
Enhancer Clusters Drive Type I Interferon-Induced TRAIL Overexpression in Cancer, and Its Intracellular Protein Accumulation Fails to Induce Apoptosis
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. In Silico Identification of Enhancer-Enriched Regions
2.2. Analyses of Cancer Datasets
2.3. Cell Culture
2.4. Cytokines and Drugs
2.5. RNA Extraction, cDNA Synthesis and qPCR
2.6. ELISA
2.7. Immunofluorescence
2.8. Flow Cytometry
2.9. Cytotoxicity Assay
2.10. Real-Time Cell Death Assay
2.11. MTT Assay
2.12. Statistical Analysis
2.13. Data Availability
3. Results
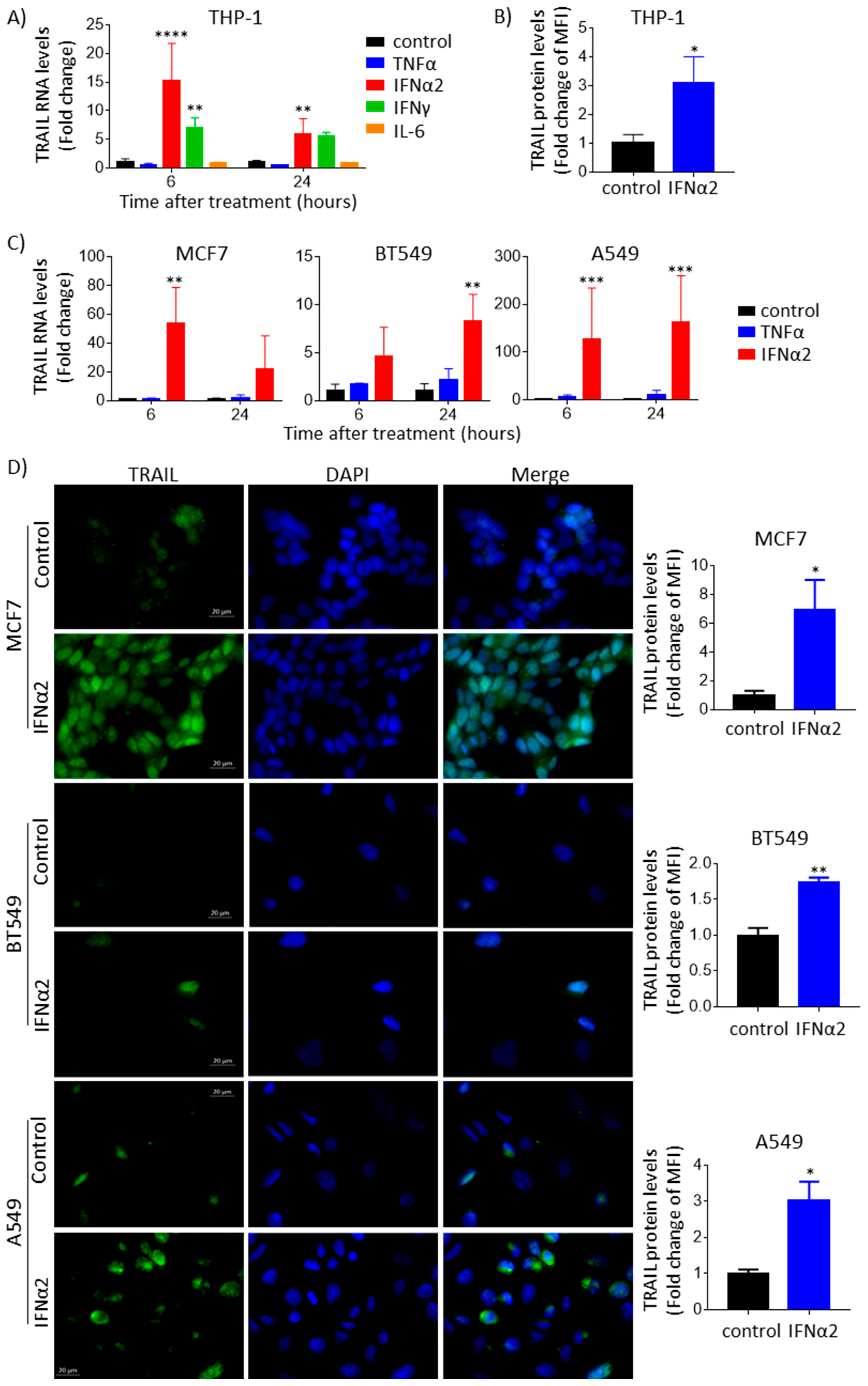
3.1. IFNα Upregulates TRAIL Gene Expression and Protein Levels across Different Cancer Types
3.2. IFNα-Induced Upregulation of TRAIL Is Linked to Chromatin Accessibility, Thus Enhancer Clusters, and Their Disruption Decreases TRAIL Expression
3.3. IFNα-Induced TRAIL Accumulates Inside Epithelial Cancer Cells and Is Not Detected Extracellularly
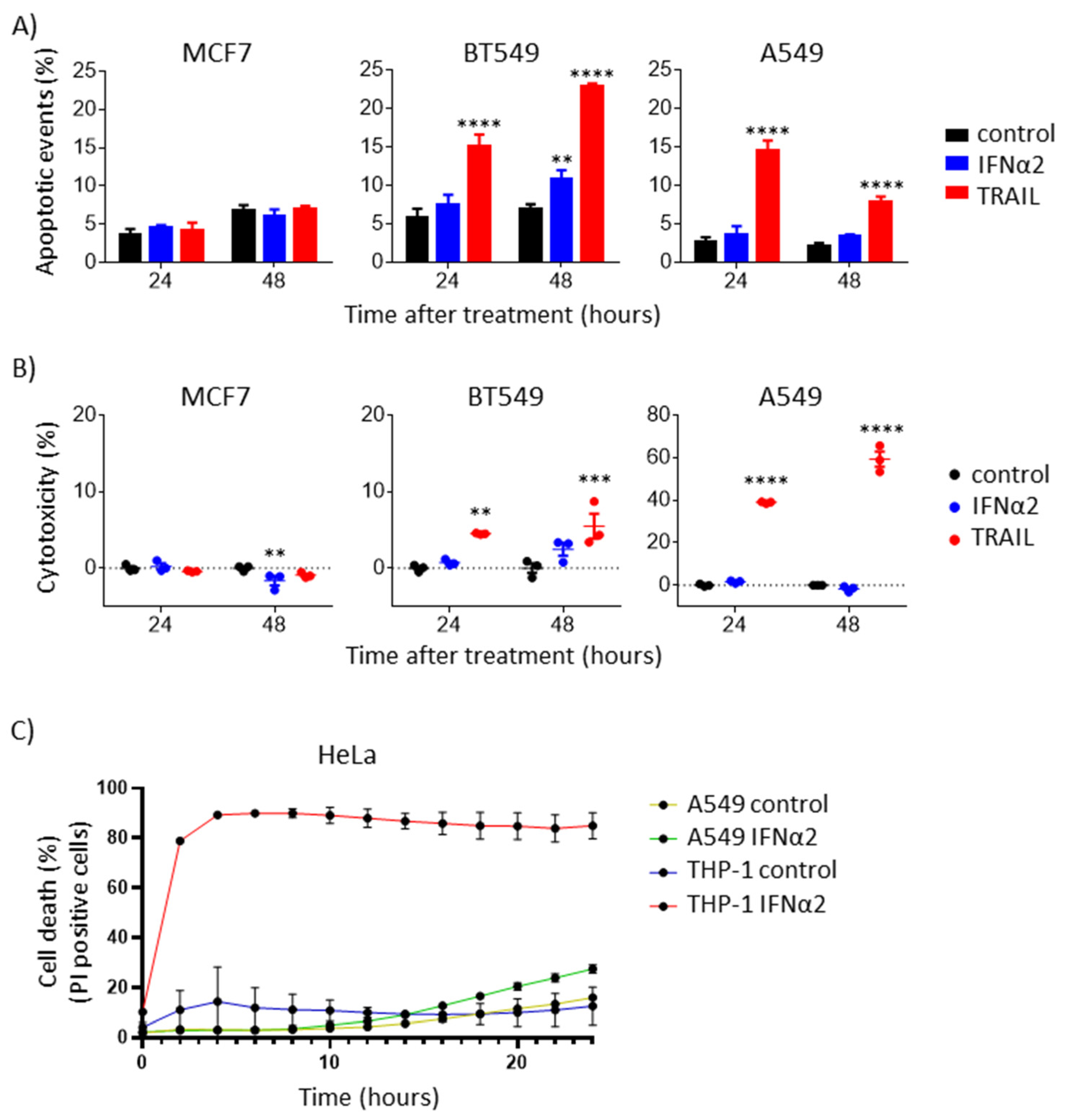
3.4. IFNα-Induced Internal TRAIL Accumulation Does Not Activate Apoptosis of Cancer Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sag, D.; Ayyildiz, Z.O.; Gunalp, S.; Wingender, G. The role of TRAIL/DRs in the modulation of immune cells and responses. Cancers 2019, 11, 1469. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Jiang, W.; Liu, Z.; Liu, S.; Liang, X. Virus infection and death receptor-mediated apoptosis. Viruses 2017, 9, 316. [Google Scholar] [CrossRef]
- Walczak, H. Death receptor–ligand systems in cancer, cell death, and inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008698. [Google Scholar] [CrossRef] [PubMed]
- Von Karstedt, S.; Montinaro, A.; Walczak, H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat. Rev. Cancer 2017, 17, 352–366. [Google Scholar] [CrossRef]
- Brincks, E.L.; Katewa, A.; Kucaba, T.A.; Griffith, T.S.; Legge, K.L. CD8 T cells utilize TRAIL to control influenza virus infection. J. Immunol. 2008, 181, 4918–4925. [Google Scholar] [CrossRef] [PubMed]
- Thomas, W.D.; Hersey, P. TNF-related apoptosis-inducing ligand (TRAIL) induces apoptosis in Fas ligand-resistant melanoma cells and mediates CD4 T cell killing of target cells. J. Immunol. 1998, 161, 2195–2200. [Google Scholar] [CrossRef]
- Cretney, E.; Takeda, K.; Yagita, H.; Glaccum, M.; Peschon, J.J.; Smyth, M.J. Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J. Immunol. 2002, 168, 1356–1361. [Google Scholar] [CrossRef]
- Papageorgiou, A.; Lashinger, L.; Millikan, R.; Grossman, H.B.; Benedict, W.; Dinney, C.P.; McConkey, D.J. Role of tumor necrosis factor-related apoptosis-inducing ligand in interferon-induced apoptosis in human bladder cancer cells. Cancer Res. 2004, 64, 8973–8979. [Google Scholar] [CrossRef]
- Clarke, N.; Jimenez-Lara, A.M.; Voltz, E.; Gronemeyer, H. Tumor suppressor IRF-1 mediates retinoid and interferon anticancer signaling to death ligand TRAIL. EMBO J. 2004, 23, 3051–3060. [Google Scholar] [CrossRef]
- Shigeno, M.; Nakao, K.; Ichikawa, T.; Suzuki, K.; Kawakami, A.; Abiru, S.; Miyazoe, S.; Nakagawa, Y.; Ishikawa, H.; Hamasaki, K. Interferon-α sensitizes human hepatoma cells to TRAIL-induced apoptosis through DR5 upregulation and NF-κB inactivation. Oncogene 2003, 22, 1653–1662. [Google Scholar] [CrossRef]
- Wang, X.; Cairns, M.J.; Yan, J. Super-enhancers in transcriptional regulation and genome organization. Nucleic Acids Res. 2019, 47, 11481–11496. [Google Scholar] [CrossRef] [PubMed]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Leonard, W.J. Chromatin accessibility and interactions in the transcriptional regulation of T cells. Front. Immunol. 2018, 9, 2738. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Bahr, C.; Von Paleske, L.; Uslu, V.V.; Remeseiro, S.; Takayama, N.; Ng, S.W.; Murison, A.; Langenfeld, K.; Petretich, M.; Scognamiglio, R. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 2018, 553, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; McKeown, M.R.; Lin, C.Y.; Monti, S.; Roemer, M.G.; Qi, J.; Rahl, P.B.; Sun, H.H.; Yeda, K.T.; Doench, J.G. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 2013, 24, 777–790. [Google Scholar] [CrossRef]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef]
- Di Benedetto, C.; Oh, J.; Choudhery, Z.; Shi, W.; Valdes, G.; Betancur, P. NSMCE2, a novel super-enhancer-regulated gene, is linked to poor prognosis and therapy resistance in breast cancer. BMC Cancer 2022, 22, 1056. [Google Scholar] [CrossRef]
- Gröschel, S.; Sanders, M.A.; Hoogenboezem, R.; de Wit, E.; Bouwman, B.A.; Erpelinck, C.; van der Velden, V.H.; Havermans, M.; Avellino, R.; van Lom, K. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 2014, 157, 369–381. [Google Scholar] [CrossRef]
- Ott, C.J.; Federation, A.J.; Schwartz, L.S.; Kasar, S.; Klitgaard, J.L.; Lenci, R.; Li, Q.; Lawlor, M.; Fernandes, S.M.; Souza, A. Enhancer architecture and essential core regulatory circuitry of chronic lymphocytic leukemia. Cancer Cell 2018, 34, 982–995.e987. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Li, J.; Li, Y.; Ma, Z.; Yu, Y.; Wang, C.-Y. Transcriptional super-enhancers control cancer stemness and metastasis genes in squamous cell carcinoma. Nat. Commun. 2021, 12, 3974. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Hitz, B.C.; Gabdank, I.; Hilton, J.A.; Kagda, M.S.; Lam, B.; Myers, Z.; Sud, P.; Jou, J.; Lin, K. New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res. 2020, 48, D882–D889. [Google Scholar] [CrossRef] [PubMed]
- Karolchik, D.; Baertsch, R.; Diekhans, M.; Furey, T.S.; Hinrichs, A.; Lu, Y.; Roskin, K.M.; Schwartz, M.; Sugnet, C.W.; Thomas, D.J. The UCSC genome browser database. Nucleic Acids Res. 2003, 31, 51–54. [Google Scholar] [CrossRef]
- Corces, M.R.; Granja, J.M.; Shams, S.; Louie, B.H.; Seoane, J.A.; Zhou, W.; Silva, T.C.; Groeneveld, C.; Wong, C.K.; Cho, S.W. The chromatin accessibility landscape of primary human cancers. Science 2018, 362, eaav1898. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H. Next-generation characterization of the cancer cell line encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Kayagaki, N.; Yamaguchi, N.; Nakayama, M.; Eto, H.; Okumura, K.; Yagita, H. Type I interferons (IFNs) regulate tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) expression on human T cells: A novel mechanism for the antitumor effects of type I IFNs. J. Exp. Med. 1999, 189, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- John, S.; Sabo, P.J.; Thurman, R.E.; Sung, M.-H.; Biddie, S.C.; Johnson, T.A.; Hager, G.L.; Stamatoyannopoulos, J.A. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat. Genet. 2011, 43, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Crawford, G.E.; Holt, I.E.; Mullikin, J.C.; Tai, D.; Green, E.D.; Wolfsberg, T.G.; Collins, F.S.; National Institutes of Health Intramural Sequencing Center. Identifying gene regulatory elements by genome-wide recovery of DNase hypersensitive sites. Proc. Natl. Acad. Sci. USA 2004, 101, 992–997. [Google Scholar] [CrossRef] [PubMed]
- Flusberg, D.A.; Sorger, P.K. Modulating cell-to-cell variability and sensitivity to death ligands by co-drugging. Phys. Biol. 2013, 10, 035002. [Google Scholar] [CrossRef]
- Varfolomeev, E.; Blankenship, J.W.; Wayson, S.M.; Fedorova, A.V.; Kayagaki, N.; Garg, P.; Zobel, K.; Dynek, J.N.; Elliott, L.O.; Wallweber, H.J. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell 2007, 131, 669–681. [Google Scholar] [CrossRef]
- Sarhan, D.; D’Arcy, P.; Wennerberg, E.; Lidén, M.; Hu, J.; Winqvist, O.; Rolny, C.; Lundqvist, A. Activated monocytes augment TRAIL-mediated cytotoxicity by human NK cells through release of IFN-γ. Eur. J. Immunol. 2013, 43, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Kalb, M.L.; Glaser, A.; Stary, G.; Koszik, F.; Stingl, G. TRAIL+ human plasmacytoid dendritic cells kill tumor cells in vitro: Mechanisms of imiquimod-and IFN-α–mediated antitumor reactivity. J. Immunol. 2012, 188, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, S.; Infante-Duarte, C.; Seeger, B.; Zipp, F. Regulation of soluble and surface-bound TRAIL in human T cells, B cells, and monocytes. Cytokine 2003, 24, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Huber, V.; Fais, S.; Iero, M.; Lugini, L.; Canese, P.; Squarcina, P.; Zaccheddu, A.; Colone, M.; Arancia, G.; Gentile, M. Human colorectal cancer cells induce T-cell death through release of proapoptotic microvesicles: Role in immune escape. Gastroenterology 2005, 128, 1796–1804. [Google Scholar] [CrossRef] [PubMed]
- Zboril, E.; Yoo, H.; Chen, L.; Liu, Z. Dynamic interactions of transcription factors and enhancer reprogramming in cancer progression. Front. Oncol. 2021, 11, 753051. [Google Scholar] [CrossRef]
- Sengupta, S.; George, R.E. Super-enhancer-driven transcriptional dependencies in cancer. Trends Cancer 2017, 3, 269–281. [Google Scholar] [CrossRef]
- Azijli, K.; Weyhenmeyer, B.; Peters, G.; De Jong, S.; Kruyt, F. Non-canonical kinase signaling by the death ligand TRAIL in cancer cells: Discord in the death receptor family. Cell Death Differ. 2013, 20, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.; Maecker, H.; Sharp, D.; Lawrence, D.; Renz, M.; Vucic, D.; Ashkenazi, A. Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. J. Biol. Chem. 2005, 280, 40599–40608. [Google Scholar] [CrossRef] [PubMed]
- Lafont, E.; Kantari-Mimoun, C.; Draber, P.; De Miguel, D.; Hartwig, T.; Reichert, M.; Kupka, S.; Shimizu, Y.; Taraborrelli, L.; Spit, M. The linear ubiquitin chain assembly complex regulates TRAIL-induced gene activation and cell death. EMBO J. 2017, 36, 1147–1166. [Google Scholar] [CrossRef] [PubMed]
- Conti, A.; Nobis, M.; Montinaro, A.; Hartwig, T.; Lemke, J.; Legler, K.; Annewanter, F.; Campbell, A.; Taraborrelli, L.; Grosse-Wilde, A. Cancer cell-autonomous TRAIL-R signaling promotes KRAS-driven cancer progression, invasion, and metastasis. Cancer Cell 2015, 27, 561–573. [Google Scholar]
- Hartwig, T.; Montinaro, A.; von Karstedt, S.; Sevko, A.; Surinova, S.; Chakravarthy, A.; Taraborrelli, L.; Draber, P.; Lafont, E.; Vargas, F.A. The TRAIL-induced cancer secretome promotes a tumor-supportive immune microenvironment via CCR2. Mol. Cell 2017, 65, 730–742.e5. [Google Scholar] [CrossRef]
- Werman, A.; Werman-Venkert, R.; White, R.; Lee, J.-K.; Werman, B.; Krelin, Y.; Voronov, E.; Dinarello, C.A.; Apte, R.N. The precursor form of IL-1α is an intracrine proinflammatory activator of transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 2434–2439. [Google Scholar] [CrossRef]
- Carriere, V.; Roussel, L.; Ortega, N.; Lacorre, D.-A.; Americh, L.; Aguilar, L.; Bouche, G.; Girard, J.-P. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 282–287. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Pai, R.C.; Fong, S.; Leung, S.; Lawrence, D.A.; Marsters, S.A.; Blackie, C.; Chang, L.; McMurtrey, A.E.; Hebert, A. Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Investig. 1999, 104, 155–162. [Google Scholar] [CrossRef]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T. Tumoricidal activity of tumor necrosis factor–related apoptosis–inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef]
- Hotte, S.J.; Hirte, H.W.; Chen, E.X.; Siu, L.L.; Le, L.H.; Corey, A.; Iacobucci, A.; MacLean, M.; Lo, L.; Fox, N.L. A phase 1 study of mapatumumab (fully human monoclonal antibody to TRAIL-R1) in patients with advanced solid malignancies. Clin. Cancer Res. 2008, 14, 3450–3455. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Eckhardt, S.G.; Kurzrock, R.; Ebbinghaus, S.; O’Dwyer, P.J.; Gordon, M.S.; Novotny, W.; Goldwasser, M.A.; Tohnya, T.M.; Lum, B.L. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J. Clin. Oncol. 2010, 28, 2839–2846. [Google Scholar] [CrossRef] [PubMed]
- Forero-Torres, A.; Infante, J.R.; Waterhouse, D.; Wong, L.; Vickers, S.; Arrowsmith, E.; He, A.R.; Hart, L.; Trent, D.; Wade, J. Phase 2, multicenter, open-label study of tigatuzumab (CS-1008), a humanized monoclonal antibody targeting death receptor 5, in combination with gemcitabine in chemotherapy-naive patients with unresectable or metastatic pancreatic cancer. Cancer Med. 2013, 2, 925–932. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Mita, M.; Meropol, N.J.; von Mehren, M.; Patnaik, A.; Padavic, K.; Hill, M.; Mays, T.; McCoy, T.; Fox, N.L. Phase I pharmacokinetic and biologic correlative study of mapatumumab, a fully human monoclonal antibody with agonist activity to tumor necrosis factor–related apoptosis-inducing ligand receptor-1. J. Clin. Oncol. 2007, 25, 1390–1396. [Google Scholar] [CrossRef] [PubMed]
- Spierings, D.C.; de Vries, E.G.; Timens, W.; Groen, H.J.; Boezen, H.M.; de Jong, S. Expression of TRAIL and TRAIL death receptors in stage III non-small cell lung cancer tumors. Clin. Cancer Res. 2003, 9, 3397–3405. [Google Scholar] [PubMed]
- Sträter, J.; Hinz, U.; Walczak, H.; Mechtersheimer, G.; Koretz, K.; Herfarth, C.; Möller, P.; Lehnert, T. Expression of TRAIL and TRAIL receptors in colon carcinoma: TRAIL-R1 is an independent prognostic parameter. Clin. Cancer Res. 2002, 8, 3734–3740. [Google Scholar]
- Montinaro, A.; Areso Zubiaur, I.; Saggau, J.; Kretz, A.-L.; Ferreira, R.M.; Hassan, O.; Kitzig, E.; Müller, I.; El-Bahrawy, M.A.; von Karstedt, S. Potent pro-apoptotic combination therapy is highly effective in a broad range of cancers. Cell Death Differ. 2022, 29, 492–503. [Google Scholar] [CrossRef]
- Lemke, J.; von Karstedt, S.; Abd El Hay, M.; Conti, A.; Arce, F.; Montinaro, A.; Papenfuss, K.; El-Bahrawy, M.A.; Walczak, H. Selective CDK9 inhibition overcomes TRAIL resistance by concomitant suppression of cFlip and Mcl-1. Cell Death Differ. 2014, 21, 491–502. [Google Scholar] [CrossRef]
- Fulda, S.; Wick, W.; Weller, M.; Debatin, K.-M. Smac agonists sensitize for Apo2L/TRAIL-or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat. Med. 2002, 8, 808–815. [Google Scholar] [CrossRef]
- Serrano-Saenz, S.; Palacios, C.; Delgado-Bellido, D.; López-Jiménez, L.; Garcia-Diaz, A.; Soto-Serrano, Y.; Casal, J.I.; Bartolomé, R.A.; Fernández-Luna, J.L.; López-Rivas, A. PIM kinases mediate resistance of glioblastoma cells to TRAIL by a p62/SQSTM1-dependent mechanism. Cell Death Dis. 2019, 10, 51. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Benedetto, C.; Khan, T.; Serrano-Saenz, S.; Rodriguez-Lemus, A.; Klomsiri, C.; Beutel, T.-M.; Thach, A.; Walczak, H.; Betancur, P. Enhancer Clusters Drive Type I Interferon-Induced TRAIL Overexpression in Cancer, and Its Intracellular Protein Accumulation Fails to Induce Apoptosis. Cancers 2023, 15, 967. https://doi.org/10.3390/cancers15030967
Di Benedetto C, Khan T, Serrano-Saenz S, Rodriguez-Lemus A, Klomsiri C, Beutel T-M, Thach A, Walczak H, Betancur P. Enhancer Clusters Drive Type I Interferon-Induced TRAIL Overexpression in Cancer, and Its Intracellular Protein Accumulation Fails to Induce Apoptosis. Cancers. 2023; 15(3):967. https://doi.org/10.3390/cancers15030967
Chicago/Turabian StyleDi Benedetto, Carolina, Taimoor Khan, Santiago Serrano-Saenz, Anthony Rodriguez-Lemus, Chananat Klomsiri, Tim-Mathis Beutel, Alysia Thach, Henning Walczak, and Paola Betancur. 2023. "Enhancer Clusters Drive Type I Interferon-Induced TRAIL Overexpression in Cancer, and Its Intracellular Protein Accumulation Fails to Induce Apoptosis" Cancers 15, no. 3: 967. https://doi.org/10.3390/cancers15030967
APA StyleDi Benedetto, C., Khan, T., Serrano-Saenz, S., Rodriguez-Lemus, A., Klomsiri, C., Beutel, T.-M., Thach, A., Walczak, H., & Betancur, P. (2023). Enhancer Clusters Drive Type I Interferon-Induced TRAIL Overexpression in Cancer, and Its Intracellular Protein Accumulation Fails to Induce Apoptosis. Cancers, 15(3), 967. https://doi.org/10.3390/cancers15030967