
Molecular Heterogeneity in BRAF-Mutant Gliomas: Diagnostic, Prognostic, and Therapeutic Implications
 , , , , and
, , , , and 
Abstract
Simple Summary
Abstract
1. Introduction
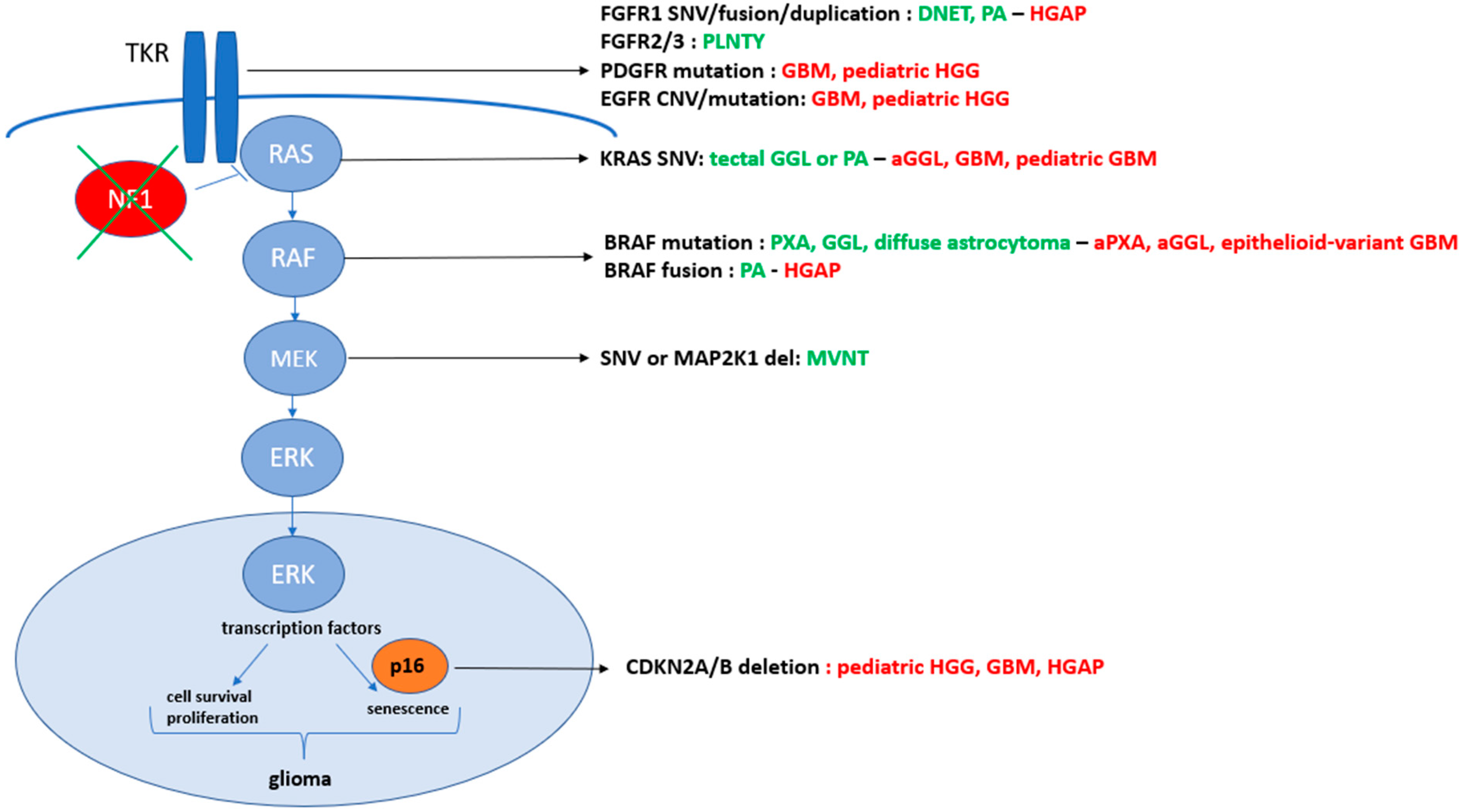
2. BRAF Mutations
3. Specific Glial Tumor Types Are Associated to BRAF Mutations

4. The Concept of Oncogenic-Stress-Induced Senescence Might Be Related to BRAF p.V600E
5. Prognosis of BRAF-Mutant Gliomas
6. Additional Molecular or Passenger Alterations Lead to the Disruption of the Balance between Cell Senescence and Proliferation in BRAF p.V600E Glioma Mutants
6.1. CDKN2A Deletion and Its Impact on CDK4/6 Function
6.2. Telomerase Reverse Transcriptase (TERT) Activation and ATP-Dependent Helicase (ATRX) Mutations Are Mutually Exclusive
6.3. BRAF/ERK and Pi3K/AKT/mTOR Pathways Cooperate in the Tumorigenesis of Gliomas
6.4. NF1 Status in BRAF p.V600E Glioma Mutants
6.5. Epigenetic and Hypermutator Phenotypes in BRAF p.V600E Glioma Mutants
7. How to Treat Those MAPK-Activated Gliomas with BRAF Mutations?
7.1. Conventional Therapies
7.2. Therapies Targeting BRAF Altered Gliomas
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. WHO Classification of Tumours Editorial Board. Central Nervous System Tumours, 5th ed.; WHO classification of tumours series; International Agency for Research on Cancer: Lyon, France, 2021; Volume 6, Available online: https://publications.iarc.fr/601 (accessed on 2 August 2021).
- De Blank, P.; Fouladi, M.; Huse, J.T. Molecular markers and targeted therapy in pediatric low-grade glioma. J. Neuro-Oncol. 2020, 150, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Ducreux, M.; Chamseddine, A.; Laurent-Puig, P.; Smolenschi, C.; Hollebecque, A.; Dartigues, P.; Samallin, E.; Boige, V.; Malka, D.; Gelli, M. Molecular targeted therapy of BRAF-mutant colorectal cancer. Ther. Adv. Med. Oncol. 2019, 11, 175883591985649. [Google Scholar] [CrossRef] [PubMed]
- Maurer, G.; Tarkowski, B.; Baccarini, M. Raf kinases in cancer–roles and therapeutic opportunities. Oncogene 2011, 30, 3477–3488. [Google Scholar] [CrossRef]
- Jacob, K.; Quang-Khuong, D.-A.; Jones, D.T.; Witt, H.; Lambert, S.; Albrecht, S.; Witt, O.; Vezina, C.; Shirinian, M.; Faury, D.; et al. Genetic Aberrations Leading to MAPK Pathway Activation Mediate Oncogene-Induced Senescence in Sporadic Pilocytic Astrocytomas. Clin. Cancer Res. 2011, 17, 4650–4660. [Google Scholar] [CrossRef] [PubMed]
- Horbinski, C. To BRAF or Not to BRAF: Is That Even a Question Anymore? J. Neuropathol. Exp. Neurol. 2013, 72, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Lind, K.T.; Chatwin, H.V.; DeSisto, J.; Coleman, P.; Sanford, B.; Donson, A.M.; Davies, K.D.; Willard, N.; A Ewing, C.; Knox, A.J.; et al. Novel RAF Fusions in Pediatric Low-Grade Gliomas Demonstrate MAPK Pathway Activation. J. Neuropathol. Exp. Neurol. 2021, 80, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Ryall, S.; Zapotocky, M.; Fukuoka, K.; Nobre, L.; Stucklin, A.G.; Bennett, J.; Siddaway, R.; Li, C.; Pajovic, S.; Arnoldo, A.; et al. Integrated Molecular and Clinical Analysis of 1,000 Pediatric Low-Grade Gliomas. Cancer Cell 2020, 7, 569–583.e5. [Google Scholar] [CrossRef] [PubMed]
- Garnett, M.J.; Marais, R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell 2004, 6, 313–319. [Google Scholar] [CrossRef]
- Ellison, D.W.; Hawkins, C.; Jones, D.T.W.; Onar-Thomas, A.; Pfister, S.F.; Reifenberger, G.; Louis, D.N. cIMPACT-NOW update 4: Diffuse gliomas characterized by MYB, MYBL1, or FGFR1 alterations or BRAFV600E mutation. Acta Neuropathol. 2019, 137, 683–687. [Google Scholar] [CrossRef]
- Khater, F.; Langlois, S.; Cassart, P.; Roy, A.-M.; Lajoie, M.; Healy, J.; Richer, C.; St-Onge, P.; Piché, N.; Perreault, S.; et al. Recurrent somatic BRAF insertion (p.V504_R506dup): A tumor marker and a potential therapeutic target in pilocytic astrocytoma. Oncogene 2018, 38, 2994–3002. [Google Scholar] [CrossRef]
- Pratt, D.; Camelo-Piragua, S.; McFadden, K.; Leung, D.; Mody, R.; Chinnaiyan, A.; Koschmann, C.; Venneti, S. BRAF activating mutations involving the beta3-alphaC loop in V600E-negative anaplastic pleomorphic xanthoastrocytoma. Acta Neuropathol. Commun. 2018, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, C.; Gloger, J.; Anacker, J.; Said, H.M.; Gerngras, S.; Kühnel, S.; Meyer, C.; Rapp, U.R.; Kämmerer, U.; Vordermark, D.; et al. RAF expression in human astrocytic tumors. Int. J. Mol. Med. 2009, 23, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Vaubel, R.; Zschernack, V.; Tran, Q.T.; Jenkins, S.; Caron, A.; Milosevic, D.; Smadbeck, J.; Vasmatzis, G.; Kandels, D.; Gnekow, A.; et al. Biology and grading of pleomorphic xanthoastrocytoma—What have we learned about it? Brain Pathol. 2021, 31, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.J.; Gong, H.; Chen, K.; Joseph, N.M.; van Ziffle, J.; Bastian, B.C.; Grenert, J.P.; Kline, C.N.; Mueller, S.; Banerjee, A.; et al. The genetic landscape of anaplastic pleomorphic xanthoastrocytoma. Brain Pathol. 2019, 29, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Wesseling, P.; Aldape, K.; Brat, D.J.; Capper, D.; Cree, I.A.; Eberhart, C.; Figarella-Branger, D.; Fouladi, M.; Fuller, G.N.; et al. cIMPACT-NOW update 6: New entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 2020, 30, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Penman, C.L.; Efaulkner, C.; Lowis, S.P.; Kurian, K.M. Current Understanding of BRAF Alterations in Diagnosis, Prognosis, and Therapeutic Targeting in Pediatric Low-Grade Gliomas. Front. Oncol. 2015, 5, 54. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-H.; Nonoguchi, N.; Paulus, W.; Brokinkel, B.; Keyvani, K.; Sure, U.; Wrede, K.; Mariani, L.; Giangaspero, F.; Tanaka, Y.; et al. Frequent BRAF Gain in Low-Grade Diffuse Gliomas with 1p/19q Loss. Brain Pathol. 2012, 22, 834–840. [Google Scholar] [CrossRef]
- Reinhardt, A.; Stichel, D.; Schrimpf, D.; Sahm, F.; Korshunov, A.; Reuss, D.E.; Koelsche, C.; Huang, K.; Wefers, A.K.; Hovestadt, V.; et al. Anaplastic astrocytoma with piloid features, a novel molecular class of IDH wildtype glioma with recurrent MAPK pathway, CDKN2A/B and ATRX alterations. Acta Neuropathol. 2018, 136, 273–291. [Google Scholar] [CrossRef]
- Capper, D.; Rodriguez, F.J.; Varlet, P.; Jones, D.T.W. High-grade astrocytoma with piloid features. In WHO Classification of Tumours Editorial Board. Central Nervous System Tumours, 5th ed.; WHO classification of tumours series; International Agency for Research on Cancer: Lyon, France, 2021; Volume 6. [Google Scholar]
- Pekmezci, M.; Villanueva-Meyer, J.E.; Goode, B.; Van Ziffle, J.; Onodera, C.; Grenert, J.P.; Bastian, B.C.; Chamyan, G.; Maher, O.M.; Khatib, Z.; et al. The genetic landscape of ganglioglioma. Acta Neuropathol. Commun. 2018, 6, 47. [Google Scholar] [CrossRef]
- Mistry, M.; Zhukova, N.; Merico, D.; Rakopoulos, P.; Krishnatry, R.; Shago, M.; Stavropoulos, J.; Alon, N.; Pole, J.D.; Ray, P.N.; et al. BRAF Mutation and CDKN2A Deletion Define a Clinically Distinct Subgroup of Childhood Secondary High-Grade Glioma. J. Clin. Oncol. 2015, 33, 1015–1022. [Google Scholar] [CrossRef]
- Andrews, L.J.; Thornton, Z.A.; Saincher, S.S.; Yao, I.Y.; Dawson, S.; McGuinness, L.A.; Jones, H.E.; Jefferies, S.; Jefferies, S.C.; Cheng, H.-Y.; et al. Prevalence of BRAFV600 in glioma and use of BRAF Inhibitors in patients with BRAFV600 mutation-positive glioma: Systematic review. Neuro-Oncol. 2022, 24, 528–540. [Google Scholar] [CrossRef] [PubMed]
- McNulty, S.N.; Schwetye, K.E.; Ferguson, C.; Storer, C.E.; Ansstas, G.; Kim, A.H.; Gutmann, D.H.; Rubin, J.B.; Head, R.D.; Dahiya, S. BRAF mutations may identify a clinically distinct subset of glioblastoma. Sci. Rep. 2021, 11, 19999. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; McCurrach, B.; Lowe, S.W. Oncogenic Ras Provokes Premature Cell Senescence Associated with Accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Blake, S.; Kusuma, F.K.; Pearson, R.B.; Kang, J.; Chan, K.T. Oncogene-induced senescence: From biology to therapy. Mech. Ageing Dev. 2020, 187, 111229. [Google Scholar] [CrossRef] [PubMed]
- Michaloglou, C.; Vredeveld, L.C.W.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; Van Der Horst, C.M.A.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Deschênes-Simard, X.; Gaumont-Leclerc, M.-F.; Bourdeau, V.; Lessard, F.; Moiseeva, O.; Forest, V.; Igelmann, S.; Mallette, F.A.; Saba-El-Leil, M.K.; Meloche, S.; et al. Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes Dev. 2013, 27, 900–915. [Google Scholar] [CrossRef] [PubMed]
- Garnett, S.; Dutchak, K.L.; McDonough, R.V.; Dankort, D. p53 loss does not permit escape from BRAFV600E-induced senescence in a mouse model of lung cancer. Oncogene 2017, 36, 6325–6335. [Google Scholar] [CrossRef]
- Raabe, E.H.; Lim, K.S.; Kim, J.M.; Meeker, A.; Mao, X.-G.; Nikkhah, G.; Maciaczyk, J.; Kahlert, U.; Jain, D.; Bar, E.; et al. BRAF Activation Induces Transformation and Then Senescence in Human Neural Stem Cells: A Pilocytic Astrocytoma Model. Clin. Cancer Res. 2011, 17, 3590–3599. [Google Scholar] [CrossRef]
- Palazzo, A.; Hernandez-Vargas, H.; Goehrig, D.; Médard, J.-J.; Vindrieux, D.; Flaman, J.-M.; Bernard, D. Transformed cells after senescence give rise to more severe tumor phenotypes than transformed non-senescent cells. Cancer Lett. 2022, 546, 215850. [Google Scholar] [CrossRef]
- Lassaletta, A.; Zapotocky, M.; Mistry, M.; Ramaswamy, V.; Honnorat, M.; Krishnatry, R.; Stucklin, A.S.G.; Zhukova, N.; Arnoldo, A.; Ryall, S.; et al. Therapeutic and Prognostic Implications of BRAF V600E in Pediatric Low-Grade Gliomas. J. Clin. Oncol. 2017, 35, 2934–2941. [Google Scholar] [CrossRef]
- Schiffman, J.D.; Hodgson, J.G.; VandenBerg, S.R.; Flaherty, P.; Polley, M.-Y.C.; Yu, M.; Fisher, P.G.; Rowitch, D.H.; Ford, J.M.; Berger, M.S.; et al. Oncogenic BRAF Mutation with CDKN2A Inactivation Is Characteristic of a Subset of Pediatric Malignant Astrocytomas. Cancer Res. 2010, 70, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Mackay, A.; Burford, A.; Molinari, V.; Jones, D.T.; Izquierdo, E.; Brouwer-Visser, J.; Giangaspero, F.; Haberler, C.; Pietsch, T.; Jacques, T.S.; et al. Molecular, Pathological, Radiological, and Immune Profiling of Non-brainstem Pediatric High-Grade Glioma from the HERBY Phase II Randomized Trial. Cancer Cell 2018, 33, 829–842.e5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.-Q.; Shi, Z.; Chen, H.; Chung, N.Y.-F.; Yin, Z.; Li, K.K.-W.; Chan, D.T.-M.; Poon, W.S.; Wu, J.; Zhou, L.; et al. Biomarker-based prognostic stratification of young adult glioblastoma. Oncotarget 2016, 7, 5030–5041. [Google Scholar] [CrossRef] [PubMed]
- Rudà, R.; Capper, D.; Waldman, A.D.; Pallud, J.; Minniti, G.; Kaley, T.J.; Bouffet, E.; Tabatabai, G.; Aronica, E.; Jakola, A.S.; et al. EANO—EURACAN—SNO Guidelines on circumscribed astrocytic gliomas, glioneuronal, and neuronal tumors. Neuro-Oncology 2022, 24, 2015–2034. [Google Scholar] [CrossRef]
- Dasgupta, T.; Olow, A.K.; Yang, X.; Hashizume, R.; Nicolaides, T.P.; Tom, M.; Aoki, Y.; Berger, M.S.; Weiss, W.A.; Stalpers, L.J.A.; et al. Survival advantage combining a BRAF inhibitor and radiation in BRAF V600E-mutant glioma. J. Neuro-Oncol. 2016, 126, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Studebaker, A.; Bondra, K.; Seum, S.; Shen, C.; Phelps, D.A.; Chronowski, C.; Leasure, J.; Smith, P.D.; Kurmasheva, R.T.; Mo, X.; et al. Inhibition of MEK confers hypersensitivity to X-radiation in the context of BRAF mutation in a model of childhood astrocytoma: Synergism Between MEK Inhibition and Radiation Therapy. Pediatr. Blood Cancer 2015, 62, 1768–1774. [Google Scholar] [CrossRef]
- Ahrendsen, J.T.; Sinai, C.; Meredith, D.M.; Malinowski, S.W.; Cooney, T.M.; Bandopadhayay, P.; Ligon, K.L.; Alexandrescu, S. Molecular Alterations in Pediatric Low-GradeGliomas That Led to Death. J. Neuropathol. Exp. Neurol. 2021, 80, 1052–1059. [Google Scholar]
- Dono, A.; Vu, J.; Anapolsky, M.; Hines, G.; Takayasu, T.; Yan, Y.; Tandon, N.; Zhu, J.-J.; Bhattacharjee, M.B.; Ballester, L.Y. Additional genetic alterations in BRAF-mutant gliomas correlate with histologic diagnoses. J. Neuro-Oncol. 2020, 149, 463–472. [Google Scholar] [CrossRef]
- Coutant, M.; Lhermitte, B.; Guérin, E.; Chammas, A.; Reita, D.; Sebastia, C.; Douzal, V.; Gabor, F.; Salmon, A.; Chenard, M.; et al. Retrospective and integrative analyses of molecular characteristics and their specific imaging parameters in pediatric grade 1 gliomas. Pediatr. Blood Cancer 2022, 69, e29575. [Google Scholar] [CrossRef]
- Rosenberg, T.; Yeo, K.K.; Mauguen, A.; Alexandrescu, S.; Prabhu, S.P.; Tsai, J.W.; Malinowski, S.; Joshirao, M.; Parikh, K.; Sait, S.F.; et al. Upfront molecular targeted therapy for the treatment of BRAF-mutant pediatric high-grade glioma. Neuro-Oncol. 2022, 24, 1964–1975. [Google Scholar] [CrossRef]
- Lucas, C.-H.G.; Davidson, C.J.; Alashari, M.; Putnam, A.R.; Whipple, N.S.; Bruggers, C.S.; Mendez, J.S.; Cheshier, S.H.; Walker, J.B.; Ramani, B.; et al. Targeted Next-Generation Sequencing Reveals Divergent Clonal Evolution in Components of Composite Pleomorphic Xanthoastrocytoma-Ganglioglioma. J. Neuropathol. Exp. Neurol. 2022, 81, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.J.; Brosnan-Cashman, J.A.; Allen, S.J.; Vizcaino, M.A.; Giannini, C.; Camelo-Piragua, S.; Webb, M.; Matsushita, M.; Wadhwani, N.; Tabbarah, A.; et al. Alternative lengthening of telomeres, ATRX loss and H3-K27M mutations in histologically defined pilocytic astrocytoma with anaplasia. Brain Pathol. 2019, 29, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Li, X.; Kong, S.; Shang, S.; Qi, Y. CDK4/6 inhibition suppresses tumour growth and enhances the effect of temozolomide in glioma cells. J. Cell. Mol. Med. 2020, 24, 5135–5145. [Google Scholar] [CrossRef] [PubMed]
- DeSisto, J.; Lucas, J.T.; Xu, K.; Donson, A.; Lin, T.; Sanford, B.; Wu, G.; Tran, Q.T.; Hedges, D.; Hsu, C.-Y.; et al. Comprehensive molecular characterization of pediatric radiation-induced high-grade glioma. Nat. Commun. 2021, 12, 5531. [Google Scholar] [CrossRef]
- Liu, R.; Bishop, J.; Zhu, G.; Zhang, T.; Ladenson, P.W.; Xing, M. Mortality Risk Stratification by Combining BRAF V600E and TERT Promoter Mutations in Papillary Thyroid Cancer: Genetic Duet of BRAF and TERT Promoter Mutations in Thyroid Cancer Mortality. JAMA Oncol. 2016, 3, 202–208. [Google Scholar] [CrossRef]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef]
- Koelsche, C.; Sahm, F.; Capper, D.; Reuss, D.; Sturm, D.; Jones, D.T.W.; Kool, M.; Northcott, P.A.; Wiestler, B.; Böhmer, K.; et al. Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathol. 2013, 126, 907–915. [Google Scholar] [CrossRef]
- Korshunov, A.; Chavez, L.; Sharma, T.; Ryzhova, M.; Schrimpf, D.; Stichel, D.; Capper, D.; Sturm, D.; Kool, M.; Habel, A.; et al. Epithelioid glioblastomas stratify into established diagnostic subsets upon integrated molecular analysis. Brain Pathol. 2018, 28, 656–662. [Google Scholar] [CrossRef]
- Liu, R.; Zhang, T.; Zhu, G.; Xing, M. Regulation of mutant TERT by BRAF V600E/MAP kinase pathway through FOS/GABP in human cancer. Nat. Commun. 2018, 9, 579. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Tan, J.; Shen, X.; Jiang, K.; Wang, C.; Zhu, G.; Xing, M. Therapeutic targeting of FOS in mutant TERT cancers through removing TERT suppression of apoptosis via regulating survivin and TRAIL-R2. Proc. Natl. Acad. Sci. USA 2021, 118, e2022779118. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, J.; Johannessen, T.C.; Ohba, S.; Chow, T.T.; Jones, L.; Pandita, A.; Pieper, R.O. Mutant IDH1 Cooperates with ATRX Loss to Drive the Alternative Lengthening of Telomere Phenotype in Glioma. Cancer Res. 2018, 78, 2966–2977. [Google Scholar] [CrossRef]
- Murakami, C.; Yoshida, Y.; Yamazaki, T.; Yamazaki, A.; Nakata, S.; Hokama, Y.; Ishiuchi, S.; Akimoto, J.; Shishido-Hara, Y.; Yoshimoto, Y.; et al. Clinicopathological characteristics of circumscribed high-grade astrocytomas with an unusual combination of BRAF V600E, ATRX, and CDKN2A/B alterations. Brain Tumor Pathol. 2019, 36, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Phillips, J.; Onar-Thomas, A.; Romero, E.; Zheng, S.; Wiencke, J.K.; McBride, S.M.; Cowdrey, C.; Prados, M.D.; Weiss, W.A.; et al. PTEN promoter methylation and activation of the PI3K/Akt/mTOR pathway in pediatric gliomas and influence on clinical outcome. Neuro-Oncol. 2012, 14, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Hütt-Cabezas, M.; Karajannis, M.A.; Zagzag, D.; Shah, S.; Horkayne-Szakaly, I.; Rushing, E.J.; Cameron, J.D.; Jain, D.; Eberhart, C.G.; Raabe, E.H.; et al. Activation of mTORC1/mTORC2 signaling in pediatric low-grade glioma and pilocytic astrocytoma reveals mTOR as a therapeutic target. Neuro-Oncol. 2013, 15, 1604–1614. [Google Scholar] [CrossRef] [PubMed]
- Prabowo, A.S.; Iyer, A.M.; Veersema, T.J.; Anink, J.J.; Meeteren, A.Y.N.S.-V.; Spliet, W.G.M.; van Rijen, P.C.; Ferrier, C.H.; Capper, D.; Thom, M.; et al. BRAF V600E Mutation Is Associated with mTOR Signaling Activation in Glioneuronal Tumors. Brain Pathol. 2014, 24, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Cases-Cunillera, S.; van Loo, K.M.J.; Pitsch, J.; Quatraccioni, A.; Sivalingam, S.; Salomoni, P.; Borger, V.; Dietrich, D.; Schoch, S.; Becker, A.J. Heterogeneity and excitability of BRAFV600E-induced tumors is determined by Akt/mTOR-signaling state and Trp53-loss. Neuro-Oncology 2022, 24, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537.e5. [Google Scholar] [CrossRef]
- Vredeveld, L.C.W.; Possik, P.A.; Smit, M.A.; Meissl, K.; Michaloglou, C.; Horlings, H.M.; Ajouaou, A.; Kortman, P.C.; Dankort, D.; McMahon, M.; et al. Abrogation of BRAFV600E-induced senescence by PI3K pathway activation contributes to melanomagenesis. Genes Dev. 2012, 26, 1055–1069. [Google Scholar] [CrossRef]
- Schreck, K.C.; Morin, A.; Zhao, G.; Allen, A.N.; Flannery, P.; Glantz, M.; Green, A.L.; Jones, C.; Jones, K.L.; Kilburn, L.B.; et al. Deconvoluting Mechanisms of Acquired Resistance to RAF Inhibitors in BRAFV600E-Mutant Human Glioma. Clin. Cancer Res. 2021, 27, 6197–6208. [Google Scholar] [CrossRef]
- Yao, Z.; Torres, N.M.; Tao, A.; Gao, Y.; Luo, L.; Li, Q.; de Stanchina, E.; Abdel-Wahab, O.; Solit, D.B.; Poulikakos, P.I.; et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell 2015, 28, 370–383. [Google Scholar] [CrossRef]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.W.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA Hypomethylation Are Major Drivers of Gene Expression in K27M Mutant Pediatric High-Grade Gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Z.; Cui, Y.; Liu, Y.; Fang, J.; Xu, L.; He, Y.; Du, J.; Su, Y.; Su, W.; et al. Evaluation of EZH2 expression, BRAF V600E mutation, and CDKN2A/B deletions in epithelioid glioblastoma and anaplastic pleomorphic xanthoastrocytoma. J. Neuro-Oncol. 2019, 144, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J. Clin. Oncol. 2016, 34, 2206–2211. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Stein, A.; Bent, M.V.D.; De Greve, J.; Wick, A.; de Vos, F.Y.F.L.; von Bubnoff, N.; E van Linde, M.; Lai, A.; Prager, G.W.; et al. Dabrafenib plus trametinib in patients with BRAFV600E-mutant low-grade and high-grade glioma (ROAR): A multicentre, open-label, single-arm, phase 2, basket trial. Lancet Oncol. 2022, 23, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Kaley, T.; Touat, M.; Subbiah, V.; Hollebecque, A.; Rodon, J.; Lockhart, A.C.; Keedy, V.; Bielle, F.; Hofheinz, R.-D.; Joly, F.; et al. BRAF Inhibition in BRAFV600-Mutant Gliomas: Results From the VE-BASKET Study. J. Clin. Oncol. 2018, 36, 3477. [Google Scholar]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, C.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Schreck, K.C.; Grossman, S.A.; Pratilas, C.A. BRAF Mutations and the Utility of RAF and MEK Inhibitors in Primary Brain Tumors. Cancers 2019, 11, 1262. [Google Scholar] [CrossRef]
- Kilburn, L.B.; Jabado, N.; Franson, A.; Chi, S.N.; Fisher, M.J.; Hargrave, D.R.; Hansford, J.R.; Ziegler, D.S.; Landi, D.; Kang, H.J.; et al. FIREFLY-1: A phase 2 study of the pan-RAF inhibitor DAY101 in pediatric patients with low-grade glioma. J. Clin. Oncol. 2021, 39 (Suppl. S15), TPS10056. [Google Scholar] [CrossRef]
- Cook, F.A.; Cook, S.J. Inhibition of RAF dimers: It takes two to tango. Biochem. Soc. Trans. 2021, 49, 237–251. [Google Scholar] [CrossRef]
- Haushild, A.; Gob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller Jr, W.H.; Kaempgen, E.; et al. Dabrafnib in BRAF-mutated metastatic melanoma: A multicenter, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Migliorini, D.; Aguiar, D.; Vargas, M.I.; Lobrinus, A.; Dietrich, P.Y. BRAF/MEK double blockade in refractory anaplastic pleomorphic xanthoastrocytoma. Neurology 2017, 88, 1291–1293. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.F.; Carter, T.; Kitchen, N.; Mulholland, P.; Kong, B.Y.; Carlino, M.S.; Menzies, A.M.; Roque, A.; Odia, Y.; Martin-Liberal, J.; et al. DaBRAFenib and trametinib in BRAFV600E mutated glioma. CNS Oncol. 2017, 6, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Nobre, L.; Zapotocky, M.; Ramaswamy, V.; Ryall, S.; Bennett, J.; Alderete, D.; Guill, J.B.; Baroni, L.; Bartels, U.; Bavle, A.; et al. Outcomes of BRAF V600E Pediatric Gliomas Treated With Targeted BRAF Inhibition. JCO Precis. Oncol. 2020, 4, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Bouffet, E.; Hansford, J.; Garré, M.L.; Hara, J.; Plant-Fox, A.; Aerts, I.; Locatelli, F.; der Lugt, J.V.; Papusha, L.; Sahm, F.; et al. Primary analysis of a phase II trial of daBRAFenib plus trametinib (dab + tram) in BRAFV600- mutant pediatric low-grade glioma (pLGG). J. Clin. Oncol. 2022, 40 (Suppl. S15), LBA2022. [Google Scholar] [CrossRef]
- Wang, J.; Yao, Z.; Jonsson, P.; Allen, A.N.; Qin, A.C.R.; Uddin, S.; Dunkel, I.J.; Petriccione, M.; Manova, K.; Haque, S.; et al. A Secondary Mutation in BRAF Confers Resistance to RAF Inhibition in a BRAFV600E-Mutant Brain Tumor. Cancer Discov. 2018, 8, 1130–1141. [Google Scholar] [CrossRef]

{kind=link}
| Alterations | LGG vs. HGG | Cell Phenotype | Outcome | Therapeutic Resistance | |
|---|---|---|---|---|---|
| ARAF/CRAF | amplification | HGG | no data | Worst | no data |
| CDKN2A/B | deletion | LGG HGG | progression progression | Worst | no data |
| CDK4/6 | overexpression | HGG | progression | no data | resistance to temozolomide |
| TERT | mutation or amplification | HGG | progression | no data | no data |
| ATRX | mutation | HGG | progression | no data | no data |
| mTor | activation | LGG HGG | progression | no data | no data |
| PTEN | deletion | LGG HGG | progression progression | Worst no data | resistance to chemotherapy no data |
| NF1 | deletion | LGG HGG | no data no data | resistance to targeted drugs in class III mutation | |
| EZH2 | overexpression | HGG | progression | Worst | no data |
| immune CD8 cells | presence | HGG | no data | better | higher response to radiotherapy and chemotherapy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lhermitte, B.; Wolf, T.; Chenard, M.P.; Coca, A.; Todeschi, J.; Proust, F.; Hirsch, E.; Schott, R.; Noel, G.; Guerin, E.; et al. Molecular Heterogeneity in BRAF-Mutant Gliomas: Diagnostic, Prognostic, and Therapeutic Implications. Cancers 2023, 15, 1268. https://doi.org/10.3390/cancers15041268
Lhermitte B, Wolf T, Chenard MP, Coca A, Todeschi J, Proust F, Hirsch E, Schott R, Noel G, Guerin E, et al. Molecular Heterogeneity in BRAF-Mutant Gliomas: Diagnostic, Prognostic, and Therapeutic Implications. Cancers. 2023; 15(4):1268. https://doi.org/10.3390/cancers15041268
Chicago/Turabian StyleLhermitte, Benoit, Thibaut Wolf, Marie Pierre Chenard, Andres Coca, Julien Todeschi, François Proust, Edouard Hirsch, Roland Schott, Georges Noel, Eric Guerin, and et al. 2023. "Molecular Heterogeneity in BRAF-Mutant Gliomas: Diagnostic, Prognostic, and Therapeutic Implications" Cancers 15, no. 4: 1268. https://doi.org/10.3390/cancers15041268
APA StyleLhermitte, B., Wolf, T., Chenard, M. P., Coca, A., Todeschi, J., Proust, F., Hirsch, E., Schott, R., Noel, G., Guerin, E., Reita, D., Chammas, A., Salmon, A., Martin, S., Dontenwill, M., & Entz-Werlé, N. (2023). Molecular Heterogeneity in BRAF-Mutant Gliomas: Diagnostic, Prognostic, and Therapeutic Implications. Cancers, 15(4), 1268. https://doi.org/10.3390/cancers15041268