
Therapeutic Strategies to Overcome Fibrotic Barriers to Nanomedicine in the Pancreatic Tumor Microenvironment
 ,
,  ,
,  and
and 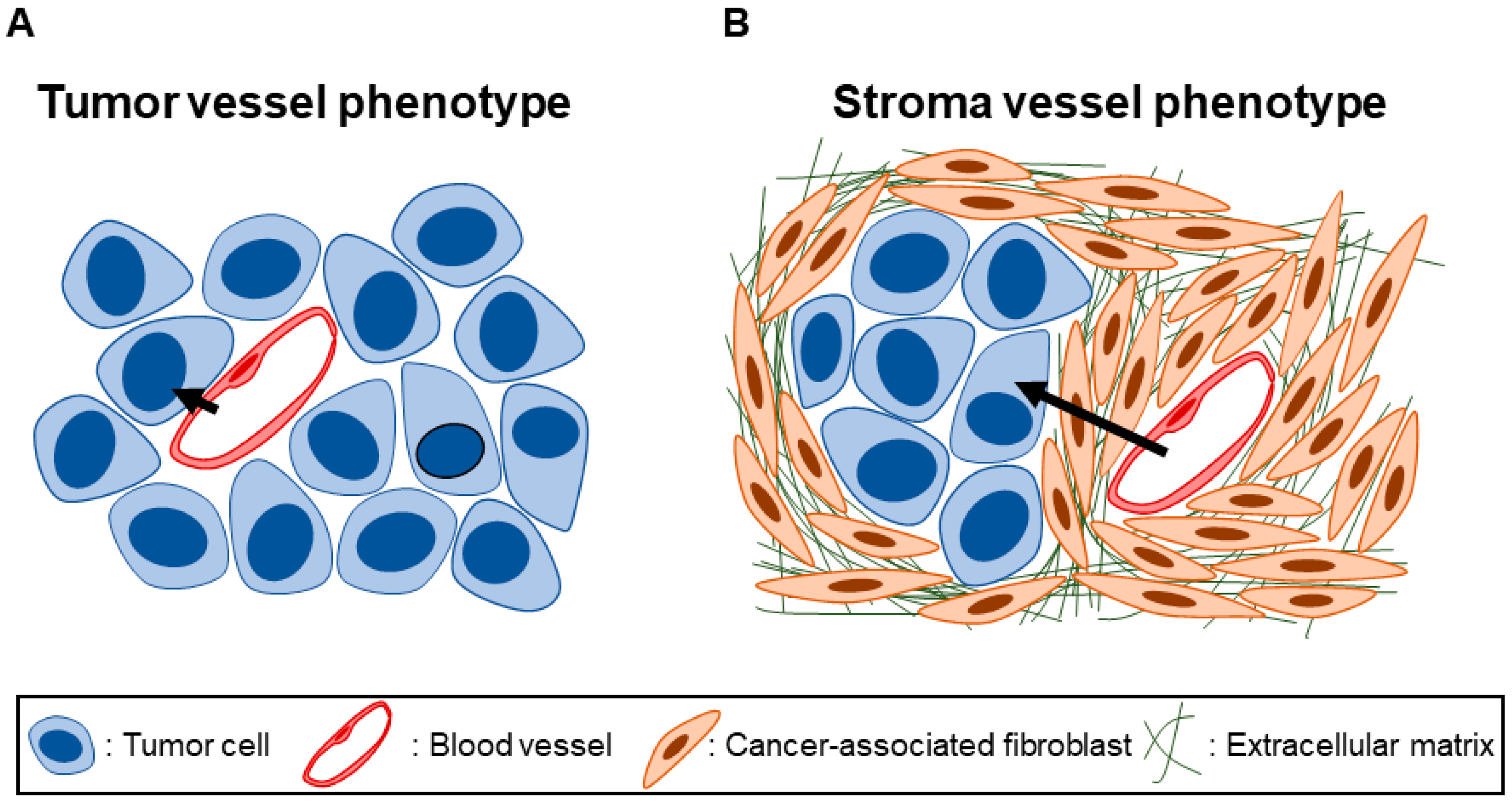{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
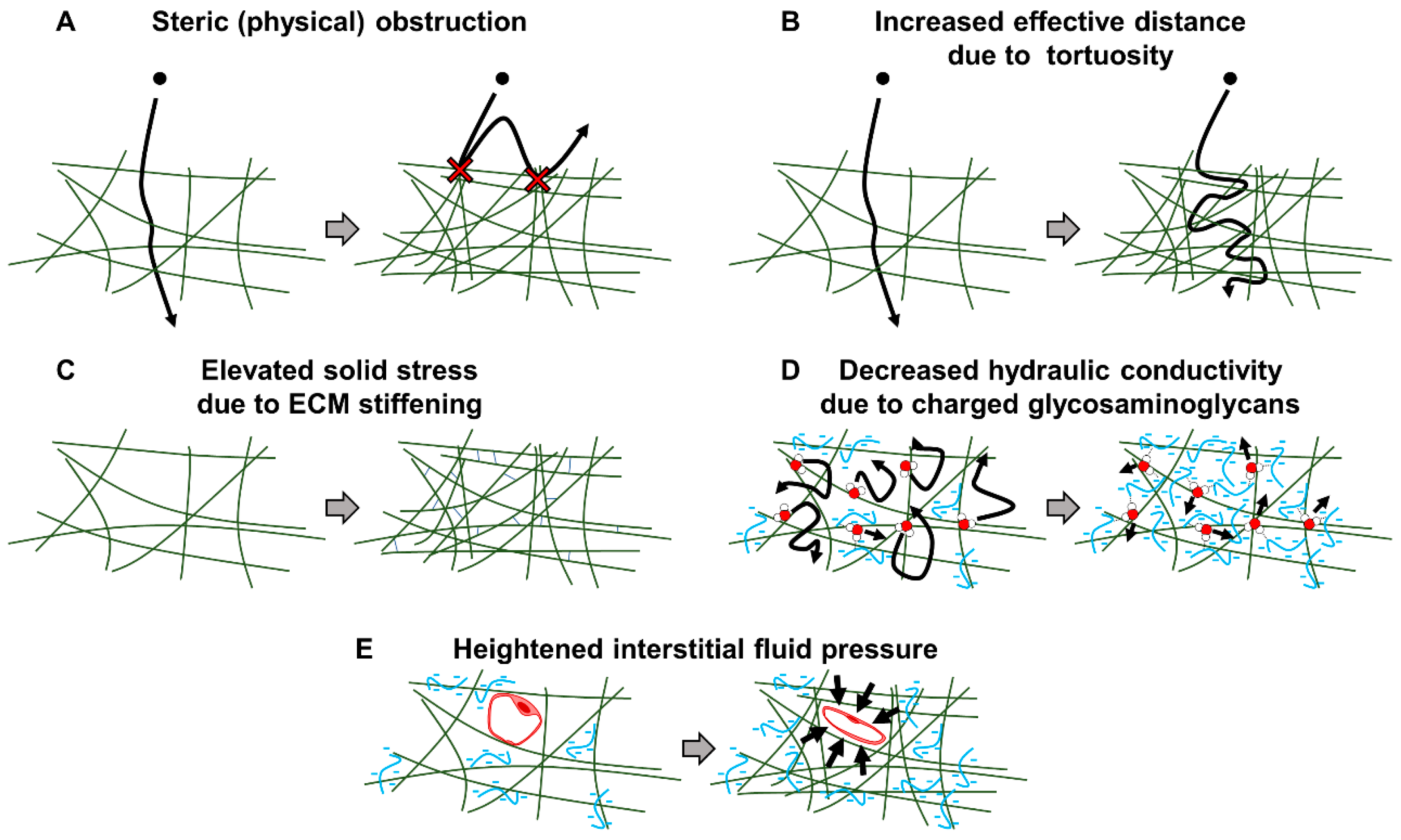
2. Fibrotic Barriers to Nanomedicine in the PDAC Tumor Microenvironment
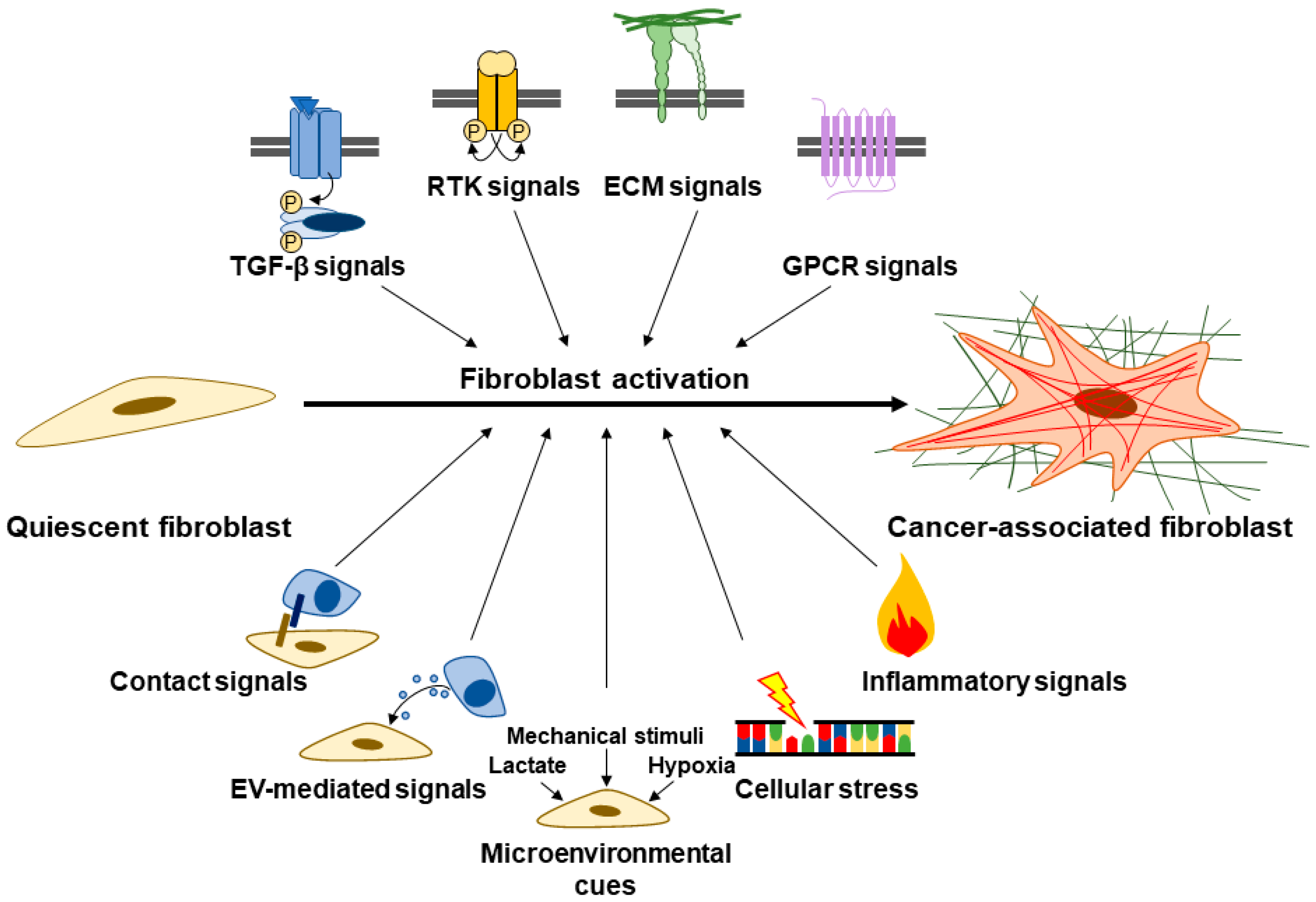
2.1. Fibroblasts: The Key Cellular Mediator of Fibrosis
2.2. The ECM in PDAC
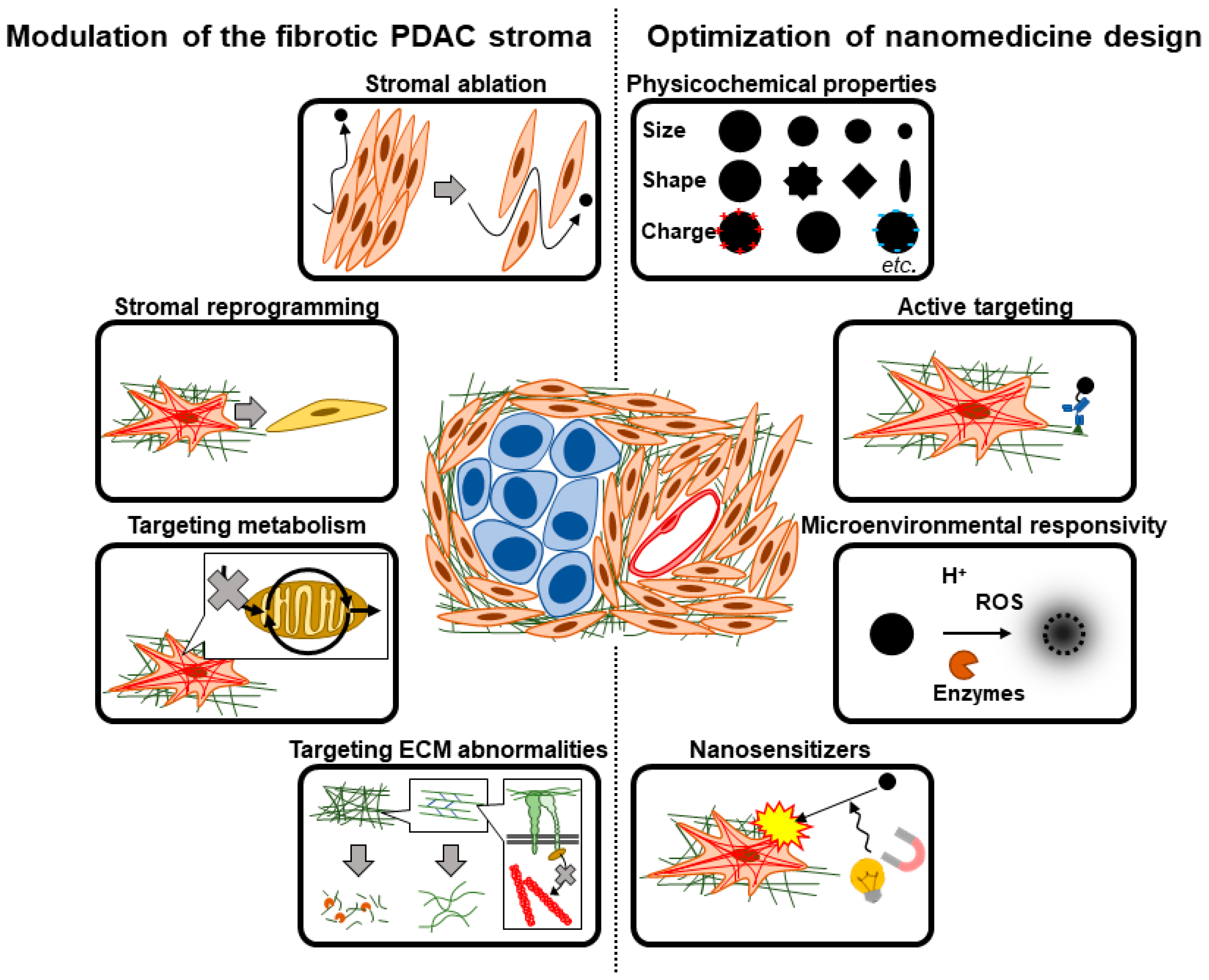
3. Therapeutic Strategies Targeting the Fibrotic Barriers to Nanomedicine
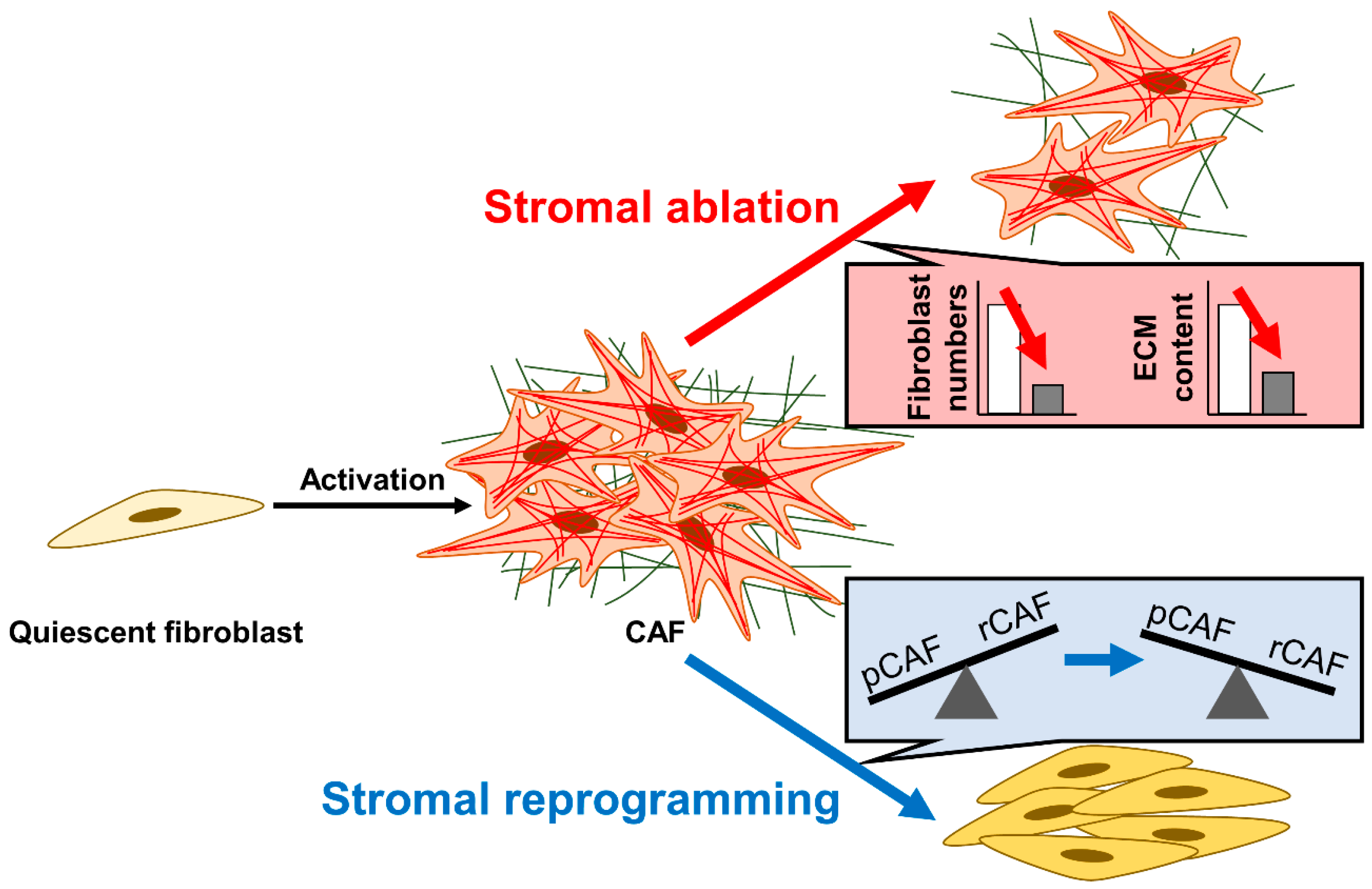
3.1. Strategy #1: Stromal Ablation—Reducing Fibrotic Stromal Content
3.2. Strategy #2: Stromal Reprogramming—Targeting Fibroblast Abnormalities
3.3. Strategy #3: Targeting Fibroblast Metabolism
3.4. Strategy #4: Targeting ECM Abnormalities
3.4.1. Strategy #4–1: Therapeutic Enzymatic Degradation of ECM Components
3.4.2. Strategy #4–2: Targeting ECM Remodeling
3.4.3. Strategy #4–3: Targeting ECM Signaling
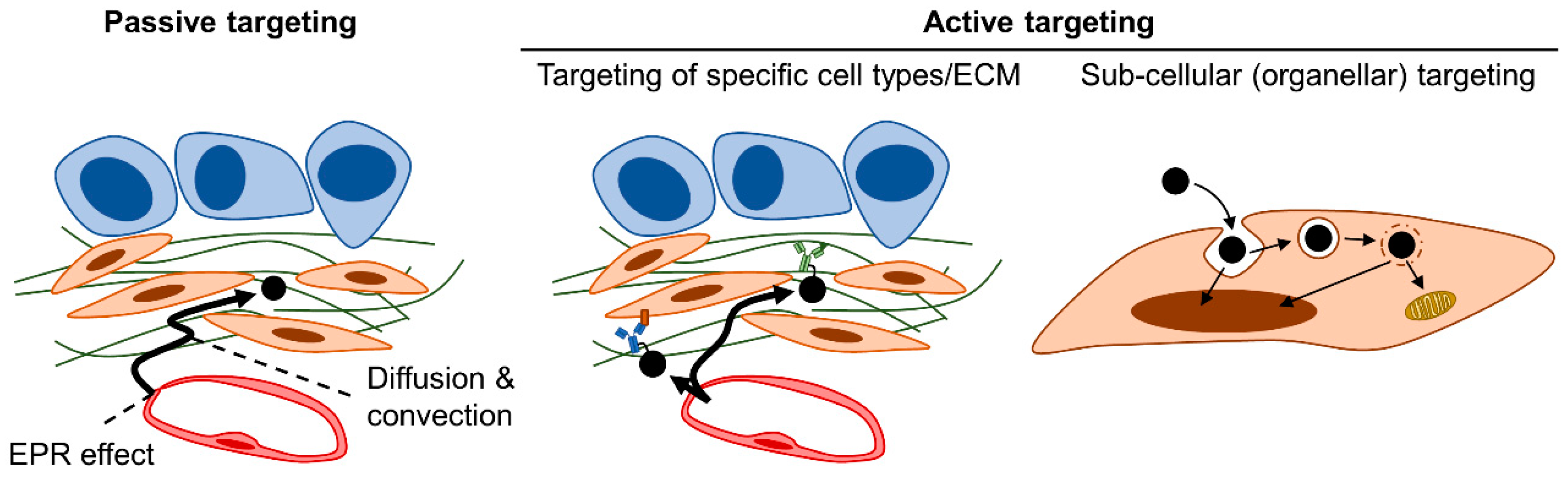
4. Therapeutic Strategies Optimizing Nanomedicine Design to Overcome Fibrotic Barriers to Nanomedicine
4.1. Strategy #5: Optimizing the Physicochemical Properties of Nanomedicine
4.2. Strategy #6: Active Targeting of Nanomedicine
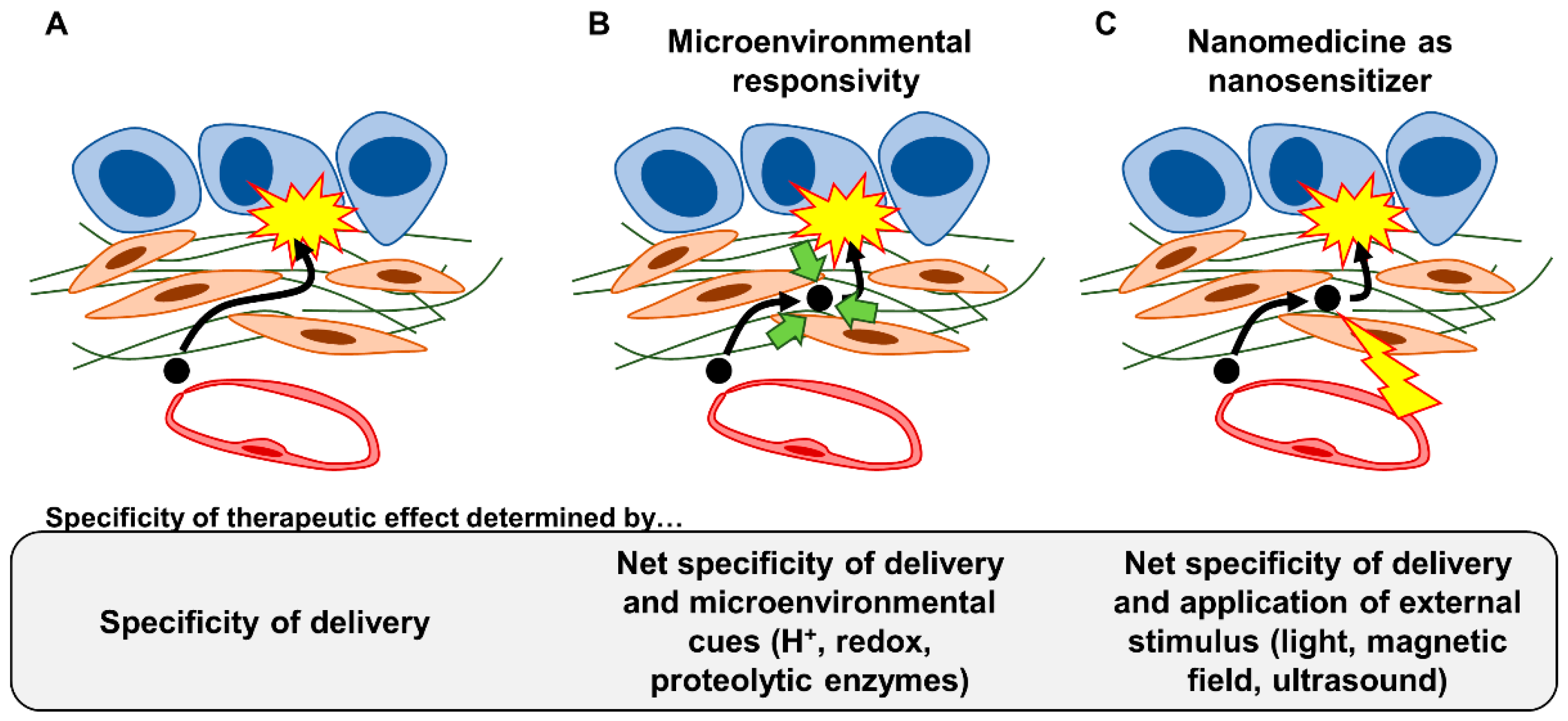
4.3. Strategy #7: Installing Microenvironmental Responsivity in Nanomedicine
4.4. Strategy #8: Utilizing Nanomedicine as Nanosensitizers to Physically Manipulate the Fibrotic Stroma
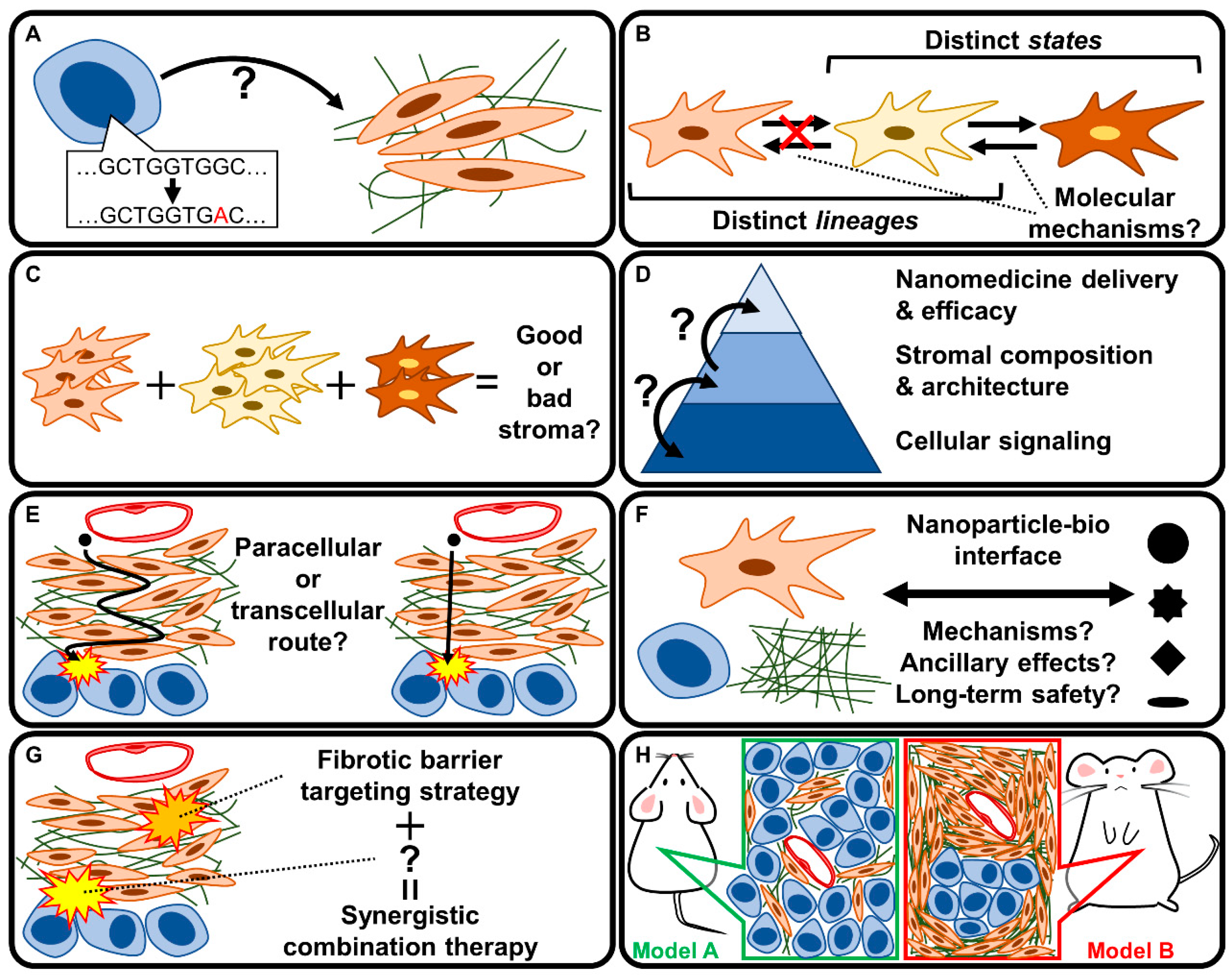
5. Discussion: Key Unknowns and Future Directions
5.1. How Does the PDAC Genotype Affect the Fibrotic Phenotype?
5.2. Are CAF Subpopulations Interconvertible?
5.3. What Distinguishes Good from Bad Stroma?
5.4. How Does Stromal Tissue Architecture Affect Nanomedicine Delivery?
5.5. How Do Nanomedicines Penetrate the PDAC Stroma?
5.6. What Are the Mechanisms Governing Nanoparticle-Bio Interactions?
5.7. How Can Therapeutic Strategies Targeting Fibrotic Stroma Be Utilized in Combination Therapies?
5.8. Which Experimental Model Should Be Used to Study Fibrotic Barriers in PDAC?
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic Cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef]
- Maitra, A.; Hruban, R.H. Pancreatic Cancer. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 157–188. [Google Scholar] [CrossRef]
- Hidalgo, M. Pancreatic Cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef] [PubMed]
- Grossberg, A.J.; Chu, L.C.; Deig, C.R.; Fishman, E.K.; Hwang, W.L.; Maitra, A.; Marks, D.L.; Mehta, A.; Nabavizadeh, N.; Simeone, D.M.; et al. Multidisciplinary Standards of Care and Recent Progress in Pancreatic Ductal Adenocarcinoma. CA Cancer J. Clin. 2020, 70, 375–403. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with Nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Arciero, V.; Luo, J.; Parmar, A.; Dai, W.F.; Beca, J.M.; Raphael, M.J.; Isaranuwatchai, W.; Habbous, S.; Tadrous, M.; Earle, C.C.; et al. Real-World Cost-Effectiveness of First-Line Gemcitabine Plus Nab-Paclitaxel vs FOLFIRINOX in Patients With Advanced Pancreatic Cancer. JNCI Cancer Spectr. 2022, 6, pkac047. [Google Scholar] [CrossRef]
- Klein-Brill, A.; Amar-Farkash, S.; Lawrence, G.; Collisson, E.A.; Aran, D. Comparison of FOLFIRINOX vs Gemcitabine Plus Nab-Paclitaxel as First-Line Chemotherapy for Metastatic Pancreatic Ductal Adenocarcinoma. JAMA Netw. Open 2022, 5, e2216199. [Google Scholar] [CrossRef]
- Riedl, J.M.; Posch, F.; Horvath, L.; Gantschnigg, A.; Renneberg, F.; Schwarzenbacher, E.; Moik, F.; Barth, D.A.; Rossmann, C.H.; Stotz, M.; et al. Gemcitabine/Nab-Paclitaxel versus FOLFIRINOX for Palliative First-Line Treatment of Advanced Pancreatic Cancer: A Propensity Score Analysis. Eur. J. Cancer 2021, 151, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Perri, G.; Prakash, L.; Qiao, W.; Varadhachary, G.R.; Wolff, R.; Fogelman, D.; Overman, M.; Pant, S.; Javle, M.; Koay, E.J.; et al. Response and Survival Associated With First-Line FOLFIRINOX vs Gemcitabine and Nab-Paclitaxel Chemotherapy for Localized Pancreatic Ductal Adenocarcinoma. JAMA Surg. 2020, 155, 832–839. [Google Scholar] [CrossRef]
- Wang-Gillam, A.; Li, C.-P.; Bodoky, G.; Dean, A.; Shan, Y.-S.; Jameson, G.; Macarulla, T.; Lee, K.-H.; Cunningham, D.; Blanc, J.F.; et al. Nanoliposomal Irinotecan with Fluorouracil and Folinic Acid in Metastatic Pancreatic Cancer after Previous Gemcitabine-Based Therapy (NAPOLI-1): A Global, Randomised, Open-Label, Phase 3 Trial. Lancet 2016, 387, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Dougan, S.K.; Aguirre, A.J.; Maitra, A. Translational Advances in Pancreatic Ductal Adenocarcinoma Therapy. Nat. Cancer 2022, 3, 272–286. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an Emerging Platform for Cancer Therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef]
- Maeda, H.; Nakamura, H.; Fang, J. The EPR Effect for Macromolecular Drug Delivery to Solid Tumors: Improvement of Tumor Uptake, Lowering of Systemic Toxicity, and Distinct Tumor Imaging In Vivo. Adv. Drug Deliv. Rev. 2013, 65, 71–79. [Google Scholar] [CrossRef]
- Nel, A.; Ruoslahti, E.; Meng, H. New Insights into “Permeability” as in the Enhanced Permeability and Retention Effect of Cancer Nanotherapeutics. ACS Nano 2017, 11, 9567–9569. [Google Scholar] [CrossRef] [PubMed]
- Sindhwani, S.; Syed, A.M.; Ngai, J.; Kingston, B.R.; Maiorino, L.; Rothschild, J.; MacMillan, P.; Zhang, Y.; Rajesh, N.U.; Hoang, T.; et al. The Entry of Nanoparticles into Solid Tumours. Nat. Mater. 2020, 19, 566–575. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Nichols, J.W.; Toh, K.; Nomoto, T.; Cabral, H.; Miura, Y.; Christie, R.J.; Yamada, N.; Ogura, T.; Kano, M.R.; et al. Vascular Bursts Enhance Permeability of Tumour Blood Vessels and Improve Nanoparticle Delivery. Nat. Nanotechnol. 2016, 11, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of Nanoparticle Design for Overcoming Biological Barriers to Drug Delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Nakamura, Y.; Mochida, A.; Choyke, P.L.; Kobayashi, H. Nanodrug Delivery: Is the Enhanced Permeability and Retention Effect Sufficient for Curing Cancer? Bioconjug. Chem. 2016, 27, 2225–2238. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of Nanoparticle Delivery to Tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Danhier, F. To Exploit the Tumor Microenvironment: Since the EPR Effect Fails in the Clinic, What Is the Future of Nanomedicine? J. Control. Release 2016, 244, 108–121. [Google Scholar] [CrossRef]
- Park, J.; Choi, Y.; Chang, H.; Um, W.; Ryu, J.H.; Kwon, I.C. Alliance with EPR Effect: Combined Strategies to Improve the EPR Effect in the Tumor Microenvironment. Theranostics 2019, 9, 8073–8090. [Google Scholar] [CrossRef]
- Jain, R.K.; Stylianopoulos, T. Delivering Nanomedicine to Solid Tumors. Nat. Rev. Clin. Oncol. 2010, 7, 653–664. [Google Scholar] [CrossRef]
- Stirland, D.L.; Matsumoto, Y.; Toh, K.; Kataoka, K.; Bae, Y.H. Analyzing Spatiotemporal Distribution of Uniquely Fluorescent Nanoparticles in Xenograft Tumors. J. Control. Release 2016, 227, 38–44. [Google Scholar] [CrossRef]
- Fang, J.; Islam, W.; Maeda, H. Exploiting the Dynamics of the EPR Effect and Strategies to Improve the Therapeutic Effects of Nanomedicines by Using EPR Effect Enhancers. Adv. Drug Deliv. Rev. 2020, 157, 142–160. [Google Scholar] [CrossRef]
- Kano, M.R. Nanotechnology and Tumor Microcirculation. Adv. Drug Deliv. Rev. 2014, 74, 2–11. [Google Scholar] [CrossRef]
- Ojha, T.; Pathak, V.; Shi, Y.; Hennink, W.E.; Moonen, C.T.W.; Storm, G.; Kiessling, F.; Lammers, T. Pharmacological and Physical Vessel Modulation Strategies to Improve EPR-Mediated Drug Targeting to Tumors. Adv. Drug Deliv. Rev. 2017, 119, 44–60. [Google Scholar] [CrossRef]
- Tanaka, H.Y.; Kano, M.R. Stromal Barriers to Nanomedicine Penetration in the Pancreatic Tumor Microenvironment. Cancer Sci. 2018, 109, 2085–2092. [Google Scholar] [CrossRef]
- Miao, L.; Lin, C.M.; Huang, L. Stromal Barriers and Strategies for the Delivery of Nanomedicine to Desmoplastic Tumors. J. Control. Release 2015, 219, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.R.; Baker, D.; Farren, M.; Pommier, A.; Swann, R.; Wang, X.; Mistry, S.; McDaid, K.; Kendrew, J.; Womack, C.; et al. Tumor Stromal Architecture Can Define the Intrinsic Tumor Response to VEGF-Targeted Therapy. Clin. Cancer Res. 2013, 19, 6943–6956. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.Y.; Kurihara, T.; Nakazawa, T.; Matsusaki, M.; Masamune, A.; Kano, M.R. Heterotypic 3D Pancreatic Cancer Model with Tunable Proportion of Fibrotic Elements. Biomaterials 2020, 251, 120077. [Google Scholar] [CrossRef]
- Tanaka, H.Y.; Kitahara, K.; Sasaki, N.; Nakao, N.; Sato, K.; Narita, H.; Shimoda, H.; Matsusaki, M.; Nishihara, H.; Masamune, A.; et al. Pancreatic Stellate Cells Derived from Human Pancreatic Cancer Demonstrate Aberrant SPARC-Dependent ECM Remodeling in 3D Engineered Fibrotic Tissue of Clinically Relevant Thickness. Biomaterials 2019, 192, 355–367. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Sakai, S.; Iwata, C.; Tanaka, H.Y.; Cabral, H.; Morishita, Y.; Miyazono, K.; Kano, M.R. Increased Fibrosis and Impaired Intratumoral Accumulation of Macromolecules in a Murine Model of Pancreatic Cancer Co-Administered with FGF-2. J. Control. Release 2016, 230, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From Mechanisms to Medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of Fibrosis: Therapeutic Translation for Fibrotic Disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in Fibrosis: Novel Roles and Mediators. Front. Pharmacol. 2014, 5, 123. [Google Scholar] [CrossRef]
- Wynn, T. Cellular and Molecular Mechanisms of Fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef]
- Foster, D.S.; Jones, R.E.; Ransom, R.C.; Longaker, M.T.; Norton, J.A. The Evolving Relationship of Wound Healing and Tumor Stroma. JCI Insight 2018, 3, e99911. [Google Scholar] [CrossRef] [PubMed]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, Definitions, and Functions in Health and Disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Neilson, E.G. Origin and Functional Heterogeneity of Fibroblasts. FASEB J. 2020, 34, 3519–3536. [Google Scholar] [CrossRef]
- Cirri, P.; Chiarugi, P. Cancer-Associated-Fibroblasts and Tumour Cells: A Diabolic Liaison Driving Cancer Progression. Cancer Metastasis Rev. 2012, 31, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.; Qiu, W.; Haviv, I. Genetic Changes in Tumour Microenvironments. J. Pathol. 2011, 223, 450–458. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds That Do Not Heal. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef]
- Masamune, A.; Kikuta, K.; Satoh, M.; Suzuki, N.; Shimosegawa, T. Protease-Activated Receptor-2-Mediated Proliferation and Collagen Production of Rat Pancreatic Stellate Cells. J. Pharmacol. Exp. Ther. 2005, 312, 651–658. [Google Scholar] [CrossRef]
- Pang, W.; Su, J.; Wang, Y.; Feng, H.; Dai, X.; Yuan, Y.; Chen, X.; Yao, W. Pancreatic Cancer-Secreted MiR-155 Implicates in the Conversion from Normal Fibroblasts to Cancer-Associated Fibroblasts. Cancer Sci. 2015, 106, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Masamune, A.; Yoshida, N.; Hamada, S.; Takikawa, T.; Nabeshima, T.; Shimosegawa, T. Exosomes Derived from Pancreatic Cancer Cells Induce Activation and Profibrogenic Activities in Pancreatic Stellate Cells. Biochem. Biophys. Res. Commun. 2018, 495, 71–77. [Google Scholar] [CrossRef]
- Wu, F.; Yang, J.; Liu, J.; Wang, Y.; Mu, J.; Zeng, Q.; Deng, S.; Zhou, H. Signaling Pathways in Cancer-Associated Fibroblasts and Targeted Therapy for Cancer. Signal Transduct. Target. Ther. 2021, 6, 218. [Google Scholar] [CrossRef]
- Masamune, A.; Kikuta, K.; Watanabe, T.; Satoh, K.; Hirota, M.; Shimosegawa, T. Hypoxia Stimulates Pancreatic Stellate Cells to Induce Fibrosis and Angiogenesis in Pancreatic Cancer. Am. J. Physiol.-Gastrointest. Liver Physiol. 2008, 295, G709–G717. [Google Scholar] [CrossRef]
- Bhagat, T.D.; Von Ahrens, D.; Dawlaty, M.; Zou, Y.; Baddour, J.; Achreja, A.; Zhao, H.; Yang, L.; Patel, B.; Kwak, C.; et al. Lactate-Mediated Epigenetic Reprogramming Regulates Formation of Human Pancreatic Cancer-Associated Fibroblasts. eLife 2019, 8, e50663. [Google Scholar] [CrossRef]
- Kalli, M.; Papageorgis, P.; Gkretsi, V.; Stylianopoulos, T. Solid Stress Facilitates Fibroblasts Activation to Promote Pancreatic Cancer Cell Migration. Ann. Biomed. Eng. 2018, 46, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Romac, J.M.-J.; Vigna, S.R.; Liddle, R.A. Piezo1-Mediated Stellate Cell Activation Causes Pressure-Induced Pancreatic Fibrosis in Mice. JCI Insight 2022, 7, e158288. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.-H.; Chang, Y.; Reed, N.I.; Sheppard, D. α-Smooth Muscle Actin Is an Inconsistent Marker of Fibroblasts Responsible for Force-Dependent TGFβ Activation or Collagen Production across Multiple Models of Organ Fibrosis. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2016, 310, L824–L836. [Google Scholar] [CrossRef]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef]
- Helms, E.; Onate, M.K.; Sherman, M.H. Fibroblast Heterogeneity in the Pancreatic Tumor Microenvironment. Cancer Discov. 2020, 10, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Boyd, L.N.C.; Andini, K.D.; Peters, G.J.; Kazemier, G.; Giovannetti, E. Heterogeneity and Plasticity of Cancer-Associated Fibroblasts in the Pancreatic Tumor Microenvironment. Semin. Cancer Biol. 2022, 82, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Brekken, R.A. Recent Advances in Understanding Cancer-Associated Fibroblasts in Pancreatic Cancer. Am. J. Physiol.-Cell Physiol. 2020, 319, C233–C243. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The Biology and Function of Fibroblasts in Cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Erkan, M.; Adler, G.; Apte, M.V.; Bachem, M.G.; Buchholz, M.; Detlefsen, S.; Esposito, I.; Friess, H.; Gress, T.M.; Habisch, H.-J.; et al. StellaTUM: Current Consensus and Discussion on Pancreatic Stellate Cell Research. Gut 2012, 61, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Omary, M.B.; Lugea, A.; Lowe, A.W.; Pandol, S.J. The Pancreatic Stellate Cell: A Star on the Rise in Pancreatic Diseases. J. Clin. Investig. 2007, 117, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Pothula, S.P.; Xu, Z.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Key Role of Pancreatic Stellate Cells in Pancreatic Cancer. Cancer Lett. 2016, 381, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Haber, P.S.; Keogh, G.W.; Apte, M.V.; Moran, C.S.; Stewart, N.L.; Crawford, D.H.G.; Pirola, R.C.; McCaughan, G.W.; Ramm, G.A.; Wilson, J.S. Activation of Pancreatic Stellate Cells in Human and Experimental Pancreatic Fibrosis. Am. J. Pathol. 1999, 155, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Helms, E.J.; Berry, M.W.; Chaw, R.C.; DuFort, C.C.; Sun, D.; Onate, M.K.; Oon, C.; Bhattacharyya, S.; Sanford-Crane, H.; Horton, W.; et al. Mesenchymal Lineage Heterogeneity Underlies Nonredundant Functions of Pancreatic Cancer-Associated Fibroblasts. Cancer Discov. 2022, 12, 484–501. [Google Scholar] [CrossRef]
- Han, L.; Wu, Y.; Fang, K.; Sweeney, S.; Roesner, U.K.; Parrish, M.; Patel, K.; Walter, T.; Piermattei, J.; Trimboli, A.; et al. The Splanchnic Mesenchyme Is the Tissue of Origin for Pancreatic Fibroblasts during Homeostasis and Tumorigenesis. Nat. Commun. 2023, 14, 1. [Google Scholar] [CrossRef]
- Waghray, M.; Yalamanchili, M.; Dziubinski, M.; Zeinali, M.; Erkkinen, M.; Yang, H.; Schradle, K.A.; Urs, S.; Pasca Di Magliano, M.; Welling, T.H.; et al. GM-CSF Mediates Mesenchymal-Epithelial Cross-Talk in Pancreatic Cancer. Cancer Discov. 2016, 6, 886–899. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of Endothelial to Mesenchymal Transition as a Source for Carcinoma-Associated Fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Wang, Z.; Zhang, Y.; Pradhan, R.N.; Ganguly, D.; Chandra, R.; Murimwa, G.; Wright, S.; Gu, X.; Maddipati, R.; et al. Mesothelial Cell-Derived Antigen-Presenting Cancer-Associated Fibroblasts Induce Expansion of Regulatory T Cells in Pancreatic Cancer. Cancer Cell 2022, 40, 656–673.e7. [Google Scholar] [CrossRef]
- Iwamoto, C.; Ohuchida, K.; Shinkawa, T.; Okuda, S.; Otsubo, Y.; Okumura, T.; Sagara, A.; Koikawa, K.; Ando, Y.; Shindo, K.; et al. Bone Marrow-Derived Macrophages Converted into Cancer-Associated Fibroblast-Like Cells Promote Pancreatic Cancer Progression. Cancer Lett. 2021, 512, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Ando, R.; Sakai, A.; Iida, T.; Kataoka, K.; Mizutani, Y.; Enomoto, A. Good and Bad Stroma in Pancreatic Cancer: Relevance of Functional States of Cancer-Associated Fibroblasts. Cancers 2022, 14, 3315. [Google Scholar] [CrossRef]
- Hynes, R.O.; Naba, A. Overview of the Matrisome--an Inventory of Extracellular Matrix Constituents and Functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [PubMed]
- Sorushanova, A.; Delgado, L.M.; Wu, Z.; Shologu, N.; Kshirsagar, A.; Raghunath, R.; Mullen, A.M.; Bayon, Y.; Pandit, A.; Raghunath, M.; et al. The Collagen Suprafamily: From Biosynthesis to Advanced Biomaterial Development. Adv. Mater. 2019, 31, e1801651. [Google Scholar] [CrossRef]
- Tian, C.; Clauser, K.R.; Öhlund, D.; Rickelt, S.; Huang, Y.; Gupta, M.; Mani, D.R.; Carr, S.A.; Tuveson, D.A.; Hynes, R.O. Proteomic Analyses of ECM during Pancreatic Ductal Adenocarcinoma Progression Reveal Different Contributions by Tumor and Stromal Cells. Proc. Natl. Acad. Sci. USA 2019, 116, 19609–19618. [Google Scholar] [CrossRef]
- Tian, C.; Öhlund, D.; Rickelt, S.; Lidström, T.; Huang, Y.; Hao, L.; Zhao, R.T.; Franklin, O.; Bhatia, S.N.; Tuveson, D.A.; et al. Cancer Cell-Derived Matrisome Proteins Promote Metastasis in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2020, 80, 1461–1474. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the Extracellular Matrix in Development and Disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Butcher, D.T.; Alliston, T.; Weaver, V.M. A Tense Situation: Forcing Tumour Progression. Nat. Rev. Cancer 2009, 9, 108–122. [Google Scholar] [CrossRef]
- McKleroy, W.; Lee, T.-H.; Atabai, K. Always Cleave up Your Mess: Targeting Collagen Degradation to Treat Tissue Fibrosis. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2013, 304, L709–L721. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of Extracellular Matrix Remodelling in Tumour Progression and Metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The Extracellular Matrix: A Dynamic Niche in Cancer Progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Hastings, J.F.; Skhinas, J.N.; Fey, D.; Croucher, D.R.; Cox, T.R. The Extracellular Matrix as a Key Regulator of Intracellular Signalling Networks. Br. J. Pharmacol. 2019, 176, 82–92. [Google Scholar] [CrossRef]
- Lukashev, M.E.; Werb, Z. ECM Signalling: Orchestrating Cell Behaviour and Misbehaviour. Trends Cell Biol. 1998, 8, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The Extracellular Matrix Modulates the Hallmarks of Cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef]
- Kumar, A.; Placone, J.K.; Engler, A.J. Understanding the Extracellular Forces That Determine Cell Fate and Maintenance. Development 2017, 144, 4261–4270. [Google Scholar] [CrossRef]
- Watt, F.M.; Huck, W.T.S. Role of the Extracellular Matrix in Regulating Stem Cell Fate. Nat. Rev. Mol. Cell Biol. 2013, 14, 467–473. [Google Scholar] [CrossRef]
- Sainio, A.; Järveläinen, H. Extracellular Matrix-Cell Interactions: Focus on Therapeutic Applications. Cell. Signal. 2020, 66, 109487. [Google Scholar] [CrossRef]
- Boudreau, N.; Bissell, M.J. Extracellular Matrix Signaling: Integration of Form and Function in Normal and Malignant Cells. Curr. Opin. Cell Biol. 1998, 10, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Kechagia, J.Z.; Ivaska, J.; Roca-Cusachs, P. Integrins as Biomechanical Sensors of the Microenvironment. Nat. Rev. Mol. Cell Biol. 2019, 20, 457–473. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.; Kukkurainen, S.; Hytönen, V.P.; Wehrle-Haller, B. Cell Adhesion by Integrins. Physiol. Rev. 2019, 99, 1655–1699. [Google Scholar] [CrossRef]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin Ligands at a Glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef]
- Hamidi, H.; Ivaska, J. Every Step of the Way: Integrins in Cancer Progression and Metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef]
- Kanchanawong, P.; Calderwood, D.A. Organization, Dynamics and Mechanoregulation of Integrin-Mediated Cell–ECM Adhesions. Nat. Rev. Mol. Cell Biol. 2023, 24, 142–161. [Google Scholar] [CrossRef] [PubMed]
- Ross, T.D.; Coon, B.G.; Yun, S.; Baeyens, N.; Tanaka, K.; Ouyang, M.; Schwartz, M.A. Integrins in Mechanotransduction. Curr. Opin. Cell Biol. 2013, 25, 613–618. [Google Scholar] [CrossRef]
- DiPersio, C.M.; Van De Water, L. Integrin Regulation of CAF Differentiation and Function. Cancers 2019, 11, 715. [Google Scholar] [CrossRef]
- Abyaneh, H.S.; Regenold, M.; McKee, T.D.; Allen, C.; Gauthier, M.A. Towards Extracellular Matrix Normalization for Improved Treatment of Solid Tumors. Theranostics 2020, 10, 1960–1980. [Google Scholar] [CrossRef]
- Xu, X.; Wu, Y.; Qian, X.; Wang, Y.; Wang, J.; Li, J.; Li, Y.; Zhang, Z. Nanomedicine Strategies to Circumvent Intratumor Extracellular Matrix Barriers for Cancer Therapy. Adv. Healthc. Mater. 2022, 11, e2101428. [Google Scholar] [CrossRef]
- Laklai, H.; Miroshnikova, Y.A.; Pickup, M.W.; Collisson, E.A.; Kim, G.E.; Barrett, A.S.; Hill, R.C.; Lakins, J.N.; Schlaepfer, D.D.; Mouw, J.K.; et al. Genotype Tunes Pancreatic Ductal Adenocarcinoma Tissue Tension to Induce Matricellular Fibrosis and Tumor Progression. Nat. Med. 2016, 22, 497–505. [Google Scholar] [CrossRef]
- Kelleher, F.C. Hedgehog Signaling and Therapeutics in Pancreatic Cancer. Carcinogenesis 2011, 32, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.A.; Bednar, F.; Zhang, Y.; Brisset, J.-C.; Galbán, S.; Galbán, C.J.; Rakshit, S.; Flannagan, K.S.; Adsay, N.V.; Pasca di Magliano, M. Oncogenic Kras Is Required for Both the Initiation and Maintenance of Pancreatic Cancer in Mice. J. Clin. Investig. 2012, 122, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Tape, C.J.; Ling, S.; Dimitriadi, M.; McMahon, K.M.; Worboys, J.D.; Leong, H.S.; Norrie, I.C.; Miller, C.J.; Poulogiannis, G.; Lauffenburger, D.A.; et al. Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell 2016, 165, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A Paracrine Requirement for Hedgehog Signalling in Cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic Hedgehog Promotes Desmoplasia in Pancreatic Cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef]
- Lauth, M.; Bergström, A.; Shimokawa, T.; Tostar, U.; Jin, Q.; Fendrich, V.; Guerra, C.; Barbacid, M.; Toftgård, R. DYRK1B-Dependent Autocrine-to-Paracrine Shift of Hedgehog Signaling by Mutant RAS. Nat. Struct. Mol. Biol. 2010, 17, 718–725. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D Cooperate to Promote Chromosomal Instability and Widely Metastatic Pancreatic Ductal Adenocarcinoma in Mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, H.; Hsiao, C.-H.; Chow, D.S.-L.; Koay, E.J.; Kang, Y.; Wen, X.; Huang, Q.; Ma, Y.; Bankson, J.A.; et al. Simultaneous Inhibition of Hedgehog Signaling and Tumor Proliferation Remodels Stroma and Enhances Pancreatic Cancer Therapy. Biomaterials 2018, 159, 215–228. [Google Scholar] [CrossRef]
- Wang, L.; Liu, X.; Zhou, Q.; Sui, M.; Lu, Z.; Zhou, Z.; Tang, J.; Miao, Y.; Zheng, M.; Wang, W.; et al. Terminating the Criminal Collaboration in Pancreatic Cancer: Nanoparticle-Based Synergistic Therapy for Overcoming Fibroblast-Induced Drug Resistance. Biomaterials 2017, 144, 105–118. [Google Scholar] [CrossRef]
- Meng, X.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The Master Regulator of Fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N. Transforming Growth Factor-β in Tissue Fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liao, S.; Diop-Frimpong, B.; Chen, W.; Goel, S.; Naxerova, K.; Ancukiewicz, M.; Boucher, Y.; Jain, R.K.; Xu, L. TGF-β Blockade Improves the Distribution and Efficacy of Therapeutics in Breast Carcinoma by Normalizing the Tumor Stroma. Proc. Natl. Acad. Sci. USA 2012, 109, 16618–16623. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; De Cant, B.; Mascariñas, E.; Wayne, E.A.; Diaz, A.M.; Akagi, N.; Hwang, R.; Pasche, B.; Dawson, D.W.; Fang, D.; et al. TGFβ Signaling in the Pancreatic Tumor Microenvironment Promotes Fibrosis and Immune Evasion to Facilitate Tumorigenesis. Cancer Res. 2016, 76, 2525–2539. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef]
- Melisi, D.; Ishiyama, S.; Sclabas, G.M.; Fleming, J.B.; Xia, Q.; Tortora, G.; Abbruzzese, J.L.; Chiao, P.J. LY2109761, a Novel Transforming Growth Factor Beta Receptor Type I and Type II Dual Inhibitor, as a Therapeutic Approach to Suppressing Pancreatic Cancer Metastasis. Mol. Cancer Ther. 2008, 7, 829–840. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, Y.; Gallegos, V.; Sorrelle, N.; Zaid, M.M.; Toombs, J.; Du, W.; Wright, S.; Hagopian, M.; Wang, Z.; et al. Targeting TGFβR2-Mutant Tumors Exposes Vulnerabilities to Stromal TGFβ Blockade in Pancreatic Cancer. EMBO Mol. Med. 2019, 11, e10515. [Google Scholar] [CrossRef]
- Derynck, R.; Budi, E.H. Specificity, Versatility, and Control of TGF-β Family Signaling. Sci. Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef]
- Principe, D.R.; Timbers, K.E.; Atia, L.G.; Koch, R.M.; Rana, A. TGFβ Signaling in the Pancreatic Tumor Microenvironment. Cancers 2021, 13, 5086. [Google Scholar] [CrossRef]
- Doyle, J.J.; Gerber, E.E.; Dietz, H.C. Matrix-Dependent Perturbation of TGFβ Signaling and Disease. FEBS Lett. 2012, 586, 2003–2015. [Google Scholar] [CrossRef]
- Diop-Frimpong, B.; Chauhan, V.P.; Krane, S.; Boucher, Y.; Jain, R.K. Losartan Inhibits Collagen I Synthesis and Improves the Distribution and Efficacy of Nanotherapeutics in Tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 2909–2914. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin Inhibition Enhances Drug Delivery and Potentiates Chemotherapy by Decompressing Tumour Blood Vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef]
- Murphy, J.E.; Wo, J.Y.; Ryan, D.P.; Clark, J.W.; Jiang, W.; Yeap, B.Y.; Drapek, L.C.; Ly, L.; Baglini, C.V.; Blaszkowsky, L.S.; et al. Total Neoadjuvant Therapy With FOLFIRINOX in Combination With Losartan Followed by Chemoradiotherapy for Locally Advanced Pancreatic Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 1020–1027. [Google Scholar] [CrossRef]
- Gao, J.; Ye, J.; Ying, Y.; Lin, H.; Luo, Z. Negative Regulation of TGF-β by AMPK and Implications in the Treatment of Associated Disorders. Acta Biochim. Biophys. Sin. 2018, 50, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Hou, Y.; Chen, X.; Zhang, P.; Kang, M.; Jin, Q.; Ji, J.; Gao, M. Metformin-Induced Stromal Depletion to Enhance the Penetration of Gemcitabine-Loaded Magnetic Nanoparticles for Pancreatic Cancer Targeted Therapy. J. Am. Chem. Soc. 2020, 142, 4944–4954. [Google Scholar] [CrossRef]
- Chen, X.; Jia, F.; Li, Y.; Deng, Y.; Huang, Y.; Liu, W.; Jin, Q.; Ji, J. Nitric Oxide-Induced Stromal Depletion for Improved Nanoparticle Penetration in Pancreatic Cancer Treatment. Biomaterials 2020, 246, 119999. [Google Scholar] [CrossRef]
- Pines, M.; Spector, I. Halofuginone—The Multifaceted Molecule. Molecules 2015, 20, 573–594. [Google Scholar] [CrossRef] [PubMed]
- Elahi-Gedwillo, K.Y.; Carlson, M.; Zettervall, J.; Provenzano, P.P. Antifibrotic Therapy Disrupts Stromal Barriers and Modulates the Immune Landscape in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2019, 79, 372–386. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Ebina, M.; Kondoh, Y.; Ogura, T.; Azuma, A.; Suga, M.; Taguchi, Y.; Takahashi, H.; Nakata, K.; Sato, A.; et al. Pirfenidone in Idiopathic Pulmonary Fibrosis. Eur. Respir. J. 2010, 35, 821–829. [Google Scholar] [CrossRef]
- Kozono, S.; Ohuchida, K.; Eguchi, D.; Ikenaga, N.; Fujiwara, K.; Cui, L.; Mizumoto, K.; Tanaka, M. Pirfenidone Inhibits Pancreatic Cancer Desmoplasia by Regulating Stellate Cells. Cancer Res. 2013, 73, 2345–2356. [Google Scholar] [CrossRef] [PubMed]
- Darakhshan, S.; Pour, A.B. Tranilast: A Review of Its Therapeutic Applications. Pharmacol. Res. 2015, 91, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Papageorgis, P.; Polydorou, C.; Mpekris, F.; Voutouri, C.; Agathokleous, E.; Kapnissi-Christodoulou, C.P.; Stylianopoulos, T. Tranilast-Induced Stress Alleviation in Solid Tumors Improves the Efficacy of Chemo- and Nanotherapeutics in a Size-Independent Manner. Sci. Rep. 2017, 7, 46140. [Google Scholar] [CrossRef] [PubMed]
- Pang, N.; Li, J.; Sun, A.; Yang, Z.; Cheng, S.; Qi, X.-R. Prior Anti-CAFs Break down the CAFs Barrier and Improve Accumulation of Docetaxel Micelles in Tumor. Int. J. Nanomedicine 2018, 13, 5971–5990. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, C.J.; Ruhrmund, D.W.; Pan, L.; Seiwert, S.D.; Kossen, K. Antifibrotic Activities of Pirfenidone in Animal Models. Eur. Respir. Rev. 2011, 20, 85–97. [Google Scholar] [CrossRef]
- Conti, P.; Caraffa, A.; Mastrangelo, F.; Tettamanti, L.; Ronconi, G.; Frydas, I.; Kritas, S.K.; Theoharides, T.C. Critical Role of Inflammatory Mast Cell in Fibrosis: Potential Therapeutic Effect of IL-37. Cell Prolif. 2018, 51, e12475. [Google Scholar] [CrossRef]
- Ma, Y.; Hwang, R.F.; Logsdon, C.D.; Ullrich, S.E. Dynamic Mast Cell-Stromal Cell Interactions Promote Growth of Pancreatic Cancer. Cancer Res. 2013, 73, 3927–3937. [Google Scholar] [CrossRef]
- Massó-Vallés, D.; Jauset, T.; Serrano, E.; Sodir, N.M.; Pedersen, K.; Affara, N.I.; Whitfield, J.R.; Beaulieu, M.-E.; Evan, G.I.; Elias, L.; et al. Ibrutinib Exerts Potent Antifibrotic and Antitumor Activities in Mouse Models of Pancreatic Adenocarcinoma. Cancer Res. 2015, 75, 1675–1681. [Google Scholar] [CrossRef]
- Tempero, M.; Oh, D.-Y.; Tabernero, J.; Reni, M.; Van Cutsem, E.; Hendifar, A.; Waldschmidt, D.-T.; Starling, N.; Bachet, J.-B.; Chang, H.-M.; et al. Ibrutinib in Combination with Nab-Paclitaxel and Gemcitabine for First-Line Treatment of Patients with Metastatic Pancreatic Adenocarcinoma: Phase III RESOLVE Study. Ann. Oncol. 2021, 32, 600–608. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.-C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal Response to Hedgehog Signaling Restrains Pancreatic Cancer Progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef]
- Catenacci, D.V.T.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef]
- Gargalionis, A.N.; Papavassiliou, K.A.; Papavassiliou, A.G. Mechanobiology of Solid Tumors. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166555. [Google Scholar] [CrossRef] [PubMed]
- MacCurtain, B.M.; Quirke, N.P.; Thorpe, S.D.; Gallagher, T.K. Pancreatic Ductal Adenocarcinoma: Relating Biomechanics and Prognosis. J. Clin. Med. 2021, 10, 2711. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D Receptor-Mediated Stromal Reprogramming Suppresses Pancreatitis and Enhances Pancreatic Cancer Therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Z.; Zhou, Q.; Gu, S.; Liu, X.; Huang, J.; Jiang, H.; Wang, H.; Cao, L.; Sun, J.; et al. Prodrug Nanoparticles Rationally Integrating Stroma Modification and Chemotherapy to Treat Metastatic Pancreatic Cancer. Biomaterials 2021, 278, 121176. [Google Scholar] [CrossRef] [PubMed]
- Davis-Yadley, A.H.; Malafa, M.P. Vitamins in Pancreatic Cancer: A Review of Underlying Mechanisms and Future Applications. Adv. Nutr. 2015, 6, 774–802. [Google Scholar] [CrossRef]
- Gorchs, L.; Ahmed, S.; Mayer, C.; Knauf, A.; Fernández Moro, C.; Svensson, M.; Heuchel, R.; Rangelova, E.; Bergman, P.; Kaipe, H. The Vitamin D Analogue Calcipotriol Promotes an Anti-Tumorigenic Phenotype of Human Pancreatic CAFs but Reduces T Cell Mediated Immunity. Sci. Rep. 2020, 10, 17444. [Google Scholar] [CrossRef]
- Borazanci, E.H.; Jameson, G.S.; Borad, M.J.; Ramanathan, R.K.; Korn, R.L.; Caldwell, L.; Ansaldo, K.; Hendrickson, K.; Marceau, K.; Von Hoff, D.D. A Phase II Pilot Trial of Nivolumab (N) + Albumin Bound Paclitaxel (AP) + Paricalcitol (P) + Cisplatin (C) + Gemcitabine (G) (NAPPCG) in Patients with Previously Untreated Metastatic Pancreatic Ductal Adenocarcinoma (PDAC). J. Clin. Oncol. 2018, 36, 358. [Google Scholar] [CrossRef]
- Bachem, M.G.; Schneider, E.; Gross, H.; Weidenbach, H.; Schmid, R.M.; Menke, A.; Siech, M.; Beger, H.; Grünert, A.; Adler, G. Identification, Culture, and Characterization of Pancreatic Stellate Cells in Rats and Humans. Gastroenterology 1998, 115, 421–432. [Google Scholar] [CrossRef]
- Apte, M.V.; Haber, P.S.; Applegate, T.L.; Norton, I.D.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Periacinar Stellate Shaped Cells in Rat Pancreas: Identification, Isolation, and Culture. Gut 1998, 43, 128–133. [Google Scholar] [CrossRef] [PubMed]
- McCarroll, J.A.; Phillips, P.A.; Santucci, N.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Vitamin A Inhibits Pancreatic Stellate Cell Activation: Implications for Treatment of Pancreatic Fibrosis. Gut 2006, 55, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Froeling, F.E.M.; Feig, C.; Chelala, C.; Dobson, R.; Mein, C.E.; Tuveson, D.A.; Clevers, H.; Hart, I.R.; Kocher, H.M. Retinoic Acid-Induced Pancreatic Stellate Cell Quiescence Reduces Paracrine Wnt-β-Catenin Signaling to Slow Tumor Progression. Gastroenterology 2011, 141, 1486–1497.e14. [Google Scholar] [CrossRef] [PubMed]
- Chronopoulos, A.; Robinson, B.; Sarper, M.; Cortes, E.; Auernheimer, V.; Lachowski, D.; Attwood, S.; García, R.; Ghassemi, S.; Fabry, B.; et al. ATRA Mechanically Reprograms Pancreatic Stellate Cells to Suppress Matrix Remodelling and Inhibit Cancer Cell Invasion. Nat. Commun. 2016, 7, 12630. [Google Scholar] [CrossRef] [PubMed]
- Kocher, H.M.; Basu, B.; Froeling, F.E.M.; Sarker, D.; Slater, S.; Carlin, D.; deSouza, N.M.; De Paepe, K.N.; Goulart, M.R.; Hughes, C.; et al. Phase I Clinical Trial Repurposing All-Trans Retinoic Acid as a Stromal Targeting Agent for Pancreatic Cancer. Nat. Commun. 2020, 11, 4841. [Google Scholar] [CrossRef]
- Wegner, C.S.; Gaustad, J.-V.; Andersen, L.M.K.; Simonsen, T.G.; Rofstad, E.K. Diffusion-Weighted and Dynamic Contrast-Enhanced MRI of Pancreatic Adenocarcinoma Xenografts: Associations with Tumor Differentiation and Collagen Content. J. Transl. Med. 2016, 14, 161. [Google Scholar] [CrossRef]
- Binkley, C.E.; Zhang, L.; Greenson, J.K.; Giordano, T.J.; Kuick, R.; Misek, D.; Hanash, S.; Logsdon, C.D.; Simeone, D.M. The Molecular Basis of Pancreatic Fibrosis: Common Stromal Gene Expression in Chronic Pancreatitis and Pancreatic Adenocarcinoma. Pancreas 2004, 29, 254–263. [Google Scholar] [CrossRef]
- Iacobuzio-Donahue, C.A.; Maitra, A.; Shen-Ong, G.L.; van Heek, T.; Ashfaq, R.; Meyer, R.; Walter, K.; Berg, K.; Hollingsworth, M.A.; Cameron, J.L.; et al. Discovery of Novel Tumor Markers of Pancreatic Cancer Using Global Gene Expression Technology. Am. J. Pathol. 2002, 160, 1239–1249. [Google Scholar] [CrossRef]
- Peran, I.; Dakshanamurthy, S.; McCoy, M.D.; Mavropoulos, A.; Allo, B.; Sebastian, A.; Hum, N.R.; Sprague, S.C.; Martin, K.A.; Pishvaian, M.J.; et al. Cadherin 11 Promotes Immunosuppression and Extracellular Matrix Deposition to Support Growth of Pancreatic Tumors and Resistance to Gemcitabine in Mice. Gastroenterology 2021, 160, 1359–1372.e13. [Google Scholar] [CrossRef]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of Fibroblast Heterogeneity in the Tumor Microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef]
- Ijichi, H. Multiphasic Heterogeneity of Fibroblasts in the Microenvironment of Pancreatic Ductal Adenocarcinoma: Dissection and the Sum of the Dynamics. Cancers 2022, 14, 4880. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct Populations of Inflammatory Fibroblasts and Myofibroblasts in Pancreatic Cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and Intra-Tumoural Heterogeneity in Cancer-Associated Fibroblasts of Human Pancreatic Ductal Adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef]
- Hosein, A.N.; Huang, H.; Wang, Z.; Parmar, K.; Du, W.; Huang, J.; Maitra, A.; Olson, E.; Verma, U.; Brekken, R.A. Cellular Heterogeneity during Mouse Pancreatic Ductal Adenocarcinoma Progression at Single-Cell Resolution. JCI Insight 2019, 5, 129212. [Google Scholar] [CrossRef]
- Dominguez, C.X.; Müller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15+ Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov. 2020, 10, 232–253. [Google Scholar] [CrossRef]
- Menezes, S.; Okail, M.H.; Jalil, S.M.A.; Kocher, H.M.; Cameron, A.J.M. Cancer-Associated Fibroblasts in Pancreatic Cancer: New Subtypes, New Markers, New Targets. J. Pathol. 2022, 257, 526–544. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, Y.; Xu, H.; Zhang, X.; Mao, T.; Cui, J.; Yao, J.; Wang, Y.; Jiao, F.; Xiao, X.; et al. Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Identifies a Novel Fibroblast Subtype Associated with Poor Prognosis but Better Immunotherapy Response. Cell Discov. 2021, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Xia, X.; Huang, L.-B.; An, H.; Cao, M.; Kim, G.D.; Chen, H.-N.; Zhang, W.-H.; Shu, Y.; Kong, X.; et al. Pan-Cancer Single-Cell Analysis Reveals the Heterogeneity and Plasticity of Cancer-Associated Fibroblasts in the Tumor Microenvironment. Nat. Commun. 2022, 13, 6619. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed]
- Mello, A.M.; Ngodup, T.; Lee, Y.; Donahue, K.L.; Li, J.; Rao, A.; Carpenter, E.S.; Crawford, H.C.; Pasca di Magliano, M.; Lee, K.E. Hypoxia Promotes an Inflammatory Phenotype of Fibroblasts in Pancreatic Cancer. Oncogenesis 2022, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Lefler, J.E.; MarElia-Bennett, C.B.; Thies, K.A.; Hildreth, B.E.; Sharma, S.M.; Pitarresi, J.R.; Han, L.; Everett, C.; Koivisto, C.; Cuitino, M.C.; et al. STAT3 in Tumor Fibroblasts Promotes an Immunosuppressive Microenvironment in Pancreatic Cancer. Life Sci. Alliance 2022, 5, e202201460. [Google Scholar] [CrossRef]
- Murray, E.R.; Menezes, S.; Henry, J.C.; Williams, J.L.; Alba-Castellón, L.; Baskaran, P.; Quétier, I.; Desai, A.; Marshall, J.J.T.; Rosewell, I.; et al. Disruption of Pancreatic Stellate Cell Myofibroblast Phenotype Promotes Pancreatic Tumor Invasion. Cell Rep. 2022, 38, 110227. [Google Scholar] [CrossRef]
- Steele, N.G.; Biffi, G.; Kemp, S.B.; Zhang, Y.; Drouillard, D.; Syu, L.; Hao, Y.; Oni, T.E.; Brosnan, E.; Elyada, E.; et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 2023–2037. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, Y.; Kobayashi, H.; Iida, T.; Asai, N.; Masamune, A.; Hara, A.; Esaki, N.; Ushida, K.; Mii, S.; Shiraki, Y.; et al. Meflin-Positive Cancer-Associated Fibroblasts Inhibit Pancreatic Carcinogenesis. Cancer Res. 2019, 79, 5367–5381. [Google Scholar] [CrossRef] [PubMed]
- Iida, T.; Mizutani, Y.; Esaki, N.; Ponik, S.M.; Burkel, B.M.; Weng, L.; Kuwata, K.; Masamune, A.; Ishihara, S.; Haga, H.; et al. Pharmacologic Conversion of Cancer-Associated Fibroblasts from a Protumor Phenotype to an Antitumor Phenotype Improves the Sensitivity of Pancreatic Cancer to Chemotherapeutics. Oncogene 2022, 41, 2764–2777. [Google Scholar] [CrossRef]
- Mizutani, Y.; Iida, T.; Ohno, E.; Ishikawa, T.; Kinoshita, F.; Kuwatsuka, Y.; Imai, M.; Shimizu, S.; Tsuruta, T.; Enomoto, A.; et al. Safety and Efficacy of MIKE-1 in Patients with Advanced Pancreatic Cancer: A Study Protocol for an Open-Label Phase I/II Investigator-Initiated Clinical Trial Based on a Drug Repositioning Approach That Reprograms the Tumour Stroma. BMC Cancer 2022, 22, 205. [Google Scholar] [CrossRef] [PubMed]
- Ghosh-Choudhary, S.; Liu, J.; Finkel, T. Metabolic Regulation of Cell Fate and Function. Trends Cell Biol. 2020, 30, 201–212. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of Cancer Metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Elia, I.; Haigis, M.C. Metabolites and the Tumour Microenvironment: From Cellular Mechanisms to Systemic Metabolism. Nat. Metab. 2021, 3, 21–32. [Google Scholar] [CrossRef]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human Pancreatic Cancer Tumors Are Nutrient Poor and Tumor Cells Actively Scavenge Extracellular Protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef]
- Sullivan, M.R.; Danai, L.V.; Lewis, C.A.; Chan, S.H.; Gui, D.Y.; Kunchok, T.; Dennstedt, E.A.; Vander Heiden, M.G.; Muir, A. Quantification of Microenvironmental Metabolites in Murine Cancers Reveals Determinants of Tumor Nutrient Availability. eLife 2019, 8, e44235. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.N.; Vander Heiden, M.G. Metabolism in the Tumor Microenvironment. Annu. Rev. Cancer Biol. 2020, 4, 17–40. [Google Scholar] [CrossRef]
- Li, Z.; Sun, C.; Qin, Z. Metabolic Reprogramming of Cancer-Associated Fibroblasts and Its Effect on Cancer Cell Reprogramming. Theranostics 2021, 11, 8322–8336. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.R.; Damaghi, M.; Marunaka, Y.; Spugnini, E.P.; Fais, S.; Gillies, R.J. Causes, Consequences, and Therapy of Tumors Acidosis. Cancer Metastasis Rev. 2019, 38, 205–222. [Google Scholar] [CrossRef]
- Yoshida, G.J. Metabolic Reprogramming: The Emerging Concept and Associated Therapeutic Strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The Reverse Warburg Effect: Aerobic Glycolysis in Cancer Associated Fibroblasts and the Tumor Stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic Stellate Cells Support Tumour Metabolism through Autophagic Alanine Secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef]
- Zhang, Y.; Recouvreux, M.V.; Jung, M.; Galenkamp, K.M.O.; Li, Y.; Zagnitko, O.; Scott, D.A.; Lowy, A.M.; Commisso, C. Macropinocytosis in Cancer-Associated Fibroblasts Is Dependent on CaMKK2/ARHGEF2 Signaling and Functions to Support Tumor and Stromal Cell Fitness. Cancer Discov. 2021, 11, 1808–1825. [Google Scholar] [CrossRef]
- Endo, S.; Nakata, K.; Ohuchida, K.; Takesue, S.; Nakayama, H.; Abe, T.; Koikawa, K.; Okumura, T.; Sada, M.; Horioka, K.; et al. Autophagy Is Required for Activation of Pancreatic Stellate Cells, Associated With Pancreatic Cancer Progression and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology 2017, 152, 1492–1506.e24. [Google Scholar] [CrossRef]
- Auciello, F.R.; Bulusu, V.; Oon, C.; Tait-Mulder, J.; Berry, M.; Bhattacharyya, S.; Tumanov, S.; Allen-Petersen, B.L.; Link, J.; Kendsersky, N.D.; et al. A Stromal Lysolipid-Autotaxin Signaling Axis Promotes Pancreatic Tumor Progression. Cancer Discov. 2019, 9, 617–627. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor Microenvironment Derived Exosomes Pleiotropically Modulate Cancer Cell Metabolism. eLife 2016, 5, e10250. [Google Scholar] [CrossRef]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-Associated Fibroblast Exosomes Regulate Survival and Proliferation of Pancreatic Cancer Cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef]
- Li, F.; Simon, M.C. Cancer Cells Don’t Live Alone: Metabolic Communication within Tumor Microenvironments. Dev. Cell 2020, 54, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Kay, E.J.; Zanivan, S. Metabolic Pathways Fuelling Protumourigenic Cancer-Associated Fibroblast Functions. Curr. Opin. Syst. Biol. 2021, 28, 100377. [Google Scholar] [CrossRef]
- Suzuki, T.; Otsuka, M.; Seimiya, T.; Iwata, T.; Kishikawa, T.; Koike, K. The Biological Role of Metabolic Reprogramming in Pancreatic Cancer. MedComm 2020, 1, 302–310. [Google Scholar] [CrossRef]
- Hamada, S.; Matsumoto, R.; Masamune, A. Pancreatic Stellate Cells and Metabolic Alteration: Physiology and Pathophysiology. Front. Physiol. 2022, 13, 865105. [Google Scholar] [CrossRef]
- Kay, E.J.; Koulouras, G.; Zanivan, S. Regulation of Extracellular Matrix Production in Activated Fibroblasts: Roles of Amino Acid Metabolism in Collagen Synthesis. Front. Oncol. 2021, 11, 719922. [Google Scholar] [CrossRef]
- Zhao, X.; Psarianos, P.; Ghoraie, L.S.; Yip, K.; Goldstein, D.; Gilbert, R.; Witterick, I.; Pang, H.; Hussain, A.; Lee, J.H.; et al. Metabolic Regulation of Dermal Fibroblasts Contributes to Skin Extracellular Matrix Homeostasis and Fibrosis. Nat. Metab. 2019, 1, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Tan, Z.; Banerjee, S.; Cui, H.; Ge, J.; Liu, R.-M.; Bernard, K.; Thannickal, V.J.; Liu, G. Glycolytic Reprogramming in Myofibroblast Differentiation and Lung Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1462–1474. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Targeting Glucose Metabolism for Cancer Therapy. J. Exp. Med. 2012, 209, 211–215. [Google Scholar] [CrossRef]
- Ganapathy-Kanniappan, S.; Geschwind, J.-F.H. Tumor Glycolysis as a Target for Cancer Therapy: Progress and Prospects. Mol. Cancer 2013, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Ren, X.; Hait, W.N.; Yang, J.-M. Therapeutic Targeting of Autophagy in Disease: Biology and Pharmacology. Pharmacol. Rev. 2013, 65, 1162–1197. [Google Scholar] [CrossRef]
- Malhotra, V.; Erlmann, P. The Pathway of Collagen Secretion. Annu. Rev. Cell Dev. Biol. 2015, 31, 109–124. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Nielsen, S.H.; Leeming, D.J.; Langholm, L.L.; Nielsen, M.J.; Manon-Jensen, T.; Siebuhr, A.; Gudmann, N.S.; Rønnow, S.; Sand, J.M.; et al. The Good and the Bad Collagens of Fibrosis—Their Role in Signaling and Organ Function. Adv. Drug Deliv. Rev. 2017, 121, 43–56. [Google Scholar] [CrossRef]
- Fuller, G.C. Perspectives for the Use of Collagen Synthesis Inhibitors as Antifibrotic Agents. J. Med. Chem. 1981, 24, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Liu, T.; Tu, B.; Yuan, M.; Shu, Z.; Fan, M.; Huo, S.; Guo, Y.; Wang, L.; Wang, H.; et al. Autophagy Loss Impedes Cancer-Associated Fibroblast Activation via Downregulating Proline Biosynthesis. Autophagy 2023, 19, 632–643. [Google Scholar] [CrossRef]
- Kay, E.J.; Paterson, K.; Riera-Domingo, C.; Sumpton, D.; Däbritz, J.H.M.; Tardito, S.; Boldrini, C.; Hernandez-Fernaud, J.R.; Athineos, D.; Dhayade, S.; et al. Cancer-Associated Fibroblasts Require Proline Synthesis by PYCR1 for the Deposition of pro-Tumorigenic Extracellular Matrix. Nat. Metab. 2022, 4, 693–710. [Google Scholar] [CrossRef] [PubMed]
- Masamune, A.; Watanabe, T.; Kikuta, K.; Satoh, K.; Shimosegawa, T. NADPH Oxidase Plays a Crucial Role in the Activation of Pancreatic Stellate Cells. Am. J. Physiol.-Gastrointest. Liver Physiol. 2008, 294, G99–G108. [Google Scholar] [CrossRef]
- Liu, R.-M.; Gaston Pravia, K.A. Oxidative Stress and Glutathione in TGF-β-Mediated Fibrogenesis. Free Radic. Biol. Med. 2010, 48, 1–15. [Google Scholar] [CrossRef]
- Liu, R.-M.; Desai, L.P. Reciprocal Regulation of TGF-β and Reactive Oxygen Species: A Perverse Cycle for Fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Schwörer, S.; Berisa, M.; Violante, S.; Qin, W.; Zhu, J.; Hendrickson, R.C.; Cross, J.R.; Thompson, C.B. Proline Biosynthesis Is a Vent for TGFβ-Induced Mitochondrial Redox Stress. EMBO J. 2020, 39, e103334. [Google Scholar] [CrossRef]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef] [PubMed]
- Chio, I.I.C.; Jafarnejad, S.M.; Ponz-Sarvise, M.; Park, Y.; Rivera, K.; Palm, W.; Wilson, J.; Sangar, V.; Hao, Y.; Öhlund, D.; et al. NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell 2016, 166, 963–976. [Google Scholar] [CrossRef]
- Hamada, S.; Taguchi, K.; Masamune, A.; Yamamoto, M.; Shimosegawa, T. Nrf2 Promotes Mutant K-Ras/P53-Driven Pancreatic Carcinogenesis. Carcinogenesis 2017, 38, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Hamada, S.; Matsumoto, R.; Taguchi, K.; Yamamoto, M.; Masamune, A. Nrf2 Expression in Pancreatic Stellate Cells Promotes Progression of Cancer. Am. J. Physiol.-Gastrointest. Liver Physiol. 2021, 321, G378–G388. [Google Scholar] [CrossRef]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting Cancer Metabolism in the Era of Precision Oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef]
- Lu, S.; Wang, Y. Nonmetabolic Functions of Metabolic Enzymes in Cancer Development. Cancer Commun. 2018, 38, 63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Jia, Y.; Yu, Y.; Zhang, B.; Xu, F.; Guo, H. Targeting the Tumor Biophysical Microenvironment to Reduce Resistance to Immunotherapy. Adv. Drug Deliv. Rev. 2022, 186, 114319. [Google Scholar] [CrossRef]
- Baronzio, G.; Parmar, G.; Baronzio, M. Overview of Methods for Overcoming Hindrance to Drug Delivery to Tumors, with Special Attention to Tumor Interstitial Fluid. Front. Oncol. 2015, 5, 165. [Google Scholar] [CrossRef]
- Stylianopoulos, T.; Munn, L.L.; Jain, R.K. Reengineering the Physical Microenvironment of Tumors to Improve Drug Delivery and Efficacy: From Mathematical Modeling to Bench to Bedside. Trends Cancer 2018, 4, 292–319. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.P.; Stylianopoulos, T.; Boucher, Y.; Jain, R.K. Delivery of Molecular and Nanoscale Medicine to Tumors: Transport Barriers and Strategies. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Martin, J.D.; Stylianopoulos, T. The Role of Mechanical Forces in Tumor Growth and Therapy. Annu. Rev. Biomed. Eng. 2014, 16, 321–346. [Google Scholar] [CrossRef] [PubMed]
- McKee, T.D.; Grandi, P.; Mok, W.; Alexandrakis, G.; Insin, N.; Zimmer, J.P.; Bawendi, M.G.; Boucher, Y.; Breakefield, X.O.; Jain, R.K. Degradation of Fibrillar Collagen in a Human Melanoma Xenograft Improves the Efficacy of an Oncolytic Herpes Simplex Virus Vector. Cancer Res. 2006, 66, 2509–2513. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.P.; Lanning, R.M.; Diop-Frimpong, B.; Mok, W.; Brown, E.B.; Padera, T.P.; Boucher, Y.; Jain, R.K. Multiscale Measurements Distinguish Cellular and Interstitial Hindrances to Diffusion In Vivo. Biophys. J. 2009, 97, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Payne, S.L.; Hendrix, M.J.C.; Kirschmann, D.A. Paradoxical Roles for Lysyl Oxidases in Cancer--a Prospect. J. Cell. Biochem. 2007, 101, 1338–1354. [Google Scholar] [CrossRef]
- Levick, J.R. Flow Through Interstitium and Other Fibrous Matrices. Q. J. Exp. Physiol. 1987, 72, 409–437. [Google Scholar] [CrossRef] [PubMed]
- Tavianatou, A.G.; Caon, I.; Franchi, M.; Piperigkou, Z.; Galesso, D.; Karamanos, N.K. Hyaluronan: Molecular Size-Dependent Signaling and Biological Functions in Inflammation and Cancer. FEBS J. 2019, 286, 2883–2908. [Google Scholar] [CrossRef]
- Toole, B.P. Hyaluronan: From Extracellular Glue to Pericellular Cue. Nat. Rev. Cancer 2004, 4, 528–539. [Google Scholar] [CrossRef]
- Sato, N.; Kohi, S.; Hirata, K.; Goggins, M. Role of Hyaluronan in Pancreatic Cancer Biology and Therapy: Once Again in the Spotlight. Cancer Sci. 2016, 107, 569–575. [Google Scholar] [CrossRef]
- Kim, P.K.; Halbrook, C.J.; Kerk, S.A.; Radyk, M.; Wisner, S.; Kremer, D.M.; Sajjakulnukit, P.; Andren, A.; Hou, S.W.; Trivedi, A.; et al. Hyaluronic Acid Fuels Pancreatic Cancer Cell Growth. eLife 2021, 10, e62645. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan Impairs Vascular Function and Drug Delivery in a Mouse Model of Pancreatic Cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef] [PubMed]
- DuFort, C.C.; DelGiorno, K.E.; Carlson, M.A.; Osgood, R.J.; Zhao, C.; Huang, Z.; Thompson, C.B.; Connor, R.J.; Thanos, C.D.; Scott Brockenbrough, J.; et al. Interstitial Pressure in Pancreatic Ductal Adenocarcinoma Is Dominated by a Gel-Fluid Phase. Biophys. J. 2016, 110, 2106–2119. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; McDonough, S.L.; Philip, P.A.; Hingorani, S.R.; Lacy, J.; Kortmansky, J.S.; Thumar, J.; Chiorean, E.G.; Shields, A.F.; Behl, D.; et al. Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase Versus FOLFIRINOX Alone in Patients With Metastatic Pancreatic Adenocarcinoma: SWOG S1313. J. Clin. Oncol. 2019, 37, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Tempero, M.A.; Sigal, D.; Oh, D.-Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa With Nab-Paclitaxel Plus Gemcitabine for Patients With Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2020, 38, 3185–3194. [Google Scholar] [CrossRef]
- Hakim, N.; Patel, R.; Devoe, C.; Saif, M.W. Why HALO 301 Failed and Implications for Treatment of Pancreatic Cancer. Pancreas 2019, 3, e1–e4. [Google Scholar] [CrossRef]
- Matsusaki, M.; Komeda, M.; Mura, S.; Tanaka, H.Y.; Kano, M.R.; Couvreur, P.; Akashi, M. Desmoplastic Reaction in 3D-Pancreatic Cancer Tissues Suppresses Molecular Permeability. Adv. Healthc. Mater. 2017, 6, 1700057. [Google Scholar] [CrossRef]
- Egeblad, M.; Rasch, M.G.; Weaver, V.M. Dynamic Interplay between the Collagen Scaffold and Tumor Evolution. Curr. Opin. Cell Biol. 2010, 22, 697–706. [Google Scholar] [CrossRef]
- Leitinger, B. Transmembrane Collagen Receptors. Annu. Rev. Cell Dev. Biol. 2011, 27, 265–290. [Google Scholar] [CrossRef]
- Ricard-Blum, S.; Salza, R. Matricryptins and Matrikines: Biologically Active Fragments of the Extracellular Matrix. Exp. Dermatol. 2014, 23, 457–463. [Google Scholar] [CrossRef]
- Olivares, O.; Mayers, J.R.; Gouirand, V.; Torrence, M.E.; Gicquel, T.; Borge, L.; Lac, S.; Roques, J.; Lavaut, M.-N.; Berthezène, P.; et al. Collagen-Derived Proline Promotes Pancreatic Ductal Adenocarcinoma Cell Survival under Nutrient Limited Conditions. Nat. Commun. 2017, 8, 16031. [Google Scholar] [CrossRef] [PubMed]
- Nicolas-Boluda, A.; Vaquero, J.; Vimeux, L.; Guilbert, T.; Barrin, S.; Kantari-Mimoun, C.; Ponzo, M.; Renault, G.; Deptula, P.; Pogoda, K.; et al. Tumor Stiffening Reversion through Collagen Crosslinking Inhibition Improves T Cell Migration and Anti-PD-1 Treatment. eLife 2021, 10, e58688. [Google Scholar] [CrossRef] [PubMed]
- Perez, V.M.; Kearney, J.F.; Yeh, J.J. The PDAC Extracellular Matrix: A Review of the ECM Protein Composition, Tumor Cell Interaction, and Therapeutic Strategies. Front. Oncol. 2021, 11, 751311. [Google Scholar] [CrossRef] [PubMed]
- Zinger, A.; Koren, L.; Adir, O.; Poley, M.; Alyan, M.; Yaari, Z.; Noor, N.; Krinsky, N.; Simon, A.; Gibori, H.; et al. Collagenase Nanoparticles Enhance the Penetration of Drugs into Pancreatic Tumors. ACS Nano 2019, 13, 11008–11021. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kim, J.; Yang, S.; Wang, H.; Wu, C.-J.; Sugimoto, H.; LeBleu, V.S.; Kalluri, R. Type I Collagen Deletion in αSMA+ Myofibroblasts Augments Immune Suppression and Accelerates Progression of Pancreatic Cancer. Cancer Cell 2021, 39, 548–565.e6. [Google Scholar] [CrossRef]
- Chung, H.J.; Steplewski, A.; Chung, K.Y.; Uitto, J.; Fertala, A. Collagen Fibril Formation: A New Target to Limit Fibrosis. J. Biol. Chem. 2008, 283, 25879–25886. [Google Scholar] [CrossRef]
- Prockop, D.J.; Fertala, A. Inhibition of the Self-Assembly of Collagen I into Fibrils with Synthetic Peptides. Demonstration That Assembly Is Driven by Specific Binding Sites on the Monomers. J. Biol. Chem. 1998, 273, 15598–15604. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, S.; Tavormina, J.; Tampe, D.; Zeisberg, M.; Wang, H.; Mahadevan, K.K.; Wu, C.-J.; Sugimoto, H.; Chang, C.-C.; et al. Oncogenic Collagen I Homotrimers from Cancer Cells Bind to α3β1 Integrin and Impact Tumor Microbiome and Immunity to Promote Pancreatic Cancer. Cancer Cell 2022, 40, 818–834.e9. [Google Scholar] [CrossRef]
- Ito, S.; Nagata, K. Biology of Hsp47 (Serpin H1), a Collagen-Specific Molecular Chaperone. Semin. Cell Dev. Biol. 2017, 62, 142–151. [Google Scholar] [CrossRef]
- Maitra, A.; Iacobuzio-Donahue, C.; Rahman, A.; Sohn, T.A.; Argani, P.; Meyer, R.; Yeo, C.J.; Cameron, J.L.; Goggins, M.; Kern, S.E.; et al. Immunohistochemical Validation of a Novel Epithelial and a Novel Stromal Marker of Pancreatic Ductal Adenocarcinoma Identified by Global Expression Microarrays: Sea Urchin Fascin Homolog and Heat Shock Protein 47. Am. J. Clin. Pathol. 2002, 118, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Li, Y.; Xu, Y.; Zhao, X.; Zhang, Y.; Yang, X.; Wang, Y.; Zhao, R.; Anderson, G.J.; Zhao, Y.; et al. Reversal of Pancreatic Desmoplasia by Re-Educating Stellate Cells with a Tumour Microenvironment-Activated Nanosystem. Nat. Commun. 2018, 9, 3390. [Google Scholar] [CrossRef] [PubMed]
- Ishiwatari, H.; Sato, Y.; Murase, K.; Yoneda, A.; Fujita, R.; Nishita, H.; Birukawa, N.K.; Hayashi, T.; Sato, T.; Miyanishi, K.; et al. Treatment of Pancreatic Fibrosis with SiRNA against a Collagen-Specific Chaperone in Vitamin A-Coupled Liposomes. Gut 2013, 62, 1328–1339. [Google Scholar] [CrossRef] [PubMed]
- Thomson, C.A.; Atkinson, H.M.; Ananthanarayanan, V.S. Identification of Small Molecule Chemical Inhibitors of the Collagen-Specific Chaperone Hsp47. J. Med. Chem. 2005, 48, 1680–1684. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Ogawa, K.; Takeuchi, K.; Takagi, M.; Yoshida, M.; Hirokawa, T.; Hirayama, S.; Shin-Ya, K.; Shimada, I.; Doi, T.; et al. A Small-Molecule Compound Inhibits a Collagen-Specific Molecular Chaperone and Could Represent a Potential Remedy for Fibrosis. J. Biol. Chem. 2017, 292, 20076–20085. [Google Scholar] [CrossRef]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.T.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef]
- Miller, B.W.; Morton, J.P.; Pinese, M.; Saturno, G.; Jamieson, N.B.; McGhee, E.; Timpson, P.; Leach, J.; McGarry, L.; Shanks, E.; et al. Targeting the LOX/Hypoxia Axis Reverses Many of the Features That Make Pancreatic Cancer Deadly: Inhibition of LOX Abrogates Metastasis and Enhances Drug Efficacy. EMBO Mol. Med. 2015, 7, 1063–1076. [Google Scholar] [CrossRef]
- Nabavizadeh, A.; Payen, T.; Iuga, A.C.; Sagalovskiy, I.R.; Desrouilleres, D.; Saharkhiz, N.; Palermo, C.F.; Sastra, S.A.; Oberstein, P.E.; Rosario, V.; et al. Noninvasive Young’s Modulus Visualization of Fibrosis Progression and Delineation of Pancreatic Ductal Adenocarcinoma (PDAC) Tumors Using Harmonic Motion Elastography (HME) In Vivo. Theranostics 2020, 10, 4614–4626. [Google Scholar] [CrossRef]
- Lee, J.; Condello, S.; Yakubov, B.; Emerson, R.; Caperell-Grant, A.; Hitomi, K.; Xie, J.; Matei, D. Tissue Transglutaminase Mediated Tumor-Stroma Interaction Promotes Pancreatic Cancer Progression. Clin. Cancer Res. 2015, 21, 4482–4493. [Google Scholar] [CrossRef]
- Song, M.; Hwang, H.; Im, C.Y.; Kim, S.-Y. Recent Progress in the Development of Transglutaminase 2 (TGase2) Inhibitors. J. Med. Chem. 2017, 60, 554–567. [Google Scholar] [CrossRef]
- Sun, Z.; Guo, S.S.; Fässler, R. Integrin-Mediated Mechanotransduction. J. Cell Biol. 2016, 215, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Drifka, C.R.; Loeffler, A.G.; Mathewson, K.; Keikhosravi, A.; Eickhoff, J.C.; Liu, Y.; Weber, S.M.; Kao, W.J.; Eliceiri, K.W. Highly Aligned Stromal Collagen Is a Negative Prognostic Factor Following Pancreatic Ductal Adenocarcinoma Resection. Oncotarget 2016, 7, 76197–76213. [Google Scholar] [CrossRef] [PubMed]
- Drifka, C.R.; Tod, J.; Loeffler, A.G.; Liu, Y.; Thomas, G.J.; Eliceiri, K.W.; Kao, W.J. Periductal Stromal Collagen Topology of Pancreatic Ductal Adenocarcinoma Differs from That of Normal and Chronic Pancreatitis. Mod. Pathol. 2015, 28, 1470–1480. [Google Scholar] [CrossRef]
- Park, D.; Wershof, E.; Boeing, S.; Labernadie, A.; Jenkins, R.P.; George, S.; Trepat, X.; Bates, P.A.; Sahai, E. Extracellular Matrix Anisotropy Is Determined by TFAP2C-Dependent Regulation of Cell Collisions. Nat. Mater. 2020, 19, 227–238. [Google Scholar] [CrossRef]
- Wei, D.; Cheng, X.; Du, C.; Wang, Y.; Sun, J.; Li, C.; Wu, J.; Tian, X.; Zhao, Y.; Nie, G.; et al. Stroma-Targeted Nanoparticles That Remodel Stromal Alignment to Enhance Drug Delivery and Improve the Antitumor Efficacy of Nab-Paclitaxel in Pancreatic Ductal Adenocarcinoma Models. Nano Today 2022, 45, 101533. [Google Scholar] [CrossRef]
- DuFort, C.C.; Paszek, M.J.; Weaver, V.M. Balancing Forces: Architectural Control of Mechanotransduction. Nat. Rev. Mol. Cell Biol. 2011, 12, 308–319. [Google Scholar] [CrossRef]
- Vogel, V. Mechanotransduction Involving Multimodular Proteins: Converting Force into Biochemical Signals. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 459–488. [Google Scholar] [CrossRef]
- Smith, M.L.; Gourdon, D.; Little, W.C.; Kubow, K.E.; Eguiluz, R.A.; Luna-Morris, S.; Vogel, V. Force-Induced Unfolding of Fibronectin in the Extracellular Matrix of Living Cells. PLOS Biol. 2007, 5, e268. [Google Scholar] [CrossRef]
- Baneyx, G.; Baugh, L.; Vogel, V. Fibronectin Extension and Unfolding within Cell Matrix Fibrils Controlled by Cytoskeletal Tension. Proc. Natl. Acad. Sci. USA 2002, 99, 5139–5143. [Google Scholar] [CrossRef]
- Klotzsch, E.; Smith, M.L.; Kubow, K.E.; Muntwyler, S.; Little, W.C.; Beyeler, F.; Gourdon, D.; Nelson, B.J.; Vogel, V. Fibronectin Forms the Most Extensible Biological Fibers Displaying Switchable Force-Exposed Cryptic Binding Sites. Proc. Natl. Acad. Sci. USA 2009, 106, 18267–18272. [Google Scholar] [CrossRef]
- Arnoldini, S.; Moscaroli, A.; Chabria, M.; Hilbert, M.; Hertig, S.; Schibli, R.; Béhé, M.; Vogel, V. Novel Peptide Probes to Assess the Tensional State of Fibronectin Fibers in Cancer. Nat. Commun. 2017, 8, 1793. [Google Scholar] [CrossRef]
- Cao, L.; Zeller, M.K.; Fiore, V.F.; Strane, P.; Bermudez, H.; Barker, T.H. Phage-Based Molecular Probes That Discriminate Force-Induced Structural States of Fibronectin In Vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 7251–7256. [Google Scholar] [CrossRef]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular Matrix Degradation and Remodeling in Development and Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Morad, G.; Jedinak, A.; Moses, M.A. Metalloproteinases and Their Roles in Human Cancer. Anat. Rec. 2020, 303, 1557–1572. [Google Scholar] [CrossRef] [PubMed]
- Giannandrea, M.; Parks, W.C. Diverse Functions of Matrix Metalloproteinases during Fibrosis. Dis. Model. Mech. 2014, 7, 193–203. [Google Scholar] [CrossRef]
- Su, H.; Yang, F.; Fu, R.; Trinh, B.; Sun, N.; Liu, J.; Kumar, A.; Baglieri, J.; Siruno, J.; Le, M.; et al. Collagenolysis-Dependent DDR1 Signalling Dictates Pancreatic Cancer Outcome. Nature 2022, 610, 366–372. [Google Scholar] [CrossRef]
- Taipale, J.; Keski-Oja, J. Growth Factors in the Extracellular Matrix. FASEB J. 1997, 11, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M.; Barker, T.H.; Gibbons, D.L.; Kurie, J.M. The Fibrotic Tumor Stroma. J. Clin. Investig. 2018, 128, 16–25. [Google Scholar] [CrossRef]
- Slapak, E.J.; Duitman, J.; Tekin, C.; Bijlsma, M.F.; Spek, C.A. Matrix Metalloproteases in Pancreatic Ductal Adenocarcinoma: Key Drivers of Disease Progression? Biology 2020, 9, 80. [Google Scholar] [CrossRef]
- Winer, A.; Adams, S.; Mignatti, P. Matrix Metalloproteinase Inhibitors in Cancer Therapy: Turning Past Failures into Future Successes. Mol. Cancer Ther. 2018, 17, 1147–1155. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, L.; Wan, D.; Zhou, L.; Zheng, S.; Lin, S.; Qiao, Y. Extracellular Matrix and Its Therapeutic Potential for Cancer Treatment. Signal Transduct. Target. Ther. 2021, 6, 153. [Google Scholar] [CrossRef]
- Mantoni, T.S.; Lunardi, S.; Al-Assar, O.; Masamune, A.; Brunner, T.B. Pancreatic Stellate Cells Radioprotect Pancreatic Cancer Cells through β1-Integrin Signaling. Cancer Res. 2011, 71, 3453–3458. [Google Scholar] [CrossRef] [PubMed]
- Martins Cavaco, A.C.; Rezaei, M.; Caliandro, M.F.; Martins Lima, A.; Stehling, M.; Dhayat, S.A.; Haier, J.; Brakebusch, C.; Eble, J.A. The Interaction between Laminin-332 and α3β1 Integrin Determines Differentiation and Maintenance of CAFs, and Supports Invasion of Pancreatic Duct Adenocarcinoma Cells. Cancers 2018, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Kuninty, P.R.; Bansal, R.; De Geus, S.W.L.; Mardhian, D.F.; Schnittert, J.; van Baarlen, J.; Storm, G.; Bijlsma, M.F.; van Laarhoven, H.W.; Metselaar, J.M.; et al. ITGA5 Inhibition in Pancreatic Stellate Cells Attenuates Desmoplasia and Potentiates Efficacy of Chemotherapy in Pancreatic Cancer. Sci. Adv. 2019, 5, eaax2770. [Google Scholar] [CrossRef]
- Franco-Barraza, J.; Francescone, R.; Luong, T.; Shah, N.; Madhani, R.; Cukierman, G.; Dulaimi, E.; Devarajan, K.; Egleston, B.L.; Nicolas, E.; et al. Matrix-Regulated Integrin αvβ5 Maintains α5β1-Dependent Desmoplastic Traits Prognostic of Neoplastic Recurrence. eLife 2017, 6, e20600. [Google Scholar] [CrossRef] [PubMed]
- Turaga, R.C.; Sharma, M.; Mishra, F.; Krasinskas, A.; Yuan, Y.; Yang, J.J.; Wang, S.; Liu, C.; Li, S.; Liu, Z.-R. Modulation of Cancer-Associated Fibrotic Stroma by an Integrin αvβ3 Targeting Protein for Pancreatic Cancer Treatment. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 161–179. [Google Scholar] [CrossRef]
- Horioka, K.; Ohuchida, K.; Sada, M.; Zheng, B.; Moriyama, T.; Fujita, H.; Manabe, T.; Ohtsuka, T.; Shimamoto, M.; Miyazaki, T.; et al. Suppression of CD51 in Pancreatic Stellate Cells Inhibits Tumor Growth by Reducing Stroma and Altering Tumor-Stromal Interaction in Pancreatic Cancer. Int. J. Oncol. 2016, 48, 1499–1508. [Google Scholar] [CrossRef]
- Schnittert, J.; Bansal, R.; Mardhian, D.F.; van Baarlen, J.; Östman, A.; Prakash, J. Integrin α11 in Pancreatic Stellate Cells Regulates Tumor Stroma Interaction in Pancreatic Cancer. FASEB J. 2019, 33, 6609–6621. [Google Scholar] [CrossRef]
- Ley, K.; Rivera-Nieves, J.; Sandborn, W.J.; Shattil, S. Integrin-Based Therapeutics: Biological Basis, Clinical Use and New Drugs. Nat. Rev. Drug Discov. 2016, 15, 173–183. [Google Scholar] [CrossRef]
- Slack, R.J.; Macdonald, S.J.F.; Roper, J.A.; Jenkins, R.G.; Hatley, R.J.D. Emerging Therapeutic Opportunities for Integrin Inhibitors. Nat. Rev. Drug Discov. 2022, 21, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.-Y.; Li, J.; Xie, Y.; Zhu, J.; Nguyen, T.T.H.; Zhang, Y.; Zhu, J.; Springer, T.A. A General Chemical Principle for Creating Closure-Stabilizing Integrin Inhibitors. Cell 2022, 185, 3533–3550.e27. [Google Scholar] [CrossRef]
- Tschumperlin, D.J.; Ligresti, G.; Hilscher, M.B.; Shah, V.H. Mechanosensing and Fibrosis. J. Clin. Investig. 2018, 128, 74–84. [Google Scholar] [CrossRef]
- Wen, D.; Gao, Y.; Ho, C.; Yu, L.; Zhang, Y.; Lyu, G.; Hu, D.; Li, Q.; Zhang, Y. Focusing on Mechanoregulation Axis in Fibrosis: Sensing, Transduction and Effecting. Front. Mol. Biosci. 2022, 9, 804680. [Google Scholar] [CrossRef] [PubMed]
- Tschumperlin, D.J.; Lagares, D. Mechano-Therapeutics: Targeting Mechanical Signaling in Fibrosis and Tumor Stroma. Pharmacol. Ther. 2020, 212, 107575. [Google Scholar] [CrossRef]
- Schiller, H.B.; Fässler, R. Mechanosensitivity and Compositional Dynamics of Cell–Matrix Adhesions. EMBO Rep. 2013, 14, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Horton, E.R.; Byron, A.; Askari, J.A.; Ng, D.H.J.; Millon-Frémillon, A.; Robertson, J.; Koper, E.J.; Paul, N.R.; Warwood, S.; Knight, D.; et al. Definition of a Consensus Integrin Adhesome and Its Dynamics during Adhesion Complex Assembly and Disassembly. Nat. Cell Biol. 2015, 17, 1577–1587. [Google Scholar] [CrossRef]
- Zhao, W.; Ajani, J.A.; Sushovan, G.; Ochi, N.; Hwang, R.; Hafley, M.; Johnson, R.L.; Bresalier, R.S.; Logsdon, C.D.; Zhang, Z.; et al. Galectin-3 Mediates Tumor Cell-Stroma Interactions by Activating Pancreatic Stellate Cells to Produce Cytokines via Integrin Signaling. Gastroenterology 2018, 154, 1524–1537.e6. [Google Scholar] [CrossRef]
- Yoshida, N.; Masamune, A.; Hamada, S.; Kikuta, K.; Takikawa, T.; Motoi, F.; Unno, M.; Shimosegawa, T. Kindlin-2 in Pancreatic Stellate Cells Promotes the Progression of Pancreatic Cancer. Cancer Lett. 2017, 390, 103–114. [Google Scholar] [CrossRef]
- Zaghdoudi, S.; Decaup, E.; Belhabib, I.; Samain, R.; Cassant-Sourdy, S.; Rochotte, J.; Brunel, A.; Schlaepfer, D.; Cros, J.; Neuzillet, C.; et al. FAK Activity in Cancer-Associated Fibroblasts Is a Prognostic Marker and a Druggable Key Metastatic Player in Pancreatic Cancer. EMBO Mol. Med. 2020, 12, e12010. [Google Scholar] [CrossRef]
- Tu, K.; Li, J.; Verma, V.K.; Liu, C.; Billadeau, D.D.; Lamprecht, G.; Xiang, X.; Guo, L.; Dhanasekaran, R.; Roberts, L.R.; et al. Vasodilator-Stimulated Phosphoprotein Promotes Activation of Hepatic Stellate Cells by Regulating Rab11-Dependent Plasma Membrane Targeting of Transforming Growth Factor Beta Receptors. Hepatology 2015, 61, 361–374. [Google Scholar] [CrossRef]
- Scaife, C.L.; Shea, J.; Emerson, L.; Boucher, K.; Firpo, M.A.; Beckerle, M.C.; Mulvihill, S.J. Prognostic Significance of PINCH Signalling in Human Pancreatic Ductal Adenocarcinoma. HPB 2010, 12, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting Focal Adhesion Kinase Renders Pancreatic Cancers Responsive to Checkpoint Immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Murphy, K.J.; Reed, D.A.; Vennin, C.; Conway, J.R.W.; Nobis, M.; Yin, J.X.; Chambers, C.R.; Pereira, B.A.; Lee, V.; Filipe, E.C.; et al. Intravital Imaging Technology Guides FAK-Mediated Priming in Pancreatic Cancer Precision Medicine According to Merlin Status. Sci. Adv. 2021, 7, eabh0363. [Google Scholar] [CrossRef]
- Yamada, T.; Tateishi, R.; Iwai, M.; Tanaka, M.; Ijichi, H.; Sano, M.; Koike, K.; Todo, T. Overcoming Resistance of Stroma-Rich Pancreatic Cancer with Focal Adhesion Kinase Inhibitor Combined with G47Δ and Immune Checkpoint Inhibitors. Mol. Ther. Oncolytics 2022, 7, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Brakebusch, C.; Fässler, R. The Integrin–Actin Connection, an Eternal Love Affair. EMBO J. 2003, 22, 2324–2333. [Google Scholar] [CrossRef]
- Hinz, B.; Celetta, G.; Tomasek, J.J.; Gabbiani, G.; Chaponnier, C. Alpha-Smooth Muscle Actin Expression Upregulates Fibroblast Contractile Activity. Mol. Biol. Cell 2001, 12, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Dugina, V.; Ballestrem, C.; Wehrle-Haller, B.; Chaponnier, C. α-Smooth Muscle Actin Is Crucial for Focal Adhesion Maturation in Myofibroblasts. Mol. Biol. Cell 2003, 14, 2508–2519. [Google Scholar] [CrossRef] [PubMed]
- Masamune, A.; Kikuta, K.; Satoh, M.; Satoh, K.; Shimosegawa, T. Rho Kinase Inhibitors Block Activation of Pancreatic Stellate Cells. Br. J. Pharmacol. 2003, 140, 1292–1302. [Google Scholar] [CrossRef]
- Rath, N.; Morton, J.P.; Julian, L.; Helbig, L.; Kadir, S.; McGhee, E.J.; Anderson, K.I.; Kalna, G.; Mullin, M.; Pinho, A.V.; et al. ROCK Signaling Promotes Collagen Remodeling to Facilitate Invasive Pancreatic Ductal Adenocarcinoma Tumor Cell Growth. EMBO Mol. Med. 2017, 9, 198–218. [Google Scholar] [CrossRef]
- Rath, N.; Munro, J.; Cutiongco, M.F.; Jagiełło, A.; Gadegaard, N.; McGarry, L.; Unbekandt, M.; Michalopoulou, E.; Kamphorst, J.J.; Sumpton, D.; et al. Rho Kinase Inhibition by AT13148 Blocks Pancreatic Ductal Adenocarcinoma Invasion and Tumor Growth. Cancer Res. 2018, 78, 3321–3336. [Google Scholar] [CrossRef] [PubMed]
- Vennin, C.; Chin, V.T.; Warren, S.C.; Lucas, M.C.; Herrmann, D.; Magenau, A.; Melenec, P.; Walters, S.N.; Del Monte-Nieto, G.; Conway, J.R.W.; et al. Transient Tissue Priming via ROCK Inhibition Uncouples Pancreatic Cancer Progression, Sensitivity to Chemotherapy, and Metastasis. Sci. Transl. Med. 2017, 9, eaai8504. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.I.; Vendramini-Costa, D.B.; Francescone, R.; Luong, T.; Franco-Barraza, J.; Shah, N.; Gardiner, J.C.; Nicolas, E.; Raghavan, K.S.; Cukierman, E. Palladin Isoforms 3 and 4 Regulate Cancer-Associated Fibroblast pro-Tumor Functions in Pancreatic Ductal Adenocarcinoma. Sci. Rep. 2021, 11, 3802. [Google Scholar] [CrossRef] [PubMed]
- Sato, D.; Tsuchikawa, T.; Mitsuhashi, T.; Hatanaka, Y.; Marukawa, K.; Morooka, A.; Nakamura, T.; Shichinohe, T.; Matsuno, Y.; Hirano, S. Stromal Palladin Expression Is an Independent Prognostic Factor in Pancreatic Ductal Adenocarcinoma. PLoS ONE 2016, 11, e0152523. [Google Scholar] [CrossRef]
- Moll, S.; Desmoulière, A.; Moeller, M.J.; Pache, J.-C.; Badi, L.; Arcadu, F.; Richter, H.; Satz, A.; Uhles, S.; Cavalli, A.; et al. DDR1 Role in Fibrosis and Its Pharmacological Targeting. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118474. [Google Scholar] [CrossRef]
- Leitinger, B. Discoidin Domain Receptor Functions in Physiological and Pathological Conditions. Int. Rev. Cell Mol. Biol. 2014, 310, 39–87. [Google Scholar] [CrossRef]
- Leitinger, B.; Hohenester, E. Mammalian Collagen Receptors. Matrix Biol. 2007, 26, 146–155. [Google Scholar] [CrossRef]
- Bansod, S.; Saifi, M.A.; Godugu, C. Inhibition of Discoidin Domain Receptors by Imatinib Prevented Pancreatic Fibrosis Demonstrated in Experimental Chronic Pancreatitis Model. Sci. Rep. 2021, 11, 12894. [Google Scholar] [CrossRef]
- Ruggeri, J.M.; Franco-Barraza, J.; Sohail, A.; Zhang, Y.; Long, D.; Pasca di Magliano, M.; Cukierman, E.; Fridman, R.; Crawford, H.C. Discoidin Domain Receptor 1 (DDR1) Is Necessary for Tissue Homeostasis in Pancreatic Injury and Pathogenesis of Pancreatic Ductal Adenocarcinoma. Am. J. Pathol. 2020, 190, 1735–1751. [Google Scholar] [CrossRef]
- Hua, S.; de Matos, M.B.C.; Metselaar, J.M.; Storm, G. Current Trends and Challenges in the Clinical Translation of Nanoparticulate Nanomedicines: Pathways for Translational Development and Commercialization. Front. Pharmacol. 2018, 9, 790. [Google Scholar] [CrossRef]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer Nanomedicine: Progress, Challenges and Opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Metselaar, J.M.; Lammers, T. Challenges in Nanomedicine Clinical Translation. Drug Deliv. Transl. Res. 2020, 10, 721–725. [Google Scholar] [CrossRef]
- Anchordoquy, T.J.; Barenholz, Y.; Boraschi, D.; Chorny, M.; Decuzzi, P.; Dobrovolskaia, M.A.; Farhangrazi, Z.S.; Farrell, D.; Gabizon, A.; Ghandehari, H.; et al. Mechanisms and Barriers in Cancer Nanomedicine: Addressing Challenges, Looking for Solutions. ACS Nano 2017, 11, 12–18. [Google Scholar] [CrossRef]
- Souri, M.; Soltani, M.; Moradi Kashkooli, F.; Kiani Shahvandi, M.; Chiani, M.; Shariati, F.S.; Mehrabi, M.R.; Munn, L.L. Towards Principled Design of Cancer Nanomedicine to Accelerate Clinical Translation. Mater. Today Bio 2022, 13, 100208. [Google Scholar] [CrossRef] [PubMed]
- Stater, E.P.; Sonay, A.Y.; Hart, C.; Grimm, J. The Ancillary Effects of Nanoparticles and Their Implications for Nanomedicine. Nat. Nanotechnol. 2021, 16, 1180–1194. [Google Scholar] [CrossRef]
- Cabral, H.; Matsumoto, Y.; Mizuno, K.; Chen, Q.; Murakami, M.; Kimura, M.; Terada, Y.; Kano, M.R.; Miyazono, K.; Uesaka, M.; et al. Accumulation of Sub-100 Nm Polymeric Micelles in Poorly Permeable Tumours Depends on Size. Nat. Nanotechnol. 2011, 6, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Hayashi, K.; Toh, K.; Kim, H.J.; Liu, X.; Chaya, H.; Fukushima, S.; Katsushima, K.; Kondo, Y.; Uchida, S.; et al. In Vivo Rendezvous of Small Nucleic Acid Drugs with Charge-Matched Block Catiomers to Target Cancers. Nat. Commun. 2019, 10, 1894. [Google Scholar] [CrossRef] [PubMed]
- Tockary, T.A.; Foo, W.; Dirisala, A.; Chen, Q.; Uchida, S.; Osawa, S.; Mochida, Y.; Liu, X.; Kinoh, H.; Cabral, H.; et al. Single-Stranded DNA-Packaged Polyplex Micelle as Adeno-Associated-Virus-Inspired Compact Vector to Systemically Target Stroma-Rich Pancreatic Cancer. ACS Nano 2019, 13, 12732–12742. [Google Scholar] [CrossRef]
- Engin, A.B.; Nikitovic, D.; Neagu, M.; Henrich-Noack, P.; Docea, A.O.; Shtilman, M.I.; Golokhvast, K.; Tsatsakis, A.M. Mechanistic Understanding of Nanoparticles’ Interactions with Extracellular Matrix: The Cell and Immune System. Part. Fibre Toxicol. 2017, 14, 22. [Google Scholar] [CrossRef]
- Tao, Z.; Muzumdar, M.D.; Detappe, A.; Huang, X.; Xu, E.S.; Yu, Y.; Mouhieddine, T.H.; Song, H.; Jacks, T.; Ghoroghchian, P.P. Differences in Nanoparticle Uptake in Transplanted and Autochthonous Models of Pancreatic Cancer. Nano Lett. 2018, 18, 2195–2208. [Google Scholar] [CrossRef]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and Challenges towards Targeted Delivery of Cancer Therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Liu, R.; Zhou, Y.; Gao, H. Size-Tunable Strategies for a Tumor Targeted Drug Delivery System. ACS Cent. Sci. 2020, 6, 100–116. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Nie, G.; Meng, H.; Xia, T.; Nel, A.; Zhao, Y. Physicochemical Properties Determine Nanomaterial Cellular Uptake, Transport, and Fate. Acc. Chem. Res. 2013, 46, 622–631. [Google Scholar] [CrossRef]
- Foroozandeh, P.; Aziz, A.A. Insight into Cellular Uptake and Intracellular Trafficking of Nanoparticles. Nanoscale Res. Lett. 2018, 13, 339. [Google Scholar] [CrossRef]
- Sabourian, P.; Yazdani, G.; Ashraf, S.S.; Frounchi, M.; Mashayekhan, S.; Kiani, S.; Kakkar, A. Effect of Physico-Chemical Properties of Nanoparticles on Their Intracellular Uptake. Int. J. Mol. Sci. 2020, 21, 8019. [Google Scholar] [CrossRef]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.-W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338.e6. [Google Scholar] [CrossRef]
- Poh, A.R.; Ernst, M. Tumor-Associated Macrophages in Pancreatic Ductal Adenocarcinoma: Therapeutic Opportunities and Clinical Challenges. Cancers 2021, 13, 2860. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Liu, Q.; Liao, Q. Tumor-Associated Macrophages in Pancreatic Ductal Adenocarcinoma: Origin, Polarization, Function, and Reprogramming. Front. Cell Dev. Biol. 2021, 8, 607209. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a Strategy for Improving Nanoparticle-Based Drug and Gene Delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [PubMed]
- Lech, M.; Anders, H.-J. Macrophages and Fibrosis: How Resident and Infiltrating Mononuclear Phagocytes Orchestrate All Phases of Tissue Injury and Repair. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Long, K.B.; Gladney, W.L.; Tooker, G.M.; Graham, K.; Fraietta, J.A.; Beatty, G.L. IFN-γ and CCL2 Cooperate to Redirect Tumor-Infiltrating Monocytes to Degrade Fibrosis and Enhance Chemotherapy Efficacy in Pancreatic Carcinoma. Cancer Discov. 2016, 6, 400–413. [Google Scholar] [CrossRef]
- Toy, R.; Peiris, P.M.; Ghaghada, K.B.; Karathanasis, E. Shaping Cancer Nanomedicine: The Effect of Particle Shape on the In Vivo Journey of Nanoparticles. Nanomed. 2014, 9, 121–134. [Google Scholar] [CrossRef]
- Liu, Y.; Tan, J.; Thomas, A.; Ou-Yang, D.; Muzykantov, V.R. The Shape of Things to Come: Importance of Design in Nanotechnology for Drug Delivery. Ther. Deliv. 2012, 3, 181–194. [Google Scholar] [CrossRef]
- Wang, H.-X.; Zuo, Z.-Q.; Du, J.-Z.; Wang, Y.-C.; Sun, R.; Cao, Z.-T.; Ye, X.-D.; Wang, J.-L.; Leong, K.W.; Wang, J. Surface Charge Critically Affects Tumor Penetration and Therapeutic Efficacy of Cancer Nanomedicines. Nano Today 2016, 11, 133–144. [Google Scholar] [CrossRef]
- Zhang, P.; Chen, D.; Li, L.; Sun, K. Charge Reversal Nano-Systems for Tumor Therapy. J. Nanobiotechnol. 2022, 20, 31. [Google Scholar] [CrossRef]
- Guo, P.; Liu, D.; Subramanyam, K.; Wang, B.; Yang, J.; Huang, J.; Auguste, D.T.; Moses, M.A. Nanoparticle Elasticity Directs Tumor Uptake. Nat. Commun. 2018, 9, 130. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.M.; Costa, D.F.; Jagielska, A.; Van Vliet, K.J.; Hammond, P.T. Stiffness of Targeted Layer-by-Layer Nanoparticles Impacts Elimination Half-Life, Tumor Accumulation, and Tumor Penetration. Proc. Natl. Acad. Sci. USA 2021, 118, e2104826118. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Feron, O.; Préat, V. To Exploit the Tumor Microenvironment: Passive and Active Tumor Targeting of Nanocarriers for Anti-Cancer Drug Delivery. J. Control. Release 2010, 148, 135–146. [Google Scholar] [CrossRef]
- Rajendran, L.; Knölker, H.-J.; Simons, K. Subcellular Targeting Strategies for Drug Design and Delivery. Nat. Rev. Drug Discov. 2010, 9, 29–42. [Google Scholar] [CrossRef]
- Saminathan, A.; Zajac, M.; Anees, P.; Krishnan, Y. Organelle-Level Precision with next-Generation Targeting Technologies. Nat. Rev. Mater. 2022, 7, 355–371. [Google Scholar] [CrossRef]
- Pearce, A.K.; O’Reilly, R.K. Insights into Active Targeting of Nanoparticles in Drug Delivery: Advances in Clinical Studies and Design Considerations for Cancer Nanomedicine. Bioconjug. Chem. 2019, 30, 2300–2311. [Google Scholar] [CrossRef] [PubMed]
- Sorbara, M.; Cordelier, P.; Bery, N. Antibody-Based Approaches to Target Pancreatic Tumours. Antibodies 2022, 11, 47. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Maier, S.H.; Li, P.; Peterhansl, J.; Belka, C.; Mayerle, J.; Mahajan, U.M. Aptamers: A Novel Targeted Theranostic Platform for Pancreatic Ductal Adenocarcinoma. Radiat. Oncol. 2020, 15, 189. [Google Scholar] [CrossRef]
- Chen, L.; Hong, W.; Ren, W.; Xu, T.; Qian, Z.; He, Z. Recent Progress in Targeted Delivery Vectors Based on Biomimetic Nanoparticles. Signal Transduct. Target. Ther. 2021, 6, 225. [Google Scholar] [CrossRef]
- Murphy, D.E.; de Jong, O.G.; Brouwer, M.; Wood, M.J.; Lavieu, G.; Schiffelers, R.M.; Vader, P. Extracellular Vesicle-Based Therapeutics: Natural versus Engineered Targeting and Trafficking. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef] [PubMed]
- MacCuaig, W.M.; Fouts, B.L.; McNally, M.W.; Grizzle, W.E.; Chuong, P.; Samykutty, A.; Mukherjee, P.; Li, M.; Jasinski, J.B.; Behkam, B.; et al. Active Targeting Significantly Outperforms Nanoparticle Size in Facilitating Tumor-Specific Uptake in Orthotopic Pancreatic Cancer. ACS Appl. Mater. Interfaces 2021, 13, 49614–49630. [Google Scholar] [CrossRef]
- Mashayekhi, V.; Mocellin, O.; Fens, M.H.A.M.; Krijger, G.C.; Brosens, L.A.A.; Oliveira, S. Targeting of Promising Transmembrane Proteins for Diagnosis and Treatment of Pancreatic Ductal Adenocarcinoma. Theranostics 2021, 11, 9022–9037. [Google Scholar] [CrossRef]
- Sakurai, Y.; Akita, H.; Harashima, H. Targeting Tumor Endothelial Cells with Nanoparticles. Int. J. Mol. Sci. 2019, 20, 5819. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Yu, D.-H.; Chen, Y.; Zhao, C.-Y.; Zhang, J.; Liu, Q.-H.; Ni, C.-R.; Zhu, M.-H. Expression of Fibroblast Activation Protein in Human Pancreatic Adenocarcinoma and Its Clinicopathological Significance. World J. Gastroenterol. 2012, 18, 840–846. [Google Scholar] [CrossRef]
- Kawase, T.; Yasui, Y.; Nishina, S.; Hara, Y.; Yanatori, I.; Tomiyama, Y.; Nakashima, Y.; Yoshida, K.; Kishi, F.; Nakamura, M.; et al. Fibroblast Activation Protein-α-Expressing Fibroblasts Promote the Progression of Pancreatic Ductal Adenocarcinoma. BMC Gastroenterol. 2015, 15, 109. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-Expressing Carcinoma-Associated Fibroblasts Synergizes with Anti-PD-L1 Immunotherapy in Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.W.; Deonarine, A.; Jones, J.O.; Denton, A.E.; Feig, C.; Lyons, S.K.; Espeli, M.; Kraman, M.; McKenna, B.; Wells, R.J.B.; et al. Depletion of Stromal Cells Expressing Fibroblast Activation Protein-α from Skeletal Muscle and Bone Marrow Results in Cachexia and Anemia. J. Exp. Med. 2013, 210, 1137–1151. [Google Scholar] [CrossRef]
- Tran, E.; Chinnasamy, D.; Yu, Z.; Morgan, R.A.; Lee, C.-C.R.; Restifo, N.P.; Rosenberg, S.A. Immune Targeting of Fibroblast Activation Protein Triggers Recognition of Multipotent Bone Marrow Stromal Cells and Cachexia. J. Exp. Med. 2013, 210, 1125–1135. [Google Scholar] [CrossRef]
- Raavé, R.; van Kuppevelt, T.H.; Daamen, W.F. Chemotherapeutic Drug Delivery by Tumoral Extracellular Matrix Targeting. J. Control. Release 2018, 274, 1–8. [Google Scholar] [CrossRef]
- Matsumura, Y. Cancer Stromal Targeting (CAST) Therapy. Adv. Drug Deliv. Rev. 2012, 64, 710–719. [Google Scholar] [CrossRef]
- Yasunaga, M.; Manabe, S.; Tarin, D.; Matsumura, Y. Cancer-Stroma Targeting Therapy by Cytotoxic Immunoconjugate Bound to the Collagen 4 Network in the Tumor Tissue. Bioconjug. Chem. 2011, 22, 1776–1783. [Google Scholar] [CrossRef]
- Leppänen, J.; Lindholm, V.; Isohookana, J.; Haapasaari, K.-M.; Karihtala, P.; Lehenkari, P.P.; Saarnio, J.; Kauppila, J.H.; Karttunen, T.J.; Helminen, O.; et al. Tenascin C, Fibronectin, and Tumor-Stroma Ratio in Pancreatic Ductal Adenocarcinoma. Pancreas 2019, 48, 43–48. [Google Scholar] [CrossRef]
- Wenger, C.; Ellenrieder, V.; Alber, B.; Lacher, U.; Menke, A.; Hameister, H.; Wilda, M.; Iwamura, T.; Beger, H.G.; Adler, G.; et al. Expression and Differential Regulation of Connective Tissue Growth Factor in Pancreatic Cancer Cells. Oncogene 1999, 18, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Hartel, M.; di Mola, F.F.; Gardini, A.; Zimmermann, A.; Di Sebastiano, P.; Guweidhi, A.; Innocenti, P.; Giese, T.; Giese, N.; Büchler, M.W.; et al. Desmoplastic Reaction Influences Pancreatic Cancer Growth Behavior. World J. Surg. 2004, 28, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Frese, K.K.; Bapiro, T.E.; Nakagawa, T.; Sternlicht, M.D.; Seeley, T.W.; Pilarsky, C.; Jodrell, D.I.; Spong, S.M.; Tuveson, D.A. CTGF Antagonism with MAb FG-3019 Enhances Chemotherapy Response without Increasing Drug Delivery in Murine Ductal Pancreas Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 12325–12330. [Google Scholar] [CrossRef]
- Infante, J.R.; Matsubayashi, H.; Sato, N.; Tonascia, J.; Klein, A.P.; Riall, T.A.; Yeo, C.; Iacobuzio-Donahue, C.; Goggins, M. Peritumoral Fibroblast SPARC Expression and Patient Outcome with Resectable Pancreatic Adenocarcinoma. J. Clin. Oncol. 2007, 25, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual Microdissection Identifies Distinct Tumor- and Stroma-Specific Subtypes of Pancreatic Ductal Adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Fukushima, N.; Maehara, N.; Matsubayashi, H.; Koopmann, J.; Su, G.H.; Hruban, R.H.; Goggins, M. SPARC/Osteonectin Is a Frequent Target for Aberrant Methylation in Pancreatic Adenocarcinoma and a Mediator of Tumor-Stromal Interactions. Oncogene 2003, 22, 5021–5030. [Google Scholar] [CrossRef] [PubMed]
- Ramu, I.; Buchholz, S.M.; Patzak, M.S.; Goetze, R.G.; Singh, S.K.; Richards, F.M.; Jodrell, D.I.; Sipos, B.; Ströbel, P.; Ellenrieder, V.; et al. SPARC Dependent Collagen Deposition and Gemcitabine Delivery in a Genetically Engineered Mouse Model of Pancreas Cancer. eBioMedicine 2019, 48, 161–168. [Google Scholar] [CrossRef]
- Desai, N.; Trieu, V.; Damascelli, B.; Soon-Shiong, P. SPARC Expression Correlates with Tumor Response to Albumin-Bound Paclitaxel in Head and Neck Cancer Patients. Transl. Oncol. 2009, 2, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Frese, K.K.; Chan, D.S.; Bapiro, T.E.; Howat, W.J.; Richards, F.M.; Ellenrieder, V.; Jodrell, D.I.; Tuveson, D.A. SPARC Independent Drug Delivery and Antitumour Effects of Nab-Paclitaxel in Genetically Engineered Mice. Gut 2014, 63, 974–983. [Google Scholar] [CrossRef]
- Hidalgo, M.; Plaza, C.; Musteanu, M.; Illei, P.; Brachmann, C.B.; Heise, C.; Pierce, D.; Lopez-Casas, P.P.; Menendez, C.; Tabernero, J.; et al. SPARC Expression Did Not Predict Efficacy of Nab-Paclitaxel plus Gemcitabine or Gemcitabine Alone for Metastatic Pancreatic Cancer in an Exploratory Analysis of the Phase III MPACT Trial. Clin. Cancer Res. 2015, 21, 4811–4818. [Google Scholar] [CrossRef]
- Le Large, T.Y.; Mantini, G.; Meijer, L.L.; Pham, T.V.; Funel, N.; van Grieken, N.C.; Kok, B.; Knol, J.; van Laarhoven, H.W.; Piersma, S.R.; et al. Microdissected Pancreatic Cancer Proteomes Reveal Tumor Heterogeneity and Therapeutic Targets. JCI Insight 2020, 5, 138290. [Google Scholar] [CrossRef]
- Corbo, C.; Molinaro, R.; Parodi, A.; Toledano Furman, N.E.; Salvatore, F.; Tasciotti, E. The Impact of Nanoparticle Protein Corona on Cytotoxicity, Immunotoxicity and Target Drug Delivery. Nanomed. 2016, 11, 81–100. [Google Scholar] [CrossRef]
- Nguyen, V.H.; Lee, B.-J. Protein Corona: A New Approach for Nanomedicine Design. Int. J. Nanomed. 2017, 12, 3137–3151. [Google Scholar] [CrossRef]
- Rampado, R.; Crotti, S.; Caliceti, P.; Pucciarelli, S.; Agostini, M. Recent Advances in Understanding the Protein Corona of Nanoparticles and in the Formulation of “Stealthy” Nanomaterials. Front. Bioeng. Biotechnol. 2020, 8, 166. [Google Scholar] [CrossRef]
- Tenzer, S.; Docter, D.; Kuharev, J.; Musyanovych, A.; Fetz, V.; Hecht, R.; Schlenk, F.; Fischer, D.; Kiouptsi, K.; Reinhardt, C.; et al. Rapid Formation of Plasma Protein Corona Critically Affects Nanoparticle Pathophysiology. Nat. Nanotechnol. 2013, 8, 772–781. [Google Scholar] [CrossRef]
- Baimanov, D.; Wang, J.; Zhang, J.; Liu, K.; Cong, Y.; Shi, X.; Zhang, X.; Li, Y.; Li, X.; Qiao, R.; et al. In Situ Analysis of Nanoparticle Soft Corona and Dynamic Evolution. Nat. Commun. 2022, 13, 5389. [Google Scholar] [CrossRef] [PubMed]
- Lundqvist, M.; Stigler, J.; Elia, G.; Lynch, I.; Cedervall, T.; Dawson, K.A. Nanoparticle Size and Surface Properties Determine the Protein Corona with Possible Implications for Biological Impacts. Proc. Natl. Acad. Sci. USA 2008, 105, 14265–14270. [Google Scholar] [CrossRef]
- Salvati, A.; Pitek, A.S.; Monopoli, M.P.; Prapainop, K.; Bombelli, F.B.; Hristov, D.R.; Kelly, P.M.; Åberg, C.; Mahon, E.; Dawson, K.A. Transferrin-Functionalized Nanoparticles Lose Their Targeting Capabilities When a Biomolecule Corona Adsorbs on the Surface. Nat. Nanotechnol. 2013, 8, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Hessmann, E.; Buchholz, S.M.; Demir, I.E.; Singh, S.K.; Gress, T.M.; Ellenrieder, V.; Neesse, A. Microenvironmental Determinants of Pancreatic Cancer. Physiol. Rev. 2020, 100, 1707–1751. [Google Scholar] [CrossRef]
- Xie, A.; Hanif, S.; Ouyang, J.; Tang, Z.; Kong, N.; Kim, N.Y.; Qi, B.; Patel, D.; Shi, B.; Tao, W. Stimuli-Responsive Prodrug-Based Cancer Nanomedicine. eBioMedicine 2020, 56, 102821. [Google Scholar] [CrossRef]
- Pham, S.H.; Choi, Y.; Choi, J. Stimuli-Responsive Nanomaterials for Application in Antitumor Therapy and Drug Delivery. Pharmaceutics 2020, 12, 630. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Huang, P.; Chen, X. Stimuli-Responsive Programmed Specific Targeting in Nanomedicine. ACS Nano 2016, 10, 2991–2994. [Google Scholar] [CrossRef]
- Li, M.; Zhao, G.; Su, W.-K.; Shuai, Q. Enzyme-Responsive Nanoparticles for Anti-Tumor Drug Delivery. Front. Chem. 2020, 8, 647. [Google Scholar] [CrossRef] [PubMed]
- Cabral, H.; Miyata, K.; Osada, K.; Kataoka, K. Block Copolymer Micelles in Nanomedicine Applications. Chem. Rev. 2018, 118, 6844–6892. [Google Scholar] [CrossRef]
- Cabane, E.; Zhang, X.; Langowska, K.; Palivan, C.G.; Meier, W. Stimuli-Responsive Polymers and Their Applications in Nanomedicine. Biointerphases 2012, 7, 9. [Google Scholar] [CrossRef]
- Joglekar, M.; Trewyn, B.G. Polymer-Based Stimuli-Responsive Nanosystems for Biomedical Applications. Biotechnol. J. 2013, 8, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Crucho, C.I.C. Stimuli-Responsive Polymeric Nanoparticles for Nanomedicine. ChemMedChem 2015, 10, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Taghizadeh, B.; Taranejoo, S.; Monemian, S.A.; Salehi Moghaddam, Z.; Daliri, K.; Derakhshankhah, H.; Derakhshani, Z. Classification of Stimuli–Responsive Polymers as Anticancer Drug Delivery Systems. Drug Deliv. 2015, 22, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Liu, S. Functional Block Copolymer Assemblies Responsive to Tumor and Intracellular Microenvironments for Site-Specific Drug Delivery and Enhanced Imaging Performance. Chem. Soc. Rev. 2013, 42, 7289–7325. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Akimoto, J.; Okano, T. Polymeric Micelles with Stimuli-Triggering Systems for Advanced Cancer Drug Targeting. J. Drug Target. 2014, 22, 584–599. [Google Scholar] [CrossRef] [PubMed]
- Tong, R.; Tang, L.; Ma, L.; Tu, C.; Baumgartner, R.; Cheng, J. Smart Chemistry in Polymeric Nanomedicine. Chem. Soc. Rev. 2014, 43, 6982–7012. [Google Scholar] [CrossRef]
- Ramasamy, T.; Ruttala, H.B.; Gupta, B.; Poudel, B.K.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Smart Chemistry-Based Nanosized Drug Delivery Systems for Systemic Applications: A Comprehensive Review. J. Control. Release 2017, 258, 226–253. [Google Scholar] [CrossRef]
- Liao, J.; Jia, Y.; Wu, Y.; Shi, K.; Yang, D.; Li, P.; Qian, Z. Physical-, Chemical-, and Biological-Responsive Nanomedicine for Cancer Therapy. WIREs Nanomed. Nanobiotechnol. 2020, 12, e1581. [Google Scholar] [CrossRef]
- Tao, J.; Yang, G.; Zhou, W.; Qiu, J.; Chen, G.; Luo, W.; Zhao, F.; You, L.; Zheng, L.; Zhang, T.; et al. Targeting Hypoxic Tumor Microenvironment in Pancreatic Cancer. J. Hematol. Oncol. 2021, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- Koong, A.C.; Mehta, V.K.; Le, Q.T.; Fisher, G.A.; Terris, D.J.; Brown, J.M.; Bastidas, A.J.; Vierra, M. Pancreatic Tumors Show High Levels of Hypoxia. Int. J. Radiat. Oncol. 2000, 48, 919–922. [Google Scholar] [CrossRef]
- High, R.A.; Randtke, E.A.; Jones, K.M.; Lindeman, L.R.; Ma, J.C.; Zhang, S.; LeRoux, L.G.; Pagel, M.D. Extracellular Acidosis Differentiates Pancreatitis and Pancreatic Cancer in Mouse Models Using AcidoCEST MRI. Neoplasia 2019, 21, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Kimbrough, C.W.; Khanal, A.; Zeiderman, M.; Khanal, B.R.; Burton, N.C.; McMasters, K.M.; Vickers, S.; Grizzle, W.E.; McNally, L.R. Targeting Acidity in Pancreatic Adenocarcinoma: Multispectral Optoacoustic Tomography Detects PH-Low Insertion Peptide Probes In Vivo. Clin. Cancer Res. 2015, 21, 4576–4585. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Monserrate, Z.; Roland, C.L.; Deng, D.; Arumugam, T.; Moshnikova, A.; Andreev, O.A.; Reshetnyak, Y.K.; Logsdon, C.D. Targeting Pancreatic Ductal Adenocarcinoma Acidic Microenvironment. Sci. Rep. 2014, 4, 4410. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, W.; Liang, C.; Shi, S.; Yu, X.; Chen, Q.; Sun, T.; Lu, Y.; Zhang, Y.; Guo, Q.; et al. Codelivery Nanosystem Targeting the Deep Microenvironment of Pancreatic Cancer. Nano Lett. 2019, 19, 3527–3534. [Google Scholar] [CrossRef]
- Wang, Q.; Cui, H.; Gan, N.; Ma, X.; Ren, W.; Wu, A. Recent Advances in Matrix Metalloproteinases-Responsive Nanoprobes for Cancer Diagnosis and Therapy. Rev. Anal. Chem. 2022, 41, 198–216. [Google Scholar] [CrossRef]
- Yokota, T.; Denham, W.; Murayama, K.; Pelham, C.; Joehl, R.; Bell, R.H. Pancreatic Stellate Cell Activation and MMP Production in Experimental Pancreatic Fibrosis. J. Surg. Res. 2002, 104, 106–111. [Google Scholar] [CrossRef]
- Schneiderhan, W.; Diaz, F.; Fundel, M.; Zhou, S.; Siech, M.; Hasel, C.; Möller, P.; Gschwend, J.E.; Seufferlein, T.; Gress, T.; et al. Pancreatic Stellate Cells Are an Important Source of MMP-2 in Human Pancreatic Cancer and Accelerate Tumor Progression in a Murine Xenograft Model and CAM Assay. J. Cell Sci. 2007, 120, 512–519. [Google Scholar] [CrossRef]
- Phillips, P.A.; McCarroll, J.A.; Park, S.; Wu, M.-J.; Pirola, R.; Korsten, M.; Wilson, J.S.; Apte, M.V. Rat Pancreatic Stellate Cells Secrete Matrix Metalloproteinases: Implications for Extracellular Matrix Turnover. Gut 2003, 52, 275–282. [Google Scholar] [CrossRef]
- Jia, M.; Zhang, D.; Zhang, C.; Li, C. Nanoparticle-Based Delivery Systems Modulate the Tumor Microenvironment in Pancreatic Cancer for Enhanced Therapy. J. Nanobiotechnol. 2021, 19, 384. [Google Scholar] [CrossRef]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The Tumour Microenvironment in Pancreatic Cancer—Clinical Challenges and Opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef]
- Sheng, Q.; Li, T.; Tang, X.; Zhao, W.; Guo, R.; Cun, X.; Zang, S.; Zhang, Z.; Li, M.; He, Q. Comprehensively Enhanced Delivery Cascade by Transformable Beaded Nanofibrils for Pancreatic Cancer Therapy. Nanoscale 2021, 13, 13328–13343. [Google Scholar] [CrossRef]
- Ji, T.; Li, S.; Zhang, Y.; Lang, J.; Ding, Y.; Zhao, X.; Zhao, R.; Li, Y.; Shi, J.; Hao, J.; et al. An MMP-2 Responsive Liposome Integrating Antifibrosis and Chemotherapeutic Drugs for Enhanced Drug Perfusion and Efficacy in Pancreatic Cancer. ACS Appl. Mater. Interfaces 2016, 8, 3438–3445. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, Z.; Liu, H.; Fetse, J.P.; Jain, A.; Lin, C.-Y.; Cheng, K. Development of a Tumor-Responsive Nanopolyplex Targeting Pancreatic Cancer Cells and Stroma. ACS Appl. Mater. Interfaces 2019, 11, 45390–45403. [Google Scholar] [CrossRef]
- Kulkarni, P.S.; Haldar, M.K.; Nahire, R.R.; Katti, P.; Ambre, A.H.; Muhonen, W.W.; Shabb, J.B.; Padi, S.K.R.; Singh, R.K.; Borowicz, P.P.; et al. MMP-9 Responsive PEG Cleavable Nanovesicles for Efficient Delivery of Chemotherapeutics to Pancreatic Cancer. Mol. Pharm. 2014, 11, 2390–2399. [Google Scholar] [CrossRef]
- Łukaszewicz-Zając, M.; Pączek, S.; Mroczko, B. A Disintegrin and Metalloproteinase (ADAM) Family—Novel Biomarkers of Selected Gastrointestinal (GI) Malignancies? Cancers 2022, 14, 2307. [Google Scholar] [CrossRef]
- Slapak, E.J.; Kong, L.; el Mandili, M.; Nieuwland, R.; Kros, A.; Bijlsma, M.F.; Spek, C.A. ADAM9-Responsive Mesoporous Silica Nanoparticles for Targeted Drug Delivery in Pancreatic Cancer. Cancers 2021, 13, 3321. [Google Scholar] [CrossRef]
- Abdel Hadi, N.; Reyes-Castellanos, G.; Carrier, A. Targeting Redox Metabolism in Pancreatic Cancer. Int. J. Mol. Sci. 2021, 22, 1534. [Google Scholar] [CrossRef]
- Datta, R.; Sivanand, S.; Lau, A.N.; Florek, L.V.; Barbeau, A.M.; Wyckoff, J.; Skala, M.C.; Vander Heiden, M.G. Interactions with Stromal Cells Promote a More Oxidized Cancer Cell Redox State in Pancreatic Tumors. Sci. Adv. 2022, 8, eabg6383. [Google Scholar] [CrossRef]
- Broekgaarden, M.; Anbil, S.; Bulin, A.-L.; Obaid, G.; Mai, Z.; Baglo, Y.; Rizvi, I.; Hasan, T. Modulation of Redox Metabolism Negates Cancer-Associated Fibroblasts-Induced Treatment Resistance in a Heterotypic 3D Culture Platform of Pancreatic Cancer. Biomaterials 2019, 222, 119421. [Google Scholar] [CrossRef] [PubMed]
- Kerr, E.M.; Gaude, E.; Turrell, F.K.; Frezza, C.; Martins, C.P. Mutant Kras Copy Number Defines Metabolic Reprogramming and Therapeutic Susceptibilities. Nature 2016, 531, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, Y.; Zhou, S.; Sun, M.; Chen, L.; Wang, J.; Xu, M.; Liu, S.; Liang, K.; Zhang, Q.; et al. Tailored Chemodynamic Nanomedicine Improves Pancreatic Cancer Treatment via Controllable Damaging Neoplastic Cells and Reprogramming Tumor Microenvironment. Nano Lett. 2020, 20, 6780–6790. [Google Scholar] [CrossRef]
- Xin, X.; Lin, F.; Wang, Q.; Yin, L.; Mahato, R.I. ROS-Responsive Polymeric Micelles for Triggered Simultaneous Delivery of PLK1 Inhibitor/MiR-34a and Effective Synergistic Therapy in Pancreatic Cancer. ACS Appl. Mater. Interfaces 2019, 11, 14647–14659. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wang, J.; Tan, H.; Weng, S.; Cheng, L.; Zhou, Z.; Wen, S. Acid- and Reduction-Sensitive Micelles for Improving the Drug Delivery Efficacy for Pancreatic Cancer Therapy. Biomater. Sci. 2018, 6, 1262–1270. [Google Scholar] [CrossRef]
- Lambin, T.; Lafon, C.; Drainville, R.A.; Pioche, M.; Prat, F. Locoregional Therapies and Their Effects on the Tumoral Microenvironment of Pancreatic Ductal Adenocarcinoma. World J. Gastroenterol. 2022, 28, 1288–1303. [Google Scholar] [CrossRef]
- Li, J.; Liu, F.; Gupta, S.; Li, C. Interventional Nanotheranostics of Pancreatic Ductal Adenocarcinoma. Theranostics 2016, 6, 1393–1402. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, W.; Jiao, R.; Lv, Z.; Lin, X.; Xiao, Y.; Zhang, K. Rational Nanomedicine Design Enhances Clinically Physical Treatment-Inspired or Combined Immunotherapy. Adv. Sci. 2022, 9, 2203921. [Google Scholar] [CrossRef]
- Chang, D.; Lim, M.; Goos, J.A.C.M.; Qiao, R.; Ng, Y.Y.; Mansfeld, F.M.; Jackson, M.; Davis, T.P.; Kavallaris, M. Biologically Targeted Magnetic Hyperthermia: Potential and Limitations. Front. Pharmacol. 2018, 9, 831. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Wang, Y.; Zhu, W.; Li, G.; Ma, X.; Zhang, Y.; Chen, S.; Tiwari, S.; Shi, K.; et al. Comprehensive Understanding of Magnetic Hyperthermia for Improving Antitumor Therapeutic Efficacy. Theranostics 2020, 10, 3793–3815. [Google Scholar] [CrossRef]
- Palzer, J.; Mues, B.; Goerg, R.; Aberle, M.; Rensen, S.S.; Olde Damink, S.W.M.; Vaes, R.D.W.; Cramer, T.; Schmitz-Rode, T.; Neumann, U.P.; et al. Magnetic Fluid Hyperthermia as Treatment Option for Pancreatic Cancer Cells and Pancreatic Cancer Organoids. Int. J. Nanomedicine 2021, 16, 2965–2981. [Google Scholar] [CrossRef] [PubMed]
- Piehler, S.; Wucherpfennig, L.; Tansi, F.L.; Berndt, A.; Quaas, R.; Teichgraeber, U.; Hilger, I. Hyperthermia Affects Collagen Fiber Architecture and Induces Apoptosis in Pancreatic and Fibroblast Tumor Hetero-Spheroids In Vitro. Nanomed. Nanotechnol. Biol. Med. 2020, 28, 102183. [Google Scholar] [CrossRef]
- Tansi, F.L.; Fröbel, F.; Maduabuchi, W.O.; Steiniger, F.; Westermann, M.; Quaas, R.; Teichgräber, U.K.; Hilger, I. Effect of Matrix-Modulating Enzymes on The Cellular Uptake of Magnetic Nanoparticles and on Magnetic Hyperthermia Treatment of Pancreatic Cancer Models In Vivo. Nanomaterials 2021, 11, 438. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Chen, Y.-C. Nanomaterials for Photohyperthermia: A Review. Curr. Pharm. Des. 2013, 19, 6622–6634. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Sun, Z.; Ren, Y.; Chen, X.; Zhang, W.; Zhu, X.; Mao, Z.; Shen, J.; Nie, S. Advances in Nanomaterials for Use in Photothermal and Photodynamic Therapeutics (Review). Mol. Med. Rep. 2019, 20, 5–15. [Google Scholar] [CrossRef]
- Nasseri, B.; Alizadeh, E.; Bani, F.; Davaran, S.; Akbarzadeh, A.; Rabiee, N.; Bahadori, A.; Ziaei, M.; Bagherzadeh, M.; Saeb, M.R.; et al. Nanomaterials for Photothermal and Photodynamic Cancer Therapy. Appl. Phys. Rev. 2022, 9, 011317. [Google Scholar] [CrossRef]
- Li, X.; Lovell, J.F.; Yoon, J.; Chen, X. Clinical Development and Potential of Photothermal and Photodynamic Therapies for Cancer. Nat. Rev. Clin. Oncol. 2020, 17, 657–674. [Google Scholar] [CrossRef]
- Raeesi, V.; Chan, W.C.W. Improving Nanoparticle Diffusion through Tumor Collagen Matrix by Photo-Thermal Gold Nanorods. Nanoscale 2016, 8, 12524–12530. [Google Scholar] [CrossRef]
- Lu, G.-H.; Shang, W.-T.; Deng, H.; Han, Z.-Y.; Hu, M.; Liang, X.-Y.; Fang, C.-H.; Zhu, X.-H.; Fan, Y.-F.; Tian, J. Targeting Carbon Nanotubes Based on IGF-1R for Photothermal Therapy of Orthotopic Pancreatic Cancer Guided by Optical Imaging. Biomaterials 2019, 195, 13–22. [Google Scholar] [CrossRef]
- Abrahamse, H.; Hamblin, M.R. New Photosensitizers for Photodynamic Therapy. Biochem. J. 2016, 473, 347–364. [Google Scholar] [CrossRef]
- Li, T.; Yan, L. Functional Polymer Nanocarriers for Photodynamic Therapy. Pharmaceuticals 2018, 11, 133. [Google Scholar] [CrossRef]
- Zhu, L.; Lin, S.; Cui, W.; Xu, Y.; Wang, L.; Wang, Z.; Yuan, S.; Zhang, Y.; Fan, Y.; Geng, J. A Nanomedicine Enables Synergistic Chemo/Photodynamic Therapy for Pancreatic Cancer Treatment. Biomater. Sci. 2022, 10, 3624–3636. [Google Scholar] [CrossRef]
- Tangutoori, S.; Spring, B.Q.; Mai, Z.; Palanisami, A.; Mensah, L.B.; Hasan, T. Simultaneous Delivery of Cytotoxic and Biologic Therapeutics Using Nanophotoactivatable Liposomes Enhances Treatment Efficacy in a Mouse Model of Pancreatic Cancer. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.W.; Greco, W.R.; Bellnier, D.A.; Vaughan, L.; Henderson, B.W. Photodynamic Therapy: A Means to Enhanced Drug Delivery to Tumors. Cancer Res. 2003, 63, 8126–8131. [Google Scholar]
- Huang, H.-C.; Rizvi, I.; Liu, J.; Anbil, S.; Kalra, A.; Lee, H.; Baglo, Y.; Paz, N.; Hayden, D.; Pereira, S.; et al. Photodynamic Priming Mitigates Chemotherapeutic Selection Pressures and Improves Drug Delivery. Cancer Res. 2018, 78, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Karimnia, V.; Stanley, M.E.; Fitzgerald, C.T.; Rizvi, I.; Slack, F.J.; Celli, J.P. Photodynamic Stromal Depletion Enhances Therapeutic Nanoparticle Delivery in 3D Pancreatic Ductal Adenocarcinoma Tumor Models. Photochem. Photobiol. 2023, 99, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Obaid, G.; Bano, S.; Mallidi, S.; Broekgaarden, M.; Kuriakose, J.; Silber, Z.; Bulin, A.-L.; Wang, Y.; Mai, Z.; Jin, W.; et al. Impacting Pancreatic Cancer Therapy in Heterotypic In Vitro Organoids and In Vivo Tumors with Specificity-Tuned, NIR-Activable Photoimmunonanoconjugates: Towards Conquering Desmoplasia? Nano Lett. 2019, 19, 7573–7587. [Google Scholar] [CrossRef]
- Huggett, M.T.; Jermyn, M.; Gillams, A.; Illing, R.; Mosse, S.; Novelli, M.; Kent, E.; Bown, S.G.; Hasan, T.; Pogue, B.W.; et al. Phase I/II Study of Verteporfin Photodynamic Therapy in Locally Advanced Pancreatic Cancer. Br. J. Cancer 2014, 110, 1698–1704. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Jiang, Z.; Chen, L.; Pan, C.; Sun, S.; Liu, C.; Li, Z.; Ren, W.; Wu, A.; Huang, P. PCN-Fe(III)-PTX Nanoparticles for MRI Guided High Efficiency Chemo-Photodynamic Therapy in Pancreatic Cancer through Alleviating Tumor Hypoxia. Nano Res. 2020, 13, 273–281. [Google Scholar] [CrossRef]
- Leenhardt, R.; Camus, M.; Mestas, J.L.; Jeljeli, M.; Abou Ali, E.; Chouzenoux, S.; Bordacahar, B.; Nicco, C.; Batteux, F.; Lafon, C.; et al. Ultrasound-Induced Cavitation Enhances the Efficacy of Chemotherapy in a 3D Model of Pancreatic Ductal Adenocarcinoma with Its Microenvironment. Sci. Rep. 2019, 9, 18916. [Google Scholar] [CrossRef]
- Li, T.; Wang, Y.-N.; Khokhlova, T.D.; D’Andrea, S.; Starr, F.; Chen, H.; McCune, J.S.; Risler, L.J.; Mashadi-Hossein, A.; Hingorani, S.R.; et al. Pulsed High-Intensity Focused Ultrasound Enhances Delivery of Doxorubicin in a Preclinical Model of Pancreatic Cancer. Cancer Res. 2015, 75, 3738–3746. [Google Scholar] [CrossRef]
- Snipstad, S.; Vikedal, K.; Maardalen, M.; Kurbatskaya, A.; Sulheim, E.; de Davies, C.L. Ultrasound and Microbubbles to Beat Barriers in Tumors: Improving Delivery of Nanomedicine. Adv. Drug Deliv. Rev. 2021, 177, 113847. [Google Scholar] [CrossRef]
- Sharma, D.; Leong, K.X.; Czarnota, G.J. Application of Ultrasound Combined with Microbubbles for Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 4393. [Google Scholar] [CrossRef]
- Tinkov, S.; Coester, C.; Serba, S.; Geis, N.A.; Katus, H.A.; Winter, G.; Bekeredjian, R. New Doxorubicin-Loaded Phospholipid Microbubbles for Targeted Tumor Therapy: In-Vivo Characterization. J. Control. Release 2010, 148, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, N.; Payne, A.; Dillon, C.; Shea, J.; Scaife, C.; Gupta, R. Focused Ultrasound-Mediated Drug Delivery to Pancreatic Cancer in a Mouse Model. J. Ther. Ultrasound 2013, 1, 11. [Google Scholar] [CrossRef]
- Kemp, S.B.; Cheng, N.; Markosyan, N.; Sor, R.; Kim, I.-K.; Hallin, J.; Shoush, J.; Quinones, L.; Brown, N.V.; Bassett, J.B.; et al. Efficacy of a Small Molecule Inhibitor of KrasG12D in Immunocompetent Models of Pancreatic Cancer. Cancer Discov. 2022, CD-22-1066. [Google Scholar] [CrossRef]
- Maddalena, M.; Mallel, G.; Nataraj, N.B.; Shreberk-Shaked, M.; Hassin, O.; Mukherjee, S.; Arandkar, S.; Rotkopf, R.; Kapsack, A.; Lambiase, G.; et al. TP53 Missense Mutations in PDAC Are Associated with Enhanced Fibrosis and an Immunosuppressive Microenvironment. Proc. Natl. Acad. Sci. USA 2021, 118, e2025631118. [Google Scholar] [CrossRef] [PubMed]
- Ijichi, H.; Chytil, A.; Gorska, A.E.; Aakre, M.E.; Fujitani, Y.; Fujitani, S.; Wright, C.V.E.; Moses, H.L. Aggressive Pancreatic Ductal Adenocarcinoma in Mice Caused by Pancreas-Specific Blockade of Transforming Growth Factor-Beta Signaling in Cooperation with Active Kras Expression. Genes Dev. 2006, 20, 3147–3160. [Google Scholar] [CrossRef]
- Dai, E.; Han, L.; Liu, J.; Xie, Y.; Kroemer, G.; Klionsky, D.J.; Zeh, H.J.; Kang, R.; Wang, J.; Tang, D. Autophagy-Dependent Ferroptosis Drives Tumor-Associated Macrophage Polarization via Release and Uptake of Oncogenic KRAS Protein. Autophagy 2020, 16, 2069–2083. [Google Scholar] [CrossRef]
- Vennin, C.; Mélénec, P.; Rouet, R.; Nobis, M.; Cazet, A.S.; Murphy, K.J.; Herrmann, D.; Reed, D.A.; Lucas, M.C.; Warren, S.C.; et al. CAF Hierarchy Driven by Pancreatic Cancer Cell P53-Status Creates a pro-Metastatic and Chemoresistant Environment via Perlecan. Nat. Commun. 2019, 10, 3637. [Google Scholar] [CrossRef]
- Shaashua, L.; Ben-Shmuel, A.; Pevsner-Fischer, M.; Friedman, G.; Levi-Galibov, O.; Nandakumar, S.; Barki, D.; Nevo, R.; Brown, L.E.; Zhang, W.; et al. BRCA Mutational Status Shapes the Stromal Microenvironment of Pancreatic Cancer Linking Clusterin Expression in Cancer Associated Fibroblasts with HSF1 Signaling. Nat. Commun. 2022, 13, 6513. [Google Scholar] [CrossRef] [PubMed]
- Herreros-Villanueva, M.; Hijona, E.; Cosme, A.; Bujanda, L. Mouse Models of Pancreatic Cancer. World J. Gastroenterol. 2012, 18, 1286–1294. [Google Scholar] [CrossRef] [PubMed]
- Deer, E.L.; Gonzalez-Hernandez, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and Genotype of Pancreatic Cancer Cell Lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef]
- Chen, S.-H.; Zhang, Y.; Van Horn, R.D.; Yin, T.; Buchanan, S.; Yadav, V.; Mochalkin, I.; Wong, S.S.; Yue, Y.G.; Huber, L.; et al. Oncogenic BRAF Deletions That Function as Homodimers and Are Sensitive to Inhibition by RAF Dimer Inhibitor LY3009120. Cancer Discov. 2016, 6, 300–315. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Trejo, C.L.; Silva, J.M.; Gu, S.; Korkola, J.E.; Heiser, L.M.; Charles, R.-P.; Rabinovich, B.A.; Hann, B.; Dankort, D.; et al. A Central Role for RAF→MEK→ERK Signaling in the Genesis of Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2012, 2, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Hutton, C.; Heider, F.; Blanco-Gomez, A.; Banyard, A.; Kononov, A.; Zhang, X.; Karim, S.; Paulus-Hock, V.; Watt, D.; Steele, N.; et al. Single-Cell Analysis Defines a Pancreatic Fibroblast Lineage That Supports Anti-Tumor Immunity. Cancer Cell 2021, 39, 1227–1244.e20. [Google Scholar] [CrossRef]
- Schoonderwoerd, M.J.A.; Hakuno, S.K.; Sassen, M.; Kuhlemaijer, E.B.; Paauwe, M.; Slingerland, M.; Fransen, M.F.; Hawinkels, L.J.A.C. Targeting Endoglin Expressing Cells in the Tumor Microenvironment Does Not Inhibit Tumor Growth in a Pancreatic Cancer Mouse Model. OncoTargets Ther. 2021, 14, 5205–5220. [Google Scholar] [CrossRef]
- Bruno, S.; Williams, R.J.; Vecchio, D.D. Epigenetic Cell Memory: The Gene’s Inner Chromatin Modification Circuit. PLOS Comput. Biol. 2022, 18, e1009961. [Google Scholar] [CrossRef]
- D’Arcangelo, E.; Wu, N.C.; Cadavid, J.L.; McGuigan, A.P. The Life Cycle of Cancer-Associated Fibroblasts within the Tumour Stroma and Its Importance in Disease Outcome. Br. J. Cancer 2020, 122, 931–942. [Google Scholar] [CrossRef]
- Balestrini, J.L.; Chaudhry, S.; Sarrazy, V.; Koehler, A.; Hinz, B. The Mechanical Memory of Lung Myofibroblasts. Integr. Biol. 2012, 4, 410–421. [Google Scholar] [CrossRef]
- Li, C.X.; Talele, N.P.; Boo, S.; Koehler, A.; Knee-Walden, E.; Balestrini, J.L.; Speight, P.; Kapus, A.; Hinz, B. MicroRNA-21 Preserves the Fibrotic Mechanical Memory of Mesenchymal Stem Cells. Nat. Mater. 2017, 16, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Killaars, A.R.; Grim, J.C.; Walker, C.J.; Hushka, E.A.; Brown, T.E.; Anseth, K.S. Extended Exposure to Stiff Microenvironments Leads to Persistent Chromatin Remodeling in Human Mesenchymal Stem Cells. Adv. Sci. 2018, 6, 1801483. [Google Scholar] [CrossRef]
- Dai, E.N.; Heo, S.-J.; Mauck, R.L. “Looping In” Mechanics: Mechanobiologic Regulation of the Nucleus and the Epigenome. Adv. Healthc. Mater. 2020, 9, 2000030. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Zhou, D.; Rucki, A.A.; Williams, J.; Zhou, J.; Mo, G.; Murphy, A.; Fujiwara, K.; Kleponis, J.; Salman, B.; et al. Cancer-Associated Fibroblasts in Pancreatic Cancer Are Reprogrammed by Tumor-Induced Alterations in Genomic DNA Methylation. Cancer Res. 2016, 76, 5395–5404. [Google Scholar] [CrossRef] [PubMed]
- Kirk, T.; Ahmed, A.; Rognoni, E. Fibroblast Memory in Development, Homeostasis and Disease. Cells 2021, 10, 2840. [Google Scholar] [CrossRef]
- O’Reilly, S. Epigenetics in Fibrosis. Mol. Asp. Med. 2017, 54, 89–102. [Google Scholar] [CrossRef]
- Mann, J.; Mann, D.A. Epigenetic Regulation of Wound Healing and Fibrosis. Curr. Opin. Rheumatol. 2013, 25, 101–107. [Google Scholar] [CrossRef]
- Gupta, R. Epigenetic Regulation and Targeting of ECM for Cancer Therapy. Am. J. Physiol.-Cell Physiol. 2022, 322, C762–C768. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting Epigenetic Regulators for Cancer Therapy: Mechanisms and Advances in Clinical Trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef]
- Ribich, S.; Harvey, D.; Copeland, R.A. Drug Discovery and Chemical Biology of Cancer Epigenetics. Cell Chem. Biol. 2017, 24, 1120–1147. [Google Scholar] [CrossRef]
- Merrell, A.J.; Stanger, B.Z. Adult Cell Plasticity In Vivo: De-Differentiation and Transdifferentiation Are Back in Style. Nat. Rev. Mol. Cell Biol. 2016, 17, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.; Boue, S.; Belmonte, J.C.I. Dedifferentiation, Transdifferentiation and Reprogramming: Three Routes to Regeneration. Nat. Rev. Mol. Cell Biol. 2011, 12, 79–89. [Google Scholar] [CrossRef]
- van der Meel, R.; Sulheim, E.; Shi, Y.; Kiessling, F.; Mulder, W.J.M.; Lammers, T. Smart Cancer Nanomedicine. Nat. Nanotechnol. 2019, 14, 1007–1017.e18. [Google Scholar] [CrossRef] [PubMed]
- Grünwald, B.T.; Devisme, A.; Andrieux, G.; Vyas, F.; Aliar, K.; McCloskey, C.W.; Macklin, A.; Jang, G.H.; Denroche, R.; Romero, J.M.; et al. Spatially Confined Sub-Tumor Microenvironments in Pancreatic Cancer. Cell 2021, 184, 5577–5592.e18. [Google Scholar] [CrossRef]
- Xavier da Silveira Dos Santos, A.; Liberali, P. From Single Cells to Tissue Self-Organization. FEBS J. 2019, 286, 1495–1513. [Google Scholar] [CrossRef]
- Sasai, Y. Cytosystems Dynamics in Self-Organization of Tissue Architecture. Nature 2013, 493, 318–326. [Google Scholar] [CrossRef]
- Gonzalez-Molina, J.; Moyano-Galceran, L.; Single, A.; Gultekin, O.; Alsalhi, S.; Lehti, K. Chemotherapy as a Regulator of Extracellular Matrix-Cell Communication: Implications in Therapy Resistance. Semin. Cancer Biol. 2022, 86, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular Matrix as a Driver of Progressive Fibrosis. J. Clin. Investig. 2018, 128, 45–53. [Google Scholar] [CrossRef]
- Stillman, N.R.; Kovacevic, M.; Balaz, I.; Hauert, S. In Silico Modelling of Cancer Nanomedicine, across Scales and Transport Barriers. Npj Comput. Mater. 2020, 6, 92. [Google Scholar] [CrossRef]
- Dogra, P.; Butner, J.D.; Chuang, Y.; Caserta, S.; Goel, S.; Brinker, C.J.; Cristini, V.; Wang, Z. Mathematical Modeling in Cancer Nanomedicine: A Review. Biomed. Microdevices 2019, 21, 40. [Google Scholar] [CrossRef]
- Bromma, K.; Bannister, A.; Kowalewski, A.; Cicon, L.; Chithrani, D.B. Elucidating the Fate of Nanoparticles among Key Cell Components of the Tumor Microenvironment for Promoting Cancer Nanotechnology. Cancer Nanotechnol. 2020, 11, 8. [Google Scholar] [CrossRef]
- Meng, H.; Nel, A.E. Use of Nano Engineered Approaches to Overcome the Stromal Barrier in Pancreatic Cancer. Adv. Drug Deliv. Rev. 2018, 130, 50–57. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, J.; Meng, H. Transcytosis—An Effective Targeting Strategy That Is Complementary to “EPR Effect” for Pancreatic Cancer Nano Drug Delivery. Theranostics 2019, 9, 8018–8025. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Dong, C.; Fan, W.; Jiang, H.; Xiang, J.; Qiu, N.; Piao, Y.; Xie, T.; Luo, Y.; Li, Z.; et al. Tumor Extravasation and Infiltration as Barriers of Nanomedicine for High Efficacy: The Current Status and Transcytosis Strategy. Biomaterials 2020, 240, 119902. [Google Scholar] [CrossRef] [PubMed]
- Walma, D.A.C.; Yamada, K.M. The Extracellular Matrix in Development. Development 2020, 147, dev175596. [Google Scholar] [CrossRef]
- Muhl, L.; Genové, G.; Leptidis, S.; Liu, J.; He, L.; Mocci, G.; Sun, Y.; Gustafsson, S.; Buyandelger, B.; Chivukula, I.V.; et al. Single-Cell Analysis Uncovers Fibroblast Heterogeneity and Criteria for Fibroblast and Mural Cell Identification and Discrimination. Nat. Commun. 2020, 11, 3953. [Google Scholar] [CrossRef]
- Qazi, T.H.; Blatchley, M.R.; Davidson, M.D.; Yavitt, F.M.; Cooke, M.E.; Anseth, K.S.; Burdick, J.A. Programming Hydrogels to Probe Spatiotemporal Cell Biology. Cell Stem Cell 2022, 29, 678–691. [Google Scholar] [CrossRef]
- Rosales, A.M.; Anseth, K.S. The Design of Reversible Hydrogels to Capture Extracellular Matrix Dynamics. Nat. Rev. Mater. 2016, 1, 15012. [Google Scholar] [CrossRef]
- Li, J.; Kataoka, K. Chemo-Physical Strategies to Advance the In Vivo Functionality of Targeted Nanomedicine: The Next Generation. J. Am. Chem. Soc. 2021, 143, 538–559. [Google Scholar] [CrossRef]
- Wolfram, J.; Ferrari, M. Clinical Cancer Nanomedicine. Nano Today 2019, 25, 85–98. [Google Scholar] [CrossRef]
- Hu, X.; Xia, F.; Lee, J.; Li, F.; Lu, X.; Zhuo, X.; Nie, G.; Ling, D. Tailor-Made Nanomaterials for Diagnosis and Therapy of Pancreatic Ductal Adenocarcinoma. Adv. Sci. 2021, 8, 2002545. [Google Scholar] [CrossRef]
- Sun, D.; Zhou, S.; Gao, W. What Went Wrong with Anticancer Nanomedicine Design and How to Make It Right. ACS Nano 2020, 14, 12281–12290. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Vaughan, H.J.; Zamboni, C.G.; Sunshine, J.C.; Green, J.J. High-Throughput Evaluation of Polymeric Nanoparticles for Tissue-Targeted Gene Expression Using Barcoded Plasmid DNA. J. Control. Release 2021, 337, 105–116. [Google Scholar] [CrossRef]
- Rui, Y.; Wilson, D.R.; Tzeng, S.Y.; Yamagata, H.M.; Sudhakar, D.; Conge, M.; Berlinicke, C.A.; Zack, D.J.; Tuesca, A.; Green, J.J. High-Throughput and High-Content Bioassay Enables Tuning of Polyester Nanoparticles for Cellular Uptake, Endosomal Escape, and Systemic In Vivo Delivery of MRNA. Sci. Adv. 2022, 8, eabk2855. [Google Scholar] [CrossRef]
- Yamankurt, G.; Berns, E.J.; Xue, A.; Lee, A.; Bagheri, N.; Mrksich, M.; Mirkin, C.A. Exploration of the Nanomedicine-Design Space with High-Throughput Screening and Machine Learning. Nat. Biomed. Eng. 2019, 3, 318–327. [Google Scholar] [CrossRef]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding Light on the Cell Biology of Extracellular Vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular Vesicles: Exosomes, Microvesicles, and Friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Martín-Taboada, M.; Corrales, P.; Medina-Gómez, G.; Vila-Bedmar, R. Tackling the Effects of Extracellular Vesicles in Fibrosis. Eur. J. Cell Biol. 2022, 101, 151221. [Google Scholar] [CrossRef]
- Brigstock, D.R. Extracellular Vesicles in Organ Fibrosis: Mechanisms, Therapies, and Diagnostics. Cells 2021, 10, 1596. [Google Scholar] [CrossRef]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The Exosome Journey: From Biogenesis to Uptake and Intracellular Signalling. Cell Commun. Signal. 2021, 19, 47. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour Exosome Integrins Determine Organotropic Metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Ferguson, S.W.; Nguyen, J. Exosomes as Therapeutics: The Implications of Molecular Composition and Exosomal Heterogeneity. J. Control. Release 2016, 228, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Abu Lila, A.S.; Kiwada, H.; Ishida, T. The Accelerated Blood Clearance (ABC) Phenomenon: Clinical Challenge and Approaches to Manage. J. Control. Release 2013, 172, 38–47. [Google Scholar] [CrossRef]
- Yang, Q.; Lai, S.K. Anti-PEG Immunity: Emergence, Characteristics, and Unaddressed Questions. WIREs Nanomed. Nanobiotechnol. 2015, 7, 655–677. [Google Scholar] [CrossRef] [PubMed]
- Hoang Thi, T.T.; Pilkington, E.H.; Nguyen, D.H.; Lee, J.S.; Park, K.D.; Truong, N.P. The Importance of Poly(Ethylene Glycol) Alternatives for Overcoming PEG Immunogenicity in Drug Delivery and Bioconjugation. Polymers 2020, 12, 298. [Google Scholar] [CrossRef]
- Shi, D.; Beasock, D.; Fessler, A.; Szebeni, J.; Ljubimova, J.Y.; Afonin, K.A.; Dobrovolskaia, M.A. To PEGylate or Not to PEGylate: Immunological Properties of Nanomedicine’s Most Popular Component, Polyethylene Glycol and Its Alternatives. Adv. Drug Deliv. Rev. 2022, 180, 114079. [Google Scholar] [CrossRef]
- Martin, J.D.; Cabral, H.; Stylianopoulos, T.; Jain, R.K. Improving Cancer Immunotherapy Using Nanomedicines: Progress, Opportunities and Challenges. Nat. Rev. Clin. Oncol. 2020, 17, 251–266. [Google Scholar] [CrossRef]
- Chakravarthy, A.; Khan, L.; Bensler, N.P.; Bose, P.; De Carvalho, D.D. TGF-β-Associated Extracellular Matrix Genes Link Cancer-Associated Fibroblasts to Immune Evasion and Immunotherapy Failure. Nat. Commun. 2018, 9, 4692. [Google Scholar] [CrossRef]
- Casadevall, A. The mRNA Vaccine Revolution Is the Dividend from Decades of Basic Science Research. J. Clin. Investig. 2021, 131, e153721. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA Vaccines—A New Era in Vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Lorentzen, C.L.; Haanen, J.B.; Met, Ö.; Svane, I.M. Clinical Advances and Ongoing Trials of mRNA Vaccines for Cancer Treatment. Lancet Oncol. 2022, 23, e450–e458. [Google Scholar] [CrossRef]
- Vonlaufen, A.; Joshi, S.; Qu, C.; Phillips, P.A.; Xu, Z.; Parker, N.R.; Toi, C.S.; Pirola, R.C.; Wilson, J.S.; Goldstein, D.; et al. Pancreatic Stellate Cells: Partners in Crime with Pancreatic Cancer Cells. Cancer Res. 2008, 68, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.-I.; Boj, S.F.; Clevers, H.; Tuveson, D.A. Preclinical Models of Pancreatic Ductal Adenocarcinoma. J. Pathol. 2016, 238, 197–204. [Google Scholar] [CrossRef]
- Heinrich, M.A.; Mostafa, A.M.R.H.; Morton, J.P.; Hawinkels, L.J.A.C.; Prakash, J. Translating Complexity and Heterogeneity of Pancreatic Tumor: 3D In Vitro to In Vivo Models. Adv. Drug Deliv. Rev. 2021, 174, 265–293. [Google Scholar] [CrossRef]
- Gündel, B.; Liu, X.; Löhr, M.; Heuchel, R. Pancreatic Ductal Adenocarcinoma: Preclinical In Vitro and Ex Vivo Models. Front. Cell Dev. Biol. 2021, 9, 741162. [Google Scholar] [CrossRef]
- Tomás-Bort, E.; Kieler, M.; Sharma, S.; Candido, J.B.; Loessner, D. 3D Approaches to Model the Tumor Microenvironment of Pancreatic Cancer. Theranostics 2020, 10, 5074–5089. [Google Scholar] [CrossRef]
- Feigin, M.E.; Tuveson, D.A. Challenges and Opportunities in Modeling Pancreatic Cancer. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Tuveson, D.; Clevers, H. Cancer Modeling Meets Human Organoid Technology. Science 2019, 364, 952–955. [Google Scholar] [CrossRef]
- Grossman, J.E.; Muthuswamy, L.; Huang, L.; Akshinthala, D.; Perea, S.; Gonzalez, R.S.; Tsai, L.L.; Cohen, J.; Bockorny, B.; Bullock, A.J.; et al. Organoid Sensitivity Correlates with Therapeutic Response in Patients with Pancreatic Cancer. Clin. Cancer Res. 2022, 28, 708–718. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanaka, H.Y.; Nakazawa, T.; Enomoto, A.; Masamune, A.; Kano, M.R. Therapeutic Strategies to Overcome Fibrotic Barriers to Nanomedicine in the Pancreatic Tumor Microenvironment. Cancers 2023, 15, 724. https://doi.org/10.3390/cancers15030724
Tanaka HY, Nakazawa T, Enomoto A, Masamune A, Kano MR. Therapeutic Strategies to Overcome Fibrotic Barriers to Nanomedicine in the Pancreatic Tumor Microenvironment. Cancers. 2023; 15(3):724. https://doi.org/10.3390/cancers15030724
Chicago/Turabian StyleTanaka, Hiroyoshi Y., Takuya Nakazawa, Atsushi Enomoto, Atsushi Masamune, and Mitsunobu R. Kano. 2023. "Therapeutic Strategies to Overcome Fibrotic Barriers to Nanomedicine in the Pancreatic Tumor Microenvironment" Cancers 15, no. 3: 724. https://doi.org/10.3390/cancers15030724
APA StyleTanaka, H. Y., Nakazawa, T., Enomoto, A., Masamune, A., & Kano, M. R. (2023). Therapeutic Strategies to Overcome Fibrotic Barriers to Nanomedicine in the Pancreatic Tumor Microenvironment. Cancers, 15(3), 724. https://doi.org/10.3390/cancers15030724