

ABL1/2 and DDR1 Drive MEKi Resistance in NRAS-Mutant Melanomas by Stabilizing RAF/MYC/ETS1 and Promoting RAF Homodimerization

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Lines
2.3. ABL1/2 Activation
2.4. Viability Assays
2.5. Apoptosis Assays
2.6. Clonogenic Assays
2.7. SiRNA Transfection
2.8. Coimmunoprecipitation (coIP)
2.9. Xenograft Assays
2.10. Statistics
3. Results
3.1. ABL1/2 Are Activated during MEKi Resistance
3.2. Targeting ABL1/2 Reverses Intrinsic and Acquired Trametinib Resistance
3.3. ABL1/2 and DDR1 Drive Acquired MEKi Resistance
3.4. SK-MEL-147MR Are Exquisitely Dependent on ABL1/2
3.5. DDR1 and CRAF→ERK Contributes to ABL1/2 Activation during Resistance
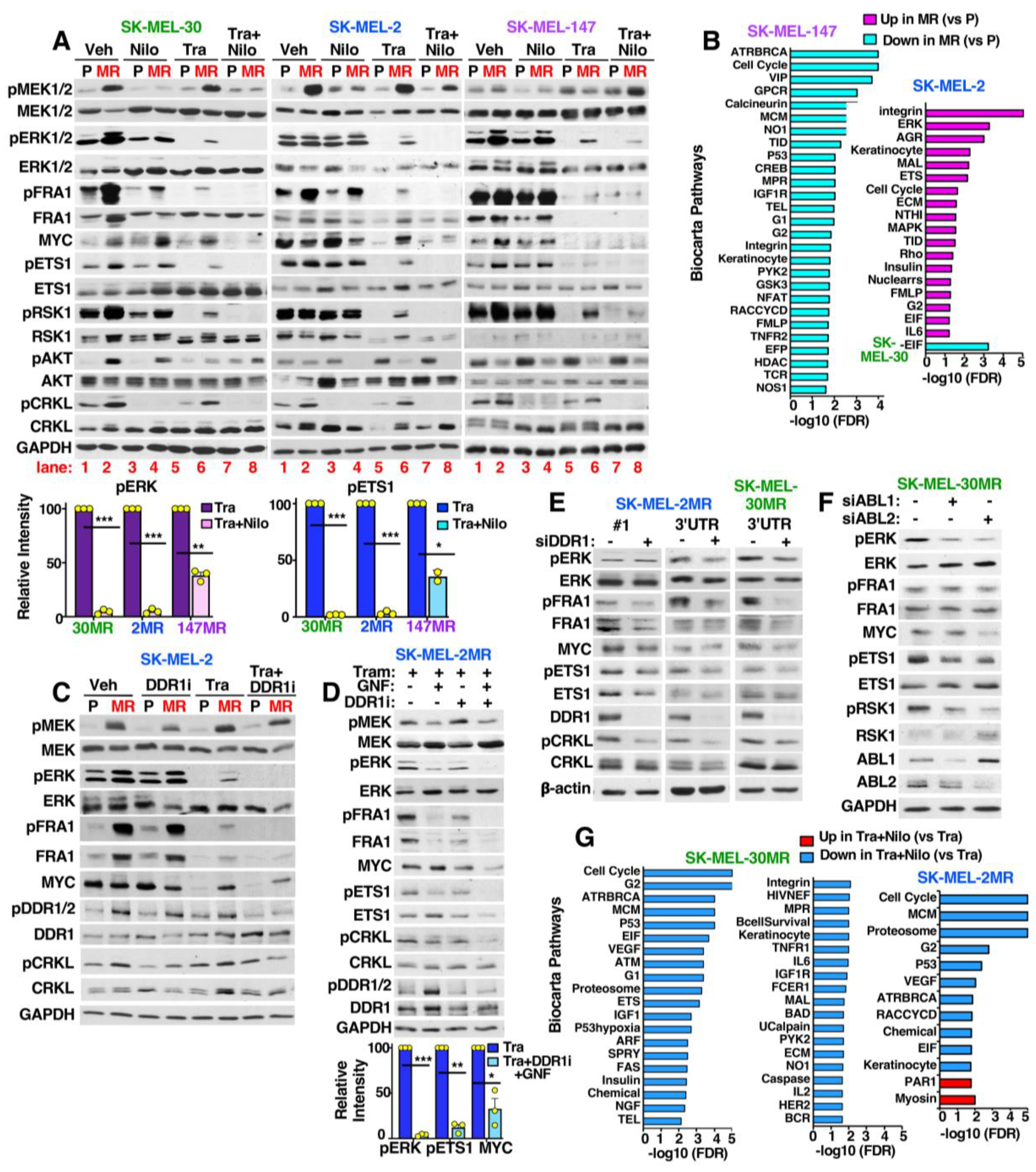
3.6. MEKi-Resistant Cells Utilize Diverse Mechanisms to Activate ERK/MYC/ETS1/RSK1 Signaling
3.7. ABL1/2 and DDR1 Are Critical Nodes Mediating ERK/MYC/ETS1/RSK1 Activation during Resistance
3.8. Targeting ABL1/2 and DDR1 Alters Expression of RAF, MYC and p27/KIP1 and Induced RAF Heterodimerization
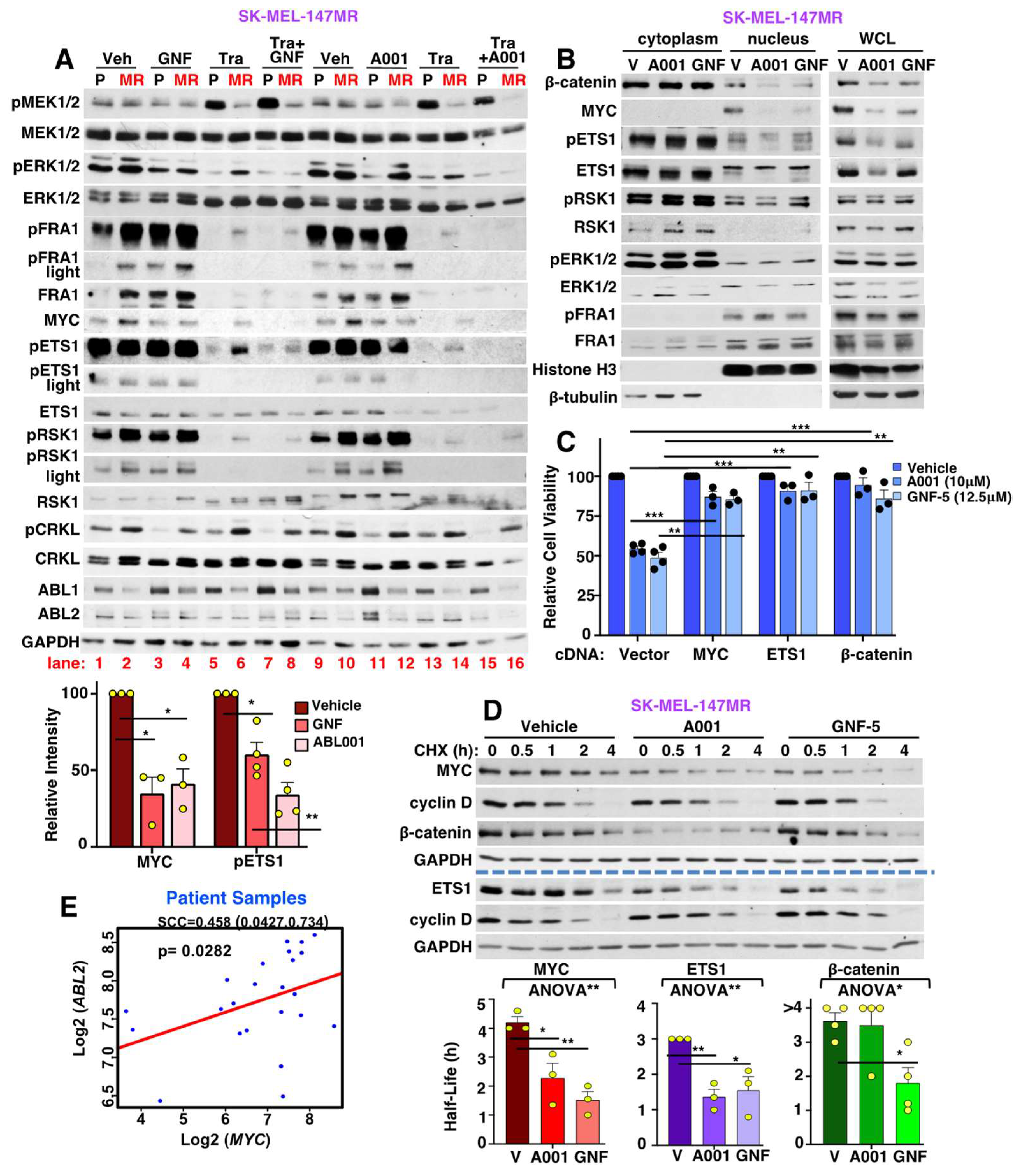
3.9. Allosteric ABL1/2 Inhibitors Prevent SK-MEL-147MR Survival by Inhibiting MYC and ETS1 Expression and Blocking β-Catenin Nuclear Localization
3.10. Targeting ABL1/2 and DDR1 Reverses Intrinsic/Adaptive Resistance by Preventing Activation of Cytoplasmic but Not Nuclear ERK Targets
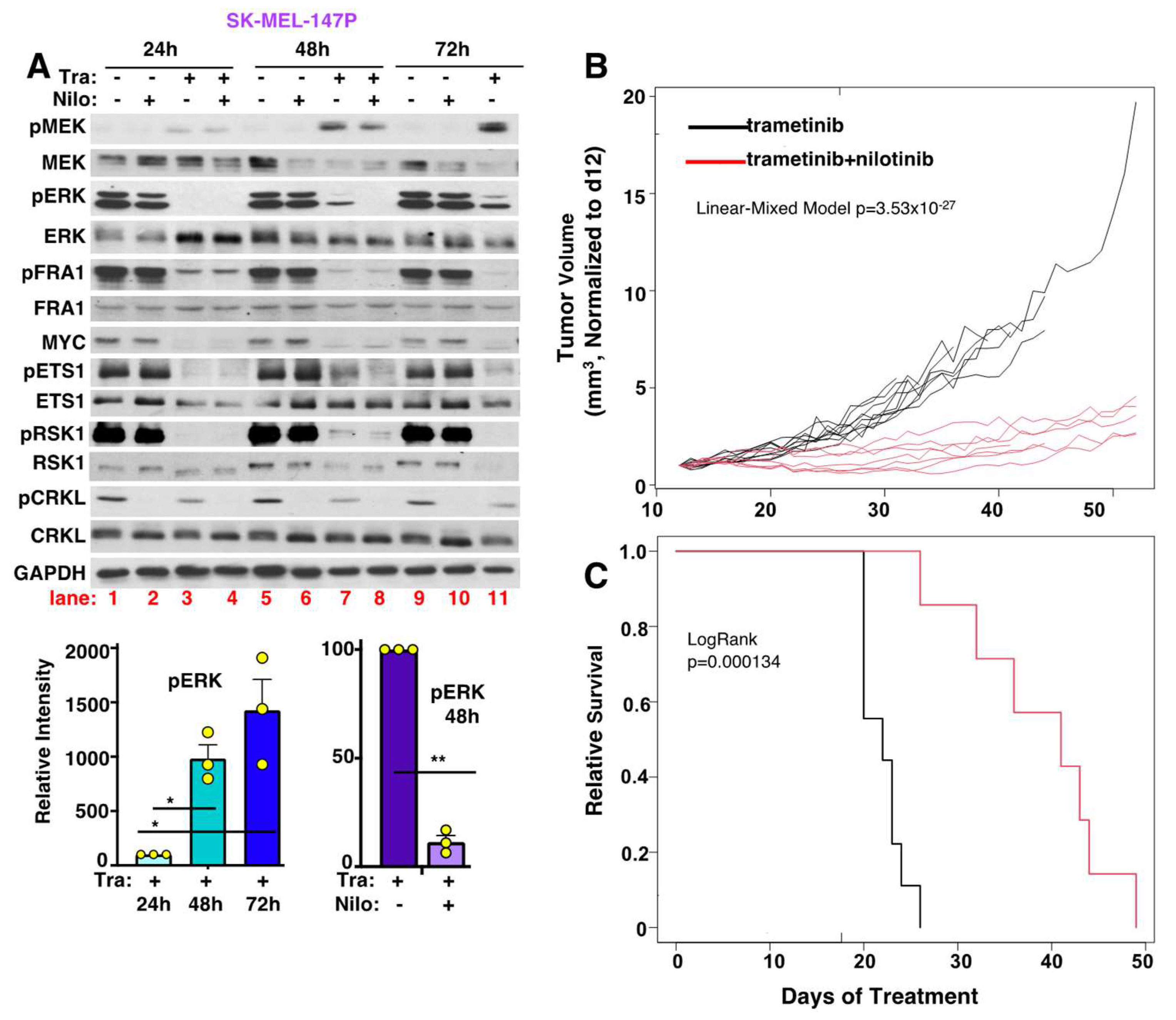
3.11. Nilotinib Reverses Intrinsic MEKi Resistance, Delays the Onset of Acquired Resistance, and Prolongs Survival, In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Munoz-Couselo, E.; Adelantado, E.Z.; Ortiz, C.; Garcia, J.S.; Perez-Garcia, J. NRAS-mutant melanoma: Current challenges and future prospect. Onco Targets Ther. 2017, 10, 3941–3947. [Google Scholar] [CrossRef]
- Gide, T.N.; Wilmott, J.S.; Scolyer, R.A.; Long, G.V. Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Metastatic Melanoma. Clin. Cancer Res. 2018, 24, 1260–1270. [Google Scholar] [CrossRef]
- Garcia-Alvarez, A.; Ortiz, C.; Munoz-Couselo, E. Current Perspectives and Novel Strategies of NRAS-Mutant Melanoma. Onco Targets Ther. 2021, 14, 3709–3719. [Google Scholar] [CrossRef]
- Boespflug, A.; Caramel, J.; Dalle, S.; Thomas, L. Treatment of NRAS-mutated advanced or metastatic melanoma: Rationale, current trials and evidence to date. Ther. Adv. Med. Oncol. 2017, 9, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Delyon, J.; Lebbe, C.; Dumaz, N. Targeted therapies in melanoma beyond BRAF: Targeting NRAS-mutated and KIT-mutated melanoma. Curr. Opin. Oncol. 2020, 32, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.; Liu, Z.; Plattner, R. EnABLing Tumor Growth and Progression: Recent progress in unraveling the functions of ABL kinases in solid tumor cells. Curr. Pharmacol. Rep. 2018, 4, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.; Fiore, L.S.; Richards, D.L.; Yang, Y.; Liu, J.; Wang, C.; Plattner, R. Abl and Arg mediate cysteine cathepsin secretion to facilitate melanoma invasion and metastasis. Sci. Signal. 2018, 11, eaao0422. [Google Scholar] [CrossRef]
- Greuber, E.K.; Smith-Pearson, P.; Wang, J.; Pendergast, A.M. Role of ABL family kinases in cancer: From leukaemia to solid tumours. Nat. Rev. Cancer 2013, 13, 559–571. [Google Scholar] [CrossRef]
- Cario, M. DDR1 and DDR2 in skin. Cell Adh. Migr. 2018, 12, 386–393. [Google Scholar] [CrossRef]
- Reger de Moura, C.; Battistella, M.; Sohail, A.; Caudron, A.; Feugeas, J.P.; Podgorniak, M.P.; Pages, C.; Mazouz Dorval, S.; Marco, O.; Menashi, S.; et al. Discoidin domain receptors: A promising target in melanoma. Pigment Cell Melanoma Res. 2019, 32, 697–707. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Lu, Q.; Nada, H.; Woo, J.; Quan, G.; Lee, K. The Journey of DDR1 and DDR2 Kinase Inhibitors as Rising Stars in the Fight Against Cancer. Int. J. Mol. Sci. 2021, 22, 6535. [Google Scholar] [CrossRef]
- Borza, C.M.; Su, Y.; Tran, T.L.; Yu, L.; Steyns, N.; Temple, K.J.; Skwark, M.J.; Meiler, J.; Lindsley, C.W.; Hicks, B.R.; et al. Discoidin domain receptor 1 kinase activity is required for regulating collagen IV synthesis. Matrix Biol. 2017, 57–58, 258–271. [Google Scholar] [CrossRef]
- Gao, Y.; Zhou, J.; Li, J. Discoidin domain receptors orchestrate cancer progression: A focus on cancer therapies. Cancer Sci. 2021, 112, 962–969. [Google Scholar] [CrossRef]
- Ganguly, S.S.; Fiore, L.S.; Sims, J.T.; Friend, J.W.; Srinivasan, D.; Thacker, M.A.; Cibull, M.L.; Wang, C.; Novak, M.; Kaetzel, D.M.; et al. c-Abl and Arg are activated in human primary melanomas, promote melanoma cell invasion via distinct pathways, and drive metastatic progression. Oncogene 2012, 31, 1804–1816. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Pendergast, A.M. Arg kinase regulates epithelial cell polarity by targeting beta1-integrin and small GTPase pathways. Curr. Biol. 2011, 21, 1534–1542. [Google Scholar] [CrossRef] [PubMed]
- White, D.; Saunders, V.; Lyons, A.B.; Branford, S.; Grigg, A.; To, L.B.; Hughes, T. In vitro sensitivity to imatinib-induced inhibition of ABL kinase activity is predictive of molecular response in patients with de novo CML. Blood 2005, 106, 2520–2526. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.J.; Rouse, C.; Xu, X.; Wang, J.; Onaitis, M.W.; Pendergast, A.M. Inactivation of ABL kinases suppresses non-small cell lung cancer metastasis. JCI Insight 2016, 1, e89647. [Google Scholar] [CrossRef][Green Version]
- Hoj, J.P.; Mayro, B.; Pendergast, A.M. The ABL2 kinase regulates an HSF1-dependent transcriptional program required for lung adenocarcinoma brain metastasis. Proc. Natl. Acad. Sci. USA 2020, 117, 33486–33495. [Google Scholar] [CrossRef]
- Jones, J.K.; Thompson, E.M. Allosteric Inhibition of ABL Kinases: Therapeutic Potential in Cancer. Mol. Cancer Ther. 2020, 19, 1763–1769. [Google Scholar] [CrossRef] [PubMed]
- Guzman, C.; Bagga, M.; Kaur, A.; Westermarck, J.; Abankwa, D. ColonyArea: An ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS ONE 2014, 9, e92444. [Google Scholar] [CrossRef]
- Wylie, A.A.; Schoepfer, J.; Jahnke, W.; Cowan-Jacob, S.W.; Loo, A.; Furet, P.; Marzinzik, A.L.; Pelle, X.; Donovan, J.; Zhu, W.; et al. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature 2017, 543, 733–737. [Google Scholar] [CrossRef]
- Jain, A.; Tripathi, R.; Turpin, C.P.; Wang, C.; Plattner, R. Abl kinase regulation by BRAF/ERK and cooperation with Akt in melanoma. Oncogene 2017, 36, 4585–4596. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Kong, X.; Kuilman, T.; Shahrabi, A.; Boshuizen, J.; Kemper, K.; Song, J.Y.; Niessen, H.W.M.; Rozeman, E.A.; Geukes Foppen, M.H.; Blank, C.U.; et al. Cancer drug addiction is relayed by an ERK2-dependent phenotype switch. Nature 2017, 550, 270–274. [Google Scholar] [CrossRef]
- Barila, D.; Superti-Furga, G. An intramolecular SH3-domain interaction regulates c-Abl activity. Nat. Genet. 1998, 18, 280–282. [Google Scholar] [CrossRef]
- Plattner, R.; Kadlec, L.; DeMali, K.A.; Kazlauskas, A.; Pendergast, A.M. c-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev. 1999, 13, 2400–2411. [Google Scholar] [CrossRef]
- Plattner, R.; Koleske, A.J.; Kazlauskas, A.; Pendergast, A.M. Bidirectional Signaling Links the Abelson Kinases to the Platelet-Derived Growth Factor Receptor. Mol. Cell Biol. 2004, 24, 2573–2583. [Google Scholar] [CrossRef]
- Srinivasan, D.; Sims, J.T.; Plattner, R. Aggressive breast cancer cells are dependent on activated Abl kinases for proliferation, anchorage-independent growth and survival. Oncogene 2008, 27, 1095–1105. [Google Scholar] [CrossRef]
- Nihira, K.; Taira, N.; Miki, Y.; Yoshida, K. TTK/Mps1 controls nuclear targeting of c-Abl by 14-3-3-coupled phosphorylation in response to oxidative stress. Oncogene 2008, 27, 7285–7295. [Google Scholar] [CrossRef]
- Madeira, F.; Tinti, M.; Murugesan, G.; Berrett, E.; Stafford, M.; Toth, R.; Cole, C.; MacKintosh, C.; Barton, G.J. 14-3-3-Pred: Improved methods to predict 14-3-3-binding phosphopeptides. Bioinformatics 2015, 31, 2276–2283. [Google Scholar] [CrossRef]
- Wang, H.; Daouti, S.; Li, W.H.; Wen, Y.; Rizzo, C.; Higgins, B.; Packman, K.; Rosen, N.; Boylan, J.F.; Heimbrook, D.; et al. Identification of the MEK1(F129L) activating mutation as a potential mechanism of acquired resistance to MEK inhibition in human cancers carrying the B-RafV600E mutation. Cancer Res. 2011, 71, 5535–5545. [Google Scholar] [CrossRef]
- Hua, K.T.; Hong, J.B.; Sheen, Y.S.; Huang, H.Y.; Huang, Y.L.; Chen, J.S.; Liao, Y.H. miR-519d Promotes Melanoma Progression by Downregulating EphA4. Cancer Res. 2018, 78, 216–229. [Google Scholar] [CrossRef]
- Sale, M.J.; Balmanno, K.; Saxena, J.; Ozono, E.; Wojdyla, K.; McIntyre, R.E.; Gilley, R.; Woroniuk, A.; Howarth, K.D.; Hughes, G.; et al. MEK1/2 inhibitor withdrawal reverses acquired resistance driven by BRAF(V600E) amplification whereas KRAS(G13D) amplification promotes EMT-chemoresistance. Nat. Commun. 2019, 10, 2030. [Google Scholar] [CrossRef]
- Miao, B.; Ji, Z.; Tan, L.; Taylor, M.; Zhang, J.; Choi, H.G.; Frederick, D.T.; Kumar, R.; Wargo, J.A.; Flaherty, K.T.; et al. EPHA2 is a mediator of vemurafenib resistance and a novel therapeutic target in melanoma. Cancer Discov. 2015, 5, 274–287. [Google Scholar] [CrossRef]
- Kong, C.; Wang, C.; Wang, L.; Ma, M.; Niu, C.; Sun, X.; Du, J.; Dong, Z.; Zhu, S.; Lu, J.; et al. NEDD9 is a positive regulator of epithelial-mesenchymal transition and promotes invasion in aggressive breast cancer. PLoS ONE 2011, 6, e22666. [Google Scholar] [CrossRef]
- Hawsawi, O.; Henderson, V.; Burton, L.J.; Dougan, J.; Nagappan, P.; Odero-Marah, V. High mobility group A2 (HMGA2) promotes EMT via MAPK pathway in prostate cancer. Biochem. Biophys. Res. Commun. 2018, 504, 196–202. [Google Scholar] [CrossRef]
- Li, Q.T.; Feng, Y.M.; Ke, Z.H.; Qiu, M.J.; He, X.X.; Wang, M.M.; Li, Y.N.; Xu, J.; Shi, L.L.; Xiong, Z.F. KCNN4 promotes invasion and metastasis through the MAPK/ERK pathway in hepatocellular carcinoma. J. Investig. Med. 2020, 68, 68–74. [Google Scholar] [CrossRef]
- Uhlitz, F.; Sieber, A.; Wyler, E.; Fritsche-Guenther, R.; Meisig, J.; Landthaler, M.; Klinger, B.; Bluthgen, N. An immediate-late gene expression module decodes ERK signal duration. Mol. Syst. Biol. 2017, 13, 928. [Google Scholar] [CrossRef]
- Kidger, A.M.; Keyse, S.M. The regulation of oncogenic Ras/ERK signalling by dual-specificity mitogen activated protein kinase phosphatases (MKPs). Semin. Cell Dev. Biol. 2016, 50, 125–132. [Google Scholar] [CrossRef]
- Tsavachidou, D.; Coleman, M.L.; Athanasiadis, G.; Li, S.; Licht, J.D.; Olson, M.F.; Weber, B.L. SPRY2 is an inhibitor of the ras/extracellular signal-regulated kinase pathway in melanocytes and melanoma cells with wild-type BRAF but not with the V599E mutant. Cancer Res. 2004, 64, 5556–5559. [Google Scholar] [CrossRef]
- Salameh, A.; Lee, A.K.; Cardo-Vila, M.; Nunes, D.N.; Efstathiou, E.; Staquicini, F.I.; Dobroff, A.S.; Marchio, S.; Navone, N.M.; Hosoya, H.; et al. PRUNE2 is a human prostate cancer suppressor regulated by the intronic long noncoding RNA PCA3. Proc. Natl. Acad. Sci. USA 2015, 112, 8403–8408. [Google Scholar] [CrossRef]
- Johnson, G.L.; Stuhlmiller, T.J.; Angus, S.P.; Zawistowski, J.S.; Graves, L.M. Molecular pathways: Adaptive kinome reprogramming in response to targeted inhibition of the BRAF-MEK-ERK pathway in cancer. Clin. Cancer Res. 2014, 20, 2516–2522. [Google Scholar] [CrossRef]
- Johnson, D.B.; Nebhan, C.A.; Noel, M.S. MEK inhibitors in non-V600 BRAF mutations and fusions. Oncotarget 2020, 11, 3900–3903. [Google Scholar] [CrossRef]
- Dorard, C.; Estrada, C.; Barbotin, C.; Larcher, M.; Garancher, A.; Leloup, J.; Beermann, F.; Baccarini, M.; Pouponnot, C.; Larue, L.; et al. RAF proteins exert both specific and compensatory functions during tumour progression of NRAS-driven melanoma. Nat. Commun. 2017, 8, 15262. [Google Scholar] [CrossRef]
- Gajos-Michniewicz, A.; Czyz, M. WNT Signaling in Melanoma. Int. J. Mol. Sci. 2020, 21, 4852. [Google Scholar] [CrossRef]
- Nagler, A.; Vredevoogd, D.W.; Alon, M.; Cheng, P.F.; Trabish, S.; Kalaora, S.; Arafeh, R.; Goldin, V.; Levesque, M.P.; Peeper, D.S.; et al. A genome-wide CRISPR screen identifies FBXO42 involvement in resistance toward MEK inhibition in NRAS-mutant melanoma. Pigment Cell Melanoma Res. 2020, 33, 334–344. [Google Scholar] [CrossRef]
- Robinson, J.P.; Rebecca, V.W.; Kircher, D.A.; Silvis, M.R.; Smalley, I.; Gibney, G.T.; Lastwika, K.J.; Chen, G.; Davies, M.A.; Grossman, D.; et al. Resistance mechanisms to genetic suppression of mutant NRAS in melanoma. Melanoma Res. 2017, 27, 545–557. [Google Scholar] [CrossRef]
- Mader, C.C.; Oser, M.; Magalhaes, M.A.; Bravo-Cordero, J.; Condeelis, J.S.; Koleske, A.J.; Gil-Henn, H. An EGFR-Src-Arg-cortactin pathway mediates functional maturation of invadopodia and breast cancer cell invasion. Cancer Res. 2011, 71, 1730–1741. [Google Scholar] [CrossRef]
- Srinivasan, D.; Plattner, R. Activation of Abl tyrosine kinases promotes invasion of aggressive breast cancer cells. Cancer Res. 2006, 66, 5648–5655. [Google Scholar] [CrossRef]
- Weigel, M.T.; Banerjee, S.; Arnedos, M.; Salter, J.; A’Hern, R.; Dowsett, M.; Martin, L.A. Enhanced expression of the PDGFR/Abl signaling pathway in aromatase inhibitor-resistant breast cancer. Ann. Oncol. 2013, 24, 126–133. [Google Scholar] [CrossRef]
- Brummer, T.; McInnes, C. RAF kinase dimerization: Implications for drug discovery and clinical outcomes. Oncogene 2020, 39, 4155–4169. [Google Scholar] [CrossRef]
- Rushworth, L.K.; Hindley, A.D.; O’Neill, E.; Kolch, W. Regulation and role of Raf-1/B-Raf heterodimerization. Mol. Cell Biol. 2006, 26, 2262–2272. [Google Scholar] [CrossRef]
- Vehlow, A.; Klapproth, E.; Jin, S.; Hannen, R.; Hauswald, M.; Bartsch, J.W.; Nimsky, C.; Temme, A.; Leitinger, B.; Cordes, N. Interaction of Discoidin Domain Receptor 1 with a 14-3-3-Beclin-1-Akt1 Complex Modulates Glioblastoma Therapy Sensitivity. Cell Rep. 2019, 26, 3672–3683. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Yamaguchi, T.; Natsume, T.; Kufe, D.; Miki, Y. JNK phosphorylation of 14-3-3 proteins regulates nuclear targeting of c-Abl in the apoptotic response to DNA damage. Nat. Cell Biol. 2005, 7, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Packer, L.M.; Rana, S.; Hayward, R.; O’Hare, T.; Eide, C.A.; Rebocho, A.; Heidorn, S.; Zabriskie, M.S.; Niculescu-Duvaz, I.; Druker, B.J.; et al. Nilotinib and MEK Inhibitors Induce Synthetic Lethality through Paradoxical Activation of RAF in Drug-Resistant Chronic Myeloid Leukemia. Cancer Cell 2011, 20, 715–727. [Google Scholar] [CrossRef]
- Rebocho, A.P.; Marais, R. ARAF acts as a scaffold to stabilize BRAF:CRAF heterodimers. Oncogene 2013, 32, 3207–3212. [Google Scholar] [CrossRef]
- Mooz, J.; Riegel, K.; Ps, H.; Sadanandam, A.; Marini, F.; Klein, M.; Werner, U.; Roth, W.; Wilken-Schmitz, A.; Tegeder, I.; et al. ARAF suppresses ERBB3 expression and metastasis in a subset of lung cancers. Sci. Adv. 2022, 8, eabk1538. [Google Scholar] [CrossRef]
- Hoang, V.T.; Nyswaner, K.; Torres-Ayuso, P.; Brognard, J. The protein kinase MAP3K19 phosphorylates MAP2Ks and thereby activates ERK and JNK kinases and increases viability of KRAS-mutant lung cancer cells. J. Biol. Chem. 2020, 295, 8470–8479. [Google Scholar] [CrossRef]
- Tripathi, R.; Liu, Z.; Jain, A.; Lyon, A.; Meeks, C.; Richards, D.; Liu, J.; He, D.; Wang, C.; Nespi, M.; et al. Combating acquired resistance to MAPK inhibitors in melanoma by targeting ABL1/2-mediated reactivation of MEK/ERK/MYC signaling. Nat. Commun. 2020, 11, 5463. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.B.; Sougnez, C.; Gabriel, S.B.; Meyerson, M.L.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Ramos, A.H.; Lichtenstein, L.; Gupta, M.; Lawrence, M.S.; Pugh, T.J.; Saksena, G.; Meyerson, M.; Getz, G. Oncotator: Cancer Variant Annotation Tool. Hum. Mutat. 2015, 36, E2423–E2429. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approacy for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Bondzi, C.; Grant, S.; Krystal, G.W. A novel assay for the measurement of Raf-1 kinase activity. Oncogene 2000, 19, 5030–5033. [Google Scholar] [CrossRef]
- Kamelgarn, M.; Chen, J.; Kuang, L.; Jin, H.; Kasarskis, E.J.; Zhu, H. ALS mutations of FUS suppress protein translation and disrupt the regulation of nonsense-mediated decay. Proc. Natl. Acad. Sci. USA 2018, 115, E11904–E11913. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- BioCarta, N.D. Biotech Software & Internet Report. Comput. Softw. J. Sci. 2001, 1, 117–120. [Google Scholar]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Matthews, L.; Gopinath, G.; Gillespie, M.; Caudy, M.; Croft, D.; de Bono, B.; Garapati, P.; Hemish, J.; Hermjakob, H.; Jassal, B.; et al. Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Res. 2009, 37, D619–D622. [Google Scholar] [CrossRef] [PubMed]
- Fiore, L.S.; Ganguly, S.S.; Sledziona, J.; Cibull, M.L.; Wang, C.; Richards, D.L.; Neltner, J.M.; Beach, C.; McCorkle, J.R.; Kaetzel, D.M.; et al. c-Abl and Arg induce cathepsin-mediated lysosomal degradation of the NM23-H1 metastasis suppressor in invasive cancer. Oncogene 2013, 33, 4508–4520. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyon, A.; Tripathi, R.; Meeks, C.; He, D.; Wu, Y.; Liu, J.; Wang, C.; Chen, J.; Zhu, H.; Mukherjee, S.; et al. ABL1/2 and DDR1 Drive MEKi Resistance in NRAS-Mutant Melanomas by Stabilizing RAF/MYC/ETS1 and Promoting RAF Homodimerization. Cancers 2023, 15, 954. https://doi.org/10.3390/cancers15030954
Lyon A, Tripathi R, Meeks C, He D, Wu Y, Liu J, Wang C, Chen J, Zhu H, Mukherjee S, et al. ABL1/2 and DDR1 Drive MEKi Resistance in NRAS-Mutant Melanomas by Stabilizing RAF/MYC/ETS1 and Promoting RAF Homodimerization. Cancers. 2023; 15(3):954. https://doi.org/10.3390/cancers15030954
Chicago/Turabian StyleLyon, Anastasia, Rakshamani Tripathi, Christina Meeks, Daheng He, Yuanyuan Wu, Jinpeng Liu, Chi Wang, Jing Chen, Haining Zhu, Sujata Mukherjee, and et al. 2023. "ABL1/2 and DDR1 Drive MEKi Resistance in NRAS-Mutant Melanomas by Stabilizing RAF/MYC/ETS1 and Promoting RAF Homodimerization" Cancers 15, no. 3: 954. https://doi.org/10.3390/cancers15030954
APA StyleLyon, A., Tripathi, R., Meeks, C., He, D., Wu, Y., Liu, J., Wang, C., Chen, J., Zhu, H., Mukherjee, S., Ganguly, S., & Plattner, R. (2023). ABL1/2 and DDR1 Drive MEKi Resistance in NRAS-Mutant Melanomas by Stabilizing RAF/MYC/ETS1 and Promoting RAF Homodimerization. Cancers, 15(3), 954. https://doi.org/10.3390/cancers15030954