
The Class IIA Histone Deacetylase (HDAC) Inhibitor TMP269 Downregulates Ribosomal Proteins and Has Anti-Proliferative and Pro-Apoptotic Effects on AML Cells
 , , ,
, , ,  ,
,  ,
,  and
and 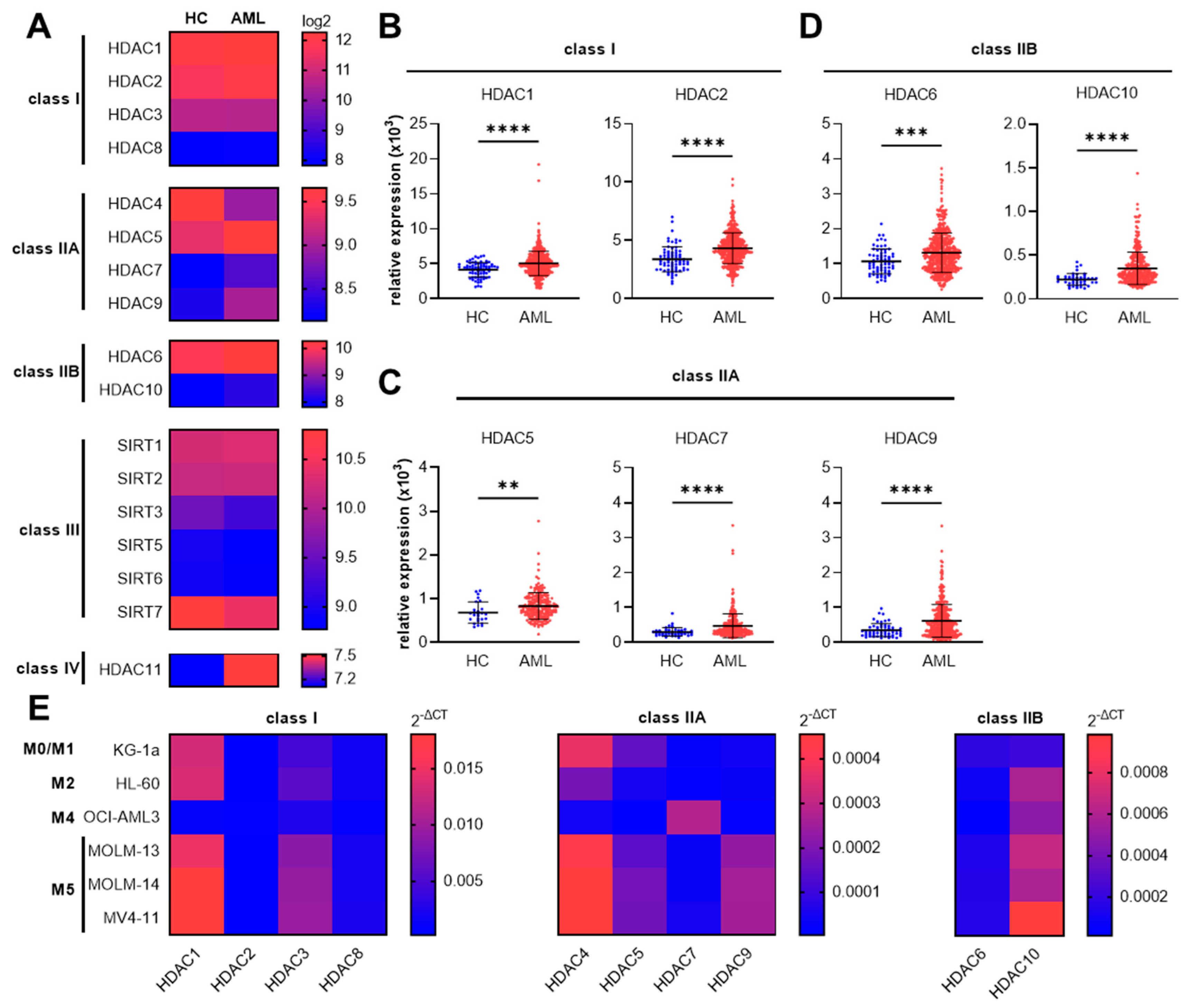{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Lines and Culture Conditions
2.3. Quantitative Real-Time PCR (RT-PCR)
2.4. Cell Proliferation Analysis
2.5. Apoptosis Assay
2.6. LDH Assay
2.7. Database Analysis
2.8. Proteomics
2.9. Bioinformatics
2.10. Western Blot Analysis
2.11. Statistical Analysis
3. Results
3.1. Increased Gene Expression of Specific Class I, Class IIA, and Class IIB HDAC Genes in AML Patients and Human AML Cell Lines
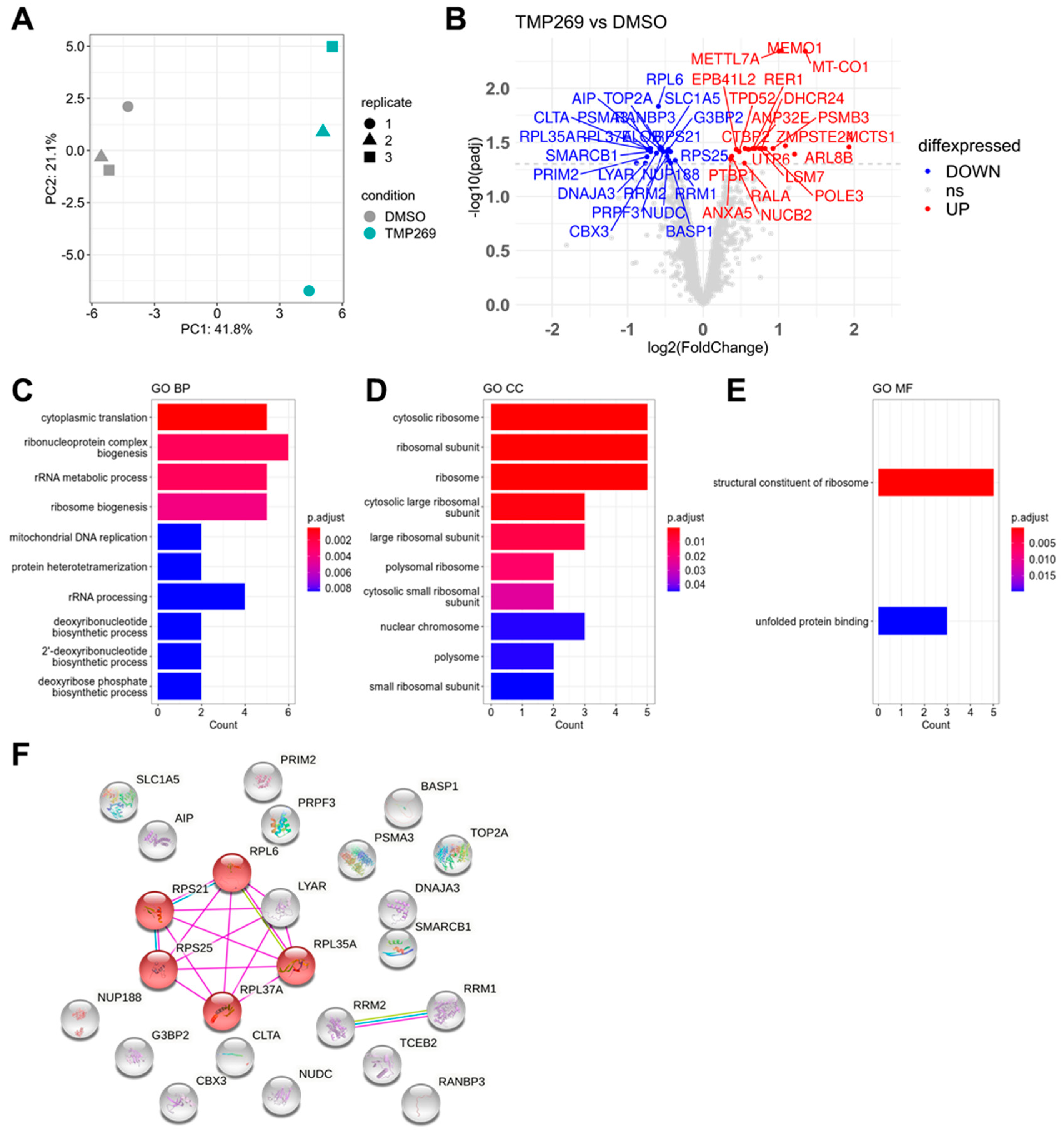
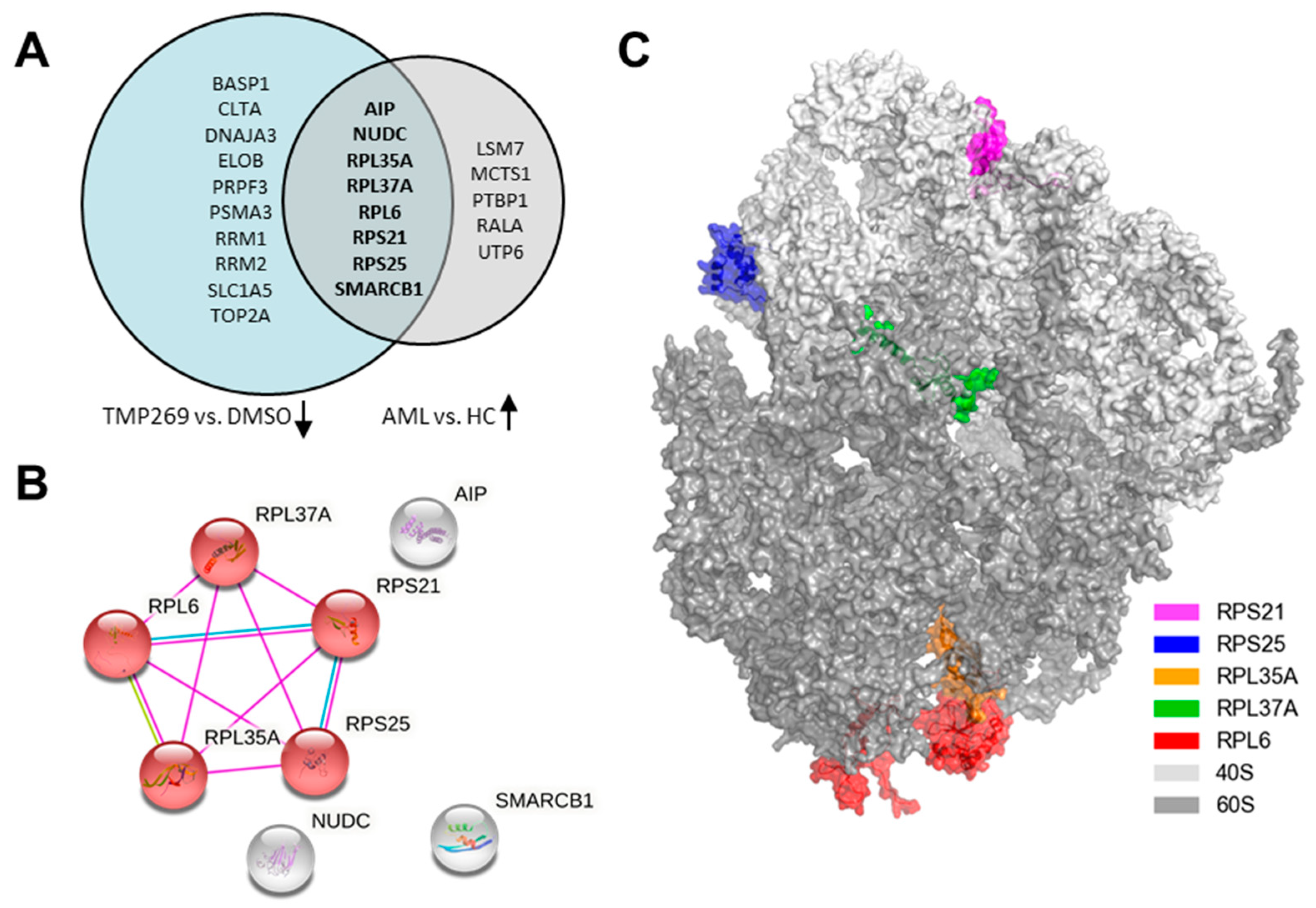
3.2. TMP269 Treatment Downregulates Ribosomal Proteins Which Are Increased in AML Patients at the Gene Expression Level
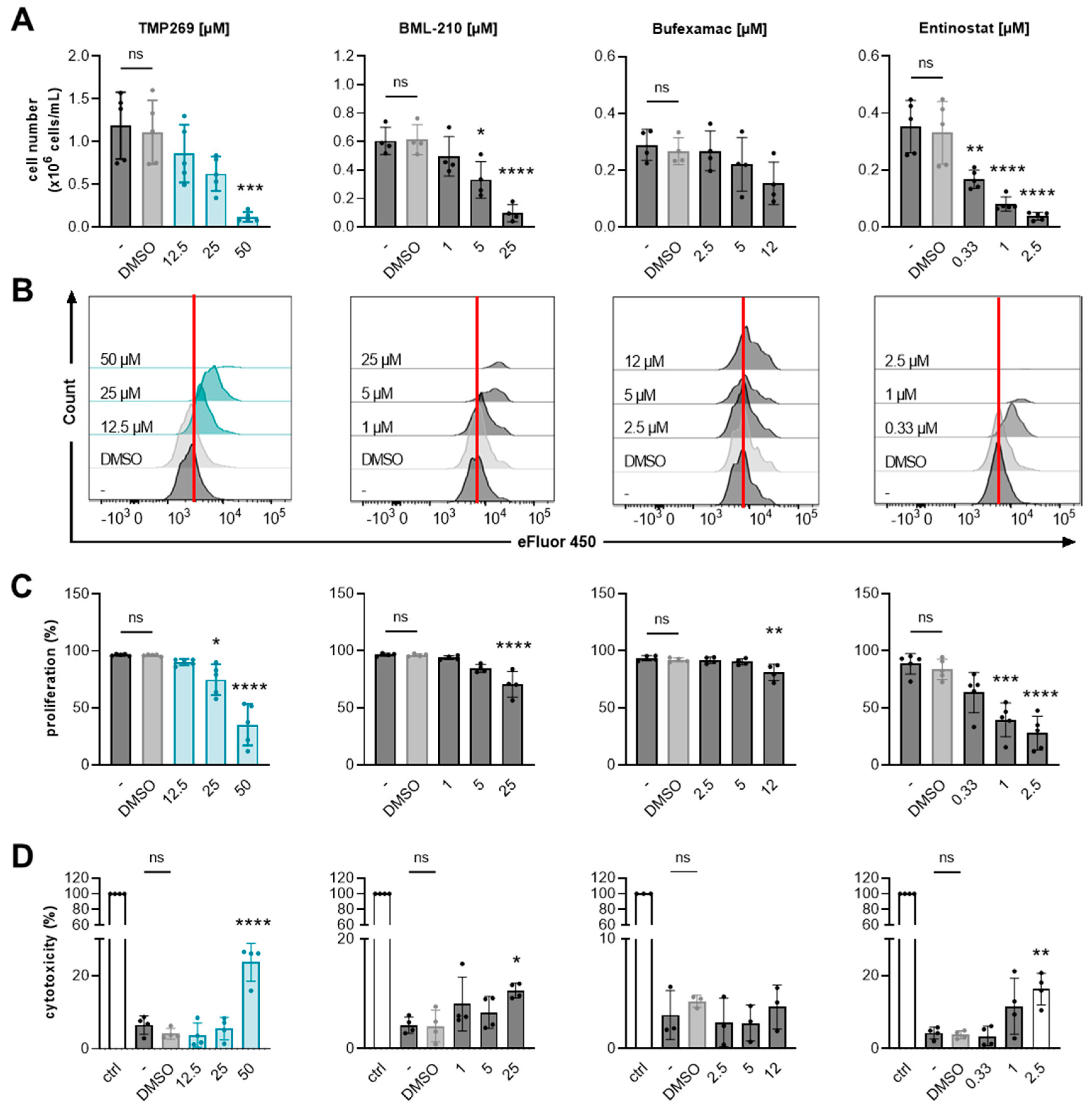
3.3. Inhibition of Class IIA HDACs by TMP269 Dampens AML Cell Growth and Reduces Cell Proliferation in a Concentration-Dependent Manner
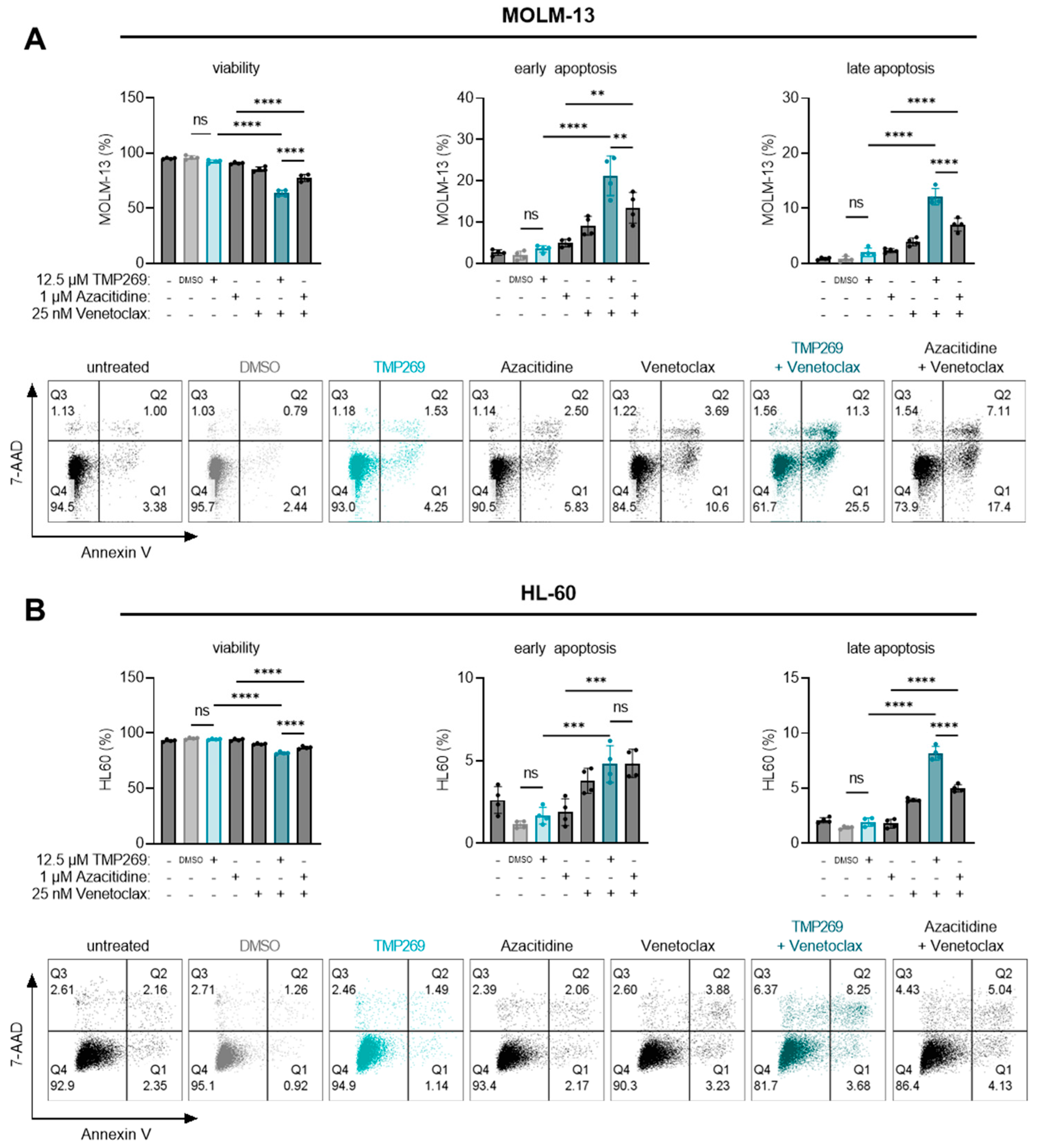
3.4. Combination Treatment of TMP269 with Venetoclax Has Additive Apoptotic Effects on AML Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- U.S. National Institutes of Health, National Cancer Institute, Surveillance, Epidemiology, and End Results Program. Cancer Stat Facts: Acute Myeloid Leukemia (AML). Available online: https://seer.cancer.gov/statfacts/html/amyl.html (accessed on 5 January 2022).
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Wei, A.H.; Pollyea, D.A.; Fathi, A.T.; Vyas, P.; DiNardo, C.D. New directions for emerging therapies in acute myeloid leukemia: The next chapter. Blood Cancer J. 2020, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Liu, H. Emerging agents and regimens for AML. J. Hematol. Oncol. 2021, 14, 49. [Google Scholar] [CrossRef]
- Di Nardo, C.D.; Pratz, K.W.; Letai, A.; Jonas, B.A.; Wei, A.H.; Thirman, M.; Arellano, M.; Frattini, M.G.; Kantarjian, H.; Popovic, R.; et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: A non-randomised, open-label, phase 1b study. Lancet Oncol. 2018, 19, 216–228. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Strickland, S.A., Jr.; Hou, J.Z.; Fiedler, W.; Lin, T.L.; Walter, R.B.; Enjeti, A.; Tiong, I.S.; Savona, M.; Lee, S.; et al. Venetoclax Combined With Low-Dose Cytarabine for Previously Untreated Patients With Acute Myeloid Leukemia: Results From a Phase Ib/II Study. J. Clin. Oncol. 2019, 37, 1277–1284. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Dohner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Pei, S.; Pollyea, D.A.; Gustafson, A.; Stevens, B.M.; Minhajuddin, M.; Fu, R.; Riemondy, K.A.; Gillen, A.E.; Sheridan, R.M.; Kim, J.; et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients with Acute Myeloid Leukemia. Cancer Discov. 2020, 10, 536–551. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.M.; Walsh, L.A.; Chan, T.A. Driver mutations of cancer epigenomes. Protein Cell 2014, 5, 265–296. [Google Scholar] [CrossRef]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef]
- Parbin, S.; Kar, S.; Shilpi, A.; Sengupta, D.; Deb, M.; Rath, S.K.; Patra, S.K. Histone deacetylases: A saga of perturbed acetylation homeostasis in cancer. J. Histochem. Cytochem. 2014, 62, 11–33. [Google Scholar] [CrossRef] [PubMed]
- Patra, S.; Panigrahi, D.P.; Praharaj, P.P.; Bhol, C.S.; Mahapatra, K.K.; Mishra, S.R.; Behera, B.P.; Jena, M.; Bhutia, S.K. Dysregulation of histone deacetylases in carcinogenesis and tumor progression: A possible link to apoptosis and autophagy. Cell Mol. Life Sci. 2019, 76, 3263–3282. [Google Scholar] [CrossRef] [PubMed]
- Chueh, A.C.; Tse, J.W.; Togel, L.; Mariadason, J.M. Mechanisms of Histone Deacetylase Inhibitor-Regulated Gene Expression in Cancer Cells. Antioxid. Redox Signal. 2015, 23, 66–84. [Google Scholar] [CrossRef]
- Hayashi, A.; Horiuchi, A.; Kikuchi, N.; Hayashi, T.; Fuseya, C.; Suzuki, A.; Konishi, I.; Shiozawa, T. Type-specific roles of histone deacetylase (HDAC) overexpression in ovarian carcinoma: HDAC1 enhances cell proliferation and HDAC3 stimulates cell migration with downregulation of E-cadherin. Int. J. Cancer 2010, 127, 1332–1346. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Ali, A.; Khan, S.; Ibrahim, M.; Alshehri, M.A.; Thirupathi, A. Inhibition of HDACs Suppresses Cell Proliferation and Cell Migration of Gastric Cancer by Regulating E2F5 Targeting BCL2. Life 2021, 11, 1425. [Google Scholar] [CrossRef] [PubMed]
- Petrella, A.; D’Acunto, C.W.; Rodriquez, M.; Festa, M.; Tosco, A.; Bruno, I.; Terracciano, S.; Taddei, M.; Paloma, L.G.; Parente, L. Effects of FR235222, a novel HDAC inhibitor, in proliferation and apoptosis of human leukaemia cell lines: Role of annexin A1. Eur. J. Cancer 2008, 44, 740–749. [Google Scholar] [CrossRef]
- Insinga, A.; Monestiroli, S.; Ronzoni, S.; Gelmetti, V.; Marchesi, F.; Viale, A.; Altucci, L.; Nervi, C.; Minucci, S.; Pelicci, P.G. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat. Med. 2005, 11, 71–76. [Google Scholar] [CrossRef]
- Guo, S.Q.; Zhang, Y.Z. Histone deacetylase inhibition: An important mechanism in the treatment of lymphoma. Cancer Biol. Med. 2012, 9, 85–89. [Google Scholar] [CrossRef]
- San Jose-Eneriz, E.; Gimenez-Camino, N.; Agirre, X.; Prosper, F. HDAC Inhibitors in Acute Myeloid Leukemia. Cancers 2019, 11, 1794. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef]
- Schaefer, E.W.; Loaiza-Bonilla, A.; Juckett, M.; DiPersio, J.F.; Roy, V.; Slack, J.; Wu, W.; Laumann, K.; Espinoza-Delgado, I.; Gore, S.D.; et al. A phase 2 study of vorinostat in acute myeloid leukemia. Haematologica 2009, 94, 1375–1382. [Google Scholar] [CrossRef]
- Young, C.S.; Clarke, K.M.; Kettyle, L.M.; Thompson, A.; Mills, K.I. Decitabine-Vorinostat combination treatment in acute myeloid leukemia activates pathways with potential for novel triple therapy. Oncotarget 2017, 8, 51429–51446. [Google Scholar] [CrossRef] [PubMed]
- Schlenk, R.F.; Krauter, J.; Raffoux, E.; Kreuzer, K.A.; Schaich, M.; Noens, L.; Pabst, T.; Vusirikala, M.; Bouscary, D.; Spencer, A.; et al. Panobinostat monotherapy and combination therapy in patients with acute myeloid leukemia: Results from two clinical trials. Haematologica 2018, 103, e25–e28. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.T.; Kim, H.N.; Lee, I.K.; Nguyen-Pham, T.N.; Ahn, J.S.; Kim, Y.K.; Lee, J.J.; Park, K.S.; Kook, H.; Kim, H.J. Improved therapeutic effect against leukemia by a combination of the histone methyltransferase inhibitor chaetocin and the histone deacetylase inhibitor trichostatin A. J. Korean Med. Sci. 2013, 28, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Momparler, R.L.; Cote, S.; Momparler, L.F.; Idaghdour, Y. Epigenetic therapy of acute myeloid leukemia using 5-aza-2’-deoxycytidine (decitabine) in combination with inhibitors of histone methylation and deacetylation. Clin. Epigenetics 2014, 6, 19. [Google Scholar] [CrossRef]
- Ramsey, J.M.; Kettyle, L.M.; Sharpe, D.J.; Mulgrew, N.M.; Dickson, G.J.; Bijl, J.J.; Austin, P.; Mayotte, N.; Cellot, S.; Lappin, T.R.; et al. Entinostat prevents leukemia maintenance in a collaborating oncogene-dependent model of cytogenetically normal acute myeloid leukemia. Stem. Cells 2013, 31, 1434–1445. [Google Scholar] [CrossRef] [PubMed]
- Gojo, I.; Jiemjit, A.; Trepel, J.B.; Sparreboom, A.; Figg, W.D.; Rollins, S.; Tidwell, M.L.; Greer, J.; Chung, E.J.; Lee, M.J.; et al. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood 2007, 109, 2781–2790. [Google Scholar] [CrossRef]
- Di Giorgio, E.; Gagliostro, E.; Brancolini, C. Selective class IIa HDAC inhibitors: Myth or reality. Cell Mol. Life Sci. 2015, 72, 73–86. [Google Scholar] [CrossRef]
- Choi, S.Y.; Kee, H.J.; Jin, L.; Ryu, Y.; Sun, S.; Kim, G.R.; Jeong, M.H. Inhibition of class IIa histone deacetylase activity by gallic acid, sulforaphane, TMP269, and panobinostat. Biomed. Pharmacother. 2018, 101, 145–154. [Google Scholar] [CrossRef]
- Haferlach, T.; Kohlmann, A.; Wieczorek, L.; Basso, G.; Kronnie, G.T.; Bene, M.C.; De Vos, J.; Hernandez, J.M.; Hofmann, W.K.; Mills, K.I.; et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: Report from the International Microarray Innovations in Leukemia Study Group. J. Clin. Oncol. 2010, 28, 2529–2537. [Google Scholar] [CrossRef]
- Kohlmann, A.; Kipps, T.J.; Rassenti, L.Z.; Downing, J.R.; Shurtleff, S.A.; Mills, K.I.; Gilkes, A.F.; Hofmann, W.K.; Basso, G.; Dell’orto, M.C.; et al. An international standardization programme towards the application of gene expression profiling in routine leukaemia diagnostics: The Microarray Innovations in LEukemia study prephase. Br. J. Haematol. 2008, 142, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Willforss, J.; Chawade, A.; Levander, F. NormalyzerDE: Online Tool for Improved Normalization of Omics Expression Data and High-Sensitivity Differential Expression Analysis. J. Proteome Res. 2019, 18, 732–740. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate—A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Zhang, X.; Smits, A.H.; van Tilburg, G.B.; Ovaa, H.; Huber, W.; Vermeulen, M. Proteome-wide identification of ubiquitin interactions using UbIA-MS. Nat. Protoc. 2018, 13, 530–550. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Frauenlob, T.; Neuper, T.; Mehinagic, M.; Dang, H.H.; Boraschi, D.; Horejs-Hoeck, J. Helicobacter pylori Infection of Primary Human Monocytes Boosts Subsequent Immune Responses to LPS. Front. Immunol. 2022, 13, 847958. [Google Scholar] [CrossRef]
- Padilha, S.L.; Souza, E.J.; Matos, M.C.; Domino, N.R. Acute myeloid leukemia: Survival analysis of patients at a university hospital of Parana. Rev. Bras. Hematol. Hemoter. 2015, 37, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Basharat, M.; Khan, S.A.; Din, N.U.; Ahmed, D. Immunophenotypic characterisation of morphologically diagnosed cases of Acute Myeloid Leukaemia (AML). Pak. J. Med. Sci. 2019, 35, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Hu, E.; Dul, E.; Sung, C.M.; Chen, Z.; Kirkpatrick, R.; Zhang, G.F.; Johanson, K.; Liu, R.; Lago, A.; Hofmann, G.; et al. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J. Pharmacol. Exp. Ther. 2003, 307, 720–728. [Google Scholar] [CrossRef]
- Vannini, A.; Volpari, C.; Filocamo, G.; Casavola, E.C.; Brunetti, M.; Renzoni, D.; Chakravarty, P.; Paolini, C.; De Francesco, R.; Gallinari, P.; et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 15064–15069. [Google Scholar] [CrossRef]
- Kato, Y.; Yoshimura, K.; Shin, T.; Verheul, H.; Hammers, H.; Sanni, T.B.; Salumbides, B.C.; Van Erp, K.; Schulick, R.; Pili, R. Synergistic in vivo antitumor effect of the histone deacetylase inhibitor MS-275 in combination with interleukin 2 in a murine model of renal cell carcinoma. Clin. Cancer Res. 2007, 13, 4538–4546. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, C.; Ikezoe, T.; Yang, J.; Takeuchi, S.; Koeffler, H.P.; Yokoyama, A. MS-275, a novel histone deacetylase inhibitor with selectivity against HDAC1, induces degradation of FLT3 via inhibition of chaperone function of heat shock protein 90 in AML cells. Leuk. Res. 2008, 32, 1382–1392. [Google Scholar] [CrossRef]
- Hess-Stumpp, H.; Bracker, T.U.; Henderson, D.; Politz, O. MS-275, a potent orally available inhibitor of histone deacetylases--the development of an anticancer agent. Int. J. Biochem. Cell Biol. 2007, 39, 1388–1405. [Google Scholar] [CrossRef]
- Lobera, M.; Madauss, K.P.; Pohlhaus, D.T.; Wright, Q.G.; Trocha, M.; Schmidt, D.R.; Baloglu, E.; Trump, R.P.; Head, M.S.; Hofmann, G.A.; et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat. Chem. Biol. 2013, 9, 319–325. [Google Scholar] [CrossRef]
- Kikuchi, S.; Suzuki, R.; Ohguchi, H.; Yoshida, Y.; Lu, D.; Cottini, F.; Jakubikova, J.; Bianchi, G.; Harada, T.; Gorgun, G.; et al. Class IIa HDAC inhibition enhances ER stress-mediated cell death in multiple myeloma. Leukemia 2015, 29, 1918–1927. [Google Scholar] [CrossRef]
- Kaletsch, A.; Pinkerneil, M.; Hoffmann, M.J.; Jaguva Vasudevan, A.A.; Wang, C.; Hansen, F.K.; Wiek, C.; Hanenberg, H.; Gertzen, C.; Gohlke, H.; et al. Effects of novel HDAC inhibitors on urothelial carcinoma cells. Clin. Epigenetics 2018, 10, 100. [Google Scholar] [CrossRef]
- El Khoury, W.; Nasr, Z. Deregulation of ribosomal proteins in human cancers. Biosci. Rep. 2021, 41, BSR20211577. [Google Scholar] [CrossRef] [PubMed]
- Jayathilaka, N.; Han, A.; Gaffney, K.J.; Dey, R.; Jarusiewicz, J.A.; Noridomi, K.; Philips, M.A.; Lei, X.; He, J.; Ye, J.; et al. Inhibition of the function of class IIa HDACs by blocking their interaction with MEF2. Nucleic Acids Res. 2012, 40, 5378–5388. [Google Scholar] [CrossRef] [PubMed]
- Savickiene, J.; Borutinskaite, V.V.; Treigyte, G.; Magnusson, K.E.; Navakauskiene, R. The novel histone deacetylase inhibitor BML-210 exerts growth inhibitory, proapoptotic and differentiation stimulating effects on the human leukemia cell lines. Eur. J. Pharmacol. 2006, 549, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Bantscheff, M.; Hopf, C.; Savitski, M.M.; Dittmann, A.; Grandi, P.; Michon, A.M.; Schlegl, J.; Abraham, Y.; Becher, I.; Bergamini, G.; et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 2011, 29, 255–265. [Google Scholar] [CrossRef]
- Savickiene, J.; Treigyte, G.; Borutinskaite, V.V.; Navakauskiene, R. Antileukemic activity of combined epigenetic agents, DNMT inhibitors zebularine and RG108 with HDAC inhibitors, against promyelocytic leukemia HL-60 cells. Cell Mol. Biol. Lett. 2012, 17, 501–525. [Google Scholar] [CrossRef]
- Vire, B.; de Walque, S.; Restouin, A.; Olive, D.; Van Lint, C.; Collette, Y. Anti-leukemia activity of MS-275 histone deacetylase inhibitor implicates 4-1BBL/4-1BB immunomodulatory functions. PLoS ONE 2009, 4, e7085. [Google Scholar] [CrossRef]
- Maggio, S.C.; Rosato, R.R.; Kramer, L.B.; Dai, Y.; Rahmani, M.; Paik, D.S.; Czarnik, A.C.; Payne, S.G.; Spiegel, S.; Grant, S. The histone deacetylase inhibitor MS-275 interacts synergistically with fludarabine to induce apoptosis in human leukemia cells. Cancer Res. 2004, 64, 2590–2600. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Yamashita, T.; Mariko, Y.; Nosaka, Y.; Tsuchiya, K.; Ando, T.; Suzuki, T.; Tsuruo, T.; Nakanishi, O. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc. Natl. Acad. Sci. USA 1999, 96, 4592–4597. [Google Scholar] [CrossRef] [PubMed]
- Borutinskaite, V.; Navakauskiene, R. The Histone Deacetylase Inhibitor BML-210 Influences Gene and Protein Expression in Human Promyelocytic Leukemia NB4 Cells via Epigenetic Reprogramming. Int. J. Mol. Sci. 2015, 16, 18252–18269. [Google Scholar] [CrossRef]
- Medler, T.R.; Craig, J.M.; Fiorillo, A.A.; Feeney, Y.B.; Harrell, J.C.; Clevenger, C.V. HDAC6 Deacetylates HMGN2 to Regulate Stat5a Activity and Breast Cancer Growth. Mol. Cancer Res. 2016, 14, 994–1008. [Google Scholar] [CrossRef]
- Cherry, E.M.; Abbott, D.; Amaya, M.; McMahon, C.; Schwartz, M.; Rosser, J.; Sato, A.; Schowinsky, J.; Inguva, A.; Minhajuddin, M.; et al. Venetoclax and azacitidine compared with induction chemotherapy for newly diagnosed patients with acute myeloid leukemia. Blood Adv. 2021, 5, 5565–5573. [Google Scholar] [CrossRef]
- Winer, E.S.; Stone, R.M. Novel therapy in Acute myeloid leukemia (AML): Moving toward targeted approaches. Ther. Adv. Hematol. 2019, 10, 2040620719860645. [Google Scholar] [CrossRef] [PubMed]
- Estey, E.; Dohner, H. Acute myeloid leukaemia. Lancet 2006, 368, 1894–1907. [Google Scholar] [CrossRef]
- Goldman, S.L.; Hassan, C.; Khunte, M.; Soldatenko, A.; Jong, Y.; Afshinnekoo, E.; Mason, C.E. Epigenetic Modifications in Acute Myeloid Leukemia: Prognosis, Treatment, and Heterogeneity. Front. Genet 2019, 10, 133. [Google Scholar] [CrossRef]
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J 2016, 6, e441. [Google Scholar] [CrossRef] [PubMed]
- Lowenberg, B.; Zittoun, R.; Kerkhofs, H.; Jehn, U.; Abels, J.; Debusscher, L.; Cauchie, C.; Peetermans, M.; Solbu, G.; Suciu, S.; et al. On the value of intensive remission-induction chemotherapy in elderly patients of 65+ years with acute myeloid leukemia: A randomized phase III study of the European Organization for Research and Treatment of Cancer Leukemia Group. J. Clin. Oncol. 1989, 7, 1268–1274. [Google Scholar] [CrossRef]
- McCurdy, S.R.; Luger, S.M. Dose intensity for induction in acute myeloid leukemia: What, when, and for whom? Haematologica 2021, 106, 2544–2554. [Google Scholar] [CrossRef]
- Wei, Y.; Cao, Y.; Sun, R.; Cheng, L.; Xiong, X.; Jin, X.; He, X.; Lu, W.; Zhao, M. Targeting Bcl-2 Proteins in Acute Myeloid Leukemia. Front. Oncol. 2020, 10, 584974. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Andersson, T.M.; Rachet, B.; Bjorkholm, M.; Lambert, P.C. Survival and cure of acute myeloid leukaemia in England, 1971-2006: A population-based study. Br. J. Haematol. 2013, 162, 509–516. [Google Scholar] [CrossRef]
- Wang, P.; Wang, Z.; Liu, J. Role of HDACs in normal and malignant hematopoiesis. Mol. Cancer 2020, 19, 5. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Schuh, A.C.; Dohner, H.; Pleyer, L.; Seymour, J.F.; Fenaux, P.; Dombret, H. Azacitidine in adult patients with acute myeloid leukemia. Crit. Rev. Oncol. Hematol. 2017, 116, 159–177. [Google Scholar] [CrossRef] [PubMed]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef]
- Yang, H.; Maddipoti, S.; Quesada, A.; Bohannan, Z.; Cabrero Calvo, M.; Colla, S.; Wei, Y.; Estecio, M.; Wierda, W.; Bueso-Ramos, C.; et al. Analysis of class I and II histone deacetylase gene expression in human leukemia. Leuk. Lymphoma 2015, 56, 3426–3433. [Google Scholar] [CrossRef]
- Clocchiatti, A.; Florean, C.; Brancolini, C. Class IIa HDACs: From important roles in differentiation to possible implications in tumourigenesis. J. Cell Mol. Med. 2011, 15, 1833–1846. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Cojocari, D.; Purkal, J.J.; Popovic, R.; Talaty, N.N.; Xiao, Y.; Solomon, L.R.; Boghaert, E.R.; Leverson, J.D.; Phillips, D.C. 5-Azacitidine Induces NOXA to Prime AML Cells for Venetoclax-Mediated Apoptosis. Clin. Cancer Res. 2020, 26, 3371–3383. [Google Scholar] [CrossRef] [PubMed]
- Verza, F.A.; Das, U.; Fachin, A.L.; Dimmock, J.R.; Marins, M. Roles of Histone Deacetylases and Inhibitors in Anticancer Therapy. Cancers 2020, 12, 1664. [Google Scholar] [CrossRef]
- Kang, J.; Brajanovski, N.; Chan, K.T.; Xuan, J.; Pearson, R.B.; Sanij, E. Ribosomal proteins and human diseases: Molecular mechanisms and targeted therapy. Signal Transduct. Target. Ther. 2021, 6, 323. [Google Scholar] [CrossRef]
- Yonezawa, K.; Sugihara, Y.; Oshima, K.; Matsuda, T.; Nadano, D. Lyar, a cell growth-regulating zinc finger protein, was identified to be associated with cytoplasmic ribosomes in male germ and cancer cells. Mol. Cell Biochem. 2014, 395, 221–229. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, Y.; Gao, H.; Wang, Y.; Cheng, Q.; Jian, S.; Ding, Q.; Gu, W.; Yao, Y.; Ma, J.; et al. LYAR Promotes Colorectal Cancer Progression by Upregulating FSCN1 Expression and Fatty Acid Metabolism. Oxid. Med. Cell Longev. 2021, 2021, 9979707. [Google Scholar] [CrossRef]
- Izumikawa, K.; Ishikawa, H.; Yoshikawa, H.; Fujiyama, S.; Watanabe, A.; Aburatani, H.; Tachikawa, H.; Hayano, T.; Miura, Y.; Isobe, T.; et al. LYAR potentiates rRNA synthesis by recruiting BRD2/4 and the MYST-type acetyltransferase KAT7 to rDNA. Nucleic Acids Res. 2019, 47, 10357–10372. [Google Scholar] [CrossRef]
- Miyazawa, N.; Yoshikawa, H.; Magae, S.; Ishikawa, H.; Izumikawa, K.; Terukina, G.; Suzuki, A.; Nakamura-Fujiyama, S.; Miura, Y.; Hayano, T.; et al. Human cell growth regulator Ly-1 antibody reactive homologue accelerates processing of preribosomal RNA. Genes Cells 2014, 19, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Atmadibrata, B.; Yu, D.; Wong, M.; Liu, B.; Ho, N.; Ling, D.; Tee, A.E.; Wang, J.; Mungrue, I.N.; et al. Upregulation of LYAR induces neuroblastoma cell proliferation and survival. Cell Death Differ. 2017, 24, 1645–1654. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.N.; Ju, G.J.; Wang, Y.X.; Wang, Y.L.; Wang, K.; Chen, J.L.; Cai, W.; Zang, Q.W. LYAR promotes the proliferation of non-small cell lung cancer and is associated with poor prognosis. Folia Histochem. Cytobiol. 2021, 59, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Zuo, M.Q.; Zhang, Y.; Li, N.; Ma, C.; Dong, M.Q.; Gao, N. Structural snapshots of human pre-60S ribosomal particles before and after nuclear export. Nat. Commun. 2020, 11, 3542. [Google Scholar] [CrossRef]
- Bai, D.; Zhang, J.; Xiao, W.; Zheng, X. Regulation of the HDM2-p53 pathway by ribosomal protein L6 in response to ribosomal stress. Nucleic Acids Res. 2014, 42, 1799–1811. [Google Scholar] [CrossRef]
- Wu, Q.; Gou, Y.; Wang, Q.; Jin, H.; Cui, L.; Zhang, Y.; He, L.; Wang, J.; Nie, Y.; Shi, Y.; et al. Downregulation of RPL6 by siRNA inhibits proliferation and cell cycle progression of human gastric cancer cell lines. PLoS ONE 2011, 6, e26401. [Google Scholar] [CrossRef]
- Zhang, J.; Ma, Q.; Han, Y.; Wen, H.; Zhang, Z.; Hao, Y.; Xiao, F.; Liang, C. Downregulated RPL6 inhibits lung cancer cell proliferation and migration and promotes cell apoptosis by regulating the AKT signaling pathway. J. Thorac. Dis. 2022, 14, 507–514. [Google Scholar] [CrossRef]
- Muhs, M.; Yamamoto, H.; Ismer, J.; Takaku, H.; Nashimoto, M.; Uchiumi, T.; Nakashima, N.; Mielke, T.; Hildebrand, P.W.; Nierhaus, K.H.; et al. Structural basis for the binding of IRES RNAs to the head of the ribosomal 40S subunit. Nucleic Acids Res. 2011, 39, 5264–5275. [Google Scholar] [CrossRef]
- Li, M.; Center, M.S. Regulation of ribosomal protein S25 in HL60 cells isolated for resistance to adriamycin. FEBS Lett. 1992, 298, 142–144. [Google Scholar] [CrossRef]
- Adilakshmi, T.; Laine, R.O. Ribosomal protein S25 mRNA partners with MTF-1 and La to provide a p53-mediated mechanism for survival or death. J. Biol. Chem. 2002, 277, 4147–4151. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.E.; Leal, J.F.; Lleonart, M.E.; Ramon, Y.C.S.; Carnero, A. Loss-of-function genetic screening identifies a cluster of ribosomal proteins regulating p53 function. Carcinogenesis 2008, 29, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Sun, D.; Liao, Y.; Shang, K.; Lu, C. RPL35A is a key promotor involved in the development and progression of gastric cancer. Cancer Cell Int. 2021, 21, 497. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.D.; Martinovsky, G.; Naumovski, L. Inhibition of cell death by ribosomal protein L35a. Cancer Lett. 2002, 180, 195–202. [Google Scholar] [CrossRef]
- Saliba, A.N.; John, A.J.; Kaufmann, S.H. Resistance to venetoclax and hypomethylating agents in acute myeloid leukemia. Cancer Drug. Resist. 2021, 4, 125–142. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urwanisch, L.; Unger, M.S.; Sieberer, H.; Dang, H.-H.; Neuper, T.; Regl, C.; Vetter, J.; Schaller, S.; Winkler, S.M.; Kerschbamer, E.; et al. The Class IIA Histone Deacetylase (HDAC) Inhibitor TMP269 Downregulates Ribosomal Proteins and Has Anti-Proliferative and Pro-Apoptotic Effects on AML Cells. Cancers 2023, 15, 1039. https://doi.org/10.3390/cancers15041039
Urwanisch L, Unger MS, Sieberer H, Dang H-H, Neuper T, Regl C, Vetter J, Schaller S, Winkler SM, Kerschbamer E, et al. The Class IIA Histone Deacetylase (HDAC) Inhibitor TMP269 Downregulates Ribosomal Proteins and Has Anti-Proliferative and Pro-Apoptotic Effects on AML Cells. Cancers. 2023; 15(4):1039. https://doi.org/10.3390/cancers15041039
Chicago/Turabian StyleUrwanisch, Laura, Michael Stefan Unger, Helene Sieberer, Hieu-Hoa Dang, Theresa Neuper, Christof Regl, Julia Vetter, Susanne Schaller, Stephan M. Winkler, Emanuela Kerschbamer, and et al. 2023. "The Class IIA Histone Deacetylase (HDAC) Inhibitor TMP269 Downregulates Ribosomal Proteins and Has Anti-Proliferative and Pro-Apoptotic Effects on AML Cells" Cancers 15, no. 4: 1039. https://doi.org/10.3390/cancers15041039
APA StyleUrwanisch, L., Unger, M. S., Sieberer, H., Dang, H.-H., Neuper, T., Regl, C., Vetter, J., Schaller, S., Winkler, S. M., Kerschbamer, E., Weichenberger, C. X., Krenn, P. W., Luciano, M., Pleyer, L., Greil, R., Huber, C. G., Aberger, F., & Horejs-Hoeck, J. (2023). The Class IIA Histone Deacetylase (HDAC) Inhibitor TMP269 Downregulates Ribosomal Proteins and Has Anti-Proliferative and Pro-Apoptotic Effects on AML Cells. Cancers, 15(4), 1039. https://doi.org/10.3390/cancers15041039