

Tumor Microenvironment in Gliomas: A Treatment Hurdle or an Opportunity to Grab?

Abstract
Simple Summary
Abstract
1. Introduction
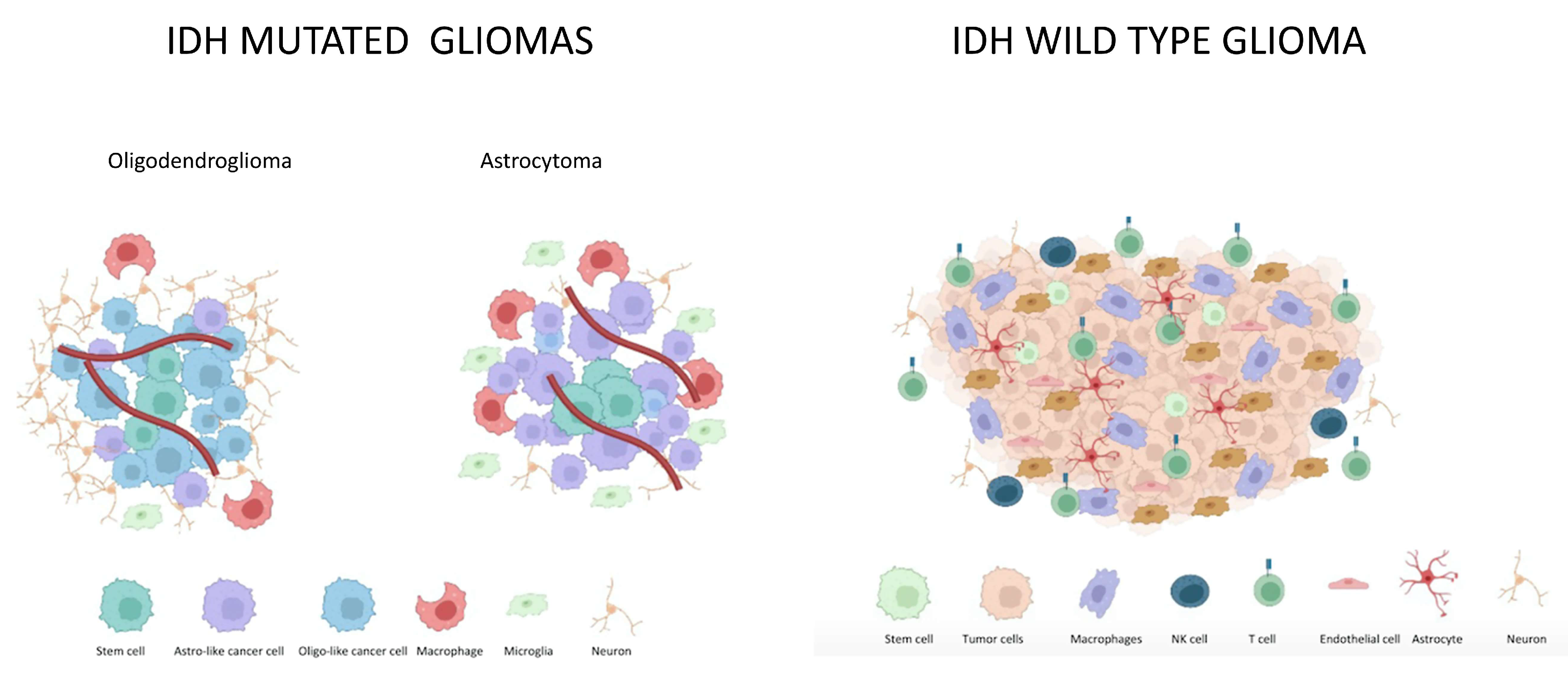
2. IDH Mutated Gliomas
2.1. Microenvironment in IDH Mutated Gliomas
2.2. Differences in Astrocytoma and Oligodendroglioma Tumor Microenvironment
2.3. Pilocytic Astrocytoma Microenvironments
3. IDH wt Gliomas
Microenvironment in IDH wt Gliomas
4. Differences in IDH-Mutated and IDH-wt Tumor Microenvironment
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Aldape, K.; Colman, H.; Figrarella-Branger, D.; Fuller, G.N.; Giannini, C.; Holland, E.C.; Jenkins, R.B.; Kleinschmidt-DeMasters, B.; Komori, T.; et al. cIMPACT-NOW update 5: Recommended grading criteria and terminologies for IDH-mutant astrocytomas. Acta Neuropathol. 2020, 139, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef]
- Wen, P.Y.; Packer, R.J. The 2021 WHO Classification of Tumors of the Central Nervous System: Clinical Implications. Neuro Oncol. 2021, 23, 1215–1217. [Google Scholar] [CrossRef]
- Di Nunno, V.; Fordellone, M.; Minniti, G.; Asioli, S.; Conti, A.; Mazzatenta, D.; Balestrini, D.; Chiodini, P.; Agati, R.; Tonon, C.; et al. Machine learning in neuro-oncology: Toward novel development fields. J. Neuro-Oncol. 2022, 159, 333–346. [Google Scholar] [CrossRef]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 2017, 355, eaai8478. [Google Scholar] [CrossRef]
- Fu, W.; Wang, W.; Li, H.; Jiao, Y.; Weng, J.; Huo, R.; Yan, Z.; Wang, J.; Xu, H.; Wang, S.; et al. High Dimensional Mass Cytometry Analysis Reveals Characteristics of the Immunosuppressive Microenvironment in Diffuse Astrocytomas. Front. Oncol. 2020, 10, 78. [Google Scholar] [CrossRef]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef]
- Di Nunno, V.; Franceschi, E.; Tosoni, A.; Gatto, L.; Bartolini, S.; Brandes, A.A. Glioblastoma Microenvironment: From an Inviolable Defense to a Therapeutic Chance. Front. Oncol. 2022, 12, 852950. [Google Scholar] [CrossRef]
- Mair, M.J.; Geurts, M.; van den Bent, M.J.; Berghoff, A.S. A basic review on systemic treatment options in WHO grade II-III gliomas. Cancer Treat. Rev. 2021, 92, 102124. [Google Scholar] [CrossRef]
- Mohile, N.A.; Messersmith, H.; Gatson, N.T.; Hottinger, A.F.; Lassman, A.; Morton, J.; Ney, D.; Nghiemphu, P.L.; Olar, A.; Olson, J.; et al. Therapy for Diffuse Astrocytic and Oligodendroglial Tumors in Adults: ASCO-SNO Guideline. J. Clin. Oncol. 2022, 40, 403–426. [Google Scholar] [CrossRef]
- Di Nunno, V.; Franceschi, E.; Gatto, L.; Tosoni, A.; Bartolini, S.; Brandes, A.A. How to treat histone 3 altered gliomas: Molecular landscape and therapeutic developments. Expert Rev. Clin. Pharmacol. 2022, 16, 17–26. [Google Scholar] [CrossRef] [PubMed]
- DeCordova, S.; Shastri, A.; Tsolaki, A.G.; Yasmin, H.; Klein, L.; Singh, S.K.; Kishore, U. Molecular Heterogeneity and Immunosuppressive Microenvironment in Glioblastoma. Front. Immunol. 2020, 11, 1402. [Google Scholar] [CrossRef]
- Di Nunno, V.; Franceschi, E.; Tosoni, A.; Gatto, L.; Maggio, I.; Lodi, R.; Angelini, D.; Bartolini, S.; Brandes, A.A. Clinical and Molecular Features of Patients with Gliomas Harboring IDH1 Non-canonical Mutations: A Systematic Review and Meta-Analysis. Adv. Ther. 2022, 39, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, E.; Biase, D.; Di Nunno, V.; Pession, A.; Tosoni, A.; Gatto, L.; Lodi, R.; Tallini, G.; Visani, M.; Bartolini, S.; et al. IDH1(105GGT) single nucleotide polymorphism improves progression free survival in patients with IDH mutated grade II and III gliomas. Pathol. Res. Pract. 2021, 221, 153445. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, E.; De Biase, D.; Di Nunno, V.; Pession, A.; Tosoni, A.; Gatto, L.; Tallini, G.; Visani, M.; Lodi, R.; Bartolini, S.; et al. IDH1 Non-Canonical Mutations and Survival in Patients with Glioma. Diagnostics 2021, 11, 342. [Google Scholar] [CrossRef]
- Wick, W.; Roth, P.; Hartmann, C.; Hau, P.; Nakamura, M.; Stockhammer, F.; Sabel, M.C.; Wick, A.; Koeppen, S.; Ketter, R.; et al. Long-term analysis of the NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with PCV or temozolomide. Neuro Oncol. 2016, 18, 1529–1537. [Google Scholar] [CrossRef]
- van den Bent, M.J.; Tesileanu, C.M.S.; Wick, W.; Sanson, M.; Brandes, A.A.; Clement, P.M.; Erridge, S.; Vogelbaum, M.A.; Nowak, A.K.; Baurain, J.F.; et al. Adjuvant and concurrent temozolomide for 1p/19q non-co-deleted anaplastic glioma (CATNON; EORTC study 26053-22054): Second interim analysis of a randomised, open-label, phase 3 study. Lancet Oncol. 2021, 22, 813–823. [Google Scholar] [CrossRef]
- van den Bent, M.J.; Brandes, A.A.; Taphoorn, M.J.; Kros, J.M.; Kouwenhoven, M.C.; Delattre, J.Y.; Bernsen, H.J.; Frenay, M.; Tijssen, C.C.; Grisold, W.; et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: Long-term follow-up of EORTC brain tumor group study 26951. J. Clin. Oncol. 2013, 31, 344–350. [Google Scholar] [CrossRef]
- van den Bent, M.J.; Afra, D.; de Witte, O.; Ben Hassel, M.; Schraub, S.; Hoang-Xuan, K.; Malmström, P.O.; Collette, L.; Piérart, M.; Mirimanoff, R.; et al. Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: The EORTC 22845 randomised trial. Lancet 2005, 366, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Jakola, A.S.; Skjulsvik, A.J.; Myrmel, K.S.; Sjåvik, K.; Unsgård, G.; Torp, S.H.; Aaberg, K.; Berg, T.; Dai, H.Y.; Johnsen, K.; et al. Surgical resection versus watchful waiting in low-grade gliomas. Ann. Oncol. 2017, 28, 1942–1948. [Google Scholar] [CrossRef] [PubMed]
- Dubbink, H.J.; Atmodimedjo, P.N.; Kros, J.M.; French, P.J.; Sanson, M.; Idbaih, A.; Wesseling, P.; Enting, R.; Spliet, W.; Tijssen, C.; et al. Molecular classification of anaplastic oligodendroglioma using next-generation sequencing: A report of the prospective randomized EORTC Brain Tumor Group 26951 phase III trial. Neuro Oncol. 2016, 18, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Zhang, P.; Cairncross, J.G.; Gilbert, M.R.; Bahary, J.P.; Dolinskas, C.A.; Chakravarti, A.; Aldape, K.D.; Bell, E.H.; Schiff, D.; et al. Phase III randomized study of radiation and temozolomide versus radiation and nitrosourea therapy for anaplastic astrocytoma: Results of NRG Oncology RTOG 9813. Neuro Oncol. 2017, 19, 252–258. [Google Scholar] [CrossRef]
- Cairncross, J.G.; Wang, M.; Jenkins, R.B.; Shaw, E.G.; Giannini, C.; Brachman, D.G.; Buckner, J.C.; Fink, K.L.; Souhami, L.; Laperriere, N.J.; et al. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J. Clin. Oncol. 2014, 32, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Cairncross, G.; Wang, M.; Shaw, E.; Jenkins, R.; Brachman, D.; Buckner, J.; Fink, K.; Souhami, L.; Laperriere, N.; Curran, W.; et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: Long-term results of RTOG 9402. J. Clin. Oncol. 2013, 31, 337–343. [Google Scholar] [CrossRef]
- Baumert, B.G.; Hegi, M.E.; van den Bent, M.J.; von Deimling, A.; Gorlia, T.; Hoang-Xuan, K.; Brandes, A.A.; Kantor, G.; Taphoorn, M.J.B.; Hassel, M.B.; et al. Temozolomide chemotherapy versus radiotherapy in high-risk low-grade glioma (EORTC 22033-26033): A randomised, open-label, phase 3 intergroup study. Lancet Oncol. 2016, 17, 1521–1532. [Google Scholar] [CrossRef]
- Tirosh, I.; Venteicher, A.S.; Hebert, C.; Escalante, L.E.; Patel, A.P.; Yizhak, K.; Fisher, J.M.; Rodman, C.; Mount, C.; Filbin, M.G.; et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 2016, 539, 309–313. [Google Scholar] [CrossRef]
- Tesileanu, C.M.S.; van den Bent, M.J.; Sanson, M.; Wick, W.; Brandes, A.A.; Clement, P.M.; Erridge, S.C.; Vogelbaum, M.A.; Nowak, A.K.; Baurain, J.F.; et al. Prognostic significance of genome-wide DNA methylation profiles within the randomized, phase 3, EORTC CATNON trial on non-1p/19q deleted anaplastic glioma. Neuro Oncol. 2021, 23, 1547–1559. [Google Scholar] [CrossRef]
- Komohara, Y.; Ohnishi, K.; Kuratsu, J.; Takeya, M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J. Pathol. 2008, 216, 15–24. [Google Scholar] [CrossRef]
- Perus, L.J.M.; Walsh, L.A. Microenvironmental Heterogeneity in Brain Malignancies. Front. Immunol. 2019, 10, 2294. [Google Scholar] [CrossRef]
- Li, F.; Liu, X.; Sampson, J.H.; Bigner, D.D.; Li, C.Y. Rapid Reprogramming of Primary Human Astrocytes into Potent Tumor-Initiating Cells with Defined Genetic Factors. Cancer Res. 2016, 76, 5143–5150. [Google Scholar] [CrossRef] [PubMed]
- Richardson, L.G.; Choi, B.D.; Curry, W.T. (R)-2-hydroxyglutarate drives immune quiescence in the tumor microenvironment of IDH-mutant gliomas. Transl. Cancer Res. 2019, 8, S167–S170. [Google Scholar] [CrossRef] [PubMed]
- Gargini, R.; Segura-Collar, B.; Sánchez-Gómez, P. Cellular Plasticity and Tumor Microenvironment in Gliomas: The Struggle to Hit a Moving Target. Cancers 2020, 12, 1622. [Google Scholar] [CrossRef] [PubMed]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; Iacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016, 17, 2445–2459. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef]
- Andersen, J.K.; Miletic, H.; Hossain, J.A. Tumor-Associated Macrophages in Gliomas-Basic Insights and Treatment Opportunities. Cancers 2022, 14, 1319. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef]
- Müller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef]
- Amankulor, N.M.; Kim, Y.; Arora, S.; Kargl, J.; Szulzewsky, F.; Hanke, M.; Margineantu, D.H.; Rao, A.; Bolouri, H.; Delrow, J.; et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017, 31, 774–786. [Google Scholar] [CrossRef]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Wilhelm, D.; Rajky, O.; Kurscheid, S.; Kresl, P.; Wöhrer, A.; Marosi, C.; Hegi, M.E.; et al. Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro Oncol. 2017, 19, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Bunse, L.; Pusch, S.; Bunse, T.; Sahm, F.; Sanghvi, K.; Friedrich, M.; Alansary, D.; Sonner, J.K.; Green, E.; Deumelandt, K.; et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 2018, 24, 1192–1203. [Google Scholar] [CrossRef] [PubMed]
- Kohanbash, G.; Carrera, D.A.; Shrivastav, S.; Ahn, B.J.; Jahan, N.; Mazor, T.; Chheda, Z.S.; Downey, K.M.; Watchmaker, P.B.; Beppler, C.; et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J. Clin. Investig. 2017, 127, 1425–1437. [Google Scholar] [CrossRef]
- Poon, C.C.; Gordon, P.M.K.; Liu, K.; Yang, R.; Sarkar, S.; Mirzaei, R.; Ahmad, S.T.; Hughes, M.L.; Yong, V.W.; Kelly, J.J.P. Differential microglia and macrophage profiles in human IDH-mutant and -wild type glioblastoma. Oncotarget 2019, 10, 3129–3143. [Google Scholar] [CrossRef]
- Wu, F.; Wang, Z.L.; Wang, K.Y.; Li, G.Z.; Chai, R.C.; Liu, Y.Q.; Jiang, H.Y.; Zhai, Y.; Feng, Y.M.; Zhao, Z.; et al. Classification of diffuse lower-grade glioma based on immunological profiling. Mol. Oncol. 2020, 14, 2081–2095. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Rodado, V.; Malta, T.M.; Seki, T.; Lita, A.; Dowdy, T.; Celiku, O.; Cavazos-Saldana, A.; Li, A.; Liu, Y.; Han, S.; et al. Metabolic reprogramming associated with aggressiveness occurs in the G-CIMP-high molecular subtypes of IDH1mut lower grade gliomas. Neuro Oncol. 2020, 22, 480–492. [Google Scholar] [CrossRef] [PubMed]
- Radin, D.P.; Tsirka, S.E. Interactions between Tumor Cells, Neurons, and Microglia in the Glioma Microenvironment. Int. J. Mol. Sci. 2020, 21, 8476. [Google Scholar] [CrossRef] [PubMed]
- Zeiner, P.S.; Preusse, C.; Golebiewska, A.; Zinke, J.; Iriondo, A.; Muller, A.; Kaoma, T.; Filipski, K.; Müller-Eschner, M.; Bernatz, S.; et al. Distribution and prognostic impact of microglia/macrophage subpopulations in gliomas. Brain Pathol. 2019, 29, 513–529. [Google Scholar] [CrossRef]
- Lv, L.; Zhang, Y.; Zhao, Y.; Wei, Q.; Zhao, Y.; Yi, Q. Effects of 1p/19q Codeletion on Immune Phenotype in Low Grade Glioma. Front. Cell. Neurosci. 2021, 15, 704344. [Google Scholar] [CrossRef]
- Yang, Y.; Schubert, M.C.; Kuner, T.; Wick, W.; Winkler, F.; Venkataramani, V. Brain Tumor Networks in Diffuse Glioma. Neurotherapeutics 2022, 19, 1832–1843. [Google Scholar] [CrossRef]
- Iwaki, T.; Iwaki, A.; Miyazono, M.; Goldman, J.E. Preferential expression of alpha B-crystallin in astrocytic elements of neuroectodermal tumors. Cancer 1991, 68, 2230–2240. [Google Scholar] [CrossRef]
- Salles, D.; Laviola, G.; Malinverni, A.C.M.; Stávale, J.N. Pilocytic Astrocytoma: A Review of General, Clinical, and Molecular Characteristics. J. Child Neurol. 2020, 35, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Di Nunno, V.; Gatto, L.; Tosoni, A.; Bartolini, S.; Franceschi, E. Implications of BRAF V600E mutation in gliomas: Molecular considerations, prognostic value and treatment evolution. Front. Oncol. 2022, 12, 1067252. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.; Han, S.J.; Sughrue, M.E.; Tihan, T.; Parsa, A.T. Immune cell infiltrate differences in pilocytic astrocytoma and glioblastoma: Evidence of distinct immunological microenvironments that reflect tumor biology. J. Neurosurg. 2011, 115, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Aichmüller, C.F.; Iskar, M.; Jones, D.T.W.; Korshunov, A.; Radlwimmer, B.; Kool, M.; Ernst, A.; Pfister, S.M.; Lichter, P.; Zapatka, M. Pilocytic astrocytoma demethylation and transcriptional landscapes link bZIP transcription factors to immune response. Neuro Oncol. 2020, 22, 1327–1338. [Google Scholar] [CrossRef]
- López-Pérez, C.A.; Franco-Mojica, X.; Villanueva-Gaona, R.; Díaz-Alba, A.; Rodríguez-Florido, M.A.; Navarro, V.G. Adult diffuse midline gliomas H3 K27-altered: Review of a redefined entity. J. Neuro-Oncol. 2022, 158, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Hernández Martínez, A.; Madurga, R.; García-Romero, N.; Ayuso-Sacido, Á. Unravelling glioblastoma heterogeneity by means of single-cell RNA sequencing. Cancer Lett. 2022, 527, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef]
- Venkataramani, V.; Yang, Y.; Schubert, M.C.; Reyhan, E.; Tetzlaff, S.K.; Wißmann, N.; Botz, M.; Soyka, S.J.; Beretta, C.A.; Pramatarov, R.L.; et al. Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell 2022, 185, 2899–2917.e2831. [Google Scholar] [CrossRef]
- Wei, Z.; Batagov, A.O.; Schinelli, S.; Wang, J.; Wang, Y.; El Fatimy, R.; Rabinovsky, R.; Balaj, L.; Chen, C.C.; Hochberg, F.; et al. Coding and noncoding landscape of extracellular RNA released by human glioma stem cells. Nat. Commun. 2017, 8, 1145. [Google Scholar] [CrossRef] [PubMed]
- Teplyuk, N.M.; Uhlmann, E.J.; Wong, A.H.; Karmali, P.; Basu, M.; Gabriely, G.; Jain, A.; Wang, Y.; Chiocca, E.A.; Stephens, R.; et al. MicroRNA-10b inhibition reduces E2F1-mediated transcription and miR-15/16 activity in glioblastoma. Oncotarget 2015, 6, 3770–3783. [Google Scholar] [CrossRef] [PubMed]
- Dapash, M.; Hou, D.; Castro, B.; Lee-Chang, C.; Lesniak, M.S. The Interplay between Glioblastoma and Its Microenvironment. Cells 2021, 10, 2257. [Google Scholar] [CrossRef]
- Di Nunno, V.; Franceschi, E.; Tosoni, A.; Gatto, L.; Bartolini, S.; Brandes, A.A. Tumor-Associated Microenvironment of Adult Gliomas: A Review. Front. Oncol. 2022, 12, 891543. [Google Scholar] [CrossRef] [PubMed]
- Tamai, S.; Ichinose, T.; Tsutsui, T.; Tanaka, S.; Garaeva, F.; Sabit, H.; Nakada, M. Tumor Microenvironment in Glioma Invasion. Brain Sci. 2022, 12, 505. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Hu, C.; Lal, B.; Zhou, W.; Ma, Y.; Ying, M.; Prinos, P.; Quiñones-Hinojosa, A.; Lim, M.; Laterra, J.; et al. Reprogramming Transcription Factors Oct4 and Sox2 Induce a BRD-Dependent Immunosuppressive Transcriptome in GBM-Propagating Cells. Cancer Res. 2021, 81, 2457–2469. [Google Scholar] [CrossRef] [PubMed]
- Nunno, V.D.; Franceschi, E.; Gatto, L.; Brandes, A.A. BET inhibitors: The promise of a new generation of immunotherapy in glioblastoma. Immunotherapy 2021, 14, 169–172. [Google Scholar] [CrossRef]
- Colquhoun, A. Cell biology-metabolic crosstalk in glioma. Int. J. Biochem. Cell Biol. 2017, 89, 171–181. [Google Scholar] [CrossRef]
- Ferreira, M.T.; Miyake, J.A.; Gomes, R.N.; Feitoza, F.; Stevannato, P.B.; da Cunha, A.S.; Serachi, F.O.; Panagopoulos, A.T.; Colquhoun, A. Cyclooxygenase Inhibition Alters Proliferative, Migratory, and Invasive Properties of Human Glioblastoma Cells In Vitro. Int. J. Mol. Sci. 2021, 22, 4297. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Mattila, S.; Tuominen, H.; Koivukangas, J.; Stenbäck, F. The terminal prostaglandin synthases mPGES-1, mPGES-2, and cPGES are all overexpressed in human gliomas. Neuropathology 2009, 29, 156–165. [Google Scholar] [CrossRef]
- Panagopoulos, A.T.; Gomes, R.N.; Almeida, F.G.; da Costa Souza, F.; Veiga, J.C.E.; Nicolaou, A.; Colquhoun, A. The prostanoid pathway contains potential prognostic markers for glioblastoma. Prostaglandins Other Lipid Mediat. 2018, 137, 52–62. [Google Scholar] [CrossRef]
- Kast, R.E. Adding high-dose celecoxib to increase effectiveness of standard glioblastoma chemoirradiation. Ann. Pharm. Fr. 2021, 79, 481–488. [Google Scholar] [CrossRef]
- Lombardi, F.; Augello, F.R.; Artone, S.; Ayroldi, E.; Giusti, I.; Dolo, V.; Cifone, M.G.; Cinque, B.; Palumbo, P. Cyclooxygenase-2 Upregulated by Temozolomide in Glioblastoma Cells Is Shuttled in Extracellular Vesicles Modifying Recipient Cell Phenotype. Front. Oncol. 2022, 12, 933746. [Google Scholar] [CrossRef]
- Lombardi, F.; Augello, F.R.; Artone, S.; Gugu, M.K.; Cifone, M.G.; Cinque, B.; Palumbo, P. Up-Regulation of Cyclooxygenase-2 (COX-2) Expression by Temozolomide (TMZ) in Human Glioblastoma (GBM) Cell Lines. Int. J. Mol. Sci. 2022, 23, 1545. [Google Scholar] [CrossRef]
- Yin, D.; Jin, G.; He, H.; Zhou, W.; Fan, Z.; Gong, C.; Zhao, J.; Xiong, H. Celecoxib reverses the glioblastoma chemo-resistance to temozolomide through mitochondrial metabolism. Aging 2021, 13, 21268–21282. [Google Scholar] [CrossRef]
- Zhao, W.; Yun, K. Propofol enhances the sensitivity of glioblastoma cells to temozolomide by inhibiting macrophage activation in tumor microenvironment to down-regulate HIF-1α expression. Exp. Cell Res. 2022, 418, 113277. [Google Scholar] [CrossRef]
- Brooks, L.J.; Clements, M.P.; Burden, J.J.; Kocher, D.; Richards, L.; Devesa, S.C.; Zakka, L.; Woodberry, M.; Ellis, M.; Jaunmuktane, Z.; et al. The white matter is a pro-differentiative niche for glioblastoma. Nat. Commun. 2021, 12, 2184. [Google Scholar] [CrossRef]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef]
- Drilon, A.E.; DuBois, S.G.; Farago, A.F.; Geoerger, B.; Grilley-Olson, J.E.; Hong, D.S.; Sohal, D.; van Tilburg, C.M.; Ziegler, D.S.; Ku, N.; et al. Activity of larotrectinib in TRK fusion cancer patients with brain metastases or primary central nervous system tumors. J. Clin. Oncol. 2019, 37, 2006. [Google Scholar] [CrossRef]
- Gatto, L.; Di Nunno, V.; Franceschi, E.; Tosoni, A.; Bartolini, S.; Brandes, A.A. Pharmacotherapeutic Treatment of Glioblastoma: Where Are We to Date? Drugs 2022, 82, 491–510. [Google Scholar] [CrossRef]
- Gatto, L.; Franceschi, E.; Tosoni, A.; Di Nunno, V.; Tonon, C.; Lodi, R.; Agati, R.; Bartolini, S.; Brandes, A.A. Beyond Imaging and Genetic Signature in Glioblastoma: Radiogenomic Holistic Approach in Neuro-Oncology. Biomedicines 2022, 10, 3205. [Google Scholar] [CrossRef] [PubMed]
- Lassman, A.B.; Sepúlveda-Sánchez, J.M.; Cloughesy, T.F.; Gil-Gil, M.J.; Puduvalli, V.K.; Raizer, J.J.; De Vos, F.Y.F.; Wen, P.Y.; Butowski, N.A.; Clement, P.M.J.; et al. Infigratinib in Patients with Recurrent Gliomas and FGFR Alterations: A Multicenter Phase II Study. Clin. Cancer Res. 2022, 28, 2270–2277. [Google Scholar] [CrossRef]
- Grossman, S.A.; Ye, X.; Chamberlain, M.; Mikkelsen, T.; Batchelor, T.; Desideri, S.; Piantadosi, S.; Fisher, J.; Fine, H.A. Talampanel with standard radiation and temozolomide in patients with newly diagnosed glioblastoma: A multicenter phase II trial. J. Clin. Oncol. 2009, 27, 4155–4161. [Google Scholar] [CrossRef]
- Lim, M.; Weller, M.; Idbaih, A.; Steinbach, J.; Finocchiaro, G.; Raval, R.R.; Ansstas, G.; Baehring, J.; Taylor, J.W.; Honnorat, J.; et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol. 2022, 24, 1935–1949. [Google Scholar] [CrossRef]
- Gueble, S.E.; Vasquez, J.C.; Bindra, R.S. The Role of PARP Inhibitors in Patients with Primary Malignant Central Nervous System Tumors. Curr. Treat. Options Oncol. 2022, 23, 1566–1589. [Google Scholar] [CrossRef]
- Banerjee, A.; Jakacki, R.I.; Onar-Thomas, A.; Wu, S.; Nicolaides, T.; Young Poussaint, T.; Fangusaro, J.; Phillips, J.; Perry, A.; Turner, D.; et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: A Pediatric Brain Tumor Consortium (PBTC) study. Neuro Oncol. 2017, 19, 1135–1144. [Google Scholar] [CrossRef]
- Fangusaro, J.; Onar-Thomas, A.; Poussaint, T.Y.; Wu, S.; Ligon, A.H.; Lindeman, N.; Campagne, O.; Banerjee, A.; Gururangan, S.; Kilburn, L.B.; et al. A phase II trial of selumetinib in children with recurrent optic pathway and hypothalamic low-grade glioma without NF1: A Pediatric Brain Tumor Consortium study. Neuro Oncol. 2021, 23, 1777–1788. [Google Scholar] [CrossRef]
- Fangusaro, J.; Onar-Thomas, A.; Young Poussaint, T.; Wu, S.; Ligon, A.H.; Lindeman, N.; Banerjee, A.; Packer, R.J.; Kilburn, L.B.; Goldman, S.; et al. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: A multicentre, phase 2 trial. Lancet Oncol. 2019, 20, 1011–1022. [Google Scholar] [CrossRef]
- Arrillaga-Romany, I.; Odia, Y.; Prabhu, V.V.; Tarapore, R.S.; Merdinger, K.; Stogniew, M.; Oster, W.; Allen, J.E.; Mehta, M.; Batchelor, T.T.; et al. Biological activity of weekly ONC201 in adult recurrent glioblastoma patients. Neuro Oncol. 2020, 22, 94–102. [Google Scholar] [CrossRef]
- Cantor, E.; Wierzbicki, K.; Tarapore, R.S.; Ravi, K.; Thomas, C.; Cartaxo, R.; Nand Yadav, V.; Ravindran, R.; Bruzek, A.K.; Wadden, J.; et al. Serial H3K27M cell-free tumor DNA (cf-tDNA) tracking predicts ONC201 treatment response and progression in diffuse midline glioma. Neuro Oncol. 2022, 24, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.N.; Malhotra, J.; Tarapore, R.S.; Malhotra, U.; Silk, A.W.; Chan, N.; Rodriguez, L.; Aisner, J.; Aiken, R.D.; Mayer, T.; et al. Safety and enhanced immunostimulatory activity of the DRD2 antagonist ONC201 in advanced solid tumor patients with weekly oral administration. J. Immunother. Cancer 2019, 7, 136. [Google Scholar] [CrossRef] [PubMed]
- Di, W.; Fan, W.; Wu, F.; Shi, Z.; Wang, Z.; Yu, M.; Zhai, Y.; Chang, Y.; Pan, C.; Li, G.; et al. Clinical characterization and immunosuppressive regulation of CD161 (KLRB1) in glioma through 916 samples. Cancer Sci. 2022, 113, 756–769. [Google Scholar] [CrossRef] [PubMed]
- Mathewson, N.D.; Ashenberg, O.; Tirosh, I.; Gritsch, S.; Perez, E.M.; Marx, S.; Jerby-Arnon, L.; Chanoch-Myers, R.; Hara, T.; Richman, A.R.; et al. Inhibitory CD161 receptor identified in glioma-infiltrating T cells by single-cell analysis. Cell 2021, 184, 1281–1298.e1226. [Google Scholar] [CrossRef]
- Gatto, L.; Franceschi, E.; Di Nunno, V.; Maggio, I.; Lodi, R.; Brandes, A.A. Engineered CAR-T and novel CAR-based therapies to fight the immune evasion of glioblastoma: Gutta cavat lapidem. Expert Rev. Anticancer. Ther. 2021, 21, 1333–1353. [Google Scholar] [CrossRef]
- Prapa, M.; Chiavelli, C.; Golinelli, G.; Grisendi, G.; Bestagno, M.; Di Tinco, R.; Dall’Ora, M.; Neri, G.; Candini, O.; Spano, C.; et al. GD2 CAR T cells against human glioblastoma. NPJ Precis. Oncol. 2021, 5, 93. [Google Scholar] [CrossRef]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; Ellingson, B.M.; Touat, M.; Maher, E.; De La Fuente, M.I.; Holdhoff, M.; Cote, G.M.; Burris, H.; Janku, F.; Young, R.J.; et al. Ivosidenib in Isocitrate Dehydrogenase 1-Mutated Advanced Glioma. J. Clin. Oncol. 2020, 38, 3398–3406. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Brem, S.; Campian, J.L.; Trusheim, J.E.; Iwamoto, F.M.; Tran, D.D.; Ansstas, G.; Cobbs, C.S.; Heth, J.A.; et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination with Extension of Survival among Patients with Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA Oncol. 2022, 9, 112. [Google Scholar] [CrossRef]
- Ahluwalia, M.S.; Reardon, D.A.; Abad, A.P.; Curry, W.T.; Wong, E.T.; Figel, S.A.; Mechtler, L.L.; Peereboom, D.M.; Hutson, A.D.; Withers, H.G.; et al. Phase IIa Study of SurVaxM Plus Adjuvant Temozolomide for Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2022. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Suginobe, N.; Nakamura, M.; Takanashi, Y.; Ban, H.; Gotoh, M. Mechanism of action of DSP-7888 (adegramotide/nelatimotide) Emulsion, a peptide-based therapeutic cancer vaccine with the potential to turn up the heat on non-immunoreactive tumors. Clin. Transl. Oncol. 2022, 25, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef] [PubMed]
- Lasek, W.; Zagożdżon, R.; Jakobisiak, M. Interleukin 12: Still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother. 2014, 63, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Li, A.W.; Lim, W.A. Engineering cytokines and cytokine circuits. Science 2020, 370, 1034–1035. [Google Scholar] [CrossRef]
- Parker, B.S.; Rautela, J.; Hertzog, P.J. Antitumour actions of interferons: Implications for cancer therapy. Nat. Rev. Cancer 2016, 16, 131–144. [Google Scholar] [CrossRef]
- Birocchi, F.; Cusimano, M.; Rossari, F.; Beretta, S.; Rancoita, P.M.V.; Ranghetti, A.; Colombo, S.; Costa, B.; Angel, P.; Sanvito, F.; et al. Targeted inducible delivery of immunoactivating cytokines reprograms glioblastoma microenvironment and inhibits growth in mouse models. Sci. Transl. Med. 2022, 14, eabl4106. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Mazzieri, R.; Politi, L.S.; Pucci, F.; Zonari, E.; Sitia, G.; Mazzoleni, S.; Moi, D.; Venneri, M.A.; Indraccolo, S.; et al. Tumor-targeted interferon-alpha delivery by Tie2-expressing monocytes inhibits tumor growth and metastasis. Cancer Cell 2008, 14, 299–311. [Google Scholar] [CrossRef]
- Escobar, G.; Barbarossa, L.; Barbiera, G.; Norelli, M.; Genua, M.; Ranghetti, A.; Plati, T.; Camisa, B.; Brombin, C.; Cittaro, D.; et al. Interferon gene therapy reprograms the leukemia microenvironment inducing protective immunity to multiple tumor antigens. Nat. Commun. 2018, 9, 2896. [Google Scholar] [CrossRef]
- Turrini, R.; Pabois, A.; Xenarios, I.; Coukos, G.; Delaloye, J.F.; Doucey, M.A. TIE-2 expressing monocytes in human cancers. Oncoimmunology 2017, 6, e1303585. [Google Scholar] [CrossRef]
- Levin, V.A.; Chan, J.; Datta, M.; Yee, J.L.; Jain, R.K. Effect of angiotensin system inhibitors on survival in newly diagnosed glioma patients and recurrent glioblastoma patients receiving chemotherapy and/or bevacizumab. J. Neuro-Oncol. 2017, 134, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Ekhator, C.; Rak, R.; Tadipatri, R.; Fonkem, E.; Grewal, J. A Single-Center Experience of Dopamine Antagonist ONC201 for Recurrent Histone H3 Lysine 27-to-Methionine (H3K27M)-Mutant Glioblastoma in Adults. Cureus 2022, 14, e28175. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Oligodendroglioma | Astrocytoma | H3—Altered Gliomas | Glioblastoma IDH wt | |
|---|---|---|---|---|
| Morphology |
|
|
|
|
| Genomic and epigenetic alterations |
|
|
|
|
| Prognosis | 8–17 years [11,12] | 6–12 years [11,12]. | 9–24 months [11,12] | 9–24 months [11,12] |
| IDH Mutated Gliomas | IDH Wildtype Gliomas | ||
|---|---|---|---|
 | Lower infiltration of immune cells than IDH wt gliomas, comprising lower microglia and macrophages percentage. |  | Higher percentage of tumor associated microglia and macrophages is related to higher glioma grade, driving ECM remodelling and angiogenesis. |
 | TME in astrocytomas reflects a predominant macrophage signature. In oligodendroglioma myeloid immune cells show mainly microglia expression states [7,8]. The 2-HG in IDH mutated gliomas interferes with recruitment and function of T cells [33]. |  | Myeloid cells in IDH wt gliomas reflect a predominant machrophage signature [33]. Infiltration by immune cells is favored by cytochines and chemokines from glioma cells (CCL2, CCL7, GDNF, CSF-1, GM-CSF, HGF, SDF-1). Also neutrophils and mast cells are recruited by GBM [32,33,34]. |
 | Astrocytoma rather than oligodendroglioma display increased percentages of PD-1+ CD8+ Tcells, TIM-3+CD4+T cells and T regulatory cells. |  | Several factors contribute to T cell-exhaustion (TGF β2, FAS-L, PD-L1, Sox2, Oct4). |
 | 2-HG is supposed to inhibit angiogenesis. Additionally through reduced HIF-1α levels, it inhibits glycolytic switch-related genes, tipically expressed in IDH wt subtypes [7,8,9,33]. |  | Stem cells located in the perivascular niche can differentiate either into cancer cells or normal cells. |
| Interactions with normal glial cells and neurons, interactions with stem cells need to be elucidate |  | Astrocytes contribute to neoangiogenesis in GBM. They can undergo neoplastic transformation [32,33,34]. | |
 | Interactions between cells involve multiple routes of communication (gap junctions, extracellular vescicles, nanotubes, microtube, paracrine signaling, extracellular RNA). | ||
| Glutamatergic neutron to brain tumorsynapses are involved in glioma invasiveness. | |||
 | |||
| Trial Name | Phase | Experimental Compounds | Setting |
|---|---|---|---|
| NCT03548571 | II/III | Dendritic Cells transfected with mRNA from autologous tumor stem cells, survivin, and hTERT [95] | Primary treated patients with IDH wild-type, MGMT-promotor methylated GBM |
| NCT04277221 | III | Autologous Dendritic Cell/Tumor Antigen (ADCTA-SSI-G1) | Recurrent GBM |
| NCT03149003 | III | DSP-7888 Dosing Emulsion [102] | Recurrent or Progressive GBM (secondary GBMexcluded) |
| NCT02761070 | III | Dose-dense temozolomide followed by Bevacizumab | Recurrent GBM |
| NCT02017717 | III | Nivolumab +/− Ipilimumab | Recurrent GBM |
| NCT02667587 | III | Nivolumab | Newly diagnosed MGMT-promotor methylated GBM (secondary GBMexcluded) |
| NCT00045968 | III | DCVax-L [103] | Newly diagnosed GBM |
| NCT03025893 | II/III | Sunitinib | Recurrent GBM |
| Trial Name | Phase | Experimental Compounds | Setting |
|---|---|---|---|
| NCT01236560 | II/III | Bevacizumab | Newly diagnosed high-grade gliomas in young patients |
| NCT00045968 | III | DCVax-L [103] | Newly diagnosed grade IV astrocytoma |
| NCT03149003 | III | DSP-7888 Dosing Emulsion [102] | Grade 4 astrocytoma. Recurrent or Progressive disease. |
| NCT04532229 | III | Nimotuzumab | Newly diagnosed diffuse intrinsic pontine glioma |
| NCT05009992 | III | ONC201 [112] | Midline glioma |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Nunno, V.; Aprile, M.; Gatto, L.; Tosoni, A.; Ranieri, L.; Bartolini, S.; Franceschi, E. Tumor Microenvironment in Gliomas: A Treatment Hurdle or an Opportunity to Grab? Cancers 2023, 15, 1042. https://doi.org/10.3390/cancers15041042
Di Nunno V, Aprile M, Gatto L, Tosoni A, Ranieri L, Bartolini S, Franceschi E. Tumor Microenvironment in Gliomas: A Treatment Hurdle or an Opportunity to Grab? Cancers. 2023; 15(4):1042. https://doi.org/10.3390/cancers15041042
Chicago/Turabian StyleDi Nunno, Vincenzo, Marta Aprile, Lidia Gatto, Alicia Tosoni, Lucia Ranieri, Stefania Bartolini, and Enrico Franceschi. 2023. "Tumor Microenvironment in Gliomas: A Treatment Hurdle or an Opportunity to Grab?" Cancers 15, no. 4: 1042. https://doi.org/10.3390/cancers15041042
APA StyleDi Nunno, V., Aprile, M., Gatto, L., Tosoni, A., Ranieri, L., Bartolini, S., & Franceschi, E. (2023). Tumor Microenvironment in Gliomas: A Treatment Hurdle or an Opportunity to Grab? Cancers, 15(4), 1042. https://doi.org/10.3390/cancers15041042