
Mechanisms of Resistance to Antibody–Drug Conjugates


Abstract
Simple Summary
Abstract
1. Introduction
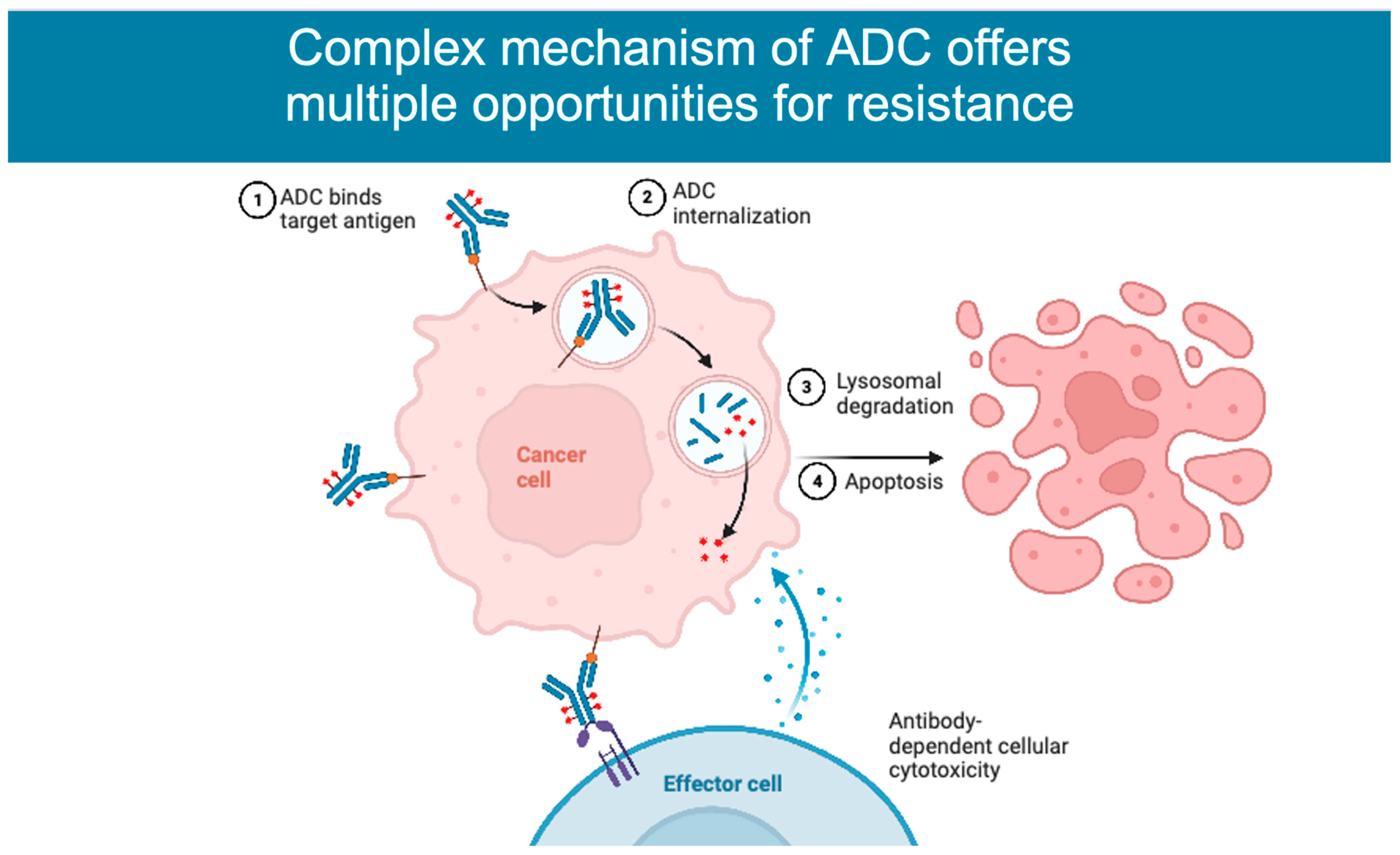
2. Mechanism of Action
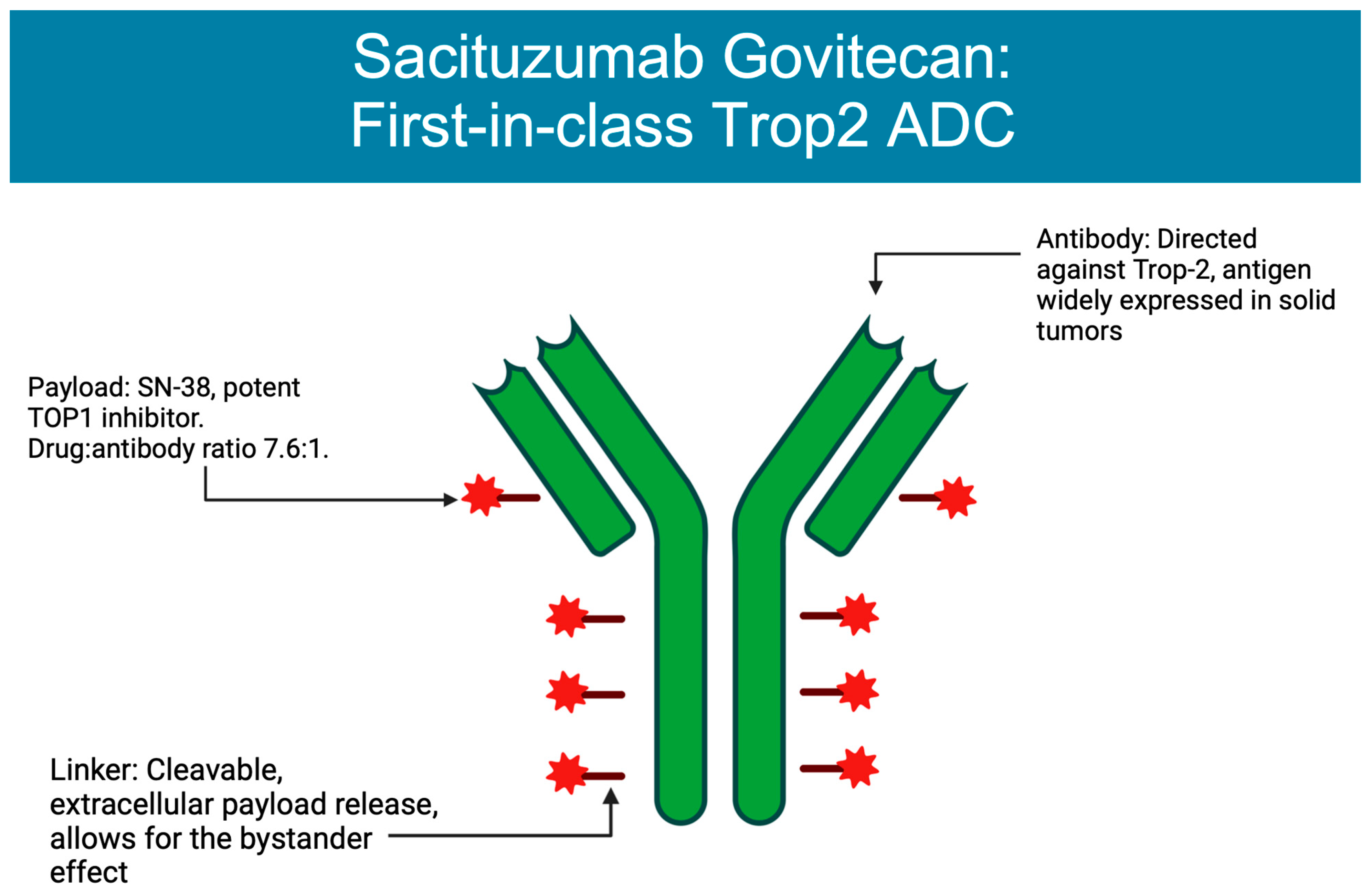
3. ADCs Approved in Breast Cancer
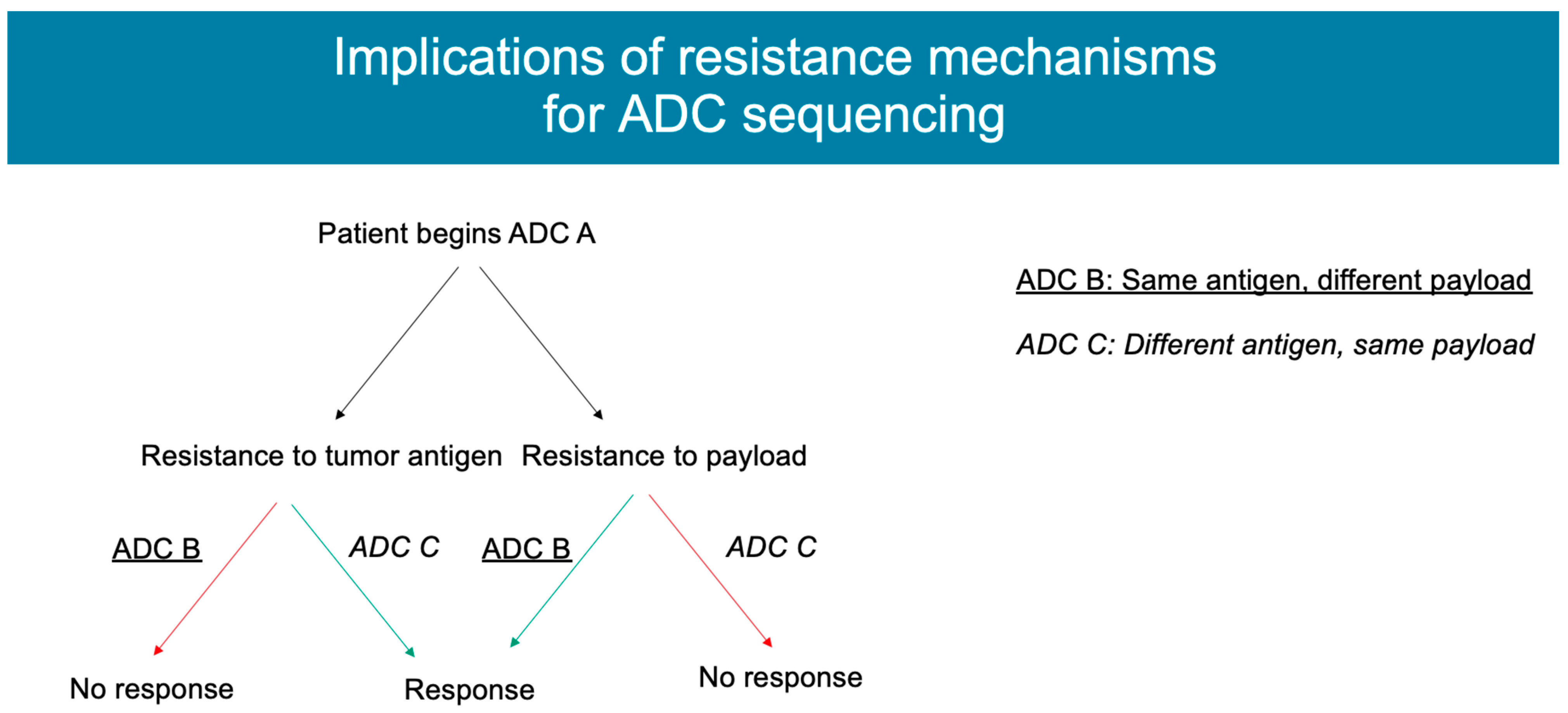
4. Mechanisms of Resistance
4.1. Antigen Expression
4.2. ADC Processing
4.3. Payload

{kind=link}
{kind=link}
{kind=link}
| Resistance Mechanism | ADC Where This Has Been Seen | Cancer Type | Examples |
|---|---|---|---|
| Antigen expression |
|
|
|
| ADC processing |
|
| |
| Payload |
|
|
|
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nagayama, A.; Ellisen, L.W.; Chabner, B.; Bardia, A. Antibody–Drug Conjugates for the Treatment of Solid Tumors: Clinical Experience and Latest Developments. Target. Oncol. 2017, 12, 719–739. [Google Scholar] [CrossRef] [PubMed]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current ADC Linker Chemistry. Pharm. Res. 2015, 32, 3526–3540. [Google Scholar] [CrossRef] [PubMed]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.C.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef]
- Sun, X.; Ponte, J.F.; Yoder, N.C.; Laleau, R.; Coccia, J.; Lanieri, L.; Qiu, Q.; Wu, R.; Hong, E.; Bogalhas, M.; et al. Effects of Drug–Antibody Ratio on Pharmacokinetics, Biodistribution, Efficacy, and Tolerability of Antibody–Maytansinoid Conjugates. Bioconjug. Chem. 2017, 28, 1371–1381. [Google Scholar] [CrossRef]
- Ogitani, Y.; Hagihara, K.; Oitate, M.; Naito, H.; Agatsuma, T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody-drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 2016, 107, 1039–1046. [Google Scholar] [CrossRef]
- Modi, S.; Park, H.; Murthy, R.K.; Iwata, H.; Tamura, K.; Tsurutani, J.; Moreno-Aspitia, A.; Doi, T.; Sagara, Y.; Redfern, C.; et al. Antitumor Activity and Safety of Trastuzumab Deruxtecan in Patients With HER2-Low-Expressing Advanced Breast Cancer: Results From a Phase Ib Study. J. Clin. Oncol. 2020, 38, 1887–1896. [Google Scholar] [CrossRef]
- Krop, I.E.; Kim, S.B.; González-Martín, A.; LoRusso, P.M.; Ferrero, J.M.; Smitt, M.; Yu, R.; Leung, A.C.; Wildiers, H. Trastuzumab emtansine versus treatment of physician’s choice for pretreated HER2-positive advanced breast cancer (TH3RESA): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 689–699. [Google Scholar] [CrossRef]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.-Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef]
- Perez, E.A.; Barrios, C.; Eiermann, W.; Toi, M.; Im, Y.H.; Conte, P.; Martin, M.; Pienkowski, T.; Pivot, X.; Burris, H., 3rd; et al. Trastuzumab Emtansine With or Without Pertuzumab Versus Trastuzumab Plus Taxane for Human Epidermal Growth Factor Receptor 2-Positive, Advanced Breast Cancer: Primary Results From the Phase III MARIANNE Study. J. Clin. Oncol. 2017, 35, 141–148. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Martin, M.; Symmans, W.F.; Jung, K.H.; Huang, C.-S.; Thompson, A.M.; Harbeck, N.; Valero, V.; Stroyakovskiy, D.; Wildiers, H.; et al. Neoadjuvant trastuzumab, pertuzumab, and chemotherapy versus trastuzumab emtansine plus pertuzumab in patients with HER2-positive breast cancer (KRISTINE): A randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2018, 19, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Hunter, F.W.; Barker, H.R.; Lipert, B.; Rothé, F.; Gebhart, G.; Piccart-Gebhart, M.J.; Sotiriou, C.; Jamieson, S.M.F. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br. J. Cancer 2020, 122, 603–612. [Google Scholar] [CrossRef] [PubMed]
- von Minckwitz, G.; Huang, C.S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A.; et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N. Engl. J. Med. 2019, 380, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Mayer, I.A.; Vahdat, L.T.; Tolaney, S.M.; Isakoff, S.J.; Diamond, J.R.; O’Shaughnessy, J.; Moroose, R.L.; Santin, A.D.; Abramson, V.G.; et al. Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2019, 380, 741–751. [Google Scholar] [CrossRef]
- Bardia, A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Rugo, H.S.; Brufsky, A.; Kalinsky, K.; Cortes, J.; O’Shaughnessy, J.; et al. Sacituzumab govitecan (SG) versus treatment of physician’s choice (TPC) in patients (pts) with previously treated, metastatic triple-negative breast cancer (mTNBC): Final results from the phase 3 ASCENT study. J. Clin. Orthod. 2022, 40, 1071. [Google Scholar] [CrossRef]
- Bardia, A.; Hurvitz, S.A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Brufsky, A.; Sardesai, S.D.; Kalinsky, K.; Zelnak, A.B.; et al. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2021, 384, 1529–1541. [Google Scholar] [CrossRef]
- García-Alonso, S.; Ocaña, A.; Pandiella, A. Resistance to Antibody-Drug Conjugates. Cancer Res. 2018, 78, 2159–2165. [Google Scholar] [CrossRef]
- Ocaña, A.; Amir, E.; Pandiella, A. HER2 heterogeneity and resistance to anti-HER2 antibody-drug conjugates. Breast Cancer Res. 2020, 22, 15. [Google Scholar] [CrossRef]
- Ferraro, E.; Drago, J.Z.; Modi, S. Implementing antibody-drug conjugates (ADCs) in HER2-positive breast cancer: State of the art and future directions. Breast Cancer Res. 2021, 23, 84. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.-B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef]
- Cortés, J.; Kim, S.-B.; Chung, W.-P.; Im, S.-A.; Park, Y.H.; Hegg, R.; Kim, M.H.; Tseng, L.-M.; Petry, V.; Chung, C.-F.; et al. Trastuzumab Deruxtecan versus Trastuzumab Emtansine for Breast Cancer. N. Engl. J. Med. 2022, 386, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, C.M.; Yamaguchi, A.; Anami, Y.; Xiong, W.; Otani, Y.; Lee, J.; Ueno, N.T.; Zhang, N.; An, Z.; Tsuchikama, K. Antibody-drug conjugates with dual payloads for combating breast tumor heterogeneity and drug resistance. Nat. Commun. 2021, 12, 3528. [Google Scholar] [CrossRef] [PubMed]
- Filho, O.M.; Viale, G.; Stein, S.; Trippa, L.; Yardley, D.A.; Mayer, I.A.; Abramson, V.G.; Arteaga, C.L.; Spring, L.M.; Waks, A.G.; et al. Impact of HER2 Heterogeneity on Treatment Response of Early-Stage HER2-Positive Breast Cancer: Phase II Neoadjuvant Clinical Trial of T-DM1 Combined with Pertuzumab. Cancer Discov. 2021, 11, 2474–2487. [Google Scholar] [CrossRef]
- Swain, S.M.; Shastry, M.; Hamilton, E. Targeting HER2-positive breast cancer: Advances and future directions. Nat. Rev. Drug Discov. 2023, 22, 101–126. [Google Scholar] [CrossRef]
- Sakai, H.; Tsurutani, J.; Iwasa, T.; Komoike, Y.; Sakai, K.; Nishio, K.; Nakagawa, K. HER2 genomic amplification in circulating tumor DNA and estrogen receptor positivity predict primary resistance to trastuzumab emtansine (T-DM1) in patients with HER2-positive metastatic breast cancer. Breast Cancer 2018, 25, 605–613. [Google Scholar] [CrossRef]
- Loganzo, F.; Sung, M.; Gerber, H.-P. Mechanisms of Resistance to Antibody-Drug Conjugates. Mol. Cancer Ther. 2016, 15, 2825–2834. [Google Scholar] [CrossRef]
- Loganzo, F.; Tan, X.; Sung, M.; Jin, G.; Myers, J.S.; Melamud, E.; Wang, F.; Diesl, V.; Follettie, M.T.; Musto, S.; et al. Tumor cells chronically treated with a trastuzumab-maytansinoid antibody-drug conjugate develop varied resistance mechanisms but respond to alternate treatments. Mol. Cancer Ther. 2015, 14, 952–963. [Google Scholar] [CrossRef]
- Sung, M.S.; Tan, X.; Hosselet, C.; Cinque, M.; Upeslacis, E.; Golas, J.; Wang, F.; Lu, B.; Tylaska, L.; King, L.; et al. Abstract 2113: Caveolae-mediated endocytosis as a novel mechanism of resistance to T-DM1 ADC. Cancer Res. 2016, 76 (Suppl. S14), 2113. [Google Scholar] [CrossRef]
- Phillips, G.D.L.; Fields, C.T.; Li, G.; Dowbenko, D.; Schaefer, G.; Miller, K.; Andre, F.; Burris, H.A.; Albain, K.S.; Harbeck, N.; et al. Dual targeting of HER2-positive cancer with trastuzumab emtansine and pertuzumab: Critical role for neuregulin blockade in antitumor response to combination therapy. Clin. Cancer Res. 2014, 20, 456–468. [Google Scholar] [CrossRef]
- Oh, S.Y.; Lee, Y.W.; Lee, E.J.; Kim, J.H.; Park, Y.J.; Heo, S.G.; Yu, M.R.; Hong, M.H.; DaSilva, J.; Daly, C.; et al. Preclinical Study of a Biparatopic METxMET Antibody-Drug Conjugate, REGN5093-M114, Overcomes MET-driven Acquired Resistance to EGFR TKIs in EGFR-mutant NSCLC. Clin. Cancer Res. 2023, 29, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Diéras, V.; Deluche, E.; Lusque, A.; Pistilli, B.; Bachelot, T.; Pierga, J.-Y.; Viret, F.; Levy, C.; Salabert, L.; Le Du, F.; et al. Abstract PD8-02: Trastuzumab deruxtecan (T-DXd) for advanced breast cancer patients (ABC), regardless HER2 status: A phase II study with biomarkers analysis (DAISY). Cancer Res. 2022, 82 (Suppl. S4), PD8-02. [Google Scholar] [CrossRef]
- Li, B.T.; Michelini, F.; Misale, S.; Cocco, E.; Baldino, L.; Cai, Y.; Shifman, S.; Tu, H.-Y.; Myers, M.L.; Xu, C.; et al. HER2-Mediated Internalization of Cytotoxic Agents in ERBB2 Amplified or Mutant Lung Cancers. Cancer Discov. 2020, 10, 674–687. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.; Tan, X.; Lu, B.; Golas, J.; Hosselet, C.; Wang, F.; Tylaska, L.; King, L.; Zhou, D.; Dushin, R.; et al. Caveolae-Mediated Endocytosis as a Novel Mechanism of Resistance to Trastuzumab Emtansine (T-DM1). Mol. Cancer Ther. 2018, 17, 243–253. [Google Scholar] [CrossRef]
- Pereira, P.M.R.; Mandleywala, K.; Monette, S.; Lumish, M.; Tully, K.M.; Panikar, S.S.; Cornejo, M.; Mauguen, A.; Ragupathi, A.; Keltee, N.C.; et al. Caveolin-1 temporal modulation enhances antibody drug efficacy in heterogeneous gastric cancer. Nat. Commun. 2022, 13, 2526. [Google Scholar] [CrossRef]
- Christianson, H.C.; Menard, J.A.; Chandran, V.I.; Bourseau-Guilmain, E.; Shevela, D.; Lidfeldt, J.; Månsson, A.-S.; Pastorekova, S.; Messinger, J.; Belting, M. Tumor antigen glycosaminoglycan modification regulates antibody-drug conjugate delivery and cytotoxicity. Oncotarget 2017, 8, 66960–66974. [Google Scholar] [CrossRef]
- Schultz, M.L.; Tecedor, L.; Chang, M.; Davidson, B.L. Clarifying lysosomal storage diseases. Trends Neurosci. 2011, 34, 401–410. [Google Scholar] [CrossRef]
- Grigoriev, I.; Splinter, D.; Keijzer, N.; Wulf, P.S.; Demmers, J.; Ohtsuka, T.; Modesti, M.; Maly, I.V.; Grosveld, F.; Hoogenraad, C.C.; et al. Rab6 regulates transport and targeting of exocytotic carriers. Dev. Cell 2007, 13, 305–314. [Google Scholar] [CrossRef]
- Callow, M.G.; Zozulya, S.; Gishizky, M.L.; Jallal, B.; Smeal, T. PAK4 mediates morphological changes through the regulation of GEF-H1. J. Cell Sci. 2005, 118 Pt 9, 1861–1872. [Google Scholar] [CrossRef]
- Tsui, C.K.; Barfield, R.M.; Fischer, C.R.; Morgens, D.W.; Li, A.; Smith, B.A.H.; Gray, M.A.; Bertozzi, C.R.; Rabuka, D.; Bassik, M.C. CRISPR-Cas9 screens identify regulators of antibody-drug conjugate toxicity. Nat. Chem. Biol. 2019, 15, 949–958. [Google Scholar] [CrossRef]
- Yu, S.-F.; Zheng, B.; Go, M.; Lau, J.; Spencer, S.; Raab, H.; Soriano, R.; Jhunjhunwala, S.; Cohen, R.; Caruso, M.; et al. A Novel Anti-CD22 Anthracycline-Based Antibody-Drug Conjugate (ADC) That Overcomes Resistance to Auristatin-Based ADCs. Clin. Cancer Res. 2015, 21, 3298–3306. [Google Scholar] [CrossRef] [PubMed]
- Takegawa, N.; Nonagase, Y.; Yonesaka, K.; Sakai, K.; Maenishi, O.; Ogitani, Y.; Tamura, T.; Nishio, K.; Nakagawa, K.; Tsurutani, J. DS-8201a, a new HER2-targeting antibody-drug conjugate incorporating a novel DNA topoisomerase I inhibitor, overcomes HER2-positive gastric cancer T-DM1 resistance. Int. J. Cancer 2017, 141, 1682–1689. [Google Scholar] [CrossRef] [PubMed]
- Conilh, L.; Sadilkova, L.; Viricel, W.; Dumontet, C. Payload diversification: A key step in the development of antibody-drug conjugates. J. Hematol. Oncol. 2023, 16, 3. [Google Scholar] [CrossRef] [PubMed]
- Coates, J.T.; Sun, S.; Leshchiner, I.; Thimmiah, N.; Martin, E.E.; McLoughlin, D.; Danysh, B.P.; Slowik, K.; Jacobs, R.A.; Rhrissorrakrai, K.; et al. Parallel Genomic Alterations of Antigen and Payload Targets Mediate Polyclonal Acquired Clinical Resistance to Sacituzumab Govitecan in Triple-Negative Breast Cancer. Cancer Discov. 2021, 11, 2436–2445. [Google Scholar] [CrossRef]
- Aldonza, M.B.D.; Hong, J.-Y.; Alinsug, M.V.; Song, J.; Lee, S.K. Multiplicity of acquired cross-resistance in paclitaxel-resistant cancer cells is associated with feedback control of TUBB3 via FOXO3a-mediated ABCB1 regulation. Oncotarget 2016, 7, 34395–34419. [Google Scholar] [CrossRef]
- Coleman, N.; Yap, T.A.; Heymach, J.V.; Meric-Bernstam, F.; Le, X. Antibody-drug conjugates in lung cancer: Dawn of a new era? NPJ Precis. Oncol. 2023, 7, 5. [Google Scholar] [CrossRef]
- Cabaud, O.; Berger, L.G.M.; Crompot, E.; Adélaide, J.; Finetti, P.; Garnier, S.; Guille, A.; Carbuccia, N.; Farina, A.; Agavnian, E.; et al. Overcoming Resistance to Anti-Nectin-4 Antibody-Drug Conjugate. Mol. Cancer Ther. 2022, 21, 1227–1235. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abelman, R.O.; Wu, B.; Spring, L.M.; Ellisen, L.W.; Bardia, A. Mechanisms of Resistance to Antibody–Drug Conjugates. Cancers 2023, 15, 1278. https://doi.org/10.3390/cancers15041278
Abelman RO, Wu B, Spring LM, Ellisen LW, Bardia A. Mechanisms of Resistance to Antibody–Drug Conjugates. Cancers. 2023; 15(4):1278. https://doi.org/10.3390/cancers15041278
Chicago/Turabian StyleAbelman, Rachel Occhiogrosso, Bogang Wu, Laura M. Spring, Leif W. Ellisen, and Aditya Bardia. 2023. "Mechanisms of Resistance to Antibody–Drug Conjugates" Cancers 15, no. 4: 1278. https://doi.org/10.3390/cancers15041278
APA StyleAbelman, R. O., Wu, B., Spring, L. M., Ellisen, L. W., & Bardia, A. (2023). Mechanisms of Resistance to Antibody–Drug Conjugates. Cancers, 15(4), 1278. https://doi.org/10.3390/cancers15041278