
Phase I Study of a Multivalent WT1 Peptide Vaccine (Galinpepimut-S) in Combination with Nivolumab in Patients with WT1-Expressing Ovarian Cancer in Second or Third Remission

 , , ,
, , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Eligibility Criteria
2.2. Treatment Plan
2.3. Vaccine Preparation
2.4. Toxicity Evaluation
2.5. Immune Response Evaluation
2.6. Statistical Considerations
3. Results
3.1. Patient Characteristics
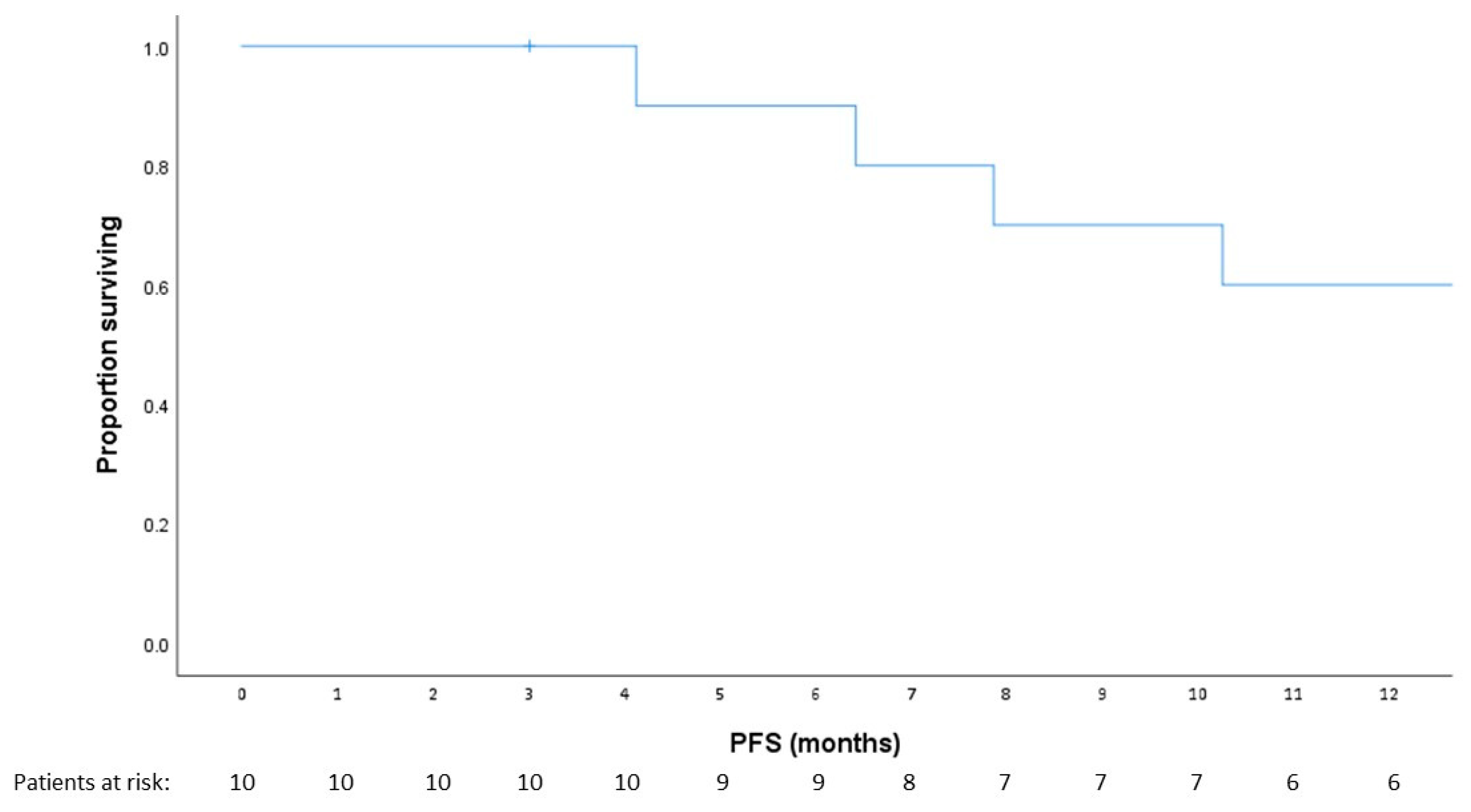
3.2. Safety and Tolerability
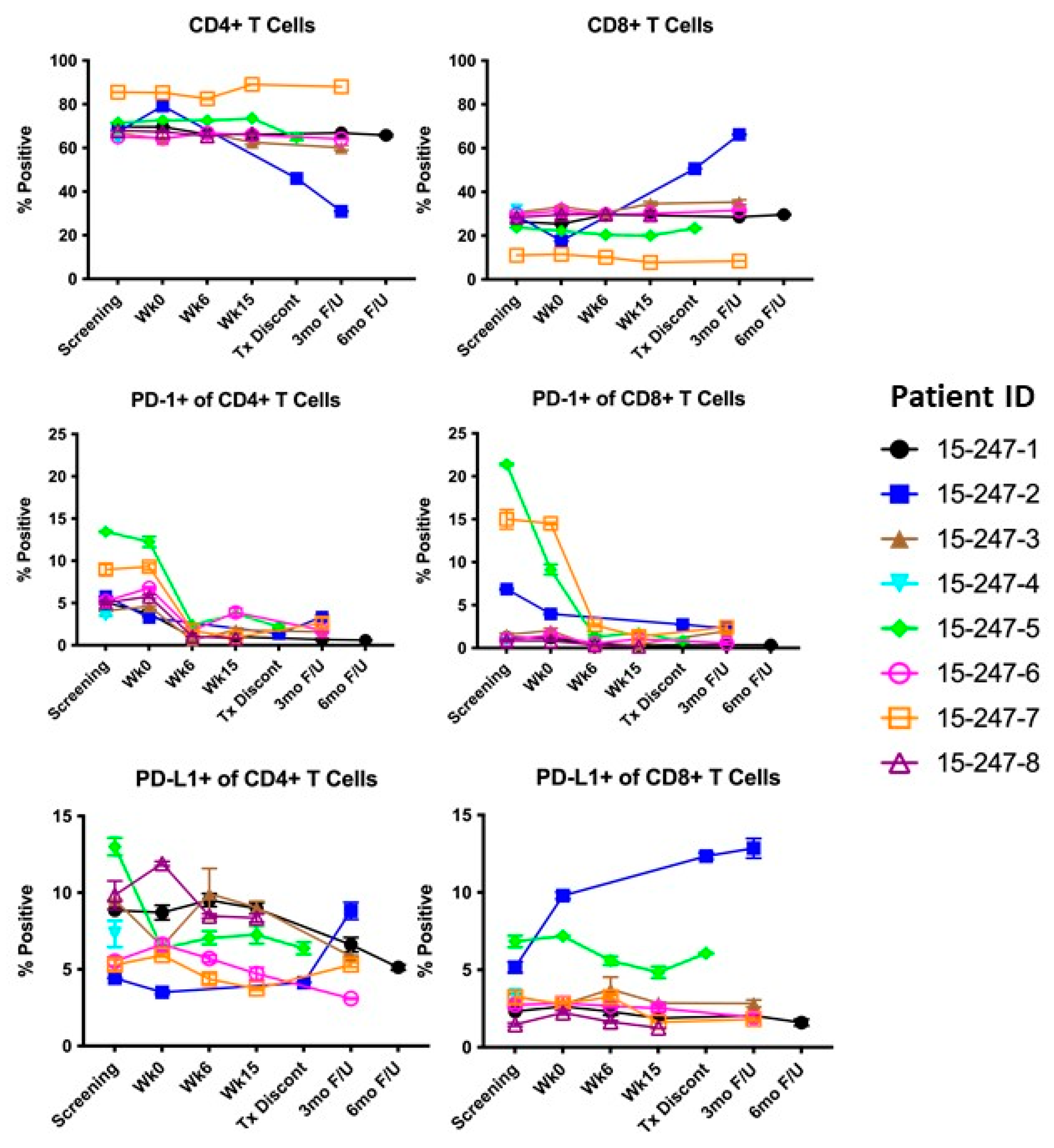
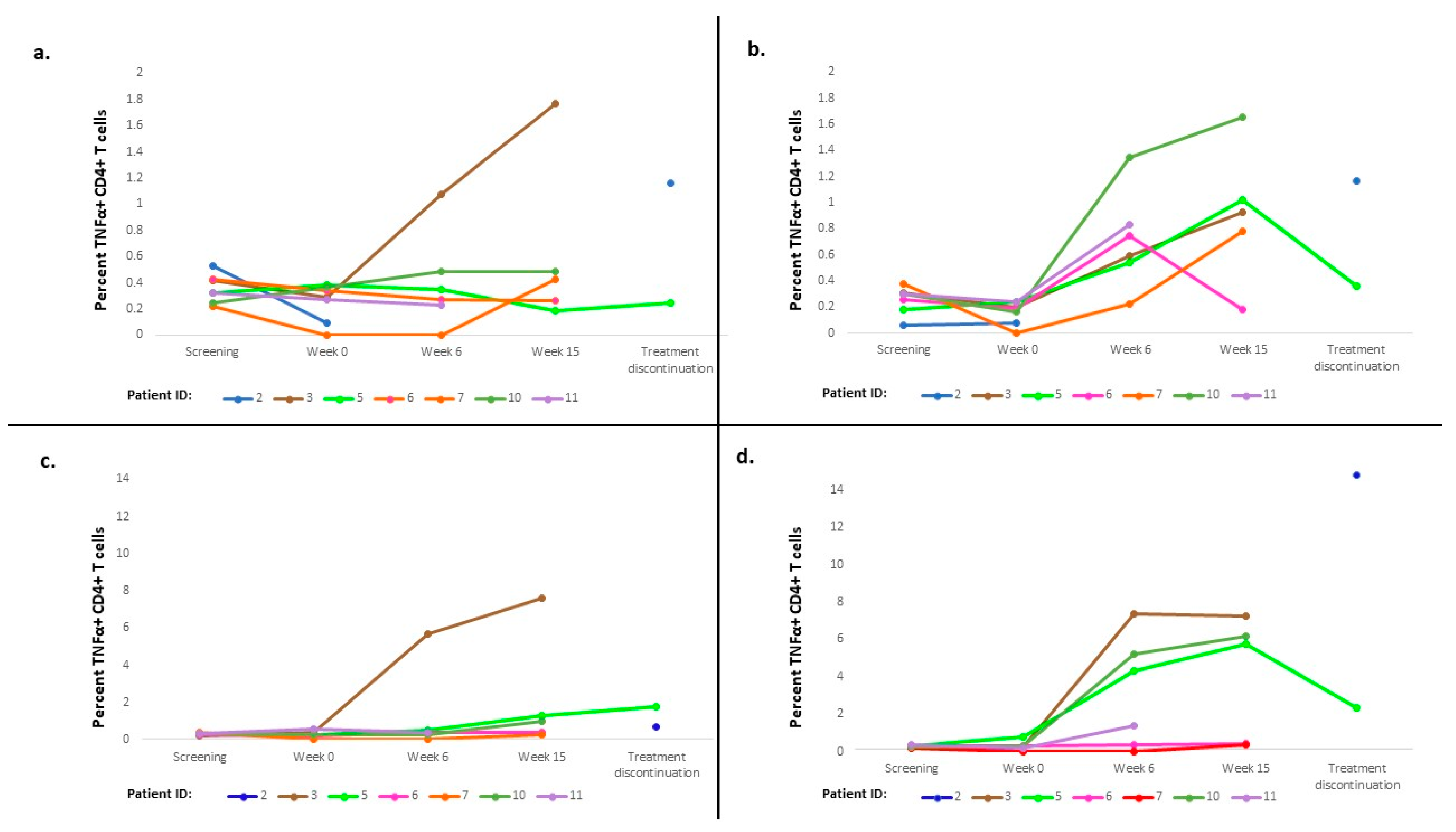
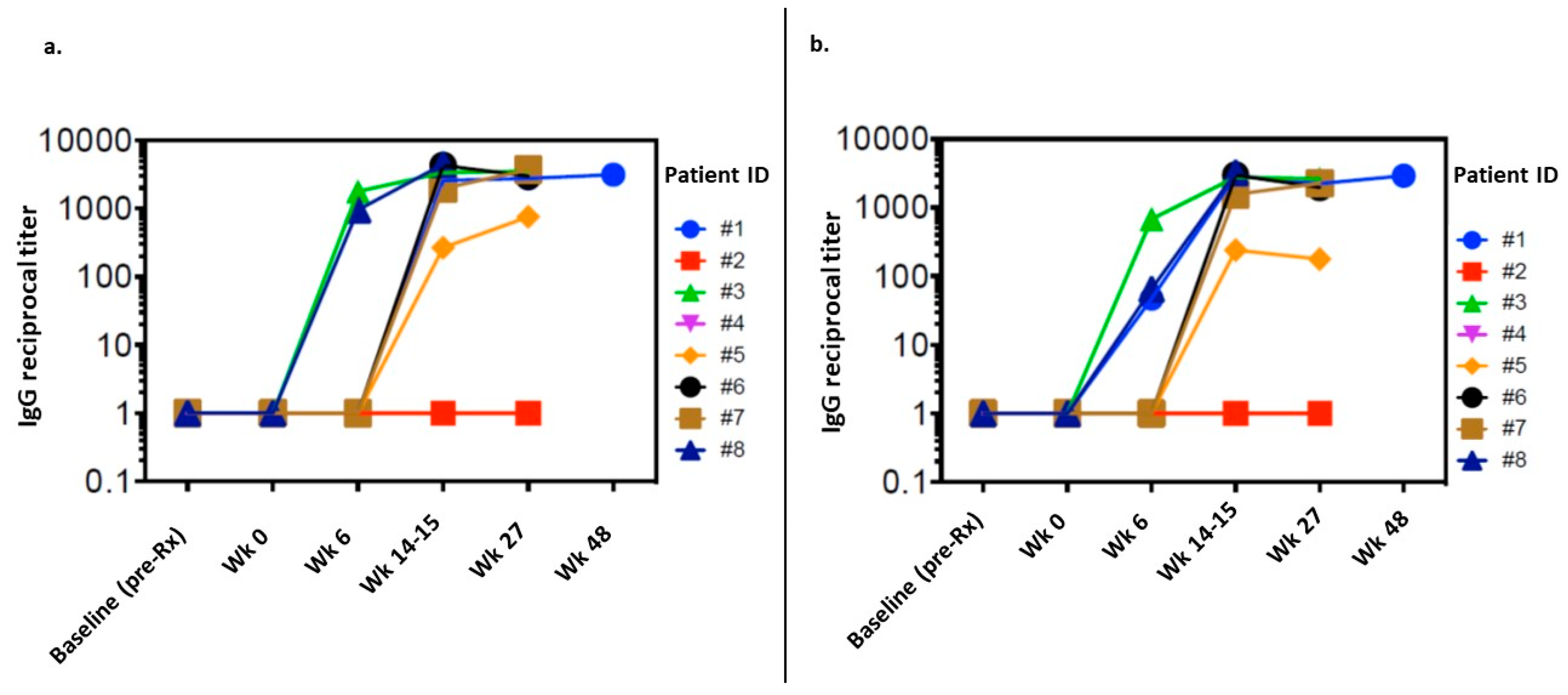
3.3. Immune Response
3.4. Exploratory Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Markman, M.; Markman, J.; Webster, K.; Zanotti, K.; Kulp, B.; Peterson, G.; Belinson, J. Duration of response to second-line, platinum-based chemotherapy for ovarian cancer: Implications for patient management and clinical trial design. J. Clin. Oncol. 2004, 22, 3120–3125. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, H.S.; Cordon-Cardo, C.; Ragupathi, G.; Livingston, P.O. Selection of tumor antigens as targets for immune attack using immunohistochemistry: Protein antigens. Clin. Cancer Res. 1998, 4, 2669–2676. [Google Scholar] [PubMed]
- Federici, M.F.; Kudryashov, V.; Saigo, P.E.; Finstad, C.L.; Lloyd, K.O. Selection of carbohydrate antigens in human epithelial ovarian cancers as targets for immunotherapy: Serous and mucinous tumors exhibit distinctive patterns of expression. Int. J. Cancer 1999, 81, 193–198. [Google Scholar] [CrossRef]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef]
- Santoiemma, P.P.; Reyes, C.; Wang, L.P.; McLane, M.W.; Feldman, M.D.; Tanyi, J.L.; Powell, D.J., Jr. Systematic evaluation of multiple immune markers reveals prognostic factors in ovarian cancer. Gynecol. Oncol. 2016, 143, 120–127. [Google Scholar] [CrossRef]
- Leffers, N.; Gooden, M.J.; de Jong, R.A.; Hoogeboom, B.N.; ten Hoor, K.A.; Hollema, H.; Boezen, H.M.; van der Zee, A.G.; Daemen, T.; Nijman, H.W. Prognostic significance of tumor-infiltrating T-lymphocytes in primary and metastatic lesions of advanced stage ovarian cancer. Cancer Immunol. Immunother. 2009, 58, 449–459. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Fujiwara, K.; Ledermann, J.A.; Oza, A.M.; Kristeleit, R.; Ray-Coquard, I.L.; Richardson, G.E.; Sessa, C.; Yonemori, K.; Banerjee, S.; et al. Avelumab alone or in combination with chemotherapy versus chemotherapy alone in platinum-resistant or platinum-refractory ovarian cancer (JAVELIN Ovarian 200): An open-label, three-arm, randomised, phase 3 study. Lancet Oncol. 2021, 22, 1034–1046. [Google Scholar] [CrossRef]
- Odunsi, K. Immunotherapy in ovarian cancer. Ann. Oncol. 2017, 28 (Suppl. S8), viii1–viii7. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Shapira-Frommer, R.; Santin, A.D.; Lisyanskaya, A.S.; Pignata, S.; Vergote, I.; Raspagliesi, F.; Sonke, G.S.; Birrer, M.; Provencher, D.M.; et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: Results from the phase II KEYNOTE-100 study. Ann. Oncol. 2019, 30, 1080–1087. [Google Scholar] [CrossRef]
- Hamanishi, J.; Takeshima, N.; Katsumata, N.; Ushijima, K.; Kimura, T.; Takeuchi, S.; Matsumoto, K.; Ito, K.; Mandai, M.; Nakai, H.; et al. Nivolumab versus gemcitabine or pegylated liposomal doxorubicin for patients with platinum-resistant ovarian cancer: Open-label, randomized trial in Japan (NINJA). J. Clin. Oncol. 2021, 39, 3671–3681. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.L.; Rovelli, R.; Sarivalasis, A.; Kandalaft, L.E. Integrating cancer vaccines in the standard-of-care of ovarian cancer: Translating preclinical models to human. Cancers 2021, 13, 4553. [Google Scholar] [CrossRef] [PubMed]
- Tornos, C.; Soslow, R.; Chen, S.; Akram, M.; Hummer, A.J.; Abu-Rustum, N.; Norton, L.; Tan, L.K. Expression of WT1, CA 125, and GCDFP-15 as useful markers in the differential diagnosis of primary ovarian carcinomas versus metastatic breast cancer to the ovary. Am. J. Surg. Pathol. 2005, 29, 1482–1489. [Google Scholar] [CrossRef] [PubMed]
- Giuntoli, R.L., 2nd; Rodriguez, G.C.; Whitaker, R.S.; Dodge, R.; Voynow, J.A. Mucin gene expression in ovarian cancers. Cancer Res. 1998, 58, 5546–5550. [Google Scholar] [PubMed]
- Odunsi, K.; Jungbluth, A.A.; Stockert, E.; Qian, F.; Gnjatic, S.; Tammela, J.; Intengan, M.; Beck, A.; Keitz, B.; Santiago, D.; et al. NY-ESO-1 and LAGE-1 cancer-testis antigens are potential targets for immunotherapy in epithelial ovarian cancer. Cancer Res. 2003, 63, 6076–6083. [Google Scholar]
- Hogdall, E.V.; Ringsholt, M.; Hogdall, C.K.; Christensen, I.J.; Johansen, J.S.; Kjaer, S.K.; Blaakaer, J.; Ostenfeld-Møller, L.; Price, P.A.; Christensen, L.H. YKL-40 tissue expression and plasma levels in patients with ovarian cancer. BMC Cancer 2009, 9, 8. [Google Scholar] [CrossRef]
- O’Cearbhaill, R.E.; Deng, W.; Chen, L.M.; Lucci, J.A., 3rd; Behbakht, K.; Spirtos, N.M.; Muller, C.Y.; Benigno, B.B.; Powell, M.A.; Berry, E.; et al. A phase II randomized, double-blind trial of a polyvalent Vaccine-KLH conjugate (NSC 748933 IND# 14384) + OPT-821 versus OPT-821 in patients with epithelial ovarian, fallopian tube, or peritoneal cancer who are in second or third complete remission: An NRG Oncology/GOG study. Gynecol. Oncol. 2019, 155, 393–399. [Google Scholar] [CrossRef]
- O’Cearbhaill, R.E.; Ragupathi, G.; Zhu, J.; Wan, Q.; Mironov, S.; Yang, G.; Spassova, M.K.; Iasonos, A.; Kravetz, S.; Ouerfelli, O.; et al. A phase I study of unimolecular pentavalent (Globo-H-GM2-sTn-TF-Tn) immunization of patients with epithelial ovarian, fallopian tube, or peritoneal cancer in first remission. Cancers 2016, 8, 46. [Google Scholar] [CrossRef]
- Zamarin, D.; Walderich, S.; Holland, A.; Zhou, Q.; Iasonos, A.E.; Torrisi, J.M.; Merghoub, T.; Chesebrough, L.F.; Mcdonnell, A.S.; Gallagher, J.M.; et al. Safety, immunogenicity, and clinical efficacy of durvalumab in combination with folate receptor alpha vaccine TPIV200 in patients with advanced ovarian cancer: A phase II trial. J. Immunother. Cancer. 2020, 8, e000829. [Google Scholar] [CrossRef]
- Sabbatini, P.J.; Ragupathi, G.; Hood, C.; Aghajanian, C.A.; Juretzka, M.; Iasonos, A.; Hensley, M.L.; Spassova, M.K.; Ouerfelli, O.; Spriggs, D.R.; et al. Pilot study of a heptavalent vaccine-keyhole limpet hemocyanin conjugate plus QS21 in patients with epithelial ovarian, fallopian tube, or peritoneal cancer. Clin. Cancer Res. 2007, 13, 4170–4177. [Google Scholar] [CrossRef] [PubMed]
- Sabbatini, P.; Tsuji, T.; Ferran, L.; Ritter, E.; Sedrak, C.; Tuballes, K.; Jungbluth, A.A.; Ritter, G.; Aghajanian, C.; Bell-McGuinn, K.; et al. Phase I trial of overlapping long peptides from a tumor self-antigen and poly-ICLC shows rapid induction of integrated immune response in ovarian cancer patients. Clin. Cancer Res. 2012, 18, 6497–6508. [Google Scholar] [CrossRef]
- Sabbatini, P.; Harter, P.; Scambia, G.; Sehouli, J.; Meier, W.; Wimberger, P.; Baumann, K.H.; Kurzeder, C.; Schmalfeldt, B.; Cibula, D.; et al. Abagovomab as maintenance therapy in patients with epithelial ovarian cancer: A phase III trial of the AGO OVAR, COGI, GINECO, and GEICO--the MIMOSA study. J. Clin. Oncol. 2013, 31, 1554–1561. [Google Scholar] [CrossRef]
- Duraiswamy, J.; Kaluza, K.M.; Freeman, G.J.; Coukos, G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013, 73, 3591–3603. [Google Scholar] [CrossRef]
- Mundlos, S.; Pelletier, J.; Darveau, A.; Bachmann, M.; Winterpacht, A.; Zabel, B. Nuclear localization of the protein encoded by the Wilms’ tumor gene WT1 in embryonic and adult tissues. Development 1993, 119, 1329–1341. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, N.S.; Bassi, D.; Uzieblo, A. WT1 is an integral component of an antibody panel to distinguish pancreaticobiliary and some ovarian epithelial neoplasms. Am. J. Clin. Pathol. 2001, 116, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.; Quenneville, L.; Yaziji, H.; Gown, A.M. Wilms tumor gene product: Sensitive and contextually specific marker of serous carcinomas of ovarian surface epithelial origin. Appl. Immunohistochem. Mol. Morphol. 2004, 12, 122–126. [Google Scholar] [CrossRef]
- Shimizu, M.; Toki, T.; Takagi, Y.; Konishi, I.; Fujii, S. Immunohistochemical detection of the Wilms’ tumor gene (WT1) in epithelial ovarian tumors. Int. J. Gynecol. Pathol. 2000, 19, 158–163. [Google Scholar] [CrossRef]
- Haber, D.A.; Sohn, R.L.; Buckler, A.J.; Pelletier, J.; Call, K.M.; Housman, D.E. Alternative splicing and genomic structure of the Wilms tumor gene WT1. Proc. Natl. Acad. Sci. USA 1991, 88, 9618–9622. [Google Scholar] [CrossRef] [PubMed]
- Zauderer, M.G.; Tsao, A.S.; Dao, T.; Panageas, K.; Lai, W.V.; Rimner, A.; Rusch, V.W.; Adusumilli, P.S.; Ginsberg, M.S.; Gomez, D.; et al. A randomized phase II trial of adjuvant galinpepimut-S, WT-1 analogue peptide vaccine, after multimodality therapy for patients with malignant pleural mesothelioma. Clin. Cancer Res. 2017, 23, 7483–7489. [Google Scholar] [CrossRef]
- Koehne, G. Targeting WT1 in hematologic malignancies? Blood 2017, 130, 1959–1960. [Google Scholar] [CrossRef]
- Maslak, P.G.; Dao, T.; Bernal, Y.; Chanel, S.M.; Zhang, R.; Frattini, M.; Rosenblat, T.; Jurcic, J.G.; Brentjens, R.J.; Arcila, M.E.; et al. Phase 2 trial of a multivalent WT1 peptide vaccine (galinpepimut-S) in acute myeloid leukemia. Blood Adv. 2018, 2, 224–234. [Google Scholar] [CrossRef]
- Maslak, P.G.; Dao, T.; Krug, L.M.; Chanel, S.; Korontsvit, T.; Zakhaleva, V.; Zhang, R.; Wolchok, J.D.; Yuan, J.; Pinilla-Ibarz, J.; et al. Vaccination with synthetic analog peptides derived from WT1 oncoprotein induces T-cell responses in patients with complete remission from acute myeloid leukemia. Blood 2010, 116, 171–179. [Google Scholar] [CrossRef]
- Brayer, J.; Lancet, J.E.; Powers, J.; List, A.; Balducci, L.; Komrokji, R.; Pinilla-Ibarz, J. WT1 vaccination in AML and MDS: A pilot trial with synthetic analog peptides. Am. J. Hematol. 2015, 90, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Dupont, J.; Wang, X.; Marshall, D.S.; Leitao, M.; Hedvat, C.V.; Hummer, A.; Thaler, H.; O’Reilly, R.J.; Soslow, R.A. Wilms Tumor Gene (WT1) and p53 expression in endometrial carcinomas: A study of 130 cases using a tissue microarray. Gynecol. Oncol. 2004, 94, 449–455. [Google Scholar] [CrossRef]
- Pinilla-Ibarz, J.; May, R.J.; Korontsvit, T.; Gomez, M.; Kappel, B.; Zakhaleva, V.; Zhang, R.H.; Scheinberg, D.A. Improved human T-cell responses against synthetic HLA-0201 analog peptides derived from the WT1 oncoprotein. Leukemia 2006, 20, 2025–2033. [Google Scholar] [CrossRef]
- Gomez-Nunez, M.; Pinilla-Ibarz, J.; Dao, T.; May, R.J.; Pao, M.; Jaggi, J.S.; Scheinberg, D.A. Peptide binding motif predictive algorithms correspond with experimental binding of leukemia vaccine candidate peptides to HLA-A*0201 molecules. Leuk. Res. 2006, 30, 1293–1298. [Google Scholar] [CrossRef]
- Borbulevych, O.Y.; Do, P.; Baker, B.M. Structures of native and affinity-enhanced WT1 epitopes bound to HLA-A*0201: Implications for WT1-based cancer therapeutics. Mol. Immunol. 2010, 47, 2519–2524. [Google Scholar] [CrossRef]
- May, R.J.; Dao, T.; Pinilla-Ibarz, J.; Korontsvit, T.; Zakhaleva, V.; Zhang, R.H.; Maslak, P.; Scheinberg, D.A. Peptide epitopes from the Wilms’ tumor 1 oncoprotein stimulate CD4+ and CD8+ T cells that recognize and kill human malignant mesothelioma tumor cells. Clin. Cancer Res. 2007, 13 Pt 1, 4547–4555. [Google Scholar] [CrossRef] [PubMed]
- Iasonos, A.; Sabbatini, P.; Spriggs, D.R.; Aghajanian, C.A.; O’Cearbhaill, R.E.; Hensley, M.L.; Thaler, H.T. Identifying clinical improvement in consolidation single-arm phase 2 trials in patients with ovarian cancer in second or greater clinical remission. Int. J. Gynecol. Cancer 2012, 22, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, S.S.; Fradley, M.G.; Cohen, J.V.; Nohria, A.; Reynolds, K.L.; Heinzerling, L.M.; Sullivan, R.J.; Damrongwatanasuk, R.; Chen, C.L.; Gupta, D.; et al. Myocarditis in patients treated with immune checkpoint inhibitors. J. Am. Coll. Cardiol. 2018, 71, 1755–1764. [Google Scholar] [CrossRef]
- Caforio, A.L.; Pankuweit, S.; Arbustini, E.; Basso, C.; Gimeno-Blanes, J.; Felix, S.B.; Fu, M.; Heliö, T.; Heymans, S.; Jahns, R.; et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart. J. 2013, 34, 2636–2648. [Google Scholar] [CrossRef]
- van Doorn, E.; Liu, H.; Huckriede, A.; Hak, E. Safety and tolerability evaluation of the use of Montanide ISA51 as vaccine adjuvant: A systematic review. Hum. Vaccin. Immunother. 2016, 12, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Tajiri, K.; Ieda, M. Cardiac complications in immune checkpoint inhibition therapy. Front. Cardiovasc. Med. 2019, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Balko, J.M.; Compton, M.L.; Chalkias, S.; Gorham, J.; Xu, Y.; Hicks, M.; Puzanov, I.; Alexander, M.R.; Bloomer, T.L.; et al. Fulminant myocarditis with combination immune checkpoint blockade. N. Engl. J. Med. 2016, 375, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, L.; Goldinger, S.M.; Hofmann, L.; Loquai, C.; Ugurel, S.; Thomas, I.; Schmidgen, M.I.; Gutzmer, R.; Utikal, J.S.; Göppner, D.; et al. Neurological, respiratory, musculoskeletal, cardiac and ocular side-effects of anti-PD-1 therapy. Eur. J. Cancer 2016, 60, 210–225. [Google Scholar] [CrossRef]
- Wang, D.Y.; Salem, J.E.; Cohen, J.V.; Chandra, S.; Menzer, C.; Ye, F.; Zhao, S.; Das, S.; Beckermann, K.E.; Ha, L.; et al. Fatal toxic fffects associated with immune checkpoint inhibitors: A systematic review and meta-analysis. JAMA Oncol. 2018, 4, 1721–1728. [Google Scholar] [CrossRef]
- Moslehi, J.J.; Salem, J.E.; Sosman, J.A.; Lebrun-Vignes, B.; Johnson, D.B. Increased reporting of fatal immune checkpoint inhibitor-associated myocarditis. Lancet 2018, 391, 933. [Google Scholar] [CrossRef]
- Krug, L.M.; Dao, T.; Brown, A.B.; Maslak, P.; Travis, W.; Bekele, S.; Korontsvit, T.; Zakhaleva, V.; Wolchok, J.; Yuan, J.; et al. WT1 peptide vaccinations induce CD4 and CD8 T cell immune responses in patients with mesothelioma and non-small cell lung cancer. Cancer Immunol. Immunother. 2010, 59, 1467–1479. [Google Scholar] [CrossRef]
- Zamarin, D.; Burger, R.A.; Sill, M.W.; Powell, D.J., Jr.; Lankes, H.A.; Feldman, M.D.; Zivanovic, O.; Gunderson, C.; Ko, E.; Mathews, C.; et al. Randomized phase II trial of nivolumab versus nivolumab and ipilimumab for recurrent or persistent ovarian cancer: An NRG Oncology Study. J. Clin. Oncol. 2020, 38, 1814–1823. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Characteristic | n (%) |
|---|---|
| Median age, years (range) | 62 (40–74) |
| Median CA-125 level at study enrollment, units/mL (range) | 7 (3–21) |
| Race | |
| Asian | 1 (9%) |
| Black | 1 (9%) |
| White | 9 (82%) |
| HLA subtype | |
| A*0201 | 4 (36%) |
| A*0101 | 2 (18%) |
| A*2402 | 2 (18%) |
| A*1101 | 1 (9%) |
| A*0301 | 1 (9%) |
| A*23 | 1 (9%) |
| Histologic subtype | |
| High-grade serous | 10 (91%) |
| Low-grade serous | 1 (9%) |
| Stage at diagnosis | |
| II | 1 (9%) |
| III | 9 (82%) |
| IV | 1 (9%) |
| Prior lines of chemotherapy | |
| 1 | 1 (9%) |
| 2 | 5 (46%) |
| 3 | 4 (36%) |
| 4 | 1 (9%) |
| Treatment-Related Adverse Event | Grade 1 n (%) | Grade 2 n (%) | Grade 3 n (%) |
|---|---|---|---|
| Injection site reaction | 7 (64%) | 0 | 0 |
| Arthralgia | 3 (27%) | 1 (9%) | 0 |
| Fatigue | 3 (27%) | 1 (9%) | 0 |
| Myositis/myocarditis | 0 | 0 | 1 (9%) |
| Rash | 1 (9%) | 1 (9%) | 0 |
| Decreased white blood cell counts | 1 (9%) | 1 (9%) | 0 |
| Decreased platelet counts | 0 | 1 (9%) | 0 |
| Hypothyroidism | 0 | 1 (9%) | 0 |
| Pneumonitis | 1 (9%) | 0 | 0 |
| Alopecia | 0 | 1 (9%) | 0 |
| Diarrhea | 1 (9%) | 0 | 0 |
| Vision changes (floaters) | 1 (9%) | 0 | 0 |
| Pruritis | 1 (9%) | 0 | 0 |
| Weight loss | 0 | 1 (9%) | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manning-Geist, B.L.; Gnjatic, S.; Aghajanian, C.; Konner, J.; Kim, S.H.; Sarasohn, D.; Soldan, K.; Tew, W.P.; Sarlis, N.J.; Zamarin, D.; et al. Phase I Study of a Multivalent WT1 Peptide Vaccine (Galinpepimut-S) in Combination with Nivolumab in Patients with WT1-Expressing Ovarian Cancer in Second or Third Remission. Cancers 2023, 15, 1458. https://doi.org/10.3390/cancers15051458
Manning-Geist BL, Gnjatic S, Aghajanian C, Konner J, Kim SH, Sarasohn D, Soldan K, Tew WP, Sarlis NJ, Zamarin D, et al. Phase I Study of a Multivalent WT1 Peptide Vaccine (Galinpepimut-S) in Combination with Nivolumab in Patients with WT1-Expressing Ovarian Cancer in Second or Third Remission. Cancers. 2023; 15(5):1458. https://doi.org/10.3390/cancers15051458
Chicago/Turabian StyleManning-Geist, Beryl L., Sacha Gnjatic, Carol Aghajanian, Jason Konner, Sarah H. Kim, Debra Sarasohn, Krysten Soldan, William P. Tew, Nicholas J. Sarlis, Dmitriy Zamarin, and et al. 2023. "Phase I Study of a Multivalent WT1 Peptide Vaccine (Galinpepimut-S) in Combination with Nivolumab in Patients with WT1-Expressing Ovarian Cancer in Second or Third Remission" Cancers 15, no. 5: 1458. https://doi.org/10.3390/cancers15051458
APA StyleManning-Geist, B. L., Gnjatic, S., Aghajanian, C., Konner, J., Kim, S. H., Sarasohn, D., Soldan, K., Tew, W. P., Sarlis, N. J., Zamarin, D., Kravetz, S., Laface, I., Rasalan-Ho, T., Qi, J., Wong, P., Sabbatini, P. J., & O’Cearbhaill, R. E. (2023). Phase I Study of a Multivalent WT1 Peptide Vaccine (Galinpepimut-S) in Combination with Nivolumab in Patients with WT1-Expressing Ovarian Cancer in Second or Third Remission. Cancers, 15(5), 1458. https://doi.org/10.3390/cancers15051458