
Genomic and Epigenetic Changes Drive Aberrant Skeletal Muscle Differentiation in Rhabdomyosarcoma

 , , ,
, , ,  ,
,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Normal Skeletal Muscle Development and Homeostasis
2.1. The Myogenic Regulatory Factor (MRF) Family
2.2. Embryonal Skeletal Muscle Development
2.3. Post-Natal Regeneration of Skeletal Muscle
2.4. Epigenetic Regulation of Muscle Development/Differentiation
3. Genetic and Epigenetic Landscape of RMS
3.1. RMS Classification
3.2. Genetic Landscape of Alveolar RMS
3.3. Genetic Landscape of PF−, Non-Alveolar RMS
3.4. Methylation Profiling of RMS
3.5. Imprinting in RMS
3.6. Genetic RMS Susceptibility due to Pathogenic Germline Variants in Cancer Genes
4. Cell Cycle Progression in Skeletal Muscle and RMS
4.1. Cell Cycle Regulation in Skeletal Muscle
4.2. Cell Cycle Regulation in RMS
4.3. Aneuploidy in RMS
5. Developmental Myogenic Heterogeneity of RMS
5.1. Cell-to-Cell Heterogeneity within the Cancer Cell Pool
5.2. Aberrant MRF Expression in RMS Tissue
5.3. Developmental Heterogeneity at the Single Cell Level within the RMS Cell Pool
5.4. Cellular Hierarchies in RMS
5.5. Differential Drug Responsiveness of RMS Cell Subsets
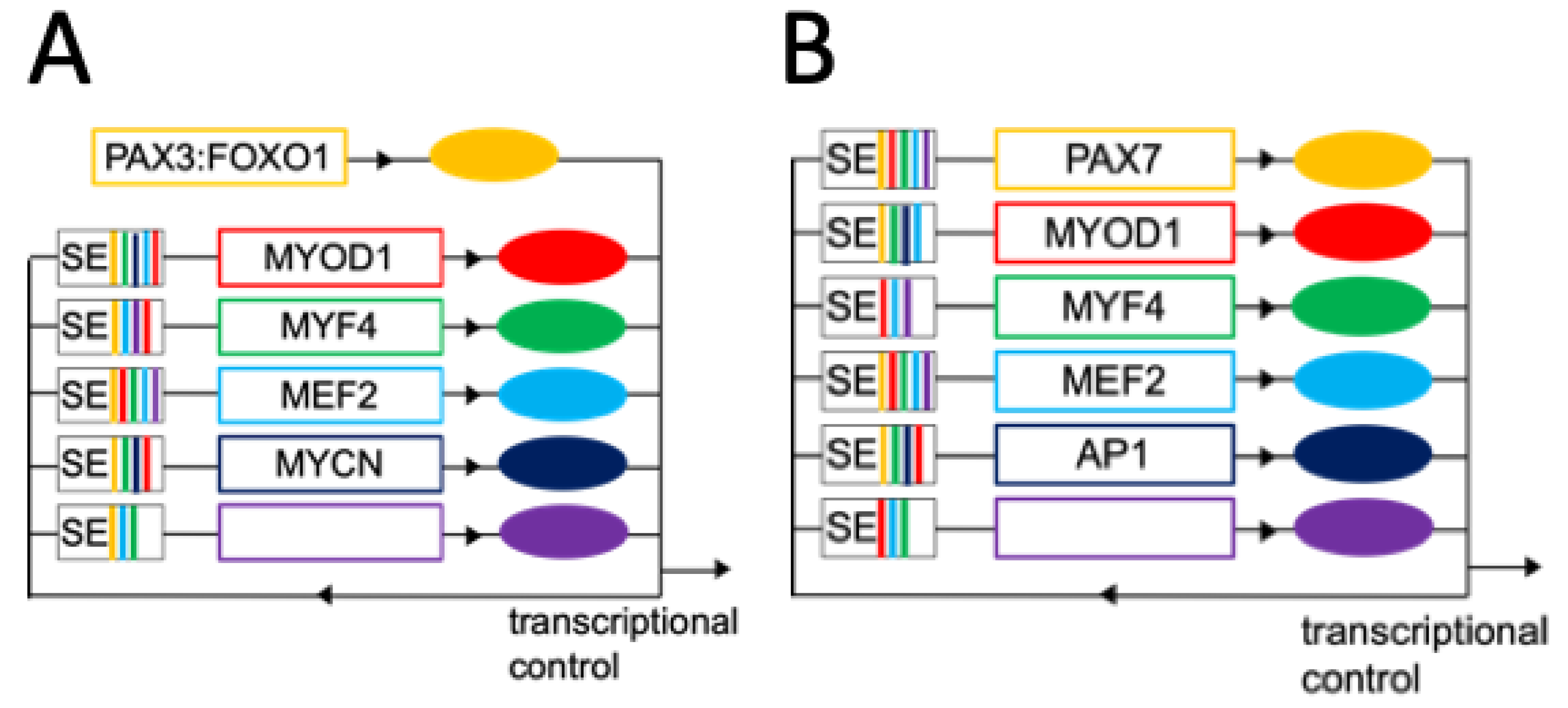
6. Aberrant Myogenic Differentiation due to Miswiring of Core Regulatory Circuits (CRCs) in RMS
6.1. Regulation of Cell Fate by CRCs
6.2. Reshaping of Skeletal Muscle CRCs in RMS
6.3. Differential Use of CRCs at the Single Cell Level
6.4. Evolving Understanding of Tumor Dependency Concepts in RMS
7. Epigenetic Regulators of Aberrant Myogenic Differentiation in RMS
7.1. Aberrant Epigenetic Control of MYOD1 Expression in RMS
7.2. Chromatin Regulatory Complex PRC2
7.3. Other Epigenetic Regulators of Aberrant Myogenic Differentiation in RMS
8. MicroRNA (miRNA)-Dependent Post-Transcriptional Dysregulation of Myogenic Differentiation in RMS
8.1. MiRNAs in Cancer
8.2. MYOmiR Family of miRNAs
8.3. Deregulation of MYOmiRs in RMS
8.4. Deregulation of Other miRNAs in RMS
8.5. Potential Avenues towards Therapeutic Targeting of miRNAs
9. Myogenic Differentiation as a Target for RMS Therapy
9.1. Differentiation Therapy in Cancer
9.2. Overcoming the Differentiation Block in RMS as a Therapeutic Principle
9.3. Targeting Cell Cycle Progression in RMS
9.4. Targeting MAPK Signaling in PF− und PF+ Cells
9.5. Targeting Cellular Hierarchies in RMS
9.6. Indirect Targeting of MYOD1 to Trigger RMS Differentiation
9.7. HDAC and EZH2 Inhibitors
9.8. Indirect Targeting of the PF Fusion Protein
9.9. Clinical Testing of Differentiating Agents in RMS
10. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Parham, D.M. Pathologic classification of rhabdomyosarcomas and correlations with molecular studies. Mod. Pathol. 2001, 14, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Rudzinski, E.R.; Kelsey, A.; Vokuhl, C.; Linardic, C.M.; Shipley, J.; Hettmer, S.; Koscielniak, E.; Hawkins, D.S.; Bisogno, G. Pathology of childhood rhabdomyosarcoma: A consensus opinion document from the Children’s Oncology Group, European Paediatric Soft Tissue Sarcoma Study Group, and the Cooperative Weichteilsarkom Studiengruppe. Pediatr. Blood Cancer 2021, 68, e28798. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.D.; Kothary, R. The myogenic kinome: Protein kinases critical to mammalian skeletal myogenesis. Skelet. Muscle 2011, 1, 29. [Google Scholar] [CrossRef] [PubMed]
- Zammit, P.S. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin. Cell Dev. Biol. 2017, 72, 19–32. [Google Scholar] [CrossRef]
- Gryder, B.E.; Pomella, S.; Sayers, C.; Wu, X.S.; Song, Y.; Chiarella, A.M.; Bagchi, S.; Chou, H.C.; Sinniah, R.S.; Walton, A.; et al. Histone hyperacetylation disrupts core gene regulatory architecture in rhabdomyosarcoma. Nat. Genet. 2019, 51, 1714–1722. [Google Scholar] [CrossRef]
- Gryder, B.E.; Wachtel, M.; Chang, K.; El Demerdash, O.; Aboreden, N.G.; Mohammed, W.; Ewert, W.; Pomella, S.; Rota, R.; Wei, J.S.; et al. Miswired Enhancer Logic Drives a Cancer of the Muscle Lineage. iScience 2020, 23, 101103. [Google Scholar] [CrossRef]
- Gryder, B.E.; Wu, L.; Woldemichael, G.M.; Pomella, S.; Quinn, T.R.; Park, P.M.C.; Cleveland, A.; Stanton, B.Z.; Song, Y.; Rota, R.; et al. Chemical genomics reveals histone deacetylases are required for core regulatory transcription. Nat. Commun. 2019, 10, 3004. [Google Scholar] [CrossRef]
- Saab, R.; Spunt, S.L.; Skapek, S.X. Myogenesis and rhabdomyosarcoma the Jekyll and Hyde of skeletal muscle. Curr. Top. Dev. Biol. 2011, 94, 197–234. [Google Scholar] [CrossRef]
- Davis, R.L.; Weintraub, H.; Lassar, A.B. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 1987, 51, 987–1000. [Google Scholar] [CrossRef]
- Wright, W.E.; Sassoon, D.A.; Lin, V.K. Myogenin, a factor regulating myogenesis, has a domain homologous to MyoD. Cell 1989, 56, 607–617. [Google Scholar] [CrossRef]
- Braun, T.; Buschhausen-Denker, G.; Bober, E.; Tannich, E.; Arnold, H.H. A novel human muscle factor related to but distinct from MyoD1 induces myogenic conversion in 10T1/2 fibroblasts. EMBO J. 1989, 8, 701–709. [Google Scholar] [CrossRef]
- Yutzey, K.E.; Rhodes, S.J.; Konieczny, S.F. Differential trans activation associated with the muscle regulatory factors MyoD1, myogenin, and MRF4. Mol. Cell. Biol. 1990, 10, 3934–3944. [Google Scholar] [CrossRef]
- Singh, K.; Dilworth, F.J. Differential modulation of cell cycle progression distinguishes members of the myogenic regulatory factor family of transcription factors. FEBS J. 2013, 280, 3991–4003. [Google Scholar] [CrossRef]
- Weintraub, H.; Tapscott, S.J.; Davis, R.L.; Thayer, M.J.; Adam, M.A.; Lassar, A.B.; Miller, A.D. Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc. Natl. Acad. Sci. USA 1989, 86, 5434–5438. [Google Scholar] [CrossRef]
- Gayraud-Morel, B.; Chretien, F.; Flamant, P.; Gomes, D.; Zammit, P.S.; Tajbakhsh, S. A role for the myogenic determination gene Myf5 in adult regenerative myogenesis. Dev. Biol. 2007, 312, 13–28. [Google Scholar] [CrossRef]
- Tapscott, S.J. The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development 2005, 132, 2685–2695. [Google Scholar] [CrossRef]
- Kassar-Duchossoy, L.; Giacone, E.; Gayraud-Morel, B.; Jory, A.; Gomes, D.; Tajbakhsh, S. Pax3/Pax7 mark a novel population of primitive myogenic cells during development. Genes. Dev. 2005, 19, 1426–1431. [Google Scholar] [CrossRef]
- Relaix, F.; Rocancourt, D.; Mansouri, A.; Buckingham, M. A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature 2005, 435, 948–953. [Google Scholar] [CrossRef]
- Summerbell, D.; Halai, C.; Rigby, P.W. Expression of the myogenic regulatory factor Mrf4 precedes or is contemporaneous with that of Myf5 in the somitic bud. Mech. Dev. 2002, 117, 331–335. [Google Scholar] [CrossRef]
- Tajbakhsh, S.; Rocancourt, D.; Cossu, G.; Buckingham, M. Redefining the genetic hierarchies controlling skeletal myogenesis: Pax-3 and Myf-5 act upstream of MyoD. Cell 1997, 89, 127–138. [Google Scholar] [CrossRef]
- Rudnicki, M.A.; Schnegelsberg, P.N.; Stead, R.H.; Braun, T.; Arnold, H.H.; Jaenisch, R. MyoD or Myf-5 is required for the formation of skeletal muscle. Cell 1993, 75, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Hasty, P.; Bradley, A.; Morris, J.H.; Edmondson, D.G.; Venuti, J.M.; Olson, E.N.; Klein, W.H. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 1993, 364, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Relaix, F.; Montarras, D.; Zaffran, S.; Gayraud-Morel, B.; Rocancourt, D.; Tajbakhsh, S.; Mansouri, A.; Cumano, A.; Buckingham, M. Pax3 and Pax7 have distinct and overlapping functions in adult muscle progenitor cells. J. Cell Biol. 2006, 172, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Seale, P.; Sabourin, L.A.; Girgis-Gabardo, A.; Mansouri, A.; Gruss, P.; Rudnicki, M.A. Pax7 is required for the specification of myogenic satellite cells. Cell 2000, 102, 777–786. [Google Scholar] [CrossRef]
- Cooper, R.N.; Tajbakhsh, S.; Mouly, V.; Cossu, G.; Buckingham, M.; Butler-Browne, G.S. In vivo satellite cell activation via Myf5 and MyoD in regenerating mouse skeletal muscle. J. Cell Sci. 1999, 112 Pt 17, 2895–2901. [Google Scholar] [CrossRef]
- Fuchtbauer, E.M.; Westphal, H. MyoD and myogenin are coexpressed in regenerating skeletal muscle of the mouse. Dev. Dyn. 1992, 193, 34–39. [Google Scholar] [CrossRef]
- Grounds, M.D.; Garrett, K.L.; Lai, M.C.; Wright, W.E.; Beilharz, M.W. Identification of skeletal muscle precursor cells in vivo by use of MyoD1 and myogenin probes. Cell Tissue Res. 1992, 267, 99–104. [Google Scholar] [CrossRef]
- Berkes, C.A.; Tapscott, S.J. MyoD and the transcriptional control of myogenesis. Semin. Cell Dev. Biol. 2005, 16, 585–595. [Google Scholar] [CrossRef]
- Segales, J.; Islam, A.B.; Kumar, R.; Liu, Q.C.; Sousa-Victor, P.; Dilworth, F.J.; Ballestar, E.; Perdiguero, E.; Munoz-Canoves, P. Chromatin-wide and transcriptome profiling integration uncovers p38alpha MAPK as a global regulator of skeletal muscle differentiation. Skelet. Muscle 2016, 6, 9. [Google Scholar] [CrossRef]
- Sincennes, M.C.; Brun, C.E.; Rudnicki, M.A. Concise Review: Epigenetic Regulation of Myogenesis in Health and Disease. Stem Cells Transl. Med. 2016, 5, 282–290. [Google Scholar] [CrossRef]
- Rudzinski, E.R.; Anderson, J.R.; Hawkins, D.S.; Skapek, S.X.; Parham, D.M.; Teot, L.A. The World Health Organization Classification of Skeletal Muscle Tumors in Pediatric Rhabdomyosarcoma: A Report From the Children’s Oncology Group. Arch. Pathol. Lab. Med. 2015, 139, 1281–1287. [Google Scholar] [CrossRef]
- Sultan, I.; Qaddoumi, I.; Yaser, S.; Rodriguez-Galindo, C.; Ferrari, A. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: An analysis of 2600 patients. J. Clin. Oncol. 2009, 27, 3391–3397. [Google Scholar] [CrossRef]
- Arnold, M.A.; Anderson, J.R.; Gastier-Foster, J.M.; Barr, F.G.; Skapek, S.X.; Hawkins, D.S.; Raney, R.B., Jr.; Parham, D.M.; Teot, L.A.; Rudzinski, E.R.; et al. Histology, Fusion Status, and Outcome in Alveolar Rhabdomyosarcoma With Low-Risk Clinical Features: A Report From the Children’s Oncology Group. Pediatr. Blood Cancer 2016, 63, 634–639. [Google Scholar] [CrossRef]
- Missiaglia, E.; Williamson, D.; Chisholm, J.; Wirapati, P.; Pierron, G.; Petel, F.; Concordet, J.P.; Thway, K.; Oberlin, O.; Pritchard-Jones, K.; et al. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J. Clin. Oncol. 2012, 30, 1670–1677. [Google Scholar] [CrossRef]
- Skapek, S.X.; Anderson, J.; Barr, F.G.; Bridge, J.A.; Gastier-Foster, J.M.; Parham, D.M.; Rudzinski, E.R.; Triche, T.; Hawkins, D.S. PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: A children’s oncology group report. Pediatr. Blood Cancer 2013, 60, 1411–1417. [Google Scholar] [CrossRef]
- Williamson, D.; Missiaglia, E.; de Reynies, A.; Pierron, G.; Thuille, B.; Palenzuela, G.; Thway, K.; Orbach, D.; Lae, M.; Freneaux, P.; et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J. Clin. Oncol. 2010, 28, 2151–2158. [Google Scholar] [CrossRef]
- Hettmer, S.; Linardic, C.M.; Kelsey, A.; Rudzinski, E.R.; Vokuhl, C.; Selfe, J.; Ruhen, O.; Shern, J.F.; Khan, J.; Kovach, A.R.; et al. Molecular testing of rhabdomyosarcoma in clinical trials to improve risk stratification and outcome: A consensus view from European paediatric Soft tissue sarcoma Study Group, Children’s Oncology Group and Cooperative Weichteilsarkom-Studiengruppe. Eur. J. Cancer 2022, 172, 367–386. [Google Scholar] [CrossRef]
- Shern, J.F.; Chen, L.; Chmielecki, J.; Wei, J.S.; Patidar, R.; Rosenberg, M.; Ambrogio, L.; Auclair, D.; Wang, J.; Song, Y.K.; et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014, 4, 216–231. [Google Scholar] [CrossRef]
- Sun, W.; Chatterjee, B.; Shern, J.F.; Patidar, R.; Song, Y.; Wang, Y.; Walker, R.L.; Pawel, B.R.; Linardic, C.M.; Houghton, P.; et al. Relationship of DNA methylation to mutational changes and transcriptional organization in fusion-positive and fusion-negative rhabdomyosarcoma. Int. J. Cancer 2019, 144, 2707–2717. [Google Scholar] [CrossRef]
- Hays, D.M.; Newton, W., Jr.; Soule, E.H.; Foulkes, M.A.; Raney, R.B.; Tefft, M.; Ragab, A.; Maurer, H.M. Mortality among children with rhabdomyosarcomas of the alveolar histologic subtype. J. Pediatr. Surg. 1983, 18, 412–417. [Google Scholar] [CrossRef]
- Newton, W.A., Jr.; Soule, E.H.; Hamoudi, A.B.; Reiman, H.M.; Shimada, H.; Beltangady, M.; Maurer, H. Histopathology of childhood sarcomas, Intergroup Rhabdomyosarcoma Studies I and II: Clinicopathologic correlation. J. Clin. Oncol. 1988, 6, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Horn, C.; Jenewein, S.; Sohn-Bosser, L.; Bremer, E.; Schmitt, L. Biochemical and structural analysis of the Bacillus subtilis ABC transporter OpuA and its isolated subunits. J. Mol. Microbiol. Biotechnol. 2005, 10, 76–91. [Google Scholar] [CrossRef] [PubMed]
- Stegmaier, S.; Poremba, C.; Schaefer, K.L.; Leuschner, I.; Kazanowska, B.; Bekassy, A.N.; Bielack, S.S.; Klingebiel, T.; Koscielniak, E. Prognostic value of PAX-FKHR fusion status in alveolar rhabdomyosarcoma: A report from the cooperative soft tissue sarcoma study group (CWS). Pediatr. Blood Cancer 2011, 57, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J.; D’Cruz, C.M.; Lovell, M.A.; Biegel, J.A.; Barr, F.G. Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res. 1994, 54, 2869–2872. [Google Scholar] [PubMed]
- Douglass, E.C.; Valentine, M.; Etcubanas, E.; Parham, D.; Webber, B.L.; Houghton, P.J.; Houghton, J.A.; Green, A.A. A specific chromosomal abnormality in rhabdomyosarcoma. Cytogenet. Cell Genet. 1987, 45, 148–155. [Google Scholar] [CrossRef]
- Barr, F.G.; Galili, N.; Holick, J.; Biegel, J.A.; Rovera, G.; Emanuel, B.S. Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 1993, 3, 113–117. [Google Scholar] [CrossRef]
- Galili, N.; Davis, R.J.; Fredericks, W.J.; Mukhopadhyay, S.; Rauscher, F.J., 3rd; Emanuel, B.S.; Rovera, G.; Barr, F.G. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 1993, 5, 230–235. [Google Scholar] [CrossRef]
- Heske, C.M.; Chi, Y.Y.; Venkatramani, R.; Li, M.; Arnold, M.A.; Dasgupta, R.; Hiniker, S.M.; Hawkins, D.S.; Mascarenhas, L. Survival outcomes of patients with localized FOXO1 fusion-positive rhabdomyosarcoma treated on recent clinical trials: A report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Cancer 2021, 127, 946–956. [Google Scholar] [CrossRef]
- Kazanowska, B.; Reich, A.; Stegmaier, S.; Bekassy, A.N.; Leuschner, I.; Chybicka, A.; Koscielniak, E. Pax3-fkhr and pax7-fkhr fusion genes impact outcome of alveolar rhabdomyosarcoma in children. Fetal Pediatr. Pathol. 2007, 26, 17–31. [Google Scholar] [CrossRef]
- Sorensen, P.H.; Lynch, J.C.; Qualman, S.J.; Tirabosco, R.; Lim, J.F.; Maurer, H.M.; Bridge, J.A.; Crist, W.M.; Triche, T.J.; Barr, F.G. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: A report from the children’s oncology group. J. Clin. Oncol. 2002, 20, 2672–2679. [Google Scholar] [CrossRef]
- Shern, J.F.; Selfe, J.; Izquierdo, E.; Patidar, R.; Chou, H.C.; Song, Y.K.; Yohe, M.E.; Sindiri, S.; Wei, J.; Wen, X.; et al. Genomic Classification and Clinical Outcome in Rhabdomyosarcoma: A Report From an International Consortium. J. Clin. Oncol. 2021, 39, 2859–2871. [Google Scholar] [CrossRef]
- Clay, M.R.; Patel, A.; Tran, Q.; Hedges, D.J.; Chang, T.C.; Stewart, E.; Charville, G.; Cline, C.; Dyer, M.A.; Orr, B.A. Methylation profiling reveals novel molecular classes of rhabdomyosarcoma. Sci. Rep. 2021, 11, 22213. [Google Scholar] [CrossRef]
- Seki, M.; Nishimura, R.; Yoshida, K.; Shimamura, T.; Shiraishi, Y.; Sato, Y.; Kato, M.; Chiba, K.; Tanaka, H.; Hoshino, N.; et al. Integrated genetic and epigenetic analysis defines novel molecular subgroups in rhabdomyosarcoma. Nat. Commun. 2015, 6, 7557. [Google Scholar] [CrossRef]
- Sun, W.; Chatterjee, B.; Wang, Y.; Stevenson, H.S.; Edelman, D.C.; Meltzer, P.S.; Barr, F.G. Distinct methylation profiles characterize fusion-positive and fusion-negative rhabdomyosarcoma. Mod. Pathol. 2015, 28, 1214–1224. [Google Scholar] [CrossRef]
- Barr, F.G.; Duan, F.; Smith, L.M.; Gustafson, D.; Pitts, M.; Hammond, S.; Gastier-Foster, J.M. Genomic and clinical analyses of 2p24 and 12q13-q14 amplification in alveolar rhabdomyosarcoma: A report from the Children’s Oncology Group. Genes. Chromosomes Cancer 2009, 48, 661–672. [Google Scholar] [CrossRef]
- Hachitanda, Y.; Toyoshima, S.; Akazawa, K.; Tsuneyoshi, M. N-myc gene amplification in rhabdomyosarcoma detected by fluorescence in situ hybridization: Its correlation with histologic features. Mod. Pathol. 1998, 11, 1222–1227. [Google Scholar]
- Barr, F.G.; Qualman, S.J.; Macris, M.H.; Melnyk, N.; Lawlor, E.R.; Strzelecki, D.M.; Triche, T.J.; Bridge, J.A.; Sorensen, P.H. Genetic heterogeneity in the alveolar rhabdomyosarcoma subset without typical gene fusions. Cancer Res. 2002, 62, 4704–4710. [Google Scholar]
- Sumegi, J.; Streblow, R.; Frayer, R.W.; Dal Cin, P.; Rosenberg, A.; Meloni-Ehrig, A.; Bridge, J.A. Recurrent t(2;2) and t(2;8) translocations in rhabdomyosarcoma without the canonical PAX-FOXO1 fuse PAX3 to members of the nuclear receptor transcriptional coactivator family. Genes. Chromosomes Cancer 2010, 49, 224–236. [Google Scholar] [CrossRef]
- Wachtel, M.; Dettling, M.; Koscielniak, E.; Stegmaier, S.; Treuner, J.; Simon-Klingenstein, K.; Buhlmann, P.; Niggli, F.K.; Schafer, B.W. Gene expression signatures identify rhabdomyosarcoma subtypes and detect a novel t(2;2)(q35;p23) translocation fusing PAX3 to NCOA1. Cancer Res. 2004, 64, 5539–5545. [Google Scholar] [CrossRef]
- Davicioni, E.; Anderson, M.J.; Finckenstein, F.G.; Lynch, J.C.; Qualman, S.J.; Shimada, H.; Schofield, D.E.; Buckley, J.D.; Meyer, W.H.; Sorensen, P.H.; et al. Molecular classification of rhabdomyosarcoma--genotypic and phenotypic determinants of diagnosis: A report from the Children’s Oncology Group. Am. J. Pathol. 2009, 174, 550–564. [Google Scholar] [CrossRef]
- Casey, D.L.; Wexler, L.H.; Pitter, K.L.; Samstein, R.M.; Slotkin, E.K.; Wolden, S.L. Genomic Determinants of Clinical Outcomes in Rhabdomyosarcoma. Clin. Cancer Res. 2020, 26, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Stewart, E.; Shelat, A.A.; Qu, C.; Bahrami, A.; Hatley, M.; Wu, G.; Bradley, C.; McEvoy, J.; Pappo, A.; et al. Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell 2013, 24, 710–724. [Google Scholar] [CrossRef] [PubMed]
- Shukla, N.; Ameur, N.; Yilmaz, I.; Nafa, K.; Lau, C.Y.; Marchetti, A.; Borsu, L.; Barr, F.G.; Ladanyi, M. Oncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathways. Clin. Cancer Res. 2012, 18, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Agaram, N.P.; LaQuaglia, M.P.; Alaggio, R.; Zhang, L.; Fujisawa, Y.; Ladanyi, M.; Wexler, L.H.; Antonescu, C.R. MYOD1-mutant spindle cell and sclerosing rhabdomyosarcoma: An aggressive subtype irrespective of age. A reappraisal for molecular classification and risk stratification. Mod. Pathol. 2019, 32, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Rekhi, B.; Upadhyay, P.; Ramteke, M.P.; Dutt, A. MYOD1 (L122R) mutations are associated with spindle cell and sclerosing rhabdomyosarcomas with aggressive clinical outcomes. Mod. Pathol. 2016, 29, 1532–1540. [Google Scholar] [CrossRef]
- Alaggio, R.; Zhang, L.; Sung, Y.S.; Huang, S.C.; Chen, C.L.; Bisogno, G.; Zin, A.; Agaram, N.P.; LaQuaglia, M.P.; Wexler, L.H.; et al. A Molecular Study of Pediatric Spindle and Sclerosing Rhabdomyosarcoma: Identification of Novel and Recurrent VGLL2-related Fusions in Infantile Cases. Am. J. Surg. Pathol. 2016, 40, 224–235. [Google Scholar] [CrossRef]
- Whittle, S.; Venkatramani, R.; Schonstein, A.; Pack, S.D.; Alaggio, R.; Vokuhl, C.; Rudzinski, E.R.; Wulf, A.L.; Zin, A.; Gruver, J.R.; et al. Congenital spindle cell rhabdomyosarcoma: An international cooperative analysis. Eur. J. Cancer 2022, 168, 56–64. [Google Scholar] [CrossRef]
- Mosquera, J.M.; Sboner, A.; Zhang, L.; Kitabayashi, N.; Chen, C.L.; Sung, Y.S.; Wexler, L.H.; LaQuaglia, M.P.; Edelman, M.; Sreekantaiah, C.; et al. Recurrent NCOA2 gene rearrangements in congenital/infantile spindle cell rhabdomyosarcoma. Genes. Chromosomes Cancer 2013, 52, 538–550. [Google Scholar] [CrossRef]
- Kommoss, F.K.F.; Stichel, D.; Mora, J.; Esteller, M.; Jones, D.T.W.; Pfister, S.M.; Brack, E.; Wachtel, M.; Bode, P.K.; Sinn, H.P.; et al. Clinicopathologic and molecular analysis of embryonal rhabdomyosarcoma of the genitourinary tract: Evidence for a distinct DICER1-associated subgroup. Mod. Pathol. 2021, 34, 1558–1569. [Google Scholar] [CrossRef]
- Gardiner-Garden, M.; Frommer, M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987, 196, 261–282. [Google Scholar] [CrossRef]
- Besnard-Guerin, C.; Newsham, I.; Winqvist, R.; Cavenee, W.K. A common region of loss of heterozygosity in Wilms’ tumor and embryonal rhabdomyosarcoma distal to the D11S988 locus on chromosome 11p15.5. Hum. Genet. 1996, 97, 163–170. [Google Scholar] [CrossRef]
- Anderson, J.; Gordon, A.; McManus, A.; Shipley, J.; Pritchard-Jones, K. Disruption of imprinted genes at chromosome region 11p15.5 in paediatric rhabdomyosarcoma. Neoplasia 1999, 1, 340–348. [Google Scholar] [CrossRef]
- Shern, J.F.; Yohe, M.E.; Khan, J. Pediatric Rhabdomyosarcoma. Crit. Rev. Oncog. 2015, 20, 227–243. [Google Scholar] [CrossRef]
- Li, F.P.; Fraumeni, J.F., Jr. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann. Intern. Med. 1969, 71, 747–752. [Google Scholar] [CrossRef]
- Akhavanfard, S.; Padmanabhan, R.; Yehia, L.; Cheng, F.; Eng, C. Comprehensive germline genomic profiles of children, adolescents and young adults with solid tumors. Nat. Commun. 2020, 11, 2206. [Google Scholar] [CrossRef]
- Grobner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef]
- Kim, J.; Light, N.; Subasri, V.; Young, E.L.; Wegman-Ostrosky, T.; Barkauskas, D.A.; Hall, D.; Lupo, P.J.; Patidar, R.; Maese, L.D.; et al. Pathogenic Germline Variants in Cancer Susceptibility Genes in Children and Young Adults With Rhabdomyosarcoma. JCO Precis. Oncol. 2021, 5, 75–87. [Google Scholar] [CrossRef]
- Li, H.; Sisoudiya, S.D.; Martin-Giacalone, B.A.; Khayat, M.M.; Dugan-Perez, S.; Marquez-Do, D.A.; Scheurer, M.E.; Muzny, D.; Boerwinkle, E.; Gibbs, R.A.; et al. Germline Cancer Predisposition Variants in Pediatric Rhabdomyosarcoma: A Report From the Children’s Oncology Group. J. Natl. Cancer Inst. 2021, 113, 875–883. [Google Scholar] [CrossRef]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef]
- Wurtemberger, J.; Ripperger, T.; Vokuhl, C.; Bauer, S.; Teichert-von Luttichau, I.; Wardelmann, E.; Niemeyer, C.; Kratz, C.P.; Schlegelberger, B.; Hettmer, S. Genetic susceptibility in children, adolescents, and young adults diagnosed with soft-tissue sarcomas. Eur. J. Med. Genet. 2023, 66, 104718. [Google Scholar] [CrossRef]
- Hettmer, S.; Archer, N.M.; Somers, G.R.; Novokmet, A.; Wagers, A.J.; Diller, L.; Rodriguez-Galindo, C.; Teot, L.A.; Malkin, D. Anaplastic rhabdomyosarcoma in TP53 germline mutation carriers. Cancer 2014, 120, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.R.; Best, A.F.; Williams, G.M.; Harney, L.A.; Carr, A.G.; Harris, A.K.; Kratz, C.P.; Dehner, L.P.; Messinger, Y.H.; Rosenberg, P.S.; et al. Neoplasm Risk Among Individuals With a Pathogenic Germline Variant in DICER1. J. Clin. Oncol. 2019, 37, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Megeney, L.A.; Kablar, B.; Garrett, K.; Anderson, J.E.; Rudnicki, M.A. MyoD is required for myogenic stem cell function in adult skeletal muscle. Genes. Dev. 1996, 10, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Ustanina, S.; Carvajal, J.; Rigby, P.; Braun, T. The myogenic factor Myf5 supports efficient skeletal muscle regeneration by enabling transient myoblast amplification. Stem Cells 2007, 25, 2006–2016. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.L.; Wu, Z.; Zhang, P.; Wood, L.D.; Bhakta, K.S.; Han, J.; Feramisco, J.R.; Karin, M.; Wang, J.Y. Induction of terminal differentiation by constitutive activation of p38 MAP kinase in human rhabdomyosarcoma cells. Genes. Dev. 2000, 14, 574–584. [Google Scholar] [CrossRef]
- Pajcini, K.V.; Corbel, S.Y.; Sage, J.; Pomerantz, J.H.; Blau, H.M. Transient inactivation of Rb and ARF yields regenerative cells from postmitotic mammalian muscle. Cell Stem Cell 2010, 7, 198–213. [Google Scholar] [CrossRef]
- Kitzmann, M.; Carnac, G.; Vandromme, M.; Primig, M.; Lamb, N.J.; Fernandez, A. The muscle regulatory factors MyoD and myf-5 undergo distinct cell cycle-specific expression in muscle cells. J. Cell Biol. 1998, 142, 1447–1459. [Google Scholar] [CrossRef]
- Lindon, C.; Montarras, D.; Pinset, C. Cell cycle-regulated expression of the muscle determination factor Myf5 in proliferating myoblasts. J. Cell Biol. 1998, 140, 111–118. [Google Scholar] [CrossRef]
- Stewart, E.; McEvoy, J.; Wang, H.; Chen, X.; Honnell, V.; Ocarz, M.; Gordon, B.; Dapper, J.; Blankenship, K.; Yang, Y.; et al. Identification of Therapeutic Targets in Rhabdomyosarcoma through Integrated Genomic, Epigenomic, and Proteomic Analyses. Cancer Cell 2018, 34, 411–426e419. [Google Scholar] [CrossRef]
- Kikuchi, K.; Hettmer, S.; Aslam, M.I.; Michalek, J.E.; Laub, W.; Wilky, B.A.; Loeb, D.M.; Rubin, B.P.; Wagers, A.J.; Keller, C. Cell-cycle dependent expression of a translocation-mediated fusion oncogene mediates checkpoint adaptation in rhabdomyosarcoma. PLoS Genet. 2014, 10, e1004107. [Google Scholar] [CrossRef]
- Regina, C.; Hamed, E.; Andrieux, G.; Angenendt, S.; Schneider, M.; Ku, M.; Follo, M.; Wachtel, M.; Ke, E.; Kikuchi, K.; et al. Negative correlation of single-cell PAX3:FOXO1 expression with tumorigenicity in rhabdomyosarcoma. Life Sci. Alliance 2021, 4, e202001002. [Google Scholar] [CrossRef]
- Keller, C.; Arenkiel, B.R.; Coffin, C.M.; El-Bardeesy, N.; DePinho, R.A.; Capecchi, M.R. Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: Cooperativity of Ink4a/ARF and Trp53 loss of function. Genes. Dev. 2004, 18, 2614–2626. [Google Scholar] [CrossRef]
- Shapiro, D.N.; Parham, D.M.; Douglass, E.C.; Ashmun, R.; Webber, B.L.; Newton, W.A., Jr.; Hancock, M.L.; Maurer, H.M.; Look, A.T. Relationship of tumor-cell ploidy to histologic subtype and treatment outcome in children and adolescents with unresectable rhabdomyosarcoma. J. Clin. Oncol. 1991, 9, 159–166. [Google Scholar] [CrossRef]
- Suva, M.L.; Tirosh, I. Single-Cell RNA Sequencing in Cancer: Lessons Learned and Emerging Challenges. Mol. Cell 2019, 75, 7–12. [Google Scholar] [CrossRef]
- Cessna, M.H.; Zhou, H.; Perkins, S.L.; Tripp, S.R.; Layfield, L.; Daines, C.; Coffin, C.M. Are myogenin and myoD1 expression specific for rhabdomyosarcoma? A study of 150 cases, with emphasis on spindle cell mimics. Am. J. Surg. Pathol. 2001, 25, 1150–1157. [Google Scholar] [CrossRef]
- Dias, P.; Chen, B.; Dilday, B.; Palmer, H.; Hosoi, H.; Singh, S.; Wu, C.; Li, X.; Thompson, J.; Parham, D.; et al. Strong immunostaining for myogenin in rhabdomyosarcoma is significantly associated with tumors of the alveolar subclass. Am. J. Pathol. 2000, 156, 399–408. [Google Scholar] [CrossRef]
- Danielli, S.G.; Porpiglia, E.; De Micheli, A.J.; Navarro, N.; Zellinger, M.J.; Bechtold, I.; Kisele, S.; Volken, L.; Marques, J.G.; Kasper, S.; et al. Single-cell profiling of alveolar rhabdomyosarcoma reveals RAS pathway inhibitors as cell-fate hijackers with therapeutic relevance. Sci. Adv. 2023, 9, eade9238. [Google Scholar] [CrossRef]
- Patel, A.G.; Chen, X.; Huang, X.; Clay, M.R.; Komorova, N.; Krasin, M.J.; Pappo, A.; Tillman, H.; Orr, B.A.; McEvoy, J.; et al. The myogenesis program drives clonal selection and drug resistance in rhabdomyosarcoma. Dev. Cell 2022, 57, 1226–1240. [Google Scholar] [CrossRef]
- Wei, Y.; Qin, Q.; Yan, C.; Hayes, M.N.; Garcia, S.P.; Xi, H.; Do, D.; Jin, A.H.; Eng, T.C.; McCarthy, K.M.; et al. Single-cell analysis and functional characterization uncover the stem cell hierarchies and developmental origins of rhabdomyosarcoma. Nat. Cancer 2022, 3, 961–975. [Google Scholar] [CrossRef]
- Ignatius, M.S.; Chen, E.; Elpek, N.M.; Fuller, A.Z.; Tenente, I.M.; Clagg, R.; Liu, S.; Blackburn, J.S.; Linardic, C.M.; Rosenberg, A.E.; et al. In vivo imaging of tumor-propagating cells, regional tumor heterogeneity, and dynamic cell movements in embryonal rhabdomyosarcoma. Cancer Cell 2012, 21, 680–693. [Google Scholar] [CrossRef]
- Generali, M.; Satheesha, S.; Bode, P.K.; Wanner, D.; Schafer, B.W.; Casanova, E.A. High Frequency of Tumor Propagating Cells in Fusion-Positive Rhabdomyosarcoma. Genes 2021, 12, 1373. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jung, J.; Babikir, H.; Shamardani, K.; Jain, S.; Feng, X.; Gupta, N.; Rosi, S.; Chang, S.; Raleigh, D.; et al. A single-cell atlas of glioblastoma evolution under therapy reveals cell-intrinsic and cell-extrinsic therapeutic targets. Nat. Cancer 2022, 3, 1534–1552. [Google Scholar] [CrossRef] [PubMed]
- Filbin, M.G.; Tirosh, I.; Hovestadt, V.; Shaw, M.L.; Escalante, L.E.; Mathewson, N.D.; Neftel, C.; Frank, N.; Pelton, K.; Hebert, C.M.; et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 2018, 360, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Yang, R.; Zhan, Y.; Lai, H.D.; Ye, C.J.; Yao, X.Y.; Luo, W.Q.; Cheng, X.M.; Miao, J.J.; Wang, J.F.; et al. Single-Cell Characterization of Malignant Phenotypes and Developmental Trajectories of Adrenal Neuroblastoma. Cancer Cell 2020, 38, 716–733e716. [Google Scholar] [CrossRef]
- Zhang, L.; He, X.; Liu, X.; Zhang, F.; Huang, L.F.; Potter, A.S.; Xu, L.; Zhou, W.; Zheng, T.; Luo, Z.; et al. Single-Cell Transcriptomics in Medulloblastoma Reveals Tumor-Initiating Progenitors and Oncogenic Cascades during Tumorigenesis and Relapse. Cancer Cell 2019, 36, 302–318.e307. [Google Scholar] [CrossRef]
- Custers, L.; Khabirova, E.; Coorens, T.H.H.; Oliver, T.R.W.; Calandrini, C.; Young, M.D.; Vieira Braga, F.A.; Ellis, P.; Mamanova, L.; Segers, H.; et al. Somatic mutations and single-cell transcriptomes reveal the root of malignant rhabdoid tumours. Nat. Commun. 2021, 12, 1407. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, L.; Lin, R.Y.; Muschen, M.; Koeffler, H.P. Core transcriptional regulatory circuitries in cancer. Oncogene 2020, 39, 6633–6646. [Google Scholar] [CrossRef]
- Huang, M.; Chen, Y.; Yang, M.; Guo, A.; Xu, Y.; Xu, L.; Koeffler, H.P. dbCoRC: A database of core transcriptional regulatory circuitries modeled by H3K27ac ChIP-seq signals. Nucleic Acids Res. 2018, 46, D71–D77. [Google Scholar] [CrossRef]
- Saint-Andre, V.; Federation, A.J.; Lin, C.Y.; Abraham, B.J.; Reddy, J.; Lee, T.I.; Bradner, J.E.; Young, R.A. Models of human core transcriptional regulatory circuitries. Genome Res. 2016, 26, 385–396. [Google Scholar] [CrossRef]
- Ahn, E.H.; Mercado, G.E.; Lae, M.; Ladanyi, M. Identification of target genes of PAX3-FOXO1 in alveolar rhabdomyosarcoma. Oncol. Rep. 2013, 30, 968–978. [Google Scholar] [CrossRef]
- Cao, L.; Yu, Y.; Bilke, S.; Walker, R.L.; Mayeenuddin, L.H.; Azorsa, D.O.; Yang, F.; Pineda, M.; Helman, L.J.; Meltzer, P.S. Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res. 2010, 70, 6497–6508. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, C. Identification of a new class of PAX3-FKHR target promoters: A role of the Pax3 paired box DNA binding domain. Oncogene 2007, 26, 1595–1605. [Google Scholar] [CrossRef]
- Gryder, B.E.; Yohe, M.E.; Chou, H.C.; Zhang, X.; Marques, J.; Wachtel, M.; Schaefer, B.; Sen, N.; Song, Y.; Gualtieri, A.; et al. PAX3-FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability. Cancer Discov. 2017, 7, 884–899. [Google Scholar] [CrossRef]
- Manzella, G.; Schreck, L.D.; Breunis, W.B.; Molenaar, J.; Merks, H.; Barr, F.G.; Sun, W.; Rommele, M.; Zhang, L.; Tchinda, J.; et al. Phenotypic profiling with a living biobank of primary rhabdomyosarcoma unravels disease heterogeneity and AKT sensitivity. Nat. Commun. 2020, 11, 4629. [Google Scholar] [CrossRef]
- Garraway, L.A.; Sellers, W.R. Lineage dependency and lineage-survival oncogenes in human cancer. Nat. Rev. Cancer 2006, 6, 593–602. [Google Scholar] [CrossRef]
- Mal, A.K. Histone methyltransferase Suv39h1 represses MyoD-stimulated myogenic differentiation. EMBO J. 2006, 25, 3323–3334. [Google Scholar] [CrossRef]
- Lee, M.H.; Jothi, M.; Gudkov, A.V.; Mal, A.K. Histone methyltransferase KMT1A restrains entry of alveolar rhabdomyosarcoma cells into a myogenic differentiated state. Cancer Res. 2011, 71, 3921–3931. [Google Scholar] [CrossRef]
- Chatterjee, B.; Wolff, D.W.; Jothi, M.; Mal, M.; Mal, A.K. p38alpha MAPK disables KMT1A-mediated repression of myogenic differentiation program. Skelet. Muscle 2016, 6, 28. [Google Scholar] [CrossRef]
- Phelps, M.P.; Bailey, J.N.; Vleeshouwer-Neumann, T.; Chen, E.Y. CRISPR screen identifies the NCOR/HDAC3 complex as a major suppressor of differentiation in rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 2016, 113, 15090–15095. [Google Scholar] [CrossRef]
- Sartorelli, V.; Puri, P.L.; Hamamori, Y.; Ogryzko, V.; Chung, G.; Nakatani, Y.; Wang, J.Y.; Kedes, L. Acetylation of MyoD directed by PCAF is necessary for the execution of the muscle program. Mol. Cell 1999, 4, 725–734. [Google Scholar] [CrossRef]
- Dilworth, F.J.; Seaver, K.J.; Fishburn, A.L.; Htet, S.L.; Tapscott, S.J. In vitro transcription system delineates the distinct roles of the coactivators pCAF and p300 during MyoD/E47-dependent transactivation. Proc. Natl. Acad. Sci. USA 2004, 101, 11593–11598. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Pan, W.S.; Dai, H.; Zhang, Y.; Wu, N.H.; Shen, Y.F. CARM1 activates myogenin gene via PCAF in the early differentiation of TPA-induced rhabdomyosarcoma-derived cells. J. Cell. Biochem. 2010, 110, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Yang, J.; Gao, X.; Lu, J.Y.; Zhang, Y.; Wang, K.; Cheng, M.B.; Wu, N.H.; Zhang, Y.; Wu, Z.; et al. Sequential recruitment of PCAF and BRG1 contributes to myogenin activation in 12-O-tetradecanoylphorbol-13-acetate-induced early differentiation of rhabdomyosarcoma-derived cells. J. Biol. Chem. 2007, 282, 18872–18878. [Google Scholar] [CrossRef] [PubMed]
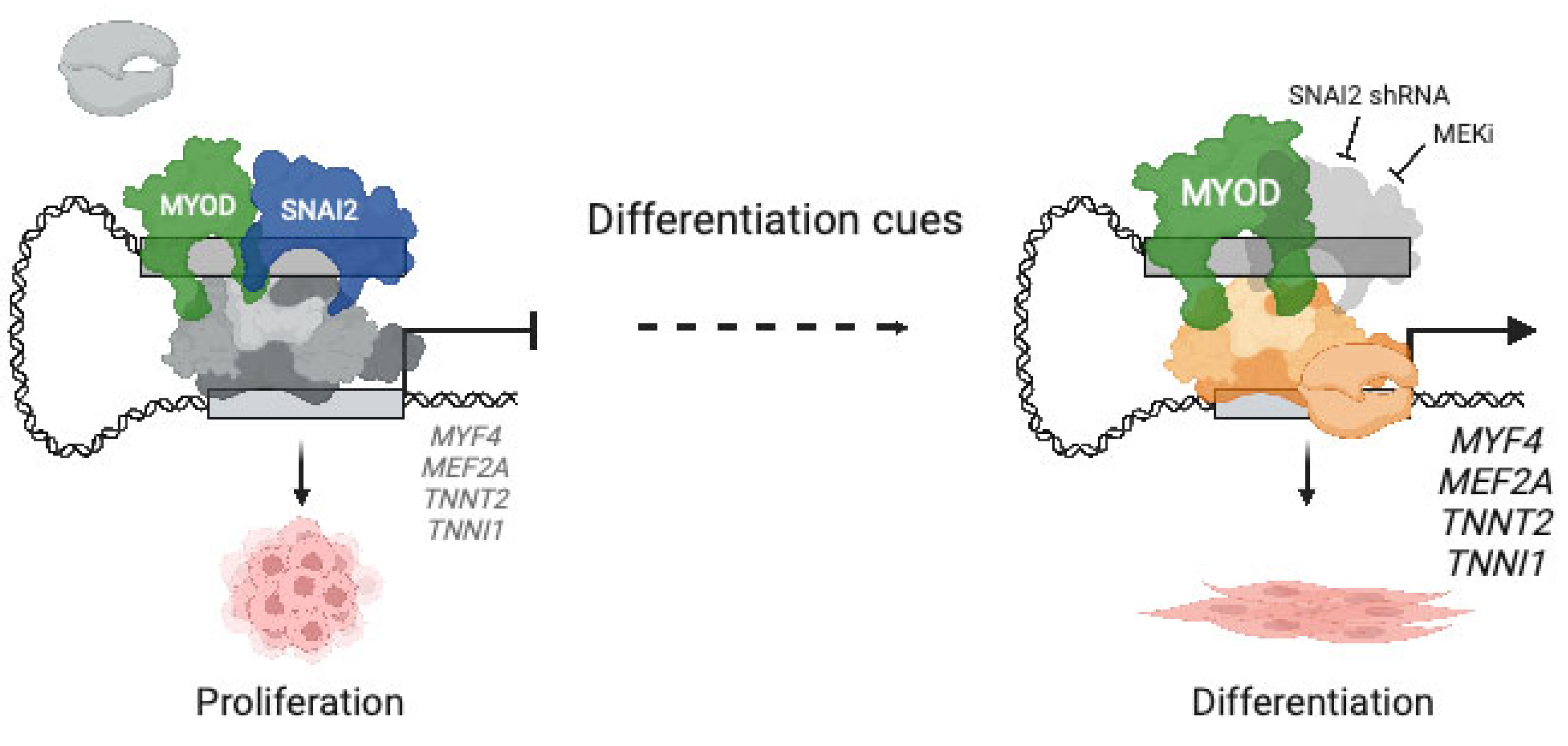
- Soleimani, V.D.; Yin, H.; Jahani-Asl, A.; Ming, H.; Kockx, C.E.; van Ijcken, W.F.; Grosveld, F.; Rudnicki, M.A. Snail regulates MyoD binding-site occupancy to direct enhancer switching and differentiation-specific transcription in myogenesis. Mol. Cell 2012, 47, 457–468. [Google Scholar] [CrossRef]
- Pomella, S.; Sreenivas, P.; Gryder, B.E.; Wang, L.; Milewski, D.; Cassandri, M.; Baxi, K.; Hensch, N.R.; Carcarino, E.; Song, Y.; et al. Interaction between SNAI2 and MYOD enhances oncogenesis and suppresses differentiation in Fusion Negative Rhabdomyosarcoma. Nat. Commun. 2021, 12, 192. [Google Scholar] [CrossRef]
- German, B.; Ellis, L. Polycomb Directed Cell Fate Decisions in Development and Cancer. Epigenomes 2022, 6, 28. [Google Scholar] [CrossRef]
- An, R.; Li, Y.Q.; Lin, Y.L.; Xu, F.; Li, M.M.; Liu, Z. EZH1/2 as targets for cancer therapy. Cancer Gene Ther. 2022, 30, 221–235. [Google Scholar] [CrossRef]
- Cho, Y.J.; Kim, S.H.; Kim, E.K.; Han, J.W.; Shin, K.H.; Hu, H.; Kim, K.S.; Choi, Y.D.; Kim, S.; Lee, Y.H.; et al. Prognostic implications of polycomb proteins ezh2, suz12, and eed1 and histone modification by H3K27me3 in sarcoma. BMC Cancer 2018, 18, 158. [Google Scholar] [CrossRef]
- Ramaglia, M.; D’Angelo, V.; Iannotta, A.; Di Pinto, D.; Pota, E.; Affinita, M.C.; Donofrio, V.; Errico, M.E.; Lombardi, A.; Indolfi, C.; et al. High EZH2 expression is correlated to metastatic disease in pediatric soft tissue sarcomas. Cancer Cell Int. 2016, 16, 59. [Google Scholar] [CrossRef]
- Karolak, M.; Tracy, I.; Shipley, J.; Walters, Z.S. Targeting EZH2 for the treatment of soft tissue sarcomas. J. Cancer Metastasis Treat. 2021, 7, 15. [Google Scholar] [CrossRef]
- Ciarapica, R.; Carcarino, E.; Adesso, L.; De Salvo, M.; Bracaglia, G.; Leoncini, P.P.; Dall’agnese, A.; Verginelli, F.; Milano, G.M.; Boldrini, R.; et al. Pharmacological inhibition of EZH2 as a promising differentiation therapy in embryonal RMS. BMC Cancer 2014, 14, 139. [Google Scholar] [CrossRef]
- Vella, S.; Pomella, S.; Leoncini, P.P.; Colletti, M.; Conti, B.; Marquez, V.E.; Strillacci, A.; Roma, J.; Gallego, S.; Milano, G.M.; et al. MicroRNA-101 is repressed by EZH2 and its restoration inhibits tumorigenic features in embryonal rhabdomyosarcoma. Clin. Epigenetics 2015, 7, 82. [Google Scholar] [CrossRef]
- Wang, H.; Garzon, R.; Sun, H.; Ladner, K.J.; Singh, R.; Dahlman, J.; Cheng, A.; Hall, B.M.; Qualman, S.J.; Chandler, D.S.; et al. NF-kappaB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell 2008, 14, 369–381. [Google Scholar] [CrossRef]
- Song, Y.L.; Yang, M.H.; Zhang, S.; Wang, H.; Kai, K.L.; Yao, C.X.; Dai, F.F.; Zhou, M.J.; Li, J.B.; Wei, Z.R.; et al. A GRIP-1-EZH2 switch binding to GATA-4 is linked to the genesis of rhabdomyosarcoma through miR-29a. Oncogene 2022, 41, 5223–5237. [Google Scholar] [CrossRef]
- Walters, Z.S.; Villarejo-Balcells, B.; Olmos, D.; Buist, T.W.; Missiaglia, E.; Allen, R.; Al-Lazikani, B.; Garrett, M.D.; Blagg, J.; Shipley, J. JARID2 is a direct target of the PAX3-FOXO1 fusion protein and inhibits myogenic differentiation of rhabdomyosarcoma cells. Oncogene 2014, 33, 1148–1157. [Google Scholar] [CrossRef]
- Wei, L.; Chiu, D.K.; Tsang, F.H.; Law, C.T.; Cheng, C.L.; Au, S.L.; Lee, J.M.; Wong, C.C.; Ng, I.O.; Wong, C.M. Histone methyltransferase G9a promotes liver cancer development by epigenetic silencing of tumor suppressor gene RARRES3. J. Hepatol. 2017, 67, 758–769. [Google Scholar] [CrossRef]
- Hua, K.T.; Wang, M.Y.; Chen, M.W.; Wei, L.H.; Chen, C.K.; Ko, C.H.; Jeng, Y.M.; Sung, P.L.; Jan, Y.H.; Hsiao, M.; et al. The H3K9 methyltransferase G9a is a marker of aggressive ovarian cancer that promotes peritoneal metastasis. Mol. Cancer 2014, 13, 189. [Google Scholar] [CrossRef]
- Casciello, F.; Al-Ejeh, F.; Kelly, G.; Brennan, D.J.; Ngiow, S.F.; Young, A.; Stoll, T.; Windloch, K.; Hill, M.M.; Smyth, M.J.; et al. G9a drives hypoxia-mediated gene repression for breast cancer cell survival and tumorigenesis. Proc. Natl. Acad. Sci. USA 2017, 114, 7077–7082. [Google Scholar] [CrossRef]
- Bhat, A.V.; Palanichamy Kala, M.; Rao, V.K.; Pignata, L.; Lim, H.J.; Suriyamurthy, S.; Chang, K.T.; Lee, V.K.; Guccione, E.; Taneja, R. Epigenetic Regulation of the PTEN-AKT-RAC1 Axis by G9a Is Critical for Tumor Growth in Alveolar Rhabdomyosarcoma. Cancer Res. 2019, 79, 2232–2243. [Google Scholar] [CrossRef]
- Shrestha, R.; Mohankumar, K.; Jin, U.H.; Martin, G.; Safe, S. The Histone Methyltransferase Gene G9A Is Regulated by Nuclear Receptor 4A1 in Alveolar Rhabdomyosarcoma Cells. Mol. Cancer Ther. 2021, 20, 612–622. [Google Scholar] [CrossRef]
- Pal, A.; Leung, J.Y.; Ang, G.C.K.; Rao, V.K.; Pignata, L.; Lim, H.J.; Hebrard, M.; Chang, K.T.; Lee, V.K.; Guccione, E.; et al. EHMT2 epigenetically suppresses Wnt signaling and is a potential target in embryonal rhabdomyosarcoma. eLife 2020, 9, e57683. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Ueda, J.; Fukuda, M.; Takeda, N.; Ohta, T.; Iwanari, H.; Sakihama, T.; Kodama, T.; Hamakubo, T.; Shinkai, Y. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes. Dev. 2005, 19, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhang, Z.; Wu, H.; Jiang, Y.; Meng, L.; Xiong, J.; Zhao, Z.; Zhou, X.; Li, J.; Li, H.; et al. Recognition of H3K9 methylation by GLP is required for efficient establishment of H3K9 methylation, rapid target gene repression, and mouse viability. Genes. Dev. 2015, 29, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Lacey, A.; Rodrigues-Hoffman, A.; Safe, S. PAX3-FOXO1A Expression in Rhabdomyosarcoma Is Driven by the Targetable Nuclear Receptor NR4A1. Cancer Res. 2017, 77, 732–741. [Google Scholar] [CrossRef]
- Nachiyappan, A.; Soon, J.L.J.; Lim, H.J.; Lee, V.K.; Taneja, R. EHMT1 promotes tumor progression and maintains stemness by regulating ALDH1A1 expression in alveolar rhabdomyosarcoma. J. Pathol. 2022, 256, 349–362. [Google Scholar] [CrossRef]
- Pless, O.; Kowenz-Leutz, E.; Knoblich, M.; Lausen, J.; Beyermann, M.; Walsh, M.J.; Leutz, A. G9a-mediated lysine methylation alters the function of CCAAT/enhancer-binding protein-beta. J. Biol. Chem. 2008, 283, 26357–26363. [Google Scholar] [CrossRef]
- Skrzypek, K.; Kusienicka, A.; Trzyna, E.; Szewczyk, B.; Ulman, A.; Konieczny, P.; Adamus, T.; Badyra, B.; Kortylewski, M.; Majka, M. SNAIL is a key regulator of alveolar rhabdomyosarcoma tumor growth and differentiation through repression of MYF5 and MYOD function. Cell Death Dis. 2018, 9, 643. [Google Scholar] [CrossRef]
- Laubscher, D.; Gryder, B.E.; Sunkel, B.D.; Andresson, T.; Wachtel, M.; Das, S.; Roschitzki, B.; Wolski, W.; Wu, X.S.; Chou, H.C.; et al. BAF complexes drive proliferation and block myogenic differentiation in fusion-positive rhabdomyosarcoma. Nat. Commun. 2021, 12, 6924. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Ramadan, F.; Saab, R.; Hussein, N.; Clezardin, P.; Cohen, P.A.; Ghayad, S.E. Non-coding RNA in rhabdomyosarcoma progression and metastasis. Front. Oncol. 2022, 12, 971174. [Google Scholar] [CrossRef]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef]
- Gebert, L.F.R.; MacRae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef]
- Liu, N.; Williams, A.H.; Kim, Y.; McAnally, J.; Bezprozvannaya, S.; Sutherland, L.B.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. An intragenic MEF2-dependent enhancer directs muscle-specific expression of microRNAs 1 and 133. Proc. Natl. Acad. Sci. USA 2007, 104, 20844–20849. [Google Scholar] [CrossRef]
- Sweetman, D.; Goljanek, K.; Rathjen, T.; Oustanina, S.; Braun, T.; Dalmay, T.; Munsterberg, A. Specific requirements of MRFs for the expression of muscle specific microRNAs, miR-1, miR-206 and miR-133. Dev. Biol. 2008, 321, 491–499. [Google Scholar] [CrossRef]
- McCarthy, J.J. MicroRNA-206: The skeletal muscle-specific myomiR. Biochim. Biophys. Acta 2008, 1779, 682–691. [Google Scholar] [CrossRef]
- Horak, M.; Novak, J.; Bienertova-Vasku, J. Muscle-specific microRNAs in skeletal muscle development. Dev. Biol. 2016, 410, 1–13. [Google Scholar] [CrossRef]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef]
- Goljanek-Whysall, K.; Mok, G.F.; Fahad Alrefaei, A.; Kennerley, N.; Wheeler, G.N.; Munsterberg, A. myomiR-dependent switching of BAF60 variant incorporation into Brg1 chromatin remodeling complexes during embryo myogenesis. Development 2014, 141, 3378–3387. [Google Scholar] [CrossRef]
- Gasparini, P.; Ferrari, A.; Casanova, M.; Limido, F.; Massimino, M.; Sozzi, G.; Fortunato, O. MiRNAs as Players in Rhabdomyosarcoma Development. Int. J. Mol. Sci. 2019, 20, 5818. [Google Scholar] [CrossRef]
- Rota, R.; Ciarapica, R.; Giordano, A.; Miele, L.; Locatelli, F. MicroRNAs in rhabdomyosarcoma: Pathogenetic implications and translational potentiality. Mol. Cancer 2011, 10, 120. [Google Scholar] [CrossRef]
- Missiaglia, E.; Shepherd, C.J.; Patel, S.; Thway, K.; Pierron, G.; Pritchard-Jones, K.; Renard, M.; Sciot, R.; Rao, P.; Oberlin, O.; et al. MicroRNA-206 expression levels correlate with clinical behaviour of rhabdomyosarcomas. Br. J. Cancer 2010, 102, 1769–1777. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.K.; Missiaglia, E.; Shields, L.; Hyde, G.; Yuan, B.; Shepherd, C.J.; Shipley, J.; Lodish, H.F. Distinct roles for miR-1 and miR-133a in the proliferation and differentiation of rhabdomyosarcoma cells. FASEB J. 2010, 24, 3427–3437. [Google Scholar] [CrossRef] [PubMed]
- Taulli, R.; Bersani, F.; Foglizzo, V.; Linari, A.; Vigna, E.; Ladanyi, M.; Tuschl, T.; Ponzetto, C. The muscle-specific microRNA miR-206 blocks human rhabdomyosarcoma growth in xenotransplanted mice by promoting myogenic differentiation. J. Clin. Investig. 2009, 119, 2366–2378. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Dong Xda, E.; Chen, X.; Wang, L.; Lu, C.; Wang, J.; Qu, J.; Tu, L. MicroRNA-1/206 targets c-Met and inhibits rhabdomyosarcoma development. J. Biol. Chem. 2009, 284, 29596–29604. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.A.; Shapiro, D.N.; Cheng, J.; Lam, P.Y.; Maas, R.L. Pax3 modulates expression of the c-Met receptor during limb muscle development. Proc. Natl. Acad. Sci. USA 1996, 93, 4213–4218. [Google Scholar] [CrossRef]
- Ginsberg, J.P.; Davis, R.J.; Bennicelli, J.L.; Nauta, L.E.; Barr, F.G. Up-regulation of MET but not neural cell adhesion molecule expression by the PAX3-FKHR fusion protein in alveolar rhabdomyosarcoma. Cancer Res. 1998, 58, 3542–3546. [Google Scholar]
- Bladt, F.; Riethmacher, D.; Isenmann, S.; Aguzzi, A.; Birchmeier, C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature 1995, 376, 768–771. [Google Scholar] [CrossRef]
- Maina, F.; Casagranda, F.; Audero, E.; Simeone, A.; Comoglio, P.M.; Klein, R.; Ponzetto, C. Uncoupling of Grb2 from the Met receptor in vivo reveals complex roles in muscle development. Cell 1996, 87, 531–542. [Google Scholar] [CrossRef]
- Chen, J.F.; Tao, Y.; Li, J.; Deng, Z.; Yan, Z.; Xiao, X.; Wang, D.Z. microRNA-1 and microRNA-206 regulate skeletal muscle satellite cell proliferation and differentiation by repressing Pax7. J. Cell Biol. 2010, 190, 867–879. [Google Scholar] [CrossRef]
- Hanna, J.A.; Garcia, M.R.; Go, J.C.; Finkelstein, D.; Kodali, K.; Pagala, V.; Wang, X.; Peng, J.; Hatley, M.E. PAX7 is a required target for microRNA-206-induced differentiation of fusion-negative rhabdomyosarcoma. Cell Death Dis. 2016, 7, e2256. [Google Scholar] [CrossRef]
- Sun, Y.; Ge, Y.; Drnevich, J.; Zhao, Y.; Band, M.; Chen, J. Mammalian target of rapamycin regulates miRNA-1 and follistatin in skeletal myogenesis. J. Cell Biol. 2010, 189, 1157–1169. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L.; Pang, Y.; Song, L.; Shang, H.; Li, Z.; Liu, Q.; Zhang, Y.; Wang, X.; Li, Q.; et al. MicroRNA-29 family inhibits rhabdomyosarcoma formation and progression by regulating GEFT function. Am. J. Transl. Res. 2020, 12, 1136–1154. [Google Scholar]
- Huang, H.J.; Liu, J.; Hua, H.; Li, S.E.; Zhao, J.; Yue, S.; Yu, T.T.; Jin, Y.C.; Cheng, S.Y. MiR-214 and N-ras regulatory loop suppresses rhabdomyosarcoma cell growth and xenograft tumorigenesis. Oncotarget 2014, 5, 2161–2175. [Google Scholar] [CrossRef]
- Diao, Y.; Guo, X.; Jiang, L.; Wang, G.; Zhang, C.; Wan, J.; Jin, Y.; Wu, Z. miR-203, a tumor suppressor frequently down-regulated by promoter hypermethylation in rhabdomyosarcoma. J. Biol. Chem. 2014, 289, 529–539. [Google Scholar] [CrossRef]
- Megiorni, F.; Cialfi, S.; McDowell, H.P.; Felsani, A.; Camero, S.; Guffanti, A.; Pizer, B.; Clerico, A.; De Grazia, A.; Pizzuti, A.; et al. Deep Sequencing the microRNA profile in rhabdomyosarcoma reveals down-regulation of miR-378 family members. BMC Cancer 2014, 14, 880. [Google Scholar] [CrossRef]
- Skrzypek, K.; Kot, M.; Konieczny, P.; Nieszporek, A.; Kusienicka, A.; Lasota, M.; Bobela, W.; Jankowska, U.; Kedracka-Krok, S.; Majka, M. SNAIL Promotes Metastatic Behavior of Rhabdomyosarcoma by Increasing EZRIN and AKT Expression and Regulating MicroRNA Networks. Cancers 2020, 12, 1870. [Google Scholar] [CrossRef]
- Dmitriev, P.; Barat, A.; Polesskaya, A.; O’Connell, M.J.; Robert, T.; Dessen, P.; Walsh, T.A.; Lazar, V.; Turki, A.; Carnac, G.; et al. Simultaneous miRNA and mRNA transcriptome profiling of human myoblasts reveals a novel set of myogenic differentiation-associated miRNAs and their target genes. BMC Genomics 2013, 14, 265. [Google Scholar] [CrossRef]
- Winkle, M.; El-Daly, S.M.; Fabbri, M.; Calin, G.A. Noncoding RNA therapeutics-challenges and potential solutions. Nat. Rev. Drug. Discov. 2021, 20, 629–651. [Google Scholar] [CrossRef]
- de The, H. Differentiation therapy revisited. Nat. Rev. Cancer 2018, 18, 117–127. [Google Scholar] [CrossRef]
- Anderson, J.; Gordon, A.; Pritchard-Jones, K.; Shipley, J. Genes, chromosomes, and rhabdomyosarcoma. Genes. Chromosomes Cancer 1999, 26, 275–285. [Google Scholar] [CrossRef]
- Coffin, C.M.; Rulon, J.; Smith, L.; Bruggers, C.; White, F.V. Pathologic features of rhabdomyosarcoma before and after treatment: A clinicopathologic and immunohistochemical analysis. Mod. Pathol. 1997, 10, 1175–1187. [Google Scholar] [PubMed]
- Jeyaraju, M.; Macatangay, R.A.; Munchel, A.T.; York, T.A.; Montgomery, E.A.; Kallen, M.E. Embryonal Rhabdomyosarcoma with Posttherapy Cytodifferentiation and Aggressive Clinical Course. Case Rep. Pathol. 2021, 2021, 1800854. [Google Scholar] [CrossRef] [PubMed]
- Saab, R.; Bills, J.L.; Miceli, A.P.; Anderson, C.M.; Khoury, J.D.; Fry, D.W.; Navid, F.; Houghton, P.J.; Skapek, S.X. Pharmacologic inhibition of cyclin-dependent kinase 4/6 activity arrests proliferation in myoblasts and rhabdomyosarcoma-derived cells. Mol. Cancer Ther. 2006, 5, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Edmonson, M.N.; Wilkinson, M.R.; Patel, A.; Wu, G.; Liu, Y.; Li, Y.; Zhang, Z.; Rusch, M.C.; Parker, M.; et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat. Genet. 2016, 48, 4–6. [Google Scholar] [CrossRef]
- Mueller, S.; Haas-Kogan, D.A. WEE1 Kinase As a Target for Cancer Therapy. J. Clin. Oncol. 2015, 33, 3485–3487. [Google Scholar] [CrossRef]
- Yohe, M.E.; Gryder, B.E.; Shern, J.F.; Song, Y.K.; Chou, H.C.; Sindiri, S.; Mendoza, A.; Patidar, R.; Zhang, X.; Guha, R.; et al. MEK inhibition induces MYOG and remodels super-enhancers in RAS-driven rhabdomyosarcoma. Sci. Transl. Med. 2018, 10, eaan4470. [Google Scholar] [CrossRef]
- Hebron, K.E.; Wan, X.; Roth, J.S.; Liewehr, D.J.; Sealover, N.E.; Frye, W.J.E.; Kim, A.; Stauffer, S.; Perkins, O.L.; Sun, W.; et al. The combination of trametinib and ganitumab is effective in RAS-mutated PAX-fusion negative rhabdomyosarcoma models. Clin. Cancer Res. 2022, 29, 472–487. [Google Scholar] [CrossRef]
- Garcia, N.; Del Pozo, V.; Yohe, M.E.; Goodwin, C.M.; Shackleford, T.J.; Wang, L.; Baxi, K.; Chen, Y.; Rogojina, A.T.; Zimmerman, S.M.; et al. Vertical Inhibition of the RAF-MEK-ERK Cascade Induces Myogenic Differentiation, Apoptosis, and Tumor Regression in H/NRAS(Q61X) Mutant Rhabdomyosarcoma. Mol. Cancer Ther. 2022, 21, 170–183. [Google Scholar] [CrossRef]
- Tenente, I.M.; Hayes, M.N.; Ignatius, M.S.; McCarthy, K.; Yohe, M.; Sindiri, S.; Gryder, B.; Oliveira, M.L.; Ramakrishnan, A.; Tang, Q.; et al. Myogenic regulatory transcription factors regulate growth in rhabdomyosarcoma. eLife 2017, 6, e19214. [Google Scholar] [CrossRef]
- Hsu, J.Y.; Danis, E.P.; Nance, S.; O’Brien, J.H.; Gustafson, A.L.; Wessells, V.M.; Goodspeed, A.E.; Talbot, J.C.; Amacher, S.L.; Jedlicka, P.; et al. SIX1 reprograms myogenic transcription factors to maintain the rhabdomyosarcoma undifferentiated state. Cell Rep. 2022, 38, 110323. [Google Scholar] [CrossRef]
- Zheng, C.; Liu, M.; Fan, H. Targeting complexes of super-enhancers is a promising strategy for cancer therapy. Oncol. Lett. 2020, 20, 2557–2566. [Google Scholar] [CrossRef]
- Bharathy, N.; Suriyamurthy, S.; Rao, V.K.; Ow, J.R.; Lim, H.J.; Chakraborty, P.; Vasudevan, M.; Dhamne, C.A.; Chang, K.T.; Min, V.L.; et al. P/CAF mediates PAX3-FOXO1-dependent oncogenesis in alveolar rhabdomyosarcoma. J. Pathol. 2016, 240, 269–281. [Google Scholar] [CrossRef]
- Ciarapica, R.; De Salvo, M.; Carcarino, E.; Bracaglia, G.; Adesso, L.; Leoncini, P.P.; Dall’Agnese, A.; Walters, Z.S.; Verginelli, F.; De Sio, L.; et al. The Polycomb group (PcG) protein EZH2 supports the survival of PAX3-FOXO1 alveolar rhabdomyosarcoma by repressing FBXO32 (Atrogin1/MAFbx). Oncogene 2014, 33, 4173–4184. [Google Scholar] [CrossRef]
- Zoroddu, S.; Marchesi, I.; Bagella, L. PRC2: An epigenetic multiprotein complex with a key role in the development of rhabdomyosarcoma carcinogenesis. Clin. Epigenetics 2021, 13, 156. [Google Scholar] [CrossRef]
- Chi, S.N.; Bourdeaut, F.; Laetsch, T.W.; Fouladi, M.; Macy, M.E.; Makin, G.; Shukla, N.N.; Wetmore, C.; Margol, A.S.; Casanova, M.; et al. Phase I study of tazemetostat, an enhancer of zeste homolog-2 inhibitor, in pediatric pts with relapsed/refractory integrase interactor 1-negative tumors. JCO 2020, 38, 10525. [Google Scholar] [CrossRef]
- Romanelli, A.; Stazi, G.; Fioravanti, R.; Zwergel, C.; Di Bello, E.; Pomella, S.; Perrone, C.; Battistelli, C.; Strippoli, R.; Tripodi, M.; et al. Design of First-in-Class Dual EZH2/HDAC Inhibitor: Biochemical Activity and Biological Evaluation in Cancer Cells. ACS Med. Chem. Lett. 2020, 11, 977–983. [Google Scholar] [CrossRef]
- Qi, S.M.; Dong, J.; Xu, Z.Y.; Cheng, X.D.; Zhang, W.D.; Qin, J.J. PROTAC: An Effective Targeted Protein Degradation Strategy for Cancer Therapy. Front. Pharmacol. 2021, 12, 692574. [Google Scholar] [CrossRef]
- Crose, L.E.; Etheridge, K.T.; Chen, C.; Belyea, B.; Talbot, L.J.; Bentley, R.C.; Linardic, C.M. FGFR4 blockade exerts distinct antitumorigenic effects in human embryonal versus alveolar rhabdomyosarcoma. Clin. Cancer Res. 2012, 18, 3780–3790. [Google Scholar] [CrossRef]
- Lagha, M.; Kormish, J.D.; Rocancourt, D.; Manceau, M.; Epstein, J.A.; Zaret, K.S.; Relaix, F.; Buckingham, M.E. Pax3 regulation of FGF signaling affects the progression of embryonic progenitor cells into the myogenic program. Genes. Dev. 2008, 22, 1828–1837. [Google Scholar] [CrossRef]
- Engert, J.C.; Berglund, E.B.; Rosenthal, N. Proliferation precedes differentiation in IGF-I-stimulated myogenesis. J. Cell Biol. 1996, 135, 431–440. [Google Scholar] [CrossRef]
- Bohm, M.; Wachtel, M.; Marques, J.G.; Streiff, N.; Laubscher, D.; Nanni, P.; Mamchaoui, K.; Santoro, R.; Schafer, B.W. Helicase CHD4 is an epigenetic coregulator of PAX3-FOXO1 in alveolar rhabdomyosarcoma. J. Clin. Investig. 2016, 126, 4237–4249. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.G.; Gryder, B.E.; Pavlovic, B.; Chung, Y.; Ngo, Q.A.; Frommelt, F.; Gstaiger, M.; Song, Y.; Benischke, K.; Laubscher, D.; et al. NuRD subunit CHD4 regulates super-enhancer accessibility in rhabdomyosarcoma and represents a general tumor dependency. eLife 2020, 9, e54993. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Abu-Zaid, A.; Jin, H.; Fang, J.; Wu, Q.; Wang, T.; Feng, H.; Quarni, W.; Shao, Y.; Maxham, L.; et al. Targeting KDM4 for treating PAX3-FOXO1-driven alveolar rhabdomyosarcoma. Sci. Transl. Med. 2022, 14, eabq2096. [Google Scholar] [CrossRef] [PubMed]
- Walters, Z.S.; Aladowicz, E.; Villarejo-Balcells, B.; Nugent, G.; Selfe, J.L.; Eve, P.; Blagg, J.; Rossanese, O.; Shipley, J. Role for the Histone Demethylase KDM4B in Rhabdomyosarcoma via CDK6 and CCNA2: Compensation by KDM4A and Apoptotic Response of Targeting Both KDM4B and KDM4A. Cancers 2021, 13, 1734. [Google Scholar] [CrossRef] [PubMed]
- Richter, G.H.S.; Hensel, T.; Schmidt, O.; Saratov, V.; von Heyking, K.; Becker-Dettling, F.; Prexler, C.; Yen, H.Y.; Steiger, K.; Fulda, S.; et al. Combined Inhibition of Epigenetic Readers and Transcription Initiation Targets the EWS-ETS Transcriptional Program in Ewing Sarcoma. Cancers 2020, 12, 304. [Google Scholar] [CrossRef]
- Bukowinski, A.; Chang, B.; Reid, J.M.; Liu, X.; Minard, C.G.; Trepel, J.B.; Lee, M.J.; Fox, E.; Weigel, B.J. A phase 1 study of entinostat in children and adolescents with recurrent or refractory solid tumors, including CNS tumors: Trial ADVL1513, Pediatric Early Phase-Clinical Trial Network (PEP-CTN). Pediatr. Blood Cancer 2021, 68, e28892. [Google Scholar] [CrossRef]
- Fouladi, M.; Park, J.R.; Stewart, C.F.; Gilbertson, R.J.; Schaiquevich, P.; Sun, J.; Reid, J.M.; Ames, M.M.; Speights, R.; Ingle, A.M.; et al. Pediatric phase I trial and pharmacokinetic study of vorinostat: A Children’s Oncology Group phase I consortium report. J. Clin. Oncol. 2010, 28, 3623–3629. [Google Scholar] [CrossRef]
- van Tilburg, C.M.; Milde, T.; Witt, R.; Ecker, J.; Hielscher, T.; Seitz, A.; Schenk, J.P.; Buhl, J.L.; Riehl, D.; Fruhwald, M.C.; et al. Phase I/II intra-patient dose escalation study of vorinostat in children with relapsed solid tumor, lymphoma, or leukemia. Clin. Epigenetics 2019, 11, 188. [Google Scholar] [CrossRef]
- Stankovic, T.; Dinic, J.; Podolski-Renic, A.; Musso, L.; Buric, S.S.; Dallavalle, S.; Pesic, M. Dual Inhibitors as a New Challenge for Cancer Multidrug Resistance Treatment. Curr. Med. Chem. 2019, 26, 6074–6106. [Google Scholar] [CrossRef]
- Bouffet, E.; Geoerger, B.; Moertel, C.; Whitlock, J.A.; Aerts, I.; Hargrave, D.; Osterloh, L.; Tan, E.; Choi, J.; Russo, M.; et al. Efficacy and Safety of Trametinib Monotherapy or in Combination with Dabrafenib in Pediatric BRAF V600–Mutant Low-Grade Glioma. J. Clin. Oncol. 2023, 41, 664–674. [Google Scholar] [CrossRef]
- Geoerger, B.; Hargrave, D.; Thomas, F.; Ndiaye, A.; Frappaz, D.; Andreiuolo, F.; Varlet, P.; Aerts, I.; Riccardi, R.; Jaspan, T.; et al. Innovative Therapies for Children with Cancer pediatric phase I study of erlotinib in brainstem glioma and relapsing/refractory brain tumors. Neuro-Oncology 2011, 13, 109–118. [Google Scholar] [CrossRef]
- Weigel, B.; Malempati, S.; Reid, J.M.; Voss, S.D.; Cho, S.Y.; Chen, H.X.; Krailo, M.; Villaluna, D.; Adamson, P.C.; Blaney, S.M. Phase 2 trial of cixutumumab in children, adolescents, and young adults with refractory solid tumors: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2014, 61, 452–456. [Google Scholar] [CrossRef]
- Akshintala, S.; Bernstein, D.; Glod, J.; Kaplan, R.N.; Shern, J.F.; Yohe, M.E.; Gross, A.M.; Derdak, J.; Dombi, E.; Palacio-Yance, I.; et al. Results of a phase I trial of ganitumab plus dasatinib in patients with rhabdomyosarcoma (RMS). J. Clin. Oncol. 2022, 40, 11561. [Google Scholar] [CrossRef]
- Akshintala, S.; Widemann, B.C.; Barkauskas, D.A.; Hall, D.; Reid, J.M.; Voss, S.D.; Kim, A.; Fox, E.; Weigel, B. Phase 2 trial of cabozantinib in children and young adults with refractory sarcomas, Wilms tumor, and rare tumors: Children’s Oncology Group Study (ADVL1622). J. Clin. Oncol. 2021, 39, 10010. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pomella, S.; Danielli, S.G.; Alaggio, R.; Breunis, W.B.; Hamed, E.; Selfe, J.; Wachtel, M.; Walters, Z.S.; Schäfer, B.W.; Rota, R.; et al. Genomic and Epigenetic Changes Drive Aberrant Skeletal Muscle Differentiation in Rhabdomyosarcoma. Cancers 2023, 15, 2823. https://doi.org/10.3390/cancers15102823
Pomella S, Danielli SG, Alaggio R, Breunis WB, Hamed E, Selfe J, Wachtel M, Walters ZS, Schäfer BW, Rota R, et al. Genomic and Epigenetic Changes Drive Aberrant Skeletal Muscle Differentiation in Rhabdomyosarcoma. Cancers. 2023; 15(10):2823. https://doi.org/10.3390/cancers15102823
Chicago/Turabian StylePomella, Silvia, Sara G. Danielli, Rita Alaggio, Willemijn B. Breunis, Ebrahem Hamed, Joanna Selfe, Marco Wachtel, Zoe S. Walters, Beat W. Schäfer, Rossella Rota, and et al. 2023. "Genomic and Epigenetic Changes Drive Aberrant Skeletal Muscle Differentiation in Rhabdomyosarcoma" Cancers 15, no. 10: 2823. https://doi.org/10.3390/cancers15102823
APA StylePomella, S., Danielli, S. G., Alaggio, R., Breunis, W. B., Hamed, E., Selfe, J., Wachtel, M., Walters, Z. S., Schäfer, B. W., Rota, R., Shipley, J. M., & Hettmer, S. (2023). Genomic and Epigenetic Changes Drive Aberrant Skeletal Muscle Differentiation in Rhabdomyosarcoma. Cancers, 15(10), 2823. https://doi.org/10.3390/cancers15102823