zhang, L.; Chen, C.-L.; Wexler, L.H.; Estilo, C.L.; Huryn, J.M.; Antonescu, C.R. Clinical and molecular heterogeneity of head and neck spindle cell and sclerosing rhabdomyosarcoma. Oral Oncol. 2016, 58, e6–e11. [Google Scholar] [CrossRef] [PubMed]
- Agaram, N.P.; LaQuaglia, M.P.; Alaggio, R.; Zhang, L.; Fujisawa, Y.; Ladanyi, M.; Wexler, L.H.; Antonescu, C.R. MYOD1-mutant spindle cell and sclerosing rhabdomyosarcoma: An aggressive subtype irrespective of age. A reappraisal for molecular classification and risk stratification. Mod. Pathol. 2019, 32, 27–36. [Google Scholar] [CrossRef]
- Tsai, J.; ChangChien, Y.; Lee, J.; Kao, Y.; Li, W.; Liang, C.; Liao, I.; Chang, Y.; Wang, J.; Tsao, C.; et al. The expanding morphological and genetic spectrum of MYOD1-mutant spindle cell/sclerosing rhabdomyosarcomas: A clinicopathological and molecular comparison of mutated and non-mutated cases. Histopathology 2019, 74, 933–943. [Google Scholar] [CrossRef]
- Gorunova, L.; Bjerkehagen, B.; Micci, F.; Heim, S.; Panagopoulos, I. Cytogenetic and Molecular Study of an Adult Sclerosing Rhabdomyosarcoma of the Extremity: MYOD1-Mutation and Clonal Evolution. Cancer Genom. Proteom. 2020, 17, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Ting, M.A.; Reuther, J.; Chandramohan, R.; Voicu, H.; Gandhi, I.; Liu, M.; Cortes-Santiago, N.; Foster, J.H.; Hicks, J.; Nuchtern, J.; et al. Genomic analysis and preclinical xenograft model development identify potential therapeutic targets for MYOD1-mutant soft-tissue sarcoma of childhood. J. Pathol. 2021, 255, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.R.; Skapek, S.X.; Hawkins, D.S. The inconvenience of convenience cohorts: Rhabdomyosarcoma and the PAX-FOXO1 biomarker. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Hettmer, S.; Linardic, C.M.; Kelsey, A.; Rudzinski, E.R.; Vokuhl, C.; Selfe, J.; Ruhen, O.; Shern, J.F.; Khan, J.; Kovach, A.R.; et al. Molecular testing of rhabdomyosarcoma in clinical trials to improve risk stratification and outcome: A consensus view from European paediatric Soft tissue sarcoma Study Group, Children’s Oncology Group and Cooperative Weichteilsarkom-Studiengruppe. Eur. J. Cancer 2022, 172, 367–386. [Google Scholar] [CrossRef] [PubMed]
- Haduong, J.H.; Heske, C.M.; Allen-Rhoades, W.; Xue, W.; Teot, L.A.; Rodeberg, D.A.; Donaldson, S.S.; Weiss, A.; Hawkins, D.S.; Venkatramani, R. An update on rhabdomyosarcoma risk stratification and the rationale for current and future Children’s Oncology Group clinical trials. Pediatr. Blood Cancer 2022, 69, e29511. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; Chou, R.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Study | No | Histological Diagnosis | Age at Diagnosis (Years) | Sex | Site | MYOD1 Mutation | Desmin IHC | Myf4 IHC | MyoD1 IHC | Other Mutations | Events | Outcome | Time from Diagnosis to Death, or Last Follow-Up (Months) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Kohsaka 2014 [25] | 1 | ERMS | 21 | F | Chest wall | p.L122R | na | na | na | no | na | DOD | na |
| 2 | ERMS | 37 | M | Pterygopalatine fossa | p.L122R | na | na | na | PIK3CA | na | DOD | na | |
| 3 | ERMS | 32 | M | Mandible | p.L122R | na | na | na | no | na | DOD | na | |
| 4 | ERMS | 41 | F | Facial | p.L122R | na | na | na | PTEN deletion | na | na | na | |
| 5 | ERMS | 32 | F | Cheek | p.L122R | na | na | na | PTEN deletion | na | na | na | |
| 6 | ERMS | 4 | F | Peritonsillar | p.L122R | na | na | na | no | na | na | na | |
| 7 | ERMS | 35 | F | Buccal | p.L122R | na | na | na | no | na | DOD | na | |
| 8 | ERMS | 24 | F | Nasopharynx | p.L122R | na | na | na | no | na | DOD | na | |
| 9 | ERMS | 13 | F | Hemidiaphragm | p.L122R | na | na | na | PIK3CA | na | DOD | na | |
| 10 | ERMS | 12 | F | Infra-temporal fossa | p.L122R | na | na | na | PIK3CA | na | DOD | na | |
| Agaram 2014 [43] | 11 | ScRMS | 34 | F | Maxilla | p.L122R | na | na | na | PIK3CA | DR | NED | 41 |
| 12 | ScRMS | 39 | M | Extremities | p.L122R | na | na | na | no | na | NED | 12 | |
| 13 | ScRMS | 76 | M | Extremities | p.L122R | na | na | na | no | DR | AWD | 17 | |
| 14 | ScRMS | 13 | F | Chest wall | p.L122R | na | na | na | PIK3CA | LR, DR | DOD | 21 | |
| 15 | ScRMS | 14 | F | Infra-temporal fossa | p.L122R | na | na | na | PIK3CA | LR, DR | DOD | 26 | |
| 16 | SpRMS | 10 | F | Paraspinal | p.L122R | na | na | na | na | LR, DR | DOD | 35 | |
| 17 | SpRMS | 2 | F | Buttock | p.L122R | na | na | na | na | DR | DOD | 12 | |
| 18 | SpRMS | 21 | M | Pelvis | p.L122R | na | na | na | na | LR, DR | AWD | 30 | |
| 19 | SpRMS | 35 | M | Extremities | p.L122R | na | na | na | na | na | AWD | 4 | |
| Szuhai 2014 [17] | 20 | SpRMS | 52 | M | Extremities | p.L122R | pos | pos | na | na | na | na | na |
| 21 | SpRMS | 32 | F | Extremities | p.L122R | pos | neg | na | na | na | na | na | |
| 22 | SpRMS | 28 | M | Extremities | p.L122R | neg | neg | na | na | na | na | na | |
| 23 | SpRMS | 71 | F | Extremities | p.L122R | pos | pos | na | na | na | na | na | |
| 24 | SpRMS | 24 | M | Pharynx | p.L122R | pos | pos | na | na | na | na | na | |
| 25 | SpRMS | 20 | F | Mouth | p.L122R | pos | pos | na | na | na | na | na | |
| 26 | SpRMS | 64 | M | Extremities | p.L122R | pos | pos | na | na | na | na | na | |
| Rekhi 2016 [44] | 27 | ScRMS | 17 | M | Extremities | p.L122R | na | na | na | na | na | na | na |
| 28 | ScRMS | 5 | F | Maxilla | p.L122R | na | na | na | na | na | AWD | 22 | |
| Alaggio 2016 [13] | 29 | ScRMS | 17 | M | Paraspinal | p.L122R | na | na | na | no | na | DOD | 24 |
| 30 | ScRMS | 10 | F | Buttock | p.L122R | na | na | na | PIK3CA, FGFR4 | na | DOD | 6 | |
| 31 | ScRMS | 8 | M | Extremities | p.L122R | na | na | na | no | na | NED | 12 | |
| 32 | ScRMS | 11 | F | Head | p.L122R | na | na | na | PIK3CA | no | on therapy | recent | |
| 33 | SpRMS | 9 | M | Head | p.L122R | na | na | na | no | LR | AWD | 36 | |
| 34 | SpRMS | 9 | F | Head | p.L122R | na | na | na | no | no | DOD | 12 | |
| Owosho 2016 [45] | 35 | Sp/ScRMS | 33 | M | Mandible | p.L122R | pos | pos | na | no | LR, DR | DOD | 65 |
| Agaram 2019 [46] | 36 | ScRMS | 4 | F | Extremities | p.L122R | na | na | na | no | na | na | na |
| 37 | ScRMS | 7 | F | Abdominal | p.L122R | na | na | na | no | LR, DR | DOD | 28 | |
| 38 | ScRMS | 9 | F | Head and neck | p.L122R | na | na | na | PIK3CA | na | na | na | |
| 39 | ScRMS | 10 | F | Head and neck | p.L122R | na | na | na | no | LR | NED | 48 | |
| 40 | SpRMS | 17 | F | Thorax | p.L122R | na | na | na | no | LR, DR | DOD | 68 | |
| 41 | SpRMS | 21 | M | Pelvis | p.L122R | na | na | na | no | LR, DR | DOD | 42 | |
| 42 | Sp/ScRMS | 21 | F | Head and neck | p.L122R | na | na | na | PIK3CA | no | NED | 30 | |
| 43 | Sp/ScRMS | 21 | F | Head and neck | p.L122R | na | na | na | na | no | na | na | |
| 44 | Sp/ScRMS | 26 | M | Extremities | p.L122R | na | na | na | PIK3CA | no | NED | 4 | |
| 45 | ScRMS | 31 | F | Head and neck | p.L122R | na | na | na | PIK3CA | LR | AWD | 12 | |
| 46 | SpRMS | 33 | M | Extremities | p.L122R | na | na | na | no | na | na | na | |
| 47 | SpRMS | 36 | M | Extremities | p.L122R | na | na | na | PIK3CA | LR, DR | DOD | 16 | |
| 48 | SpRMS | 38 | F | Extremities | p.L122R | na | na | na | no | na | na | na | |
| 49 | Sp/ScRMS | 39 | M | Extremities | p.L122R | na | na | na | no | no | NED | 60 | |
| 50 | SpRMS | 44 | F | Paraspinal | p.L122R | na | na | na | no | no | NED | 13 | |
| 51 | SpRMS | 45 | M | Liver | p.L122R | na | na | na | no | na | na | na | |
| 52 | SpRMS | 77 | M | Extremities | p.L122R | na | na | na | no | DR | DOD | 32 | |
| 53 | ScRMS | 94 | M | Extremities | p.L122R | na | na | na | no | na | na | na | |
| Tsai 2019 [47] | 54 | Sp/ScRMS | 42 | F | Head | p.L122R | pos | pos | pos | no | no | NED | 134 |
| 55 | ScRMS | 23 | F | Parapharynx | p.L122R | pos | pos | pos | no | LR, DR | DOD | 24 | |
| 56 | SpRMS | 34 | F | Mediastinum | p.L122R | pos | pos | pos | no | LR | AWD | 12 | |
| 57 | Sp/ScRMS | 64 | M | Extremities | p.L122R | pos | pos | pos | no | LR | AWD | 13 | |
| 58 | ScRMS | 22 | F | Extremities | p.L122R | pos | pos | pos | no | no | NED | 2 | |
| 59 | ScRMS | 15 | F | Parapharynx | p.L122R | pos | pos | pos | no | no | AWD | 13 | |
| 60 | SpRMS | 42 | M | Extremities | p.L122R | pos | pos | pos | no | DR | AWD | 12 | |
| 61 | SpRMS | 8 | F | Head | p.L122R | pos | pos | pos | no | DR | AWD | 6 | |
| 62 | ScRMS | 28 | M | Head | p.L122R | pos | pos | pos | no | no | AWD | 14 | |
| 63 | Sp/ScRMS | 8 | F | Extremities | p.L122R | pos | pos | pos | no | LR | AWD | 29 | |
| 64 | SpRMS | 19 | F | Pterygomandibular | p.L122R | pos | pos | pos | no | no | NED | 50 | |
| 65 | Sp/ScRMS | 16 | F | Pre-auricular | p.L122R | pos | pos | pos | no | LR | AWD | 24 | |
| Gorunova 2020 [48] | 66 | ScRMS | 3.5 | F | Extremities | p.L122R | na | na | na | na | na | na | na |
| 67 | ScRMS | 30 | M | Extremities | p.L122R | na | na | na | na | na | na | na | |
| Ting 2021 [49] | 68 | ERMS | 10 | M | Retroperitoneum | p.L122R | na | na | na | PIK3CA, MDM2 gain | LR | DOD | 24 |
| 69 | ScRMS | 20 | M | Chest wall | p.L122R | na | na | na | no | LR, DR | DOD | 10 | |
| 70 | ScRMS | 15 | F | Extremities | p.L122R | na | na | na | NRAS | LR, DR | DOD | 49 | |
| 71 | SpRMS | 15 | M | Head and neck | p.L122R | na | na | na | MDM2 gain | no | DOD | 13 | |
| 72 | ERMS | 13 | F | Head and neck | p.L122R | na | na | no | na | AWD | na |
| Children (n = 18) | Adults (n = 19) | Tot (n = 37) | |
|---|---|---|---|
| Events | |||
| na | 3 | 1 | 4 |
| No | 3 | 6 | 9 |
| Yes | 12 | 12 | 24 |
| Type of event | |||
| LR | 5 | 3 | 8 |
| DR | 1 | 4 | 5 |
| LR + DR | 6 | 5 | 11 |
| Outcome | |||
| NED | 2 | 7 | 9 |
| AWD | 5 | 7 | 12 |
| DOD | 11 | 5 | 16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Carlo, D.; Chisholm, J.; Kelsey, A.; Alaggio, R.; Bisogno, G.; Minard-Colin, V.; Jenney, M.; Dávila Fajardo, R.; Merks, J.H.M.; Shipley, J.M.; et al. Biological Role and Clinical Implications of MYOD1L122R Mutation in Rhabdomyosarcoma. Cancers 2023, 15, 1644. https://doi.org/10.3390/cancers15061644
Di Carlo D, Chisholm J, Kelsey A, Alaggio R, Bisogno G, Minard-Colin V, Jenney M, Dávila Fajardo R, Merks JHM, Shipley JM, et al. Biological Role and Clinical Implications of MYOD1L122R Mutation in Rhabdomyosarcoma. Cancers. 2023; 15(6):1644. https://doi.org/10.3390/cancers15061644
Chicago/Turabian StyleDi Carlo, Daniela, Julia Chisholm, Anna Kelsey, Rita Alaggio, Gianni Bisogno, Veronique Minard-Colin, Meriel Jenney, Raquel Dávila Fajardo, Johannes H. M. Merks, Janet M. Shipley, and et al. 2023. "Biological Role and Clinical Implications of MYOD1L122R Mutation in Rhabdomyosarcoma" Cancers 15, no. 6: 1644. https://doi.org/10.3390/cancers15061644
APA StyleDi Carlo, D., Chisholm, J., Kelsey, A., Alaggio, R., Bisogno, G., Minard-Colin, V., Jenney, M., Dávila Fajardo, R., Merks, J. H. M., Shipley, J. M., & Selfe, J. L. (2023). Biological Role and Clinical Implications of MYOD1L122R Mutation in Rhabdomyosarcoma. Cancers, 15(6), 1644. https://doi.org/10.3390/cancers15061644