
Dynamic NIR Fluorescence Imaging and Machine Learning Framework for Stratifying High vs. Low Notch-Dll4 Expressing Host Microenvironment in Triple-Negative Breast Cancer
 , ,
, ,  , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Hypothesis and Objective
3. Materials and Methods
3.1. Animals
3.2. Cell Culture and Triple-Negative Breast Cancer Xenografts
3.3. In Vivo NIR Fluorescence Imaging
3.4. Denoising and Motion Correction
3.5. Principal Component Analysis for Extraction of Spatial Patterns of Internal Organs
3.6. PCA Ranking Tumor Detection
3.7. Video Processing
3.8. Peak and Latency Estimation
3.9. Feature Design
3.10. Classification Algorithms and Feature Selection
3.11. Primary Classification
3.12. Congenic Pair Selection
3.13. Feature Selection and Secondary Classification
3.14. Data Augmentation
3.15. Training and Testing Dataset
3.16. Statistical Analysis
3.17. Data Availability
4. Results and Discussion
4.1. Dynamic Contrast-Enhanced NIR Fluorescence Imaging and Tumor Detection
4.2. Dll4 and Its Effect on the NIR Time Series
4.3. Primary Classification and Congenic Dissimilarity
4.4. Feature Selection
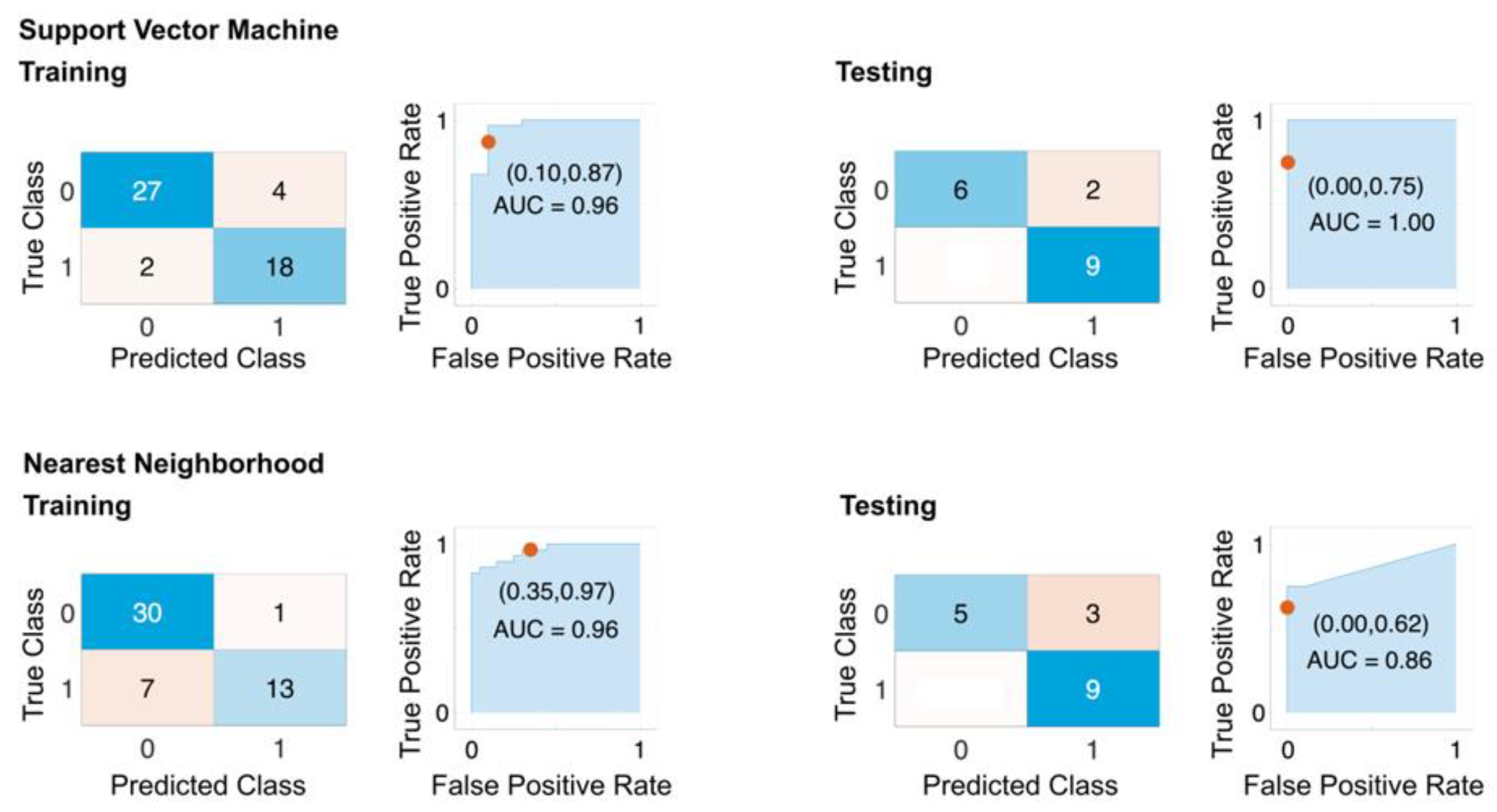
4.5. Performance of the Classification Models Based on the Selected Features
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pasha, N.; Turner, N.C. Understanding and overcoming tumor heterogeneity in metastatic breast cancer treatment. Nat. Cancer 2021, 2, 680–692. [Google Scholar] [CrossRef]
- Marisa, L.; de Reyniès, A.; Duval, A.; Selves, J.; Gaub, M.P.; Vescovo, L.; Etienne-Grimaldi, M.-C.; Schiappa, R.; Guenot, D.; Ayadi, M.; et al. Gene Expression Classification of Colon Cancer into Molecular Subtypes: Characterization, Validation, and Prognostic Value. PLoS Med. 2013, 10, e1001453. [Google Scholar] [CrossRef]
- Sohn, B.H.; Hwang, J.-E.; Jang, H.-J.; Lee, H.-S.; Oh, S.C.; Shim, J.-J.; Lee, K.-W.; Kim, E.H.; Yim, S.Y.; Lee, S.H.; et al. Clinical Significance of Four Molecular Subtypes of Gastric Cancer Identified by The Cancer Genome Atlas Project. Clin. Cancer Res. 2017, 23, 4441–4449. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Pietenpol, J.A. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J. Pathol. 2014, 232, 142–150. [Google Scholar] [CrossRef]
- Belli, C.; Trapani, D.; Viale, G.; D’Amico, P.; Duso, B.A.; Della Vigna, P.; Orsi, F.; Curigliano, G. Targeting the microenvironment in solid tumors. Cancer Treat. Rev. 2018, 65, 22–32. [Google Scholar] [CrossRef]
- Bernard, J.J.; Wellberg, E.A. The Tumor Promotional Role of Adipocytes in the Breast Cancer Microenvironment and Macroenvironment. Am. J. Pathol. 2021, 191, 1342–1352. [Google Scholar] [CrossRef]
- Jagtap, J.; Sharma, G.; Parchur, A.K.; Gogineni, V.; Bergom, C.; White, S.; Flister, M.J.; Joshi, A. Methods for detecting host genetic modifiers of tumor vascular function using dynamic near-infrared fluorescence imaging. Biomed. Opt. Express 2018, 9, 543–556. [Google Scholar] [CrossRef]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef]
- Economopoulou, P.; Kotsantis, I.; Psyrri, A. Tumor Microenvironment and Immunotherapy Response in Head and Neck Cancer. Cancers 2020, 12, 3377. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019, 79, 4557–4567. [Google Scholar] [CrossRef]
- Pitt, J.M.; Marabelle, A.; Eggermont, A.; Soria, J.-C.; Kroemer, G.; Zitvogel, L. Targeting the tumor microenvironment: Removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. 2016, 27, 1482–1492. [Google Scholar] [CrossRef]
- Ungefroren, H.; Sebens, S.; Seidl, D.; Lehnert, H.; Hass, R. Interaction of tumor cells with the microenvironment. Cell Commun. Signal. 2011, 9, 18. [Google Scholar] [CrossRef]
- Stamatelos, S.K.; Bhargava, A.; Kim, E.; Popel, A.S.; Pathak, A.P. Tumor Ensemble-Based Modeling and Visualization of Emergent Angiogenic Heterogeneity in Breast Cancer. Sci. Rep. 2019, 9, 5276. [Google Scholar] [CrossRef]
- Stamatelos, S.; Kim, E.; Pathak, A.; Popel, A. Image-based Characterization of Functional and Structural Heterogeneity of Tumor Xenografts using Blood Flow modeling, Oxygenation Modeling and Multivariate Analysis. FASEB J. 2015, 29, 787.11. [Google Scholar] [CrossRef]
- Jain, R.K. Normalizing Tumor Microenvironment to Treat Cancer: Bench to Bedside to Biomarkers. J. Clin. Oncol. 2013, 31, 2205–2218. [Google Scholar] [CrossRef]
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C569–C579. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef]
- Willett, C.G.; Duda, D.G.; di Tomaso, E.; Boucher, Y.; Ancukiewicz, M.; Sahani, D.V.; Lahdenranta, J.; Chung, D.C.; Fischman, A.J.; Lauwers, G.Y.; et al. Efficacy, Safety, and Biomarkers of Neoadjuvant Bevacizumab, Radiation Therapy, and Fluorouracil in Rectal Cancer: A Multidisciplinary Phase II Study. J. Clin. Oncol. 2009, 27, 3020–3026. [Google Scholar] [CrossRef]
- Raut, C.P.; Boucher, Y.; Duda, D.G.; Morgan, J.A.; Quek, R.; Ancukiewicz, M.; Lahdenranta, J.; Eder, J.P.; Demetri, G.D.; Jain, R.K. Effects of Sorafenib on Intra-Tumoral Interstitial Fluid Pressure and Circulating Biomarkers in Patients with Refractory Sarcomas (NCI Protocol 6948). PLoS ONE 2012, 7, e26331. [Google Scholar] [CrossRef]
- Eichler, A.F.; Chung, E.; Kodack, D.P.; Loeffler, J.S.; Fukumura, D.; Jain, R.K. The biology of brain metastases—Translation to new therapies. Nat. Rev. Clin. Oncol. 2011, 8, 344–356. [Google Scholar] [CrossRef]
- Hobbs, S.K.; Monsky, W.L.; Yuan, F.; Roberts, W.G.; Griffith, L.; Torchilin, V.P.; Jain, R.K. Regulation of transport pathways in tumor vessels: Role of tumor type and microenvironment. Proc. Natl. Acad. Sci. USA 1998, 95, 4607–4612. [Google Scholar] [CrossRef]
- Lee, I.; Demhartner, T.J.; Boucher, Y.; Jain, R.K.; Intaglietta, M. Effect of Hemodilution and Resuscitation on Tumor Interstitial Fluid Pressure, Blood Flow, and Oxygenation. Microvasc. Res. 1994, 48, 1–12. [Google Scholar] [CrossRef]
- Vakoc, B.J.; Lanning, R.M.; A Tyrrell, J.; Padera, T.; A Bartlett, L.; Stylianopoulos, T.; Munn, L.L.; Tearney, G.J.; Fukumura, D.; Jain, R.K.; et al. Three-dimensional microscopy of the tumor microenvironment in vivo using optical frequency domain imaging. Nat. Med. 2009, 15, 1219–1223. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef]
- Kazerounian, S.; Lawler, J. Integration of pro- and anti-angiogenic signals by endothelial cells. J. Cell Commun. Signal. 2017, 12, 171–179. [Google Scholar] [CrossRef]
- Brzozowa-Zasada, M. The role of Notch ligand, Delta-like ligand 4 (DLL4), in cancer angiogenesis—Implications for therapy. Eur. Surg.-Acta Chir. Austriaca 2021, 53, 274–280. [Google Scholar] [CrossRef]
- Canavese, M.; Altruda, F.; Ruzicka, T.; Schauber, J. Vascular endothelial growth factor (VEGF) in the pathogenesis of psoriasis—A possible target for novel therapies? J. Dermatol. Sci. 2010, 58, 171–176. [Google Scholar] [CrossRef]
- Akil, A.; Gutiérrez-García, A.K.; Guenter, R.; Rose, J.B.; Beck, A.W.; Chen, H.; Ren, B. Notch Signaling in Vascular Endothelial Cells, Angiogenesis, and Tumor Progression: An Update and Prospective. Front. Cell Dev. Biol. 2021, 9, 642352. [Google Scholar] [CrossRef]
- Hellström, M.; Phng, L.-K.; Hofmann, J.J.; Wallgard, E.; Coultas, L.; Lindblom, P.; Alva, J.; Nilsson, A.-K.; Karlsson, L.; Gaiano, N.; et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 2007, 445, 776–780. [Google Scholar] [CrossRef]
- Noguera-Troise, I.; Daly, C.; Papadopoulos, N.J.; Coetzee, S.; Boland, P.; Gale, N.W.; Lin, H.C.; Yancopoulos, G.D.; Thurston, G. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 2006, 444, 1032–1037. [Google Scholar] [CrossRef]
- Ding, Z.; Zu, S.; Gu, J. Evaluating the molecule-based prediction of clinical drug responses in cancer. Bioinformatics 2016, 32, 2891–2895. [Google Scholar] [CrossRef] [PubMed]
- Zohny, S.F.; Zamzami, M.A.; Al-Malki, A.L.; Trabulsi, N.H. Highly Expressed DLL4 and JAG1: Their Role in Incidence of Breast Cancer Metastasis. Arch. Med. Res. 2020, 51, 145–152. [Google Scholar] [CrossRef]
- A Charles, N.; Holland, E.C. The perivascular niche microenvironment in brain tumor progression. Cell Cycle 2010, 9, 3084–3093. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.C.; Ceresa, B.P. Cellular localization of the activated EGFR determines its effect on cell growth in MDA-MB-468 cells. Exp. Cell Res. 2008, 314, 3415–3425. [Google Scholar] [CrossRef] [PubMed]
- Scehnet, J.S.; Jiang, W.; Kumar, S.R.; Krasnoperov, V.; Trindade, A.; Benedito, R.; Djokovic, D.; Borges, C.; Ley, E.J.; Duarte, A.; et al. Inhibition of Dll4-mediated signaling induces proliferation of immature vessels and results in poor tissue perfusion. Blood 2007, 109, 4753–4760. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Lu, C.; Dong, H.H.; Huang, J.; Shen, D.-Y.; Stone, R.L.; Nick, A.M.; Shahzad, M.M.; Mora, E.; Jennings, N.B.; et al. Biological Roles of the Delta Family Notch Ligand Dll4 in Tumor and Endothelial Cells in Ovarian Cancer. Cancer Res 2011, 71, 6030–6039. [Google Scholar] [CrossRef]
- Comunanza, V.; Bussolino, F. Therapy for Cancer: Strategy of Combining Anti-Angiogenic and Target Therapies. Front. Cell Dev. Biol. 2017, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Jagtap, J.; Parchur, A.K.; Gogineni, V.R.; Ran, S.; Bergom, C.; White, S.B.; Flister, M.J.; Joshi, A. Heritable modifiers of the tumor microenvironment influence nanoparticle uptake, distribution and response to photothermal therapy. Theranostics 2020, 10, 5368–5383. [Google Scholar] [CrossRef] [PubMed]
- Flister, M.J.; Tsaih, S.-W.; Stoddard, A.; Plasterer, C.; Jagtap, J.; Parchur, A.K.; Sharma, G.; Prisco, A.R.; Lemke, A.; Murphy, D.; et al. Host genetic modifiers of nonproductive angiogenesis inhibit breast cancer. Breast Cancer Res. Treat. 2017, 165, 53–64. [Google Scholar] [CrossRef]
- Flister, M.J.; Endres, B.T.; Rudemiller, N.; Sarkis, A.B.; Santarriaga, S.; Roy, I.; Lemke, A.; Geurts, A.M.; Moreno, C.; Ran, S.; et al. CXM: A New Tool for Mapping Breast Cancer Risk in the Tumor Microenvironment. Cancer Res 2014, 74, 6419–6429. [Google Scholar] [CrossRef]
- Goh, V.; Padhani, A.; Rasheed, S. Functional imaging of colorectal cancer angiogenesis. Lancet Oncol. 2007, 8, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Choi, K.; Ryu, S.-W.; Lee, J.; Choi, C. Dynamic fluorescence imaging for multiparametric measurement of tumor vasculature. J. Biomed. Opt. 2011, 16, 046008. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Godavarty, A. Near-Infrared Fluorescence-Enhanced Optical Tomography. BioMed Res. Int. 2016, 2016, 5040814. [Google Scholar] [CrossRef]
- Meng, B.; Strawbridge, R.R.; Tichauer, K.M.; Samkoe, K.S.; Davis, S.C. Estimating paired-agent uptake in altered tumor vasculature using MRI-coupled fluorescence tomography. Proc. SPIE Int. Soc. Opt. Eng. 2020, 11216, 112160U. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.W.; Hohenstein, N.; Carpenter, C.; Pattison, A.J.; Morin, O.; Valdes, G.; Sanchez, C.T.; Perkins, J.; Solberg, T.D.; Yom, S.S. Artificial Intelligence-Guided Prediction of Dental Doses Before Planning of Radiation Therapy for Oropharyngeal Cancer: Technical Development and Initial Feasibility of Implementation. Adv. Radiat. Oncol. 2022, 7, 100886. [Google Scholar] [CrossRef] [PubMed]
- Brodin, P.; Shukla, P.; Hauze, M.; Shulte, L.; Carpenter, C.; Kumar, R.; Bodner, W.; Kalnicki, S.; Garg, M.; Tomé, W. PD-0322 Artificial intelligence organ-at-risk dose prediction for high-risk prostate cancer IMRT. Radiother. Oncol. 2022, 170, S284–S285. [Google Scholar] [CrossRef]
- Zhuk, S.; Epperlein, J.P.; Nair, R.; Thirupati, S.; Aonghusa, P.M.; Cahill, R.; O’Shea, D. Perfusion Quantification from Endoscopic Videos: Learning to Read Tumor Signatures. Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics). arXiv 2006. [Google Scholar] [CrossRef]
- Epperlein, J.P.; Zayats, M.; Tirupathi, S.; Zhuk, S.; Tchrakian, T.; Mac Aonghusa, P.; O’Shea, D.F.; Hardy, N.P.; Dalli, J.; Cahill, R.A. Practical Perfusion Quantification in Multispectral Endoscopic Video: Using the Minutes after ICG Administration to Assess Tissue Pathology. AMIA Annu. Symp. Proc. 2021, 2021, 428. [Google Scholar]
- Houston, J.P. Near Infrared Optical Lymphography for Cancer Diagnostics. Ph.D. Dissertation, Texas A&M University, Canyon, TX, USA, 2007. Available online: https://oaktrust.library.tamu.edu/handle/1969.1/4807 (accessed on 14 December 2022).
- Flister, M.J.; Hoffman, M.J.; Reddy, P.; Jacob, H.J.; Moreno, C. Congenic Mapping and Sequence Analysis of the Renin Locus. Hypertension 2013, 61, 850–856. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving Bioscience Research Reporting: The ARRIVE Guidelines for Reporting Animal Research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef]
- Rat Research Model Service Center|Genomic Sciences and Precision Medicine Center|Medical College of Wisconsin. Available online: https://www.mcw.edu/departments/genomic-sciences-and-precision-medicine-center-gspmc/about-us/what-we-do/research/rat-research-model-service-center (accessed on 7 November 2022).
- Flister, M.J.; Joshi, A.; Bergom, C.; Rui, H. Mapping Mammary Tumor Traits in the Rat. Methods Mol. Biol. 2019, 2018, 249–267. [Google Scholar] [CrossRef] [PubMed]
- Miles, K.M.; Seshadri, M.; Ciamporcero, E.; Adelaiye, R.; Gillard, B.; Sotomayor, P.; Attwood, K.; Shen, L.; Conroy, D.; Kuhnert, F.; et al. Dll4 Blockade Potentiates the Anti-Tumor Effects of VEGF Inhibition in Renal Cell Carcinoma Patient-Derived Xenografts. PLoS ONE 2014, 9, e112371. [Google Scholar] [CrossRef] [PubMed]
- Jolliffe, I. Principal Component Analysis for Special Types of Data. 2002. Available online: https://link.springer.com/content/pdf/10.1007/0-387-22440-8_13.pdf (accessed on 9 December 2022).
- Seo, J.; An, Y.; Lee, J.; Ku, T.; Kang, Y.; Ahn, C.; Choi, C. Principal component analysis of dynamic fluorescence images for diagnosis of diabetic vasculopathy. J. Biomed. Opt. 2016, 21, 46003. [Google Scholar] [CrossRef] [PubMed]
- Benson, R.C.; Kues, H.A. Fluorescence properties of indocyanine green as related to angiography. Phys. Med. Biol. 1978, 23, 159–163. [Google Scholar] [CrossRef]
- Savitzky, A.; Golay, M.J. Smoothing and Differentiation of Data by Simplified Least Squares Procedures. Z. Physiol Chem. 1951, 40, 1832. Available online: https://pubs.acs.org/sharingguidelines (accessed on 23 November 2022). [CrossRef]
- Turner, E. Predictive Variable Selection for Subgroup Identification (Doctoral dissertation, The University of Manchester (United Kingdom)). 2018. Available online: https://www.proquest.com/openview/453e7fde6f6a9539be513ad1523a27f7/1?pq-origsite=gscholar&cbl=2026366 (accessed on 22 October 2022).
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. the Journal of machine Learning research. Available online: http://scikit-learn.sourceforge.net (accessed on 28 October 2022).
- Guyon, I.; Weston, J.; Barnhill, S.; Vapnik, V. Gene Selection for Cancer Classification using Support Vector Machines. Mach. Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- Cristianini, N.; Shawe-Taylor, J. An Introduction to Support Vector Machines and Other Kernel-Based Learning Methods. 2000. Available online: https://books.google.com/books?hl=en&lr=&id=_PXJn_cxv0AC&oi=fnd&pg=PR9&dq=Cristianini,+N.,+%26+Shawe-Taylor,+J.+(2000).+An+Introduction+to+Support+Vector+Machines+and+Other+Kernel-based+Learning+Methods.+London:+Cambridge+University+Press.&ots=xTTd9A_t0f&sig=h5zo1SL2mhMYxWB-99xKF-sJoaQ (accessed on 9 December 2022).
- Morrison, S.; Sosnoff, J.J.; Heffernan, K.S.; Jae, S.Y.; Fernhall, B. Aging, hypertension and physiological tremor: The contribution of the cardioballistic impulse to tremorgenesis in older adults. J. Neurol. Sci. 2013, 326, 68–74. [Google Scholar] [CrossRef]
- One-Way ANOVA: Independent Samples: I. Available online: http://vassarstats.net/textbook/ch14pt1.html (accessed on 19 December 2022).
- Tukey, J.W. Comparing Individual Means in the Analysis of Variance. Biometrics 1949, 5, 99–114. [Google Scholar] [CrossRef]
- Li, C.; Wang, Q. Advanced NIR-II Fluorescence Imaging Technology for In Vivo Precision Tumor Theranostics. Adv. Ther. 2019, 2, 1900053. [Google Scholar] [CrossRef]
- Meng, X.; Pang, X.; Zhang, K.; Gong, C.; Yang, J.; Dong, H.; Zhang, X. Recent Advances in Near-Infrared-II Fluorescence Imaging for Deep-Tissue Molecular Analysis and Cancer Diagnosis. Small 2022, 18, 2202035. [Google Scholar] [CrossRef]
- Wu, H.; Wu, H.; He, Y.; Gan, Z.; Xu, Z.; Zhou, M.; Liu, S.; Liu, H. Synovitis in mice with inflammatory arthritis monitored with quantitative analysis of dynamic contrast-enhanced NIR fluorescence imaging using iRGD-targeted liposomes as fluorescence probes. Int. J. Nanomed. 2018, 13, 1841–1850. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Ma, J. Feature selection for hyperspectral data based on recursive support vector machines. Int. J. Remote Sens. 2009, 30, 3669–3677. [Google Scholar] [CrossRef]
- Khaire, U.M.; Dhanalakshmi, R. Stability of feature selection algorithm: A review. J. King Saud Univ.-Comput. Inf. Sci. 2022, 34, 1060–1073. [Google Scholar] [CrossRef]
- Brownlee, J. Data Preparation for Machine Learning: Data Cleaning, Feature Selection, and Data Transforms in Python. 2020. Available online: https://books.google.com/books?hl=en&lr=&id=uAPuDwAAQBAJ&oi=fnd&pg=PP1&dq=Data+Preparation+for+Machine+Learning+Data+Cleaning,+Feature+Selection,+and+Data+Transforms+in+Python&ots=Cl7GwbdUoS&sig=g-7t_iDmR16YA-r3zPDu1pHnQos (accessed on 29 December 2022).
- Alieva, M.; van Rheenen, J.; Broekman, M.L.D. Potential impact of invasive surgical procedures on primary tumor growth and metastasis. Clin. Exp. Metastasis 2018, 35, 319–331. [Google Scholar] [CrossRef]
- Lerman, C.; Trock, B.; Rimer, B.K.; Jepson, C.; Brody, D.; Boyce, A. Psychological side effects of breast cancer screening. Health Psychol. 1991, 10, 259–267. [Google Scholar] [CrossRef]
- Zhou, R.; Wang, S.; Wen, H.; Wang, M.; Wu, M. The bispecific antibody HB-32, blockade of both VEGF and DLL4 shows potent anti-angiogenic activity in vitro and anti-tumor activity in breast cancer xenograft models. Exp. Cell Res. 2019, 380, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Moore, K.N.; Gordon, M.; Chugh, R.; Diamond, J.R.; Aljumaily, R.; Mendelson, D.; Kapoun, A.M.; Xu, L.; Smith, D.C.; et al. A first-in-human phase 1a study of the bispecific anti-DLL4/anti-VEGF antibody navicixizumab (OMP-305B83) in patients with previously treated solid tumors. Investig New Drugs 2019, 37, 461–472. [Google Scholar] [CrossRef]
- Mohammadlou, M.; Salehi, S.; Baharlou, R. Development of anti DLL4 Nanobody fused to truncated form of Pseudomonas exotoxin: As a novel immunotoxin to inhibit of cell proliferation and neovascularization. Anal. Biochem. 2022, 653, 114776. [Google Scholar] [CrossRef]
- Couch, J.A.; Zhang, G.; Beyer, J.C.; de Zafra, C.L.Z.; Gupta, P.; Kamath, A.V.; Lewin-Koh, N.; Tarrant, J.; Allamneni, K.P.; Cain, G.; et al. Balancing Efficacy and Safety of an Anti-DLL4 Antibody through Pharmacokinetic Modulation. Clin. Cancer Res. 2016, 22, 1469–1479. [Google Scholar] [CrossRef]
- Yan, M.; Callahan, C.A.; Beyer, J.C.; Allamneni, K.P.; Zhang, G.; Ridgway, J.B.; Niessen, K.; Plowman, G.D. Chronic DLL4 blockade induces vascular neoplasms. Nature 2010, 463, E6–E7. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Average Classification Metrics (Ascore) | Separation Score (Sscore) | ||
|---|---|---|---|---|
| Dll4+|CG− | CG+|Dll4− | CG+|CG− | ||
| CG1|CG3 | 0.9 | 0.74 | 0.72 | 0.775 |
| CG1|CG4 | 0.92 | 0.74 | 0.75 | 0.7875 |
| CG5|CG3 | 0.9 | 0.83 | 0.61 | 0.7925 |
| CG6|CG3 | 0.9 | 0.86 | 0.778 | 0.8495 |
| CG5|CG4 | 0.92 | 0.83 | 0.8 | 0.845 |
| CG6|CG4 | 0.92 | 0.86 | 0.78 | 0.855 |
| Groups | Feature | Best Average | Best Accuracy | Best Sensitivity | Best Specificity | ||||
|---|---|---|---|---|---|---|---|---|---|
| Alg. | Value(std) | Alg. | Value(std) | Alg. | Value(std) | Alg. | Value(std) | ||
| Dll4+|Dll4− | HIF10_avg, HIF12_avg | DT | 0.8666 (0.2309) | DT | 1 (0) | DT | 0.6 (0.5026) | DT | 1 (0) |
| HIF6_avg, HIF50_avg | LR | 0.7597 (0.1412) | LR | 0.8682 (0.1468) | LR | 0.6 (0.5026) | DT | 0.9391 (0.116) | |
| Dll4+|CG3 | HIF300_avg, HIF200_avg | RBF SVM | 0.6415 (0.3196) | RBF SVM | 0.5914 (0.0983) | L SVM | 1 (0) | KNN | 0.9166 (0.1147) |
| HIF300_avg, TR_rel | LR | 0.6453 (0.1036) | LR | 0.6176 (0.1624) | LR | 0.85 (0.2665) | KNN | 0.9083 (0.1147) | |
| Dll4+|CG4 | HIF30_avg, D16_avg | DT | 0.9175 (0.0303) | DT | 0.9125 (0.0915) | L SVM | 0.95 (0.0888) | NB | 1 (0) |
| HIF8_avg, D6_avg | RBF SVM | 0.88125 (0.1254) | RBF SVM | 0.89375 (0.1174) | L SVM | 1 (0) | NB | 0.85 (0.2016) | |
| CG5|Dll4− | HIF5_avg, HIF16_avg | DT | 0.8251 (0.0698) | DT | 0.8292 (0.1093) | L SVM | 1 (0) | NB | 0.845 (0.1952) |
| HIF8_avg, HIF25_avg | KNN | 0.8044 (0.1505) | KNN | 0.8201 (0.0919) | L SVM | 1 (0) | NB | 0.7925 (0.2014) | |
| CG5|CG3 | HIF300_avg, HIF400_avg | L SVM | 0.5683 (0.4037) | LR | 0.5292 (0.1507) | L SVM | 1 (0) | KNN | 0.85 (0.1613) |
| HIF300_avg, HIF50_avg | LR | 0.5919 (0.4037) | LR | 0.5626 (0.1652) | L SVM | 1 (0) | KNN | 0.8333 (0.1324) | |
| CG5|CG4 | D18_avg, HIF4_rel | KNN | 0.7811 (0.0485) | KNN | 0.7948 (0) | L SVM | 0.8928 (0) | DT | 0.7272 (0) |
| D18_avg, HIF4_avg | KNN | 0.7811 (0.0485) | KNN | 0.7948 (0) | L SVM | 0.8928 (0) | DT | 0.7272 (0) | |
| CG6|Dll4− | D40_avg, D2_avg | KNN | 0.8685 (0.0404) | KNN | 0.8755 (0.1180) | L SVM | 0.95 (0.1574 | KNN | 0.905 (0.1422) |
| D40_avg, HIF5_rel | KNN | 0.8482 (0.01263) | KNN | 0.8440 (0.1386) | RBF SVM | 0.9375 (0.1293) | KNN | 0.8383 (0.1643) | |
| CG6|CG3 | HIF300_avg, HIF350_avg | L SVM | 0.5729 (0.3995) | KNN | 0.5318 (0.1215) | L SVM | 1 (0) | NN | 0.8583 (0.1733) |
| HIF300_avg, HIF180_avg | RBF SVM | 0.5890 (0.2399) | RBF SVM | 0.5570 (0.1259) | L SVM | 1 (0) | KNN | 0.85 (0.2222) | |
| CG6|CG4 | HIF8_avg, HIF23_avg | KNN | 0.7278 (0.1917) | KNN | 0.7437 (0.0904) | RBF SVM | 1 (0) | NB | 0.7285 (0.2853) |
| HIF6_avg, HIF50_avg | NB | 0.7280 (0.2263) | NB | 0.7468 (0.0982) | L SVM | 1 (0) | DT | 0.6071 (0.1846) | |
| SVM | KNN | |||
|---|---|---|---|---|
| Measure | Training | Testing | Training | Testing |
| Sensitivity | 0.9310 | 1.0000 | 0.8108 | 1.0000 |
| Specificity | 0.8182 | 0.8182 | 0.9286 | 0.7500 |
| Precision | 0.8710 | 0.7500 | 0.9677 | 0.6250 |
| Negative Predictive Value | 0.9000 | 1.0000 | 0.6500 | 1.0000 |
| False-Positive Rate | 0.1818 | 0.1818 | 0.0714 | 0.2500 |
| False Discovery Rate | 0.1290 | 0.2500 | 0.0323 | 0.3750 |
| False-Negative Rate | 0.0690 | 0.0000 | 0.1892 | 0.0000 |
| Accuracy | 0.8824 | 0.8824 | 0.8431 | 0.8235 |
| F1 Score | 0.9000 | 0.8571 | 0.8824 | 0.7692 |
| Matthews Correlation Coefficient | 0.7600 | 0.7833 | 0.6758 | 0.6847 |
| SVM | KKN | |||
|---|---|---|---|---|
| Measure | Training | Testing | Training | Testing |
| Sensitivity | 0.9253 | 0.9756 | 0.9702 | 0.9762 |
| Specificity | 0.9132 | 0.9125 | 0.9211 | 0.9241 |
| Precision | 0.8564 | 0.8511 | 0.867 | 0.8723 |
| Negative Predictive Value | 0.9562 | 0.9865 | 0.9832 | 0.9865 |
| False Positive Rate | 0.0868 | 0.0875 | 0.0789 | 0.0759 |
| False Discovery Rate | 0.1436 | 0.1489 | 0.133 | 0.1277 |
| False Negative Rate | 0.0747 | 0.0244 | 0.0298 | 0.0238 |
| Accuracy | 0.9175 | 0.9339 | 0.9381 | 0.9421 |
| F1 Score | 0.8895 | 0.9091 | 0.9157 | 0.9213 |
| Matthews Correlation Coefficient | 0.8254 | 0.8625 | 0.8705 | 0.8793 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shafiee, S.; Jagtap, J.; Zayats, M.; Epperlein, J.; Banerjee, A.; Geurts, A.; Flister, M.; Zhuk, S.; Joshi, A. Dynamic NIR Fluorescence Imaging and Machine Learning Framework for Stratifying High vs. Low Notch-Dll4 Expressing Host Microenvironment in Triple-Negative Breast Cancer. Cancers 2023, 15, 1460. https://doi.org/10.3390/cancers15051460
Shafiee S, Jagtap J, Zayats M, Epperlein J, Banerjee A, Geurts A, Flister M, Zhuk S, Joshi A. Dynamic NIR Fluorescence Imaging and Machine Learning Framework for Stratifying High vs. Low Notch-Dll4 Expressing Host Microenvironment in Triple-Negative Breast Cancer. Cancers. 2023; 15(5):1460. https://doi.org/10.3390/cancers15051460
Chicago/Turabian StyleShafiee, Shayan, Jaidip Jagtap, Mykhaylo Zayats, Jonathan Epperlein, Anjishnu Banerjee, Aron Geurts, Michael Flister, Sergiy Zhuk, and Amit Joshi. 2023. "Dynamic NIR Fluorescence Imaging and Machine Learning Framework for Stratifying High vs. Low Notch-Dll4 Expressing Host Microenvironment in Triple-Negative Breast Cancer" Cancers 15, no. 5: 1460. https://doi.org/10.3390/cancers15051460
APA StyleShafiee, S., Jagtap, J., Zayats, M., Epperlein, J., Banerjee, A., Geurts, A., Flister, M., Zhuk, S., & Joshi, A. (2023). Dynamic NIR Fluorescence Imaging and Machine Learning Framework for Stratifying High vs. Low Notch-Dll4 Expressing Host Microenvironment in Triple-Negative Breast Cancer. Cancers, 15(5), 1460. https://doi.org/10.3390/cancers15051460