
Individualized Multimodal Immunotherapy for Adults with IDH1 Wild-Type GBM: A Single Institute Experience
 , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Patients and Methods
2.1. Patients
2.2. Individualized Multimodal Immunotherapy
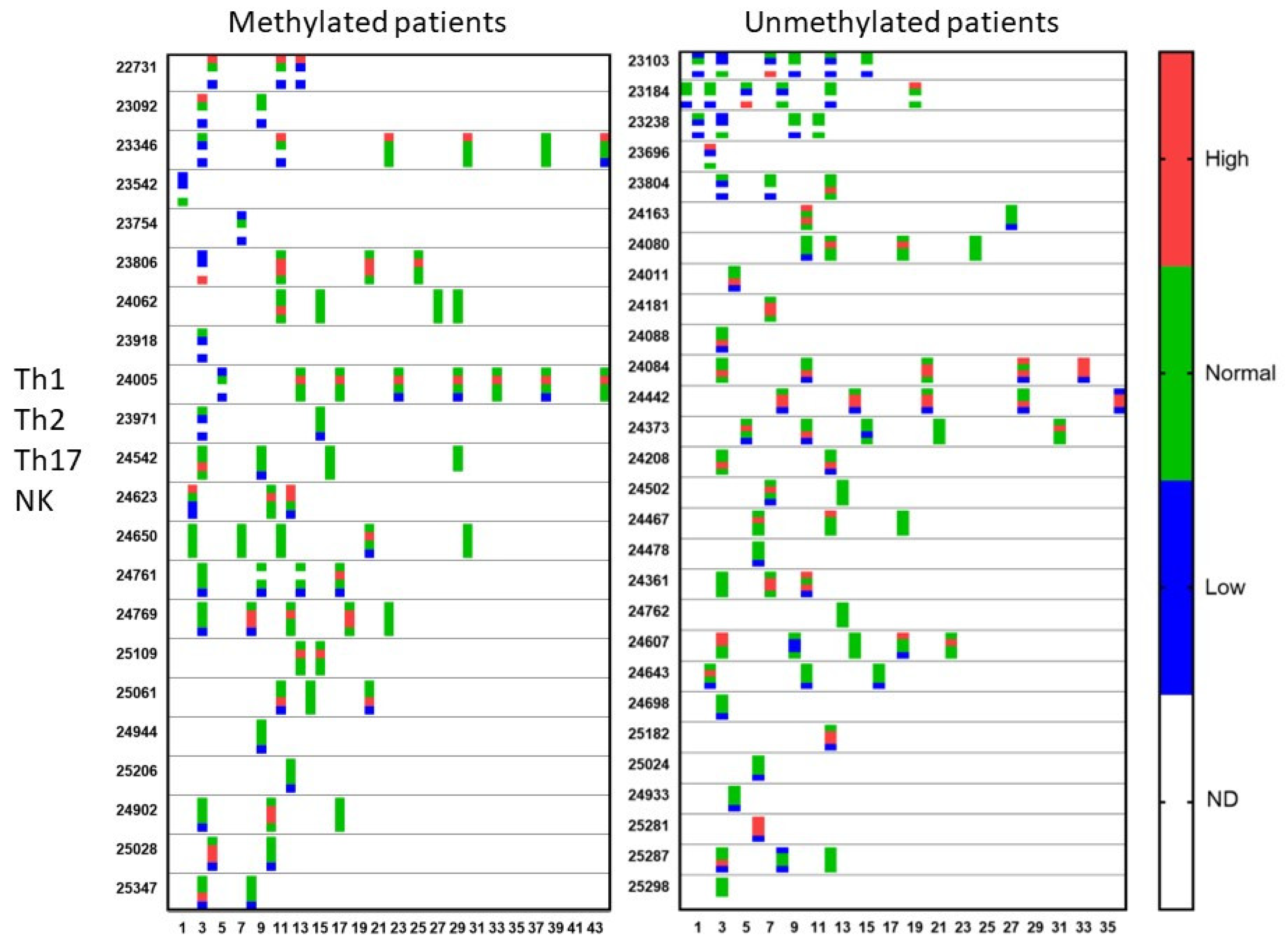
2.3. Accompanying Laboratory Tests
2.4. Statistics
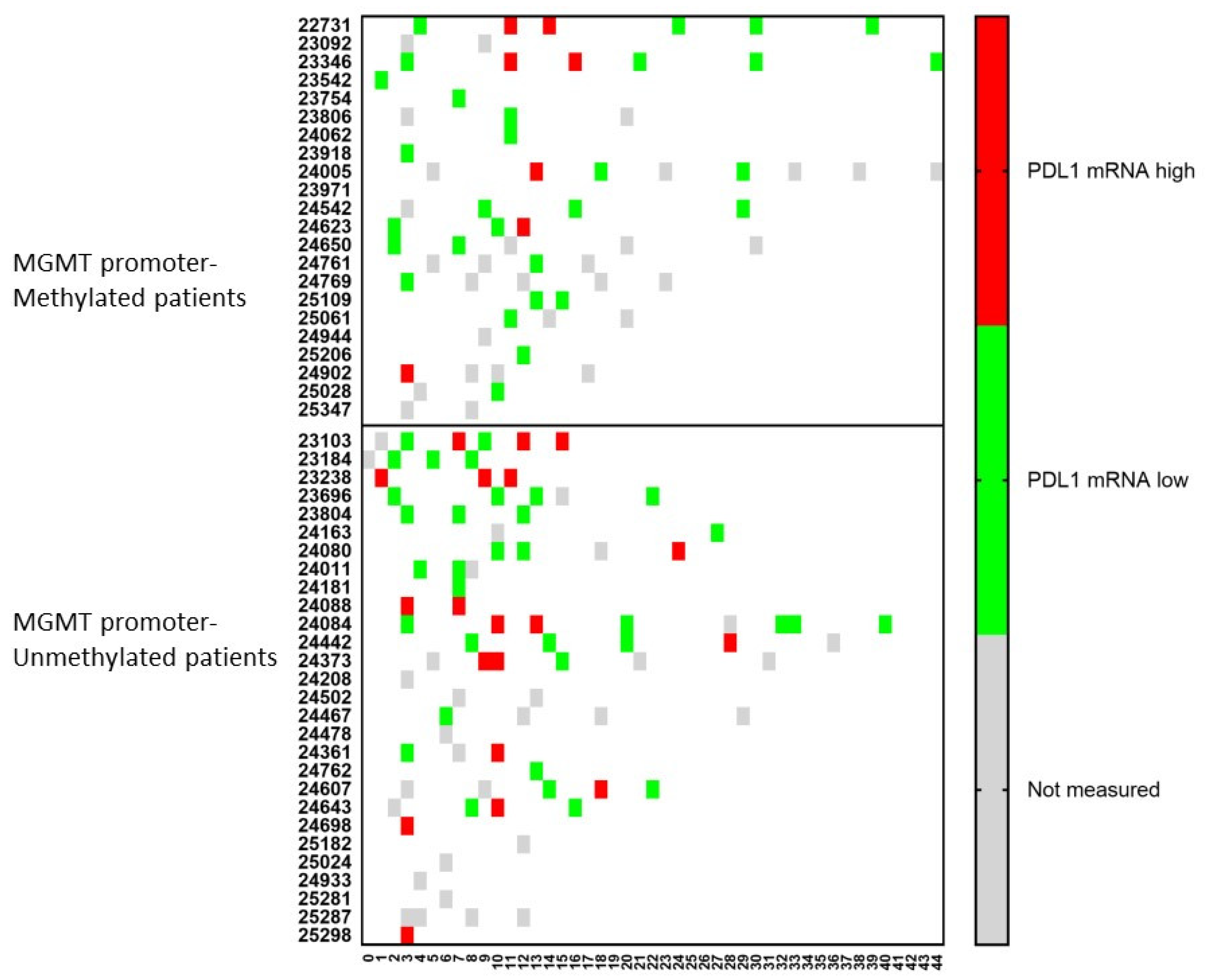
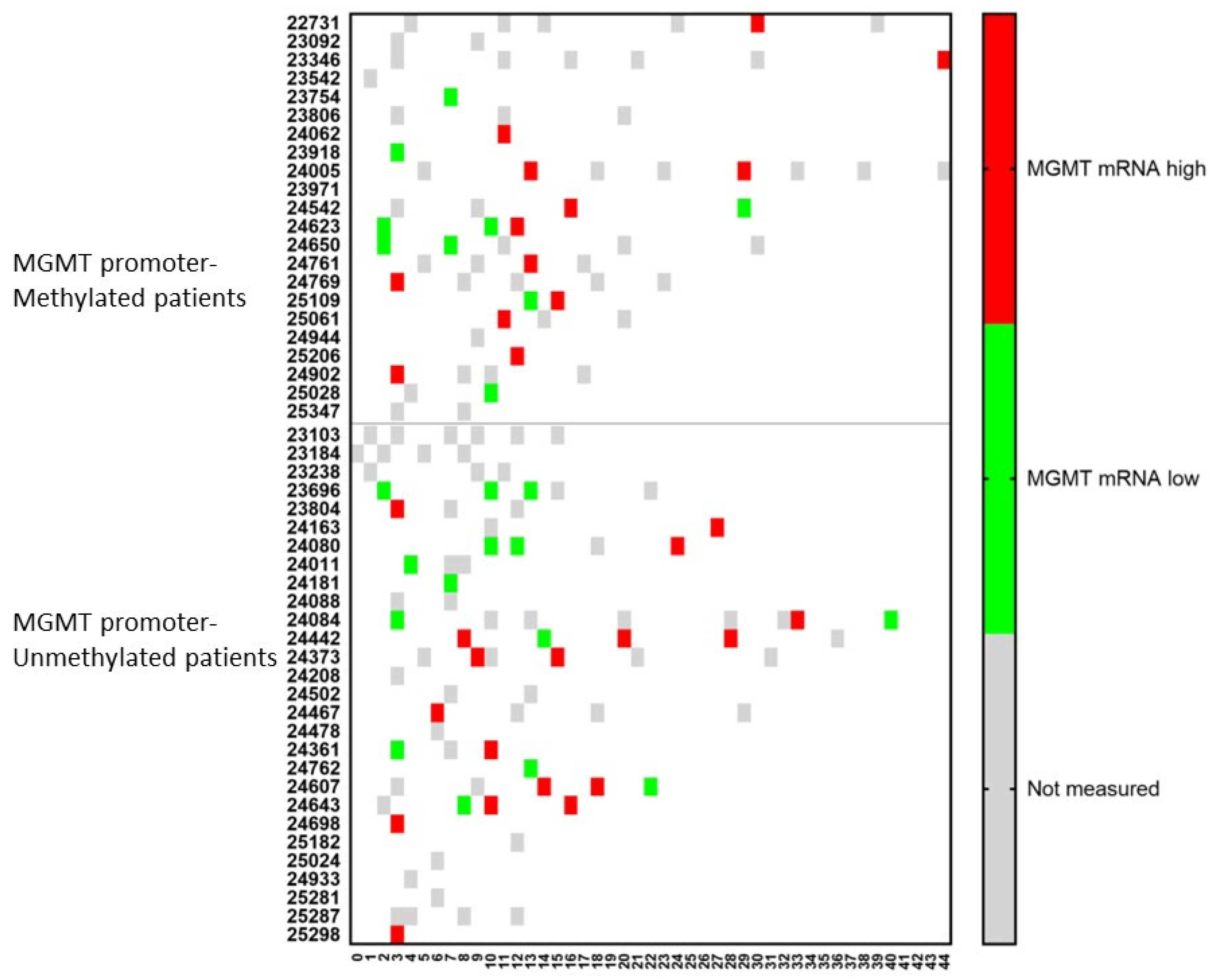
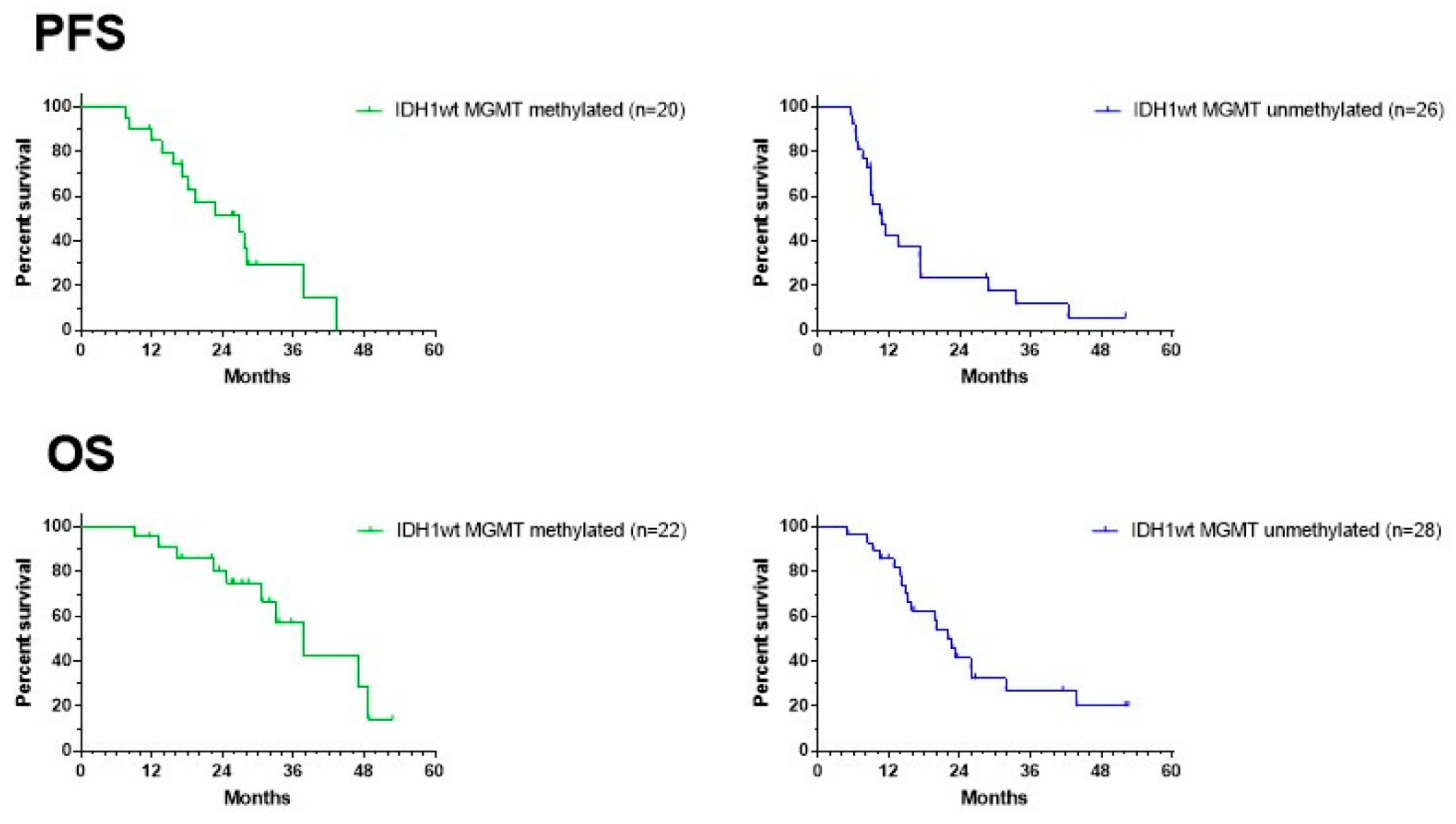
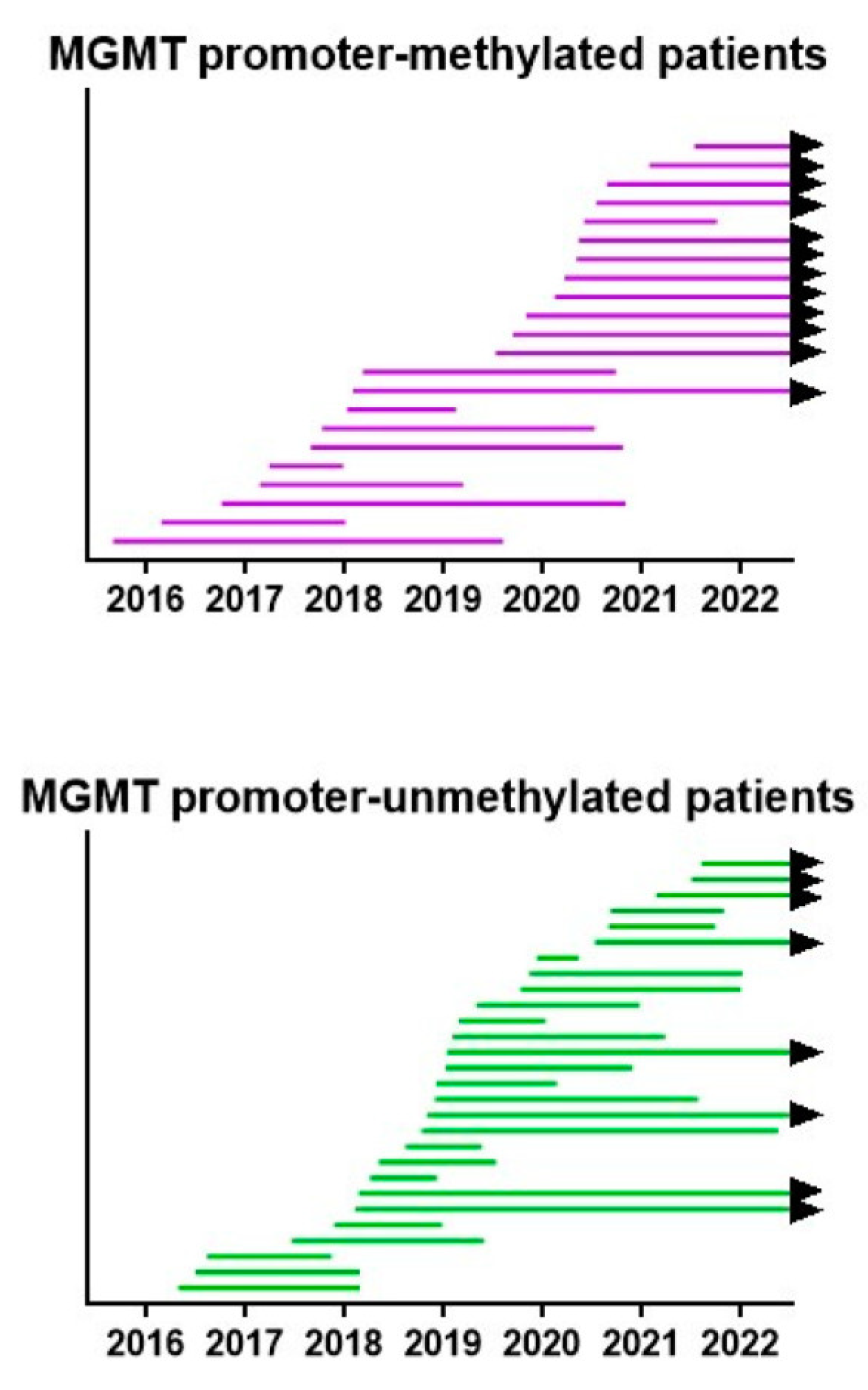
3. Results
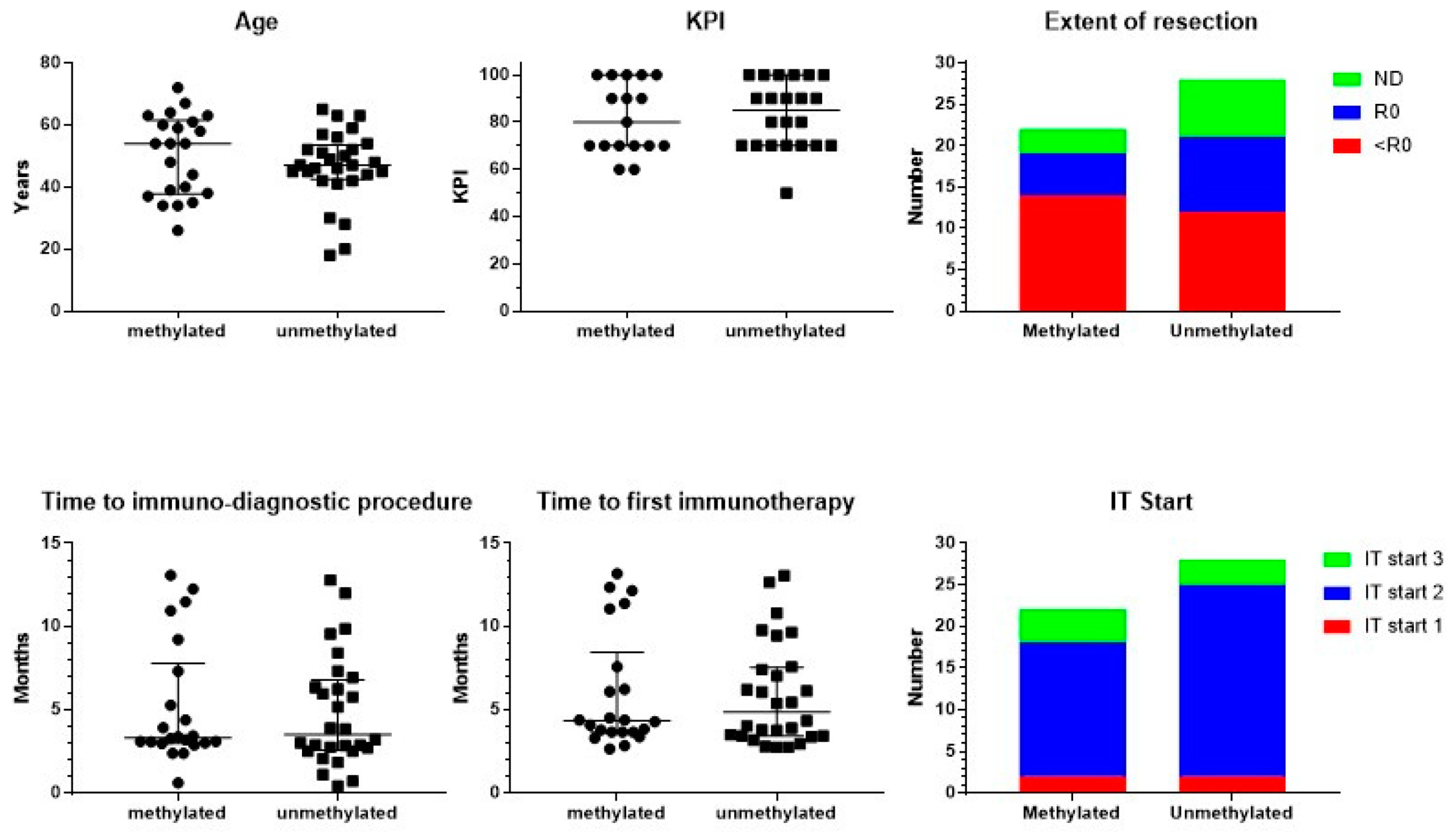
3.1. Patient Characteristics
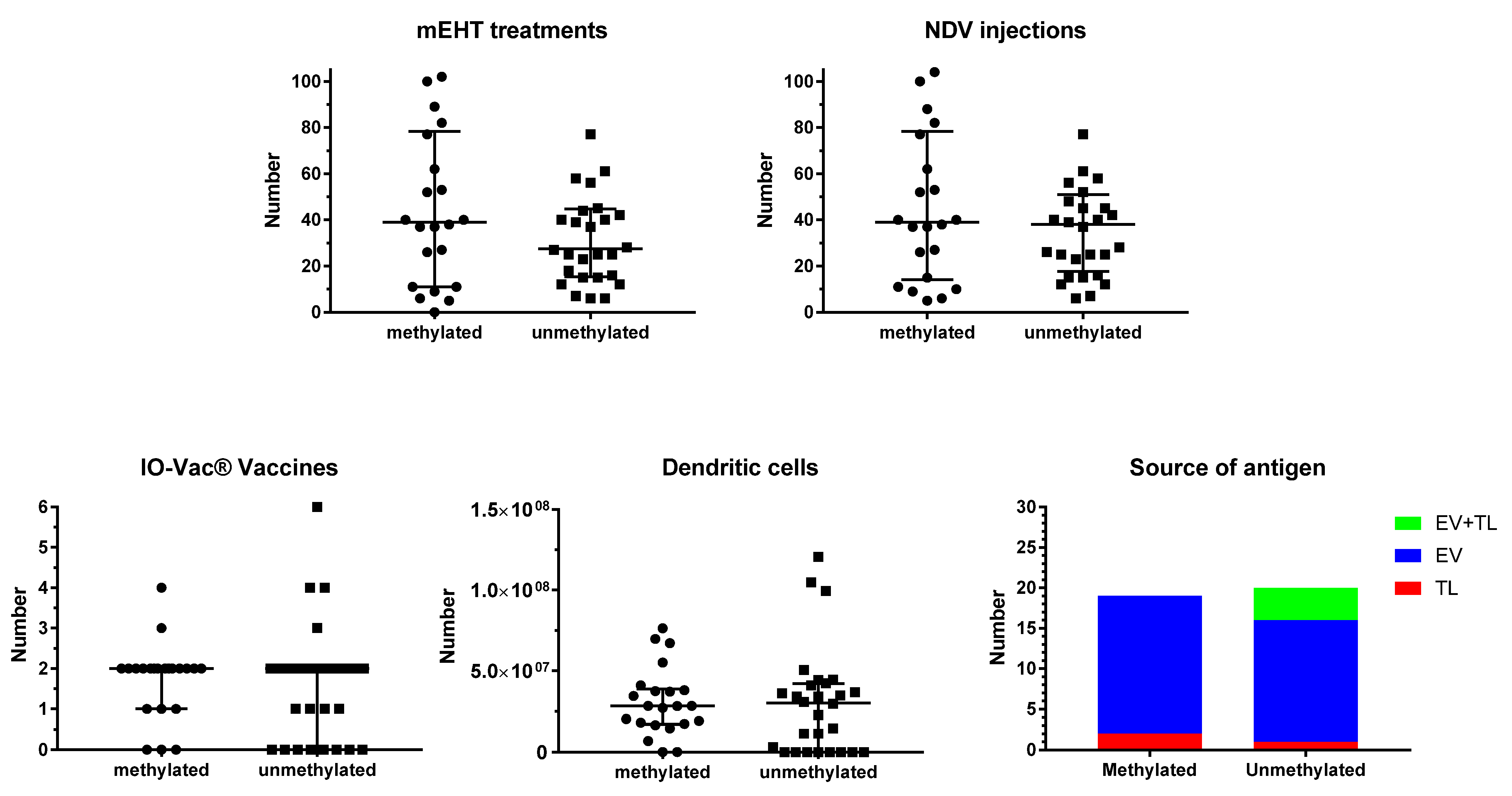
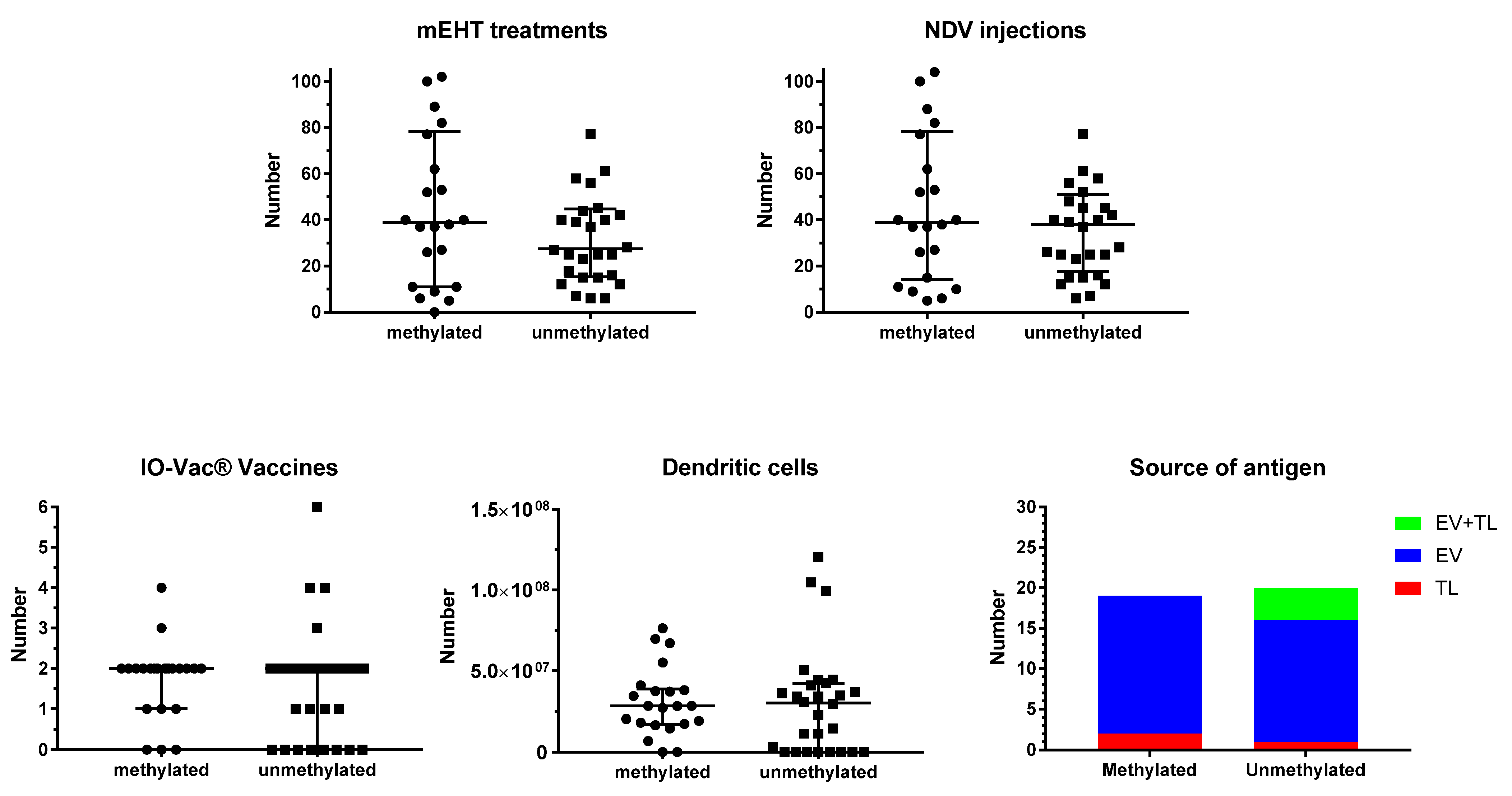
3.2. Treatment
3.3. Evolution of the Patients
3.4. Survival Outcome of the Patients
3.5. Side Effects
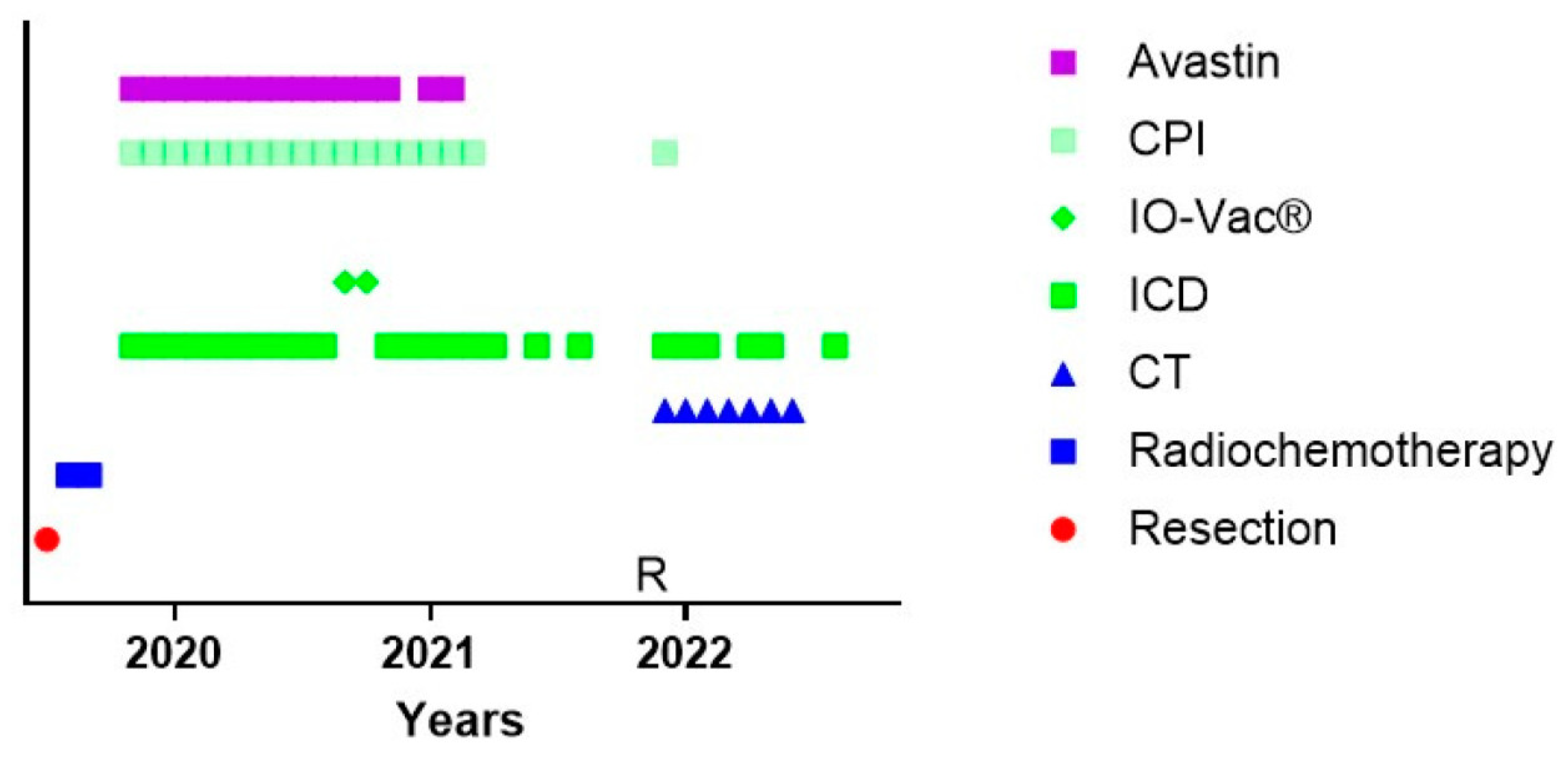
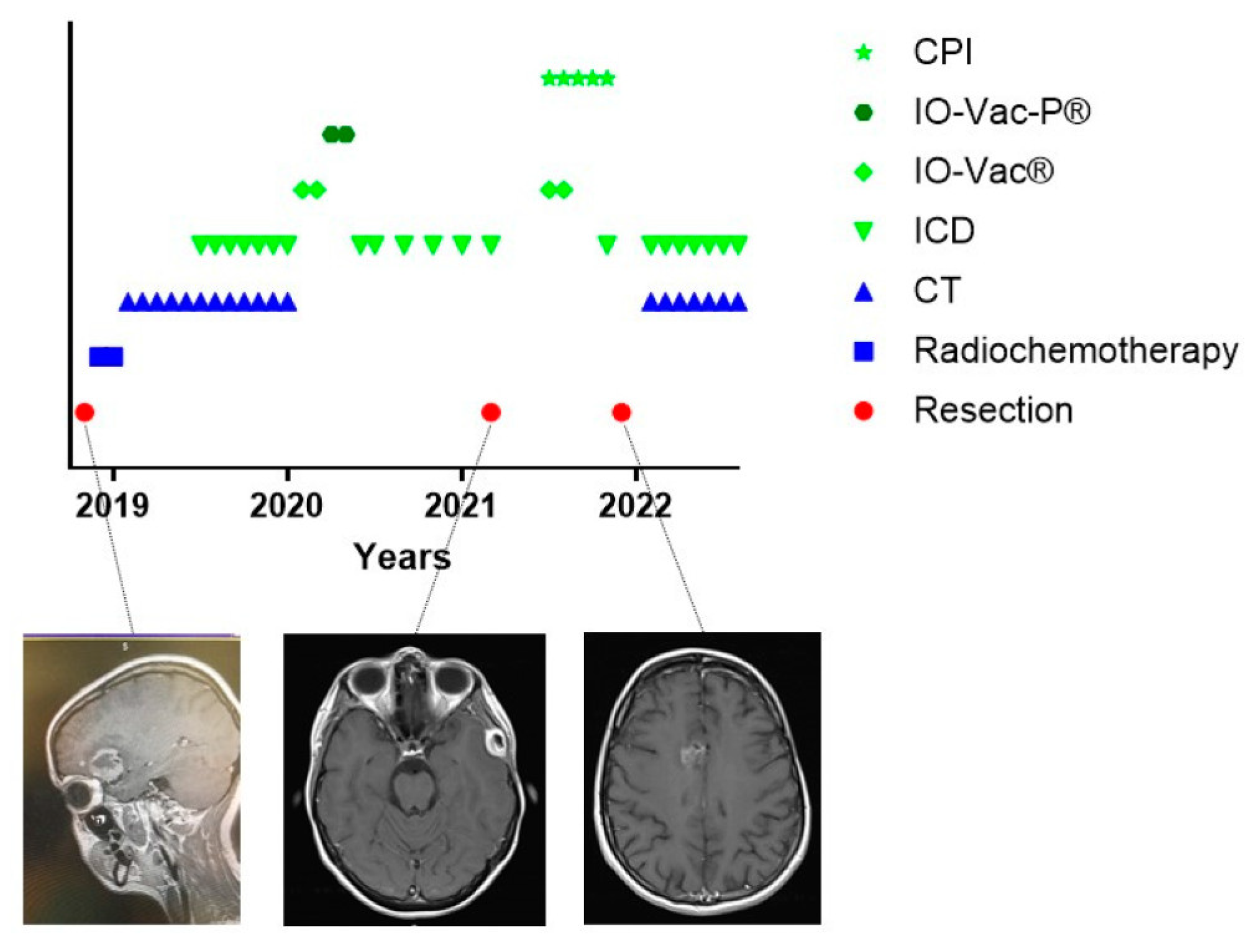
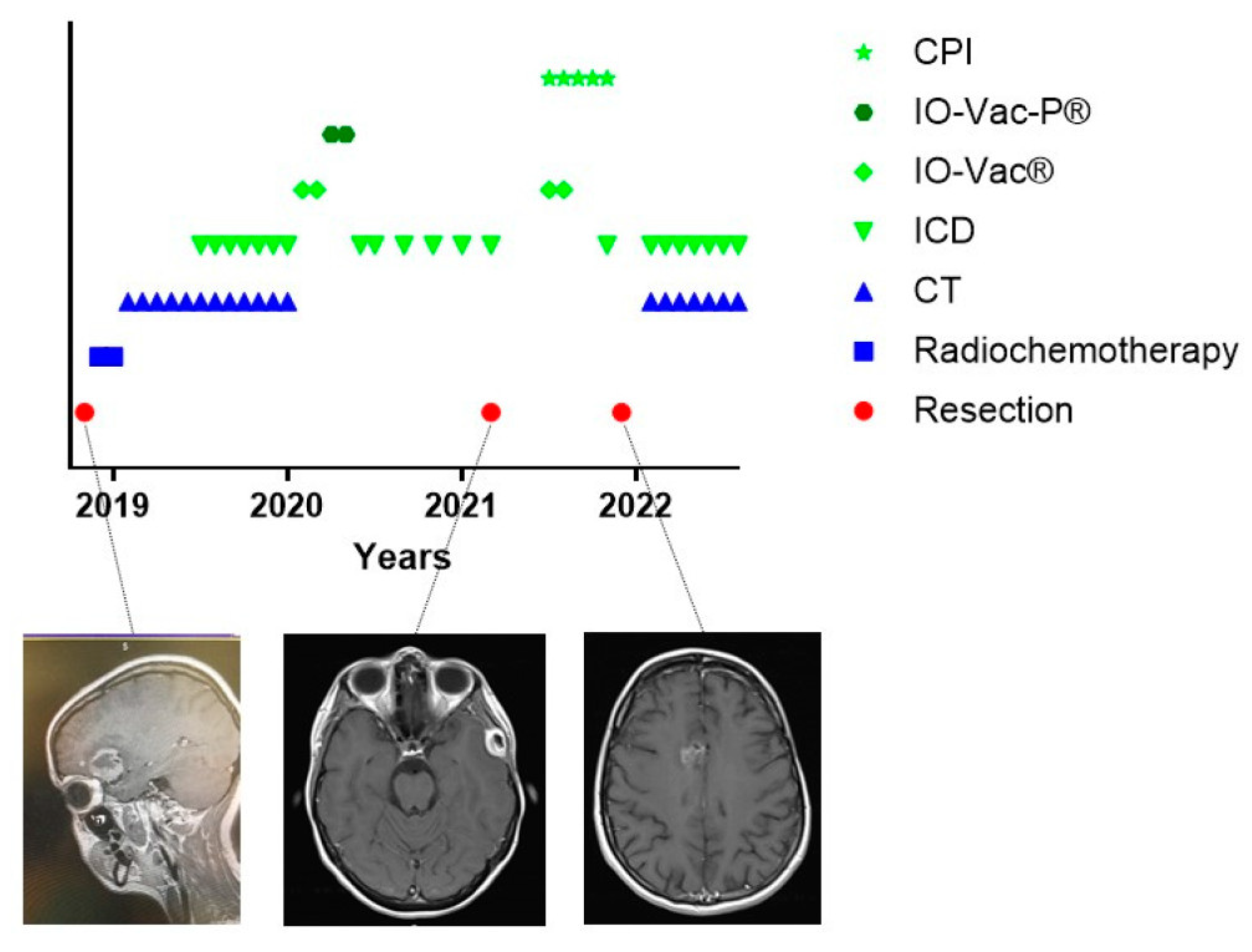
3.6. The Challenge of Keeping Tumor Control in a Dynamic Cancer Disease
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahn, B.J.; Pollack, I.F.; Okada, H. Immune-Checkpoint Blockade and Active Immunotherapy for Glioma. Cancers 2013, 5, 1379–1412. [Google Scholar] [CrossRef]
- Lieberman, N.A.P.; Vitanza, N.A.; Crane, C.A. Immunotherapy for brain tumors: Understanding early successes and limitations. Expert Rev. Neurother. 2018, 18, 251–259. [Google Scholar] [CrossRef]
- McGranahan, T.; Therkelsen, K.E.; Ahmad, S.; Nagpal, S. Current State of Immunotherapy for Treatment of Glioblastoma. Curr. Treat. Options Oncol. 2019, 20, 24. [Google Scholar] [CrossRef]
- Wang, X.; Guo, G.; Guan, H.; Yu, Y.; Lu, J.; Yu, J. Challenges and potential of PD-1/PD-L1 checkpoint blockade immunotherapy for glioblastoma. J. Exp. Clin. Cancer Res. 2019, 38, 87. [Google Scholar] [CrossRef]
- The American Cancer Society Medical and Editorial Content Team. Immunotherapy. Available online: https://www.cancer.org/content/dam/CRC/PDF/Public/6678.00.pdf (accessed on 1 September 2022).
- De Vleeschouwer, S.; Van Gool, S.W.; Van Calenbergh, F. Immunotherapy for malignant gliomas: Emphasis on strategies of active specific immunotherapy using autologous dendritic cells. Childs Nerv. Syst. 2005, 21, 7–18. [Google Scholar] [CrossRef]
- Wolff, J.E.; Wagner, S.; Reinert, C.; Gnekow, A.; Kortmann, R.D.; Kuhl, J.; Van Gool, S.W. Maintenance treatment with interferon-gamma and low-dose cyclophosphamide for pediatric high-grade glioma. J. Neurooncol. 2006, 79, 315–321. [Google Scholar] [CrossRef]
- Lampson, L.A. Monoclonal antibodies in neuro-oncology: Getting past the blood-brain barrier. MAbs 2011, 3, 153–160. [Google Scholar] [CrossRef]
- Reardon, D.A.; Akabani, G.; Coleman, R.E.; Friedman, A.H.; Friedman, H.S.; Herndon, J.E.; McLendon, R.E.; Pegram, C.N.; Provenzale, J.M.; Quinn, J.A.; et al. Salvage radioimmunotherapy with murine iodine-131-labeled antitenascin monoclonal antibody 81C6 for patients with recurrent primary and metastatic malignant brain tumors: Phase II study results. J. Clin. Oncol. 2006, 24, 115–122. [Google Scholar] [CrossRef]
- Lin, Y.; Okada, H. Cellular immunotherapy for malignant gliomas. Expert Opin. Biol. Ther. 2016, 16, 1265–1275. [Google Scholar] [CrossRef]
- Chuntova, P.; Downey, K.M.; Hegde, B.; Almeida, N.D.; Okada, H. Genetically Engineered T-Cells for Malignant Glioma: Overcoming the Barriers to Effective Immunotherapy. Front. Immunol. 2018, 9, 3062. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, Y.; Ji, N. Challenges in the Treatment of Glioblastoma by Chimeric Antigen Receptor T-Cell Immunotherapy and Possible Solutions. Front. Immunol. 2022, 13, 927132. [Google Scholar] [CrossRef]
- Smith, C.; Lineburg, K.E.; Martins, J.P.; Ambalathingal, G.R.; Neller, M.A.; Morrison, B.; Matthews, K.K.; Rehan, S.; Crooks, P.; Panikkar, A.; et al. Autologous CMV-specific T cells are a safe adjuvant immunotherapy for primary glioblastoma multiforme. J. Clin. Investig. 2020, 130, 6041–6053. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef]
- Koks, C.A.E.; Garg, A.D.; Ehrhardt, M.; Riva, M.; De Vleeschouwer, S.; Agostinis, P.; Graf, N.; Van Gool, S.W. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int. J. Cancer 2014, 136, e313–e325. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.A.; Kesari, S.; Steinberg, D.M.; Toms, S.A.; Taylor, L.P.; Lieberman, F.; Silvani, A.; Fink, K.L.; et al. Maintenance Therapy With Tumor-Treating Fields Plus Temozolomide vs Temozolomide Alone for Glioblastoma: A Randomized Clinical Trial. JAMA 2015, 314, 2535–2543. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef]
- Voloshin, T.; Kaynan, N.; Davidi, S.; Porat, Y.; Shteingauz, A.; Schneiderman, R.S.; Zeevi, E.; Munster, M.; Blat, R.; Tempel Brami, C.; et al. Tumor-treating fields (TTFields) induce immunogenic cell death resulting in enhanced antitumor efficacy when combined with anti-PD-1 therapy. Cancer Immunol. Immunother. 2020, 69, 1191–1204. [Google Scholar] [CrossRef]
- Fiorentini, G.; Sarti, D.; Milandri, C.; Dentico, P.; Mambrini, A.; Fiorentini, C.; Mattioli, G.; Casadei, V.; Guadagni, S. Modulated Electrohyperthermia in Integrative Cancer Treatment for Relapsed Malignant Glioblastoma and Astrocytoma: Retrospective Multicenter Controlled Study. Integr. Cancer Ther. 2018, 18, 1534735418812691. [Google Scholar] [CrossRef]
- Vancsik, T.; Kovago, C.; Kiss, E.; Papp, E.; Forika, G.; Benyo, Z.; Meggyeshazi, N.; Krenacs, T. Modulated electro-hyperthermia induced loco-regional and systemic tumor destruction in colorectal cancer allografts. J. Cancer 2018, 9, 41–53. [Google Scholar] [CrossRef]
- Minnaar, C.A.; Kotzen, J.A.; Ayeni, O.A.; Vangu, M.D.; Baeyens, A. Potentiation of the Abscopal Effect by Modulated Electro-Hyperthermia in Locally Advanced Cervical Cancer Patients. Front. Oncol. 2020, 10, 376. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Soyano, A.E.; Dholaria, B.; Knutson, K.L.; Lou, Y. Cancer immunotherapy beyond immune checkpoint inhibitors. J. Hematol. Oncol. 2018, 11, 8. [Google Scholar] [CrossRef]
- Mende, A.L.; Schulte, J.D.; Okada, H.; Clarke, J.L. Current Advances in Immunotherapy for Glioblastoma. Curr. Oncol. Rep. 2021, 23, 21. [Google Scholar] [CrossRef]
- Khasraw, M.; Reardon, D.A.; Weller, M.; Sampson, J.H. PD-1 inhibitors: Do they have a future in the treatment of glioblastoma? Clin. Cancer Res. 2020, 26, 5287–5296. [Google Scholar] [CrossRef]
- Bausart, M.; Preat, V.; Malfanti, A. Immunotherapy for glioblastoma: The promise of combination strategies. J. Exp. Clin. Cancer Res. 2022, 41, 35. [Google Scholar] [CrossRef]
- Schirrmacher, V.; Sprenger, T.; Stuecker, W.; Van Gool, S.W. Evidence-Based Medicine in Oncology: Commercial Versus Patient Benefit. Biomedicines 2020, 8, 237. [Google Scholar] [CrossRef]
- Dejaegher, J.; Solie, L.; Hunin, Z.; Sciot, R.; Capper, D.; Siewert, C.; Van Cauter, S.; Wilms, G.; van Loon, J.; Ectors, N.; et al. Methylation based glioblastoma subclassification is related to tumoral T cell infiltration and survival. Neuro-Oncology 2021, 23, 240–250. [Google Scholar] [CrossRef]
- Van Gool, S.W.; Makalowski, J.; Bitar, M.; Van de Vliet, P.; Schirrmacher, V.; Stuecker, W. Synergy between TMZ and individualized multimodal immunotherapy to improve overall survival of IDH1 wild-type MGMT promoter-unmethylated GBM patients. Genes Immun. 2022, 23, 255–259. [Google Scholar] [CrossRef]
- Van Gool, S.W.; Makalowski, J.; Feyen, O.; Prix, L.; Schirrmacher, V.; Stuecker, W. The induction of immunogenic cell death (ICD) during maintenance chemotherapy and subsequent multimodal immunotherapy for glioblastoma (GBM). Austin Oncol. Case Rep. 2018, 3, 1010. [Google Scholar]
- Van Gool, S.W.; Makalowski, J.; Fiore, S.; Sprenger, T.; Prix, L.; Schirrmacher, V.; Stuecker, W. Randomized controlled immunotherapy clinical trials for GBM challenged. Cancers (Basel) 2021, 13, 32. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoom, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Eng. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Giladi, M.; Schneiderman, R.S.; Voloshin, T.; Porat, Y.; Munster, M.; Blat, R.; Sherbo, S.; Bomzon, Z.; Urman, N.; Itzhaki, A.; et al. Mitotic Spindle Disruption by Alternating Electric Fields Leads to Improper Chromosome Segregation and Mitotic Catastrophe in Cancer Cells. Sci. Rep. 2015, 5, 18046. [Google Scholar] [CrossRef]
- Kirson, E.D.; Dbaly, V.; Tovarys, F.; Vymazal, J.; Soustiel, J.F.; Itzhaki, A.; Mordechovich, D.; Steinberg-Shapira, S.; Gurvich, Z.; Schneiderman, R.; et al. Alternating electric fields arrest cell proliferation in animal tumor models and human brain tumors. Proc. Natl. Acad. Sci. USA 2007, 104, 10152–10157. [Google Scholar] [CrossRef]
- Chen, D.; Le, S.B.; Hutchinson, T.E.; Calinescu, A.A.; Sebastian, M.; Jin, D.; Liu, T.; Ghiaseddin, A.; Rahman, M.; Tran, D.D. Tumor Treating Fields dually activate STING and AIM2 inflammasomes to induce adjuvant immunity in glioblastoma. J. Clin. Investig. 2022, 132, e149258. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Brem, S.; Campian, J.L.; Trusheim, J.E.; Iwamoto, F.M.; Tran, D.D.; Ansstas, G.; Cobbs, C.S.; Heth, J.A.; et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination with Extension of Survival Among Patients With Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA Oncol. 2023, 9, 112–121. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef]
- Antonopoulos, M.; Van gool, S.W.; Dionysiou, D.; Graf, N.; Stamatakos, G. Immune phenotype correlates with survival in patients with GBM treated with standard temozolomide-based therapy and immunotherapy. Anticancer Res. 2019, 39, 2043–2051. [Google Scholar] [CrossRef]
- Schirrmacher, V.; van Gool, S.; Stuecker, W. Counteracting Immunosuppression in the Tumor Microenvironment by Oncolytic Newcastle Disease Virus and Cellular Immunotherapy. Int. J. Mol. Sci. 2022, 23, 13050. [Google Scholar] [CrossRef]
- Daniel, P.; Sabri, S.; Chaddad, A.; Meehan, B.; Jean-Claude, B.; Rak, J.; Abdulkarim, B.S. Temozolomide Induced Hypermutation in Glioma: Evolutionary Mechanisms and Therapeutic Opportunities. Front. Oncol. 2019, 9, 41. [Google Scholar] [CrossRef]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef]
- Antonios, J.P.; Soto, H.; Everson, R.G.; Orpilla, J.; Moughon, D.; Shin, N.; Sedighim, S.; Yong, W.H.; Li, G.; Cloughesy, T.F.; et al. PD-1 blockade enhances the vaccination-induced immune response in glioma. JCI Insight 2016, 1, e87059. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Wang, M.; Aldape, K.D.; Stupp, R.; Hegi, M.E.; Jaeckle, K.A.; Armstrong, T.S.; Wefel, J.S.; Won, M.; Blumenthal, D.T.; et al. Dose-dense temozolomide for newly diagnosed glioblastoma: A randomized phase III clinical trial. J. Clin. Oncol. 2013, 31, 4085–4091. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef]
- Wen, P.Y.; Reardon, D.A.; Armstrong, T.S.; Phuphanich, S.; Aiken, R.D.; Landolfi, J.C.; Curry, W.T.; Zhu, J.J.; Glantz, M.; Peereboom, D.M.; et al. A Randomized Double-Blind Placebo-Controlled Phase II Trial of Dendritic Cell Vaccine ICT-107 in Newly Diagnosed Patients with Glioblastoma. Clin. Cancer Res. 2019, 25, 5799–5807. [Google Scholar] [CrossRef]
- Stummer, W.; Pichlmeier, U.; Meinel, T.; Wiestler, O.D.; Zanella, F.; Reulen, H.J. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: A randomised controlled multicentre phase III trial. Lancet Oncol. 2006, 7, 392–401. [Google Scholar] [CrossRef]
- Dix, A.R.; Brooks, W.H.; Roszman, T.L.; Morford, L.A. Immune defects observed in patients with primary malignant brain tumors. J. Neuroimmunol. 1999, 100, 216–232. [Google Scholar] [CrossRef]
- Rapp, M.; Ozcan, Z.; Steiger, H.J.; Wernet, P.; Sabel, M.C.; Sorg, R.V. Cellular immunity of patients with malignant glioma: Prerequisites for dendritic cell vaccination immunotherapy. J. Neurosurg. 2006, 105, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Dutoit, V.; Philippin, G.; Widmer, V.; Marinari, E.; Vuilleumier, A.; Migliorini, D.; Schaller, K.; Dietrich, P.Y. Impact of Radiochemotherapy on Immune Cell Subtypes in High-Grade Glioma Patients. Front. Oncol. 2020, 10, 89. [Google Scholar] [CrossRef] [PubMed]
- Parajuli, P.; Mittal, S. Role of IL-17 in Glioma Progression. J. Spine Neurosurg. 2013. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Diao, S.; Wang, Q.; Zhu, C.; Sun, X.; Yin, B.; Zhang, X.; Meng, X.; Wang, B. IL-17A promotes cell migration and invasion of glioblastoma cells via activation of PI3K/AKT signalling pathway. J. Cell Mol. Med. 2019, 23, 357–369. [Google Scholar] [CrossRef]
- Liang, H.; Yi, L.; Wang, X.; Zhou, C.; Xu, L. Interleukin-17 facilitates the immune suppressor capacity of high-grade glioma-derived CD4 (+) CD25 (+) Foxp3 (+) T cells via releasing transforming growth factor beta. Scand J. Immunol. 2014, 80, 144–150. [Google Scholar] [CrossRef]
- Narita, Y.; Arakawa, Y.; Yamasaki, F.; Nishikawa, R.; Aoki, T.; Kanamori, M.; Nagane, M.; Kumabe, T.; Hirose, Y.; Ichikawa, T.; et al. A randomized, double-blind, phase III trial of personalized peptide vaccination for recurrent glioblastoma. Neuro-Oncol. 2019, 21, 348–359. [Google Scholar] [CrossRef]
- Sedgwick, A.J.; Ghazanfari, N.; Constantinescu, P.; Mantamadiotis, T.; Barrow, A.D. The Role of NK Cells and Innate Lymphoid Cells in Brain Cancer. Front. Immunol. 2020, 11, 1549. [Google Scholar] [CrossRef]
- Wang, M.; Zhou, Z.; Wang, X.; Zhang, C.; Jiang, X. Natural killer cell awakening: Unleash cancer-immunity cycle against glioblastoma. Cell Death Dis. 2022, 13, 588. [Google Scholar] [CrossRef]
- Pellegatta, S.; Eoli, M.; Frigerio, S.; Antozzi, C.; Bruzzone, M.G.; Cantini, G.; Nava, S.; Anghileri, E.; Cuppini, L.; Cuccarini, V.; et al. The natural killer cell response and tumor debulking are associated with prolonged survival in recurrent glioblastoma patients receiving dendritic cells loaded with autologous tumor lysates. Oncoimmunology 2013, 2, e23401. [Google Scholar] [CrossRef]
- Pellegatta, S.; Eoli, M.; Cuccarini, V.; Anghileri, E.; Pollo, B.; Pessina, S.; Frigerio, S.; Servida, M.; Cuppini, L.; Antozzi, C.; et al. Survival gain in glioblastoma patients treated with dendritic cell immunotherapy is associated with increased NK but not CD8(+) T cell activation in the presence of adjuvant temozolomide. Oncoimmunology 2018, 7, e1412901. [Google Scholar] [CrossRef]
- Sampson, J.H.; Heimberger, A.B.; Archer, G.E.; Aldape, K.D.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E.; McLendon, R.E.; Mitchell, D.A.; et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2010, 28, 4722–4729. [Google Scholar] [CrossRef]
- Muller, C.; Holtschmidt, J.; Auer, M.; Heitzer, E.; Lamszus, K.; Schulte, A.; Matschke, J.; Langer-Freitag, S.; Gasch, C.; Stoupiec, M.; et al. Hematogenous dissemination of glioblastoma multiforme. Sci. Transl. Med. 2014, 6, 247ra101. [Google Scholar] [CrossRef]
- Adamczyk, L.A.; Williams, H.; Frankow, A.; Ellis, H.P.; Haynes, H.R.; Perks, C.; Holly, J.M.; Kurian, K.M. Current Understanding of Circulating Tumor Cells—Potential Value in Malignancies of the Central Nervous System. Front. Neurol. 2015, 6, 174. [Google Scholar] [CrossRef]
- Bachg, D.; Haselhorst, U. Molecular Oncology. Available online: https://www.biofocus.de/media/files/downloads/117_directoy-services-2017.pdf (accessed on 1 September 2022).
- Preusser, M.; Berghoff, A.S.; Wick, W.; Weller, M. Clinical Neuropathology mini-review 6-2015: PD-L1: Emerging biomarker in glioblastoma? Clin. Neuropathol. 2015, 34, 313–321. [Google Scholar] [CrossRef]
- Menna, G.; Manini, I.; Cesselli, D.; Skrap, M.; Olivi, A.; Ius, T.; Della Pepa, G.M. Immunoregulatory effects of glioma-associated stem cells on the glioblastoma peritumoral microenvironment: A differential PD-L1 expression from core to periphery? Neurosurg. Focus 2022, 52, E4. [Google Scholar] [CrossRef]
- Tufano, M.; D’Arrigo, P.; D’Agostino, M.; Giordano, C.; Marrone, L.; Cesaro, E.; Romano, M.F.; Romano, S. PD-L1 Expression Fluctuates Concurrently with Cyclin D in Glioblastoma Cells. Cells 2021, 10, 2366. [Google Scholar] [CrossRef]
- Jia, H.; Xie, X.; Wang, L.; Wang, L.; Che, F. IFN-gamma induces PD-L1 through p38/JNK/ERK signaling pathways and counteracts the tumor promoting effect mediated by PD-L1 in Glioblastoma. Comput. Intell. Neurosci. 2022, 2022, 5492602. [Google Scholar] [CrossRef]
- Sung, K.S.; Roh, T.H.; Moon, J.H.; Kim, E.H.; Kang, S.G.; Kim, S.H.; Chang, J.H. Treatment Results for Recurrent Glioblastoma and Alteration of Programmed Death-Ligand 1 Expression After Recurrence. World Neurosurg. 2020, 135, e459–e467. [Google Scholar] [CrossRef]
- Brandner, S.; McAleenan, A.; Kelly, C.; Spiga, F.; Cheng, H.Y.; Dawson, S.; Schmidt, L.; Faulkner, C.L.; Wragg, C.; Jefferies, S.; et al. MGMT promoter methylation testing to predict overall survival in people with glioblastoma treated with temozolomide: A comprehensive meta-analysis based on a Cochrane Systematic Review. Neuro-Oncology 2021, 23, 1457–1469. [Google Scholar] [CrossRef]
- Brandes, A.A.; Franceschi, E.; Paccapelo, A.; Tallini, G.; De Biase, D.; Ghimenton, C.; Danieli, D.; Zunarelli, E.; Lanza, G.; Silini, E.M.; et al. Role of MGMT Methylation Status at Time of Diagnosis and Recurrence for Patients with Glioblastoma: Clinical Implications. Oncologist 2017, 22, 432–437. [Google Scholar] [CrossRef]
- Felsberg, J.; Thon, N.; Eigenbrod, S.; Hentschel, B.; Sabel, M.C.; Westphal, M.; Schackert, G.; Kreth, F.W.; Pietsch, T.; Loffler, M.; et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int. J. Cancer 2011, 129, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Huang, J.; Xi, H.; Zhou, X. Efficacy and safety of dendritic cell vaccines for patients with glioblastoma: A meta-analysis of randomized controlled trials. Int. Immunopharmacol. 2020, 83, 106336. [Google Scholar] [CrossRef] [PubMed]
- Burman, B.; Pesci, G.; Zamarin, D. Newcastle Disease Virus at the Forefront of Cancer Immunotherapy. Cancers 2020, 12, 3552. [Google Scholar] [CrossRef] [PubMed]
- Roussakow, S.V. Clinical and economic evaluation of modulated electrohyperthermia concurrent to dose-dense temozolomide 21/28 days regimen in the treatment of recurrent glioblastoma: A retrospective analysis of a two-centre German cohort trial with systematic comparison and effect-to-treatment analysis. BMJ Open 2017, 7, e017387. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. Cancer Vaccines and Oncolytic Viruses Exert Profoundly Lower Side Effects in Cancer Patients than Other Systemic Therapies: A Comparative Analysis. Biomedicines 2020, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Skaga, E.; Skretteberg, M.A.; Johannesen, T.B.; Brandal, P.; Vik-Mo, E.O.; Helseth, E.; Langmoen, I.A. Real-world validity of randomized controlled phase III trials in newly diagnosed glioblastoma: To whom do the results of the trials apply? Neurooncol. Adv. 2021, 3, vdab008. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methylated | Unmethylated | Chi-Square | |||||
|---|---|---|---|---|---|---|---|
| L (%) | N (%) | H (%) | L (%) | N (%) | H (%) | ||
| White Blood cell count | 6 (27) | 15 (68) | 1 (5) | 1 (4) | 23 (82) | 4 (14) | 0.0402 |
| Lymphocytes | 16 (73) | 5 (23) | 1 (5) | 16 (59) | 11 (41) | 0 (0) | 0.2505 |
| Monocytes | 6 (27) | 15 (68) | 1 (5) | 3 (11) | 24 (89) | 0 (0) | 0.1649 |
| Neutrophils | 4 (18) | 15 (68) | 3 (14) | 1 (4) | 21 (78) | 5 (19) | 0.2442 |
| Eosinophils | 4 (18) | 17 (77) | 1 (5) | 2 (7) | 24 (89) | 1 (4) | 0.5051 |
| Basophils | 1 (5) | 21 (95) | 0 (0) | 0 (0) | 25 (93) | 2 (7) | 0.2384 |
| Hemoglobin | 5 (24) | 16 (76) | 0 (0) | 3 (11) | 23 (82) | 2 (7) | 0.2449 |
| Platelets | 9 (41) | 12 (55) | 1 (5) | 6 (21) | 22 (79) | 0 (0) | 0.1439 |
| T cells | 18 (82) | 3 (14) | 1 (5) | 19 (73) | 7 (27) | 0 (0) | 0.3151 |
| B cells | 18 (82) | 4 (18) | 0 (0) | 20 (27) | 5 (19) | 1 (4) | 0.6411 |
| NK cells | 15 (68) | 7 (32) | 0 (0) | 17 (65) | 9 (35) | 0 (0) | 0.8377 |
| Th2 skewing | 5 (23) | 15 (68) | 2 (9) | 3 (11) | 16 (57) | 9 (32) | 0.1148 |
| Th17 skewing | 1 (8) | 7 (54) | 5 (38) | 0 (0) | 13 (57) | 10 (43) | 0.4005 |
| Th1 skewing | 4 (18) | 15 (68) | 3 (14) | 1 (4) | 23 (82) | 4 (14) | 0.2288 |
| Th1/Th2 | 3 (23) | 9 (69) | 1 (8) | 8 (33) | 15 (63) | 1 (4) | 0.7591 |
| NK cell function | 16 (73) | 5 (23) | 1 (5) | 18 (67) | 9 (33) | 0 (0) | 0.3312 |
| Good (%) | Moderate (%) | Increased (%) | Good (%) | Moderate (%) | Increased (%) | ||
| Oxidative load | 5 (24) | 9 (43) | 7 (33) | 7 (28) | 7 (28) | 11 (44) | 0.5675 |
| Low (%) | Medium (%) | High (%) | Low (%) | Medium (%) | High (%) | ||
| Total anti-oxidative capacity | 3 (14) | 5 (24) | 13 (62) | 2 (8) | 3 (12) | 20 (80) | 0.3963 |
| No CCC (%) | CCC PDL1− (%) | CCC PDL1+ (%) | No CCC (%) | CCC PDL1− (%) | CCC PDL1+ (%) | ||
| CCC | 7 (33) | 13 (62) | 1 (5) | 13 (50) | 9 (35) | 4 (15) | 0.1467 |
| October 2018 (Primary Tumor) | December 2021 (Second Relapse) | ||||
|---|---|---|---|---|---|
| No | Peptide | NAF (DNA) | No | Peptide | NAF (DNA) |
| 1 | DLKNRTGFAV | 0.46 | 11 | RLASDLAEF | 0.63 |
| 2 | SLHNHMRFR | 0.26 | 12 | FAARPCAEI | 0.61 |
| 3 | HFFCDTYPLLK | 0.43 | 13 | IMENSPKDVY | 0.33 |
| 4 | RIFNLISM | 0.16 | 14 | RVALVPIKY | 0.26 |
| 5 | ALDIRAHIEEF | 0.25 | 15 | NCNGPSPNM | 0.4 |
| 6 | KVHQNIHTGEK | 0.17 | 16 | GSHGYDLSTF | 0.76 |
| 7 | KAGLKVHQNIHTGEKPH | 0.09 | 17 | RSDHYSEEL | 0.19 |
| 8 | RGANPDLKNRTGFAVIH | 0.46 | 18 | PAAPYIPGL | 0.35 |
| 9 | TCPLPSSLHNHMRFRHS | 0.26 | 2 | SLHNHMRFR | 0.44 |
| 10 | VALDIRAHIEEFKPYI | 0.25 | 19 | SVSAPAFYSPQK | 0.09 |
| 20 | KTSYIIMIGPD | 0.19 | |||
| 21 | YEVLLVTSSFVSPSESRSG | 0.61 | |||
| 8 | RGANPDLKNRTGFAVIH | 0.4 | |||
| 22 | IMENSPKDVYVVQIEAFD | 0.33 | |||
| 23 | TPYLLHFSNVSVPRVRAE | 0.34 | |||
| 24 | AEPEKMGGDGTVCSPLE | 0.43 | |||
| 25 | SVESGANDVVFIRTLG | 0.29 | |||
| 26 | PKRGSEGGLAAFVDFVD | 0.17 | |||
| 7 | KAGLKVHQNIHTGEKPH | 0.61 | |||
| 27 | AKQESLETLVLSGIGST | 0.3 | |||
| 28 | KSNHDKNVTPDEVLQTL | 0.18 | |||
| 29 | FSQKSRVTENPTEALS | 0.31 | |||
| 30 | AEPPGTPPDSHSHLDAA | 0.34 | |||
| 31 | AISWARTKRIPFLGV | 0.08 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Gool, S.W.; Makalowski, J.; Van de Vliet, P.; Van Gool, S.; Sprenger, T.; Schirrmacher, V.; Stuecker, W. Individualized Multimodal Immunotherapy for Adults with IDH1 Wild-Type GBM: A Single Institute Experience. Cancers 2023, 15, 1194. https://doi.org/10.3390/cancers15041194
Van Gool SW, Makalowski J, Van de Vliet P, Van Gool S, Sprenger T, Schirrmacher V, Stuecker W. Individualized Multimodal Immunotherapy for Adults with IDH1 Wild-Type GBM: A Single Institute Experience. Cancers. 2023; 15(4):1194. https://doi.org/10.3390/cancers15041194
Chicago/Turabian StyleVan Gool, Stefaan W., Jennifer Makalowski, Peter Van de Vliet, Stefanie Van Gool, Tobias Sprenger, Volker Schirrmacher, and Wilfried Stuecker. 2023. "Individualized Multimodal Immunotherapy for Adults with IDH1 Wild-Type GBM: A Single Institute Experience" Cancers 15, no. 4: 1194. https://doi.org/10.3390/cancers15041194
APA StyleVan Gool, S. W., Makalowski, J., Van de Vliet, P., Van Gool, S., Sprenger, T., Schirrmacher, V., & Stuecker, W. (2023). Individualized Multimodal Immunotherapy for Adults with IDH1 Wild-Type GBM: A Single Institute Experience. Cancers, 15(4), 1194. https://doi.org/10.3390/cancers15041194