
Cancer-Associated Adipocytes and Breast Cancer: Intertwining in the Tumor Microenvironment and Challenges for Cancer Therapy
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
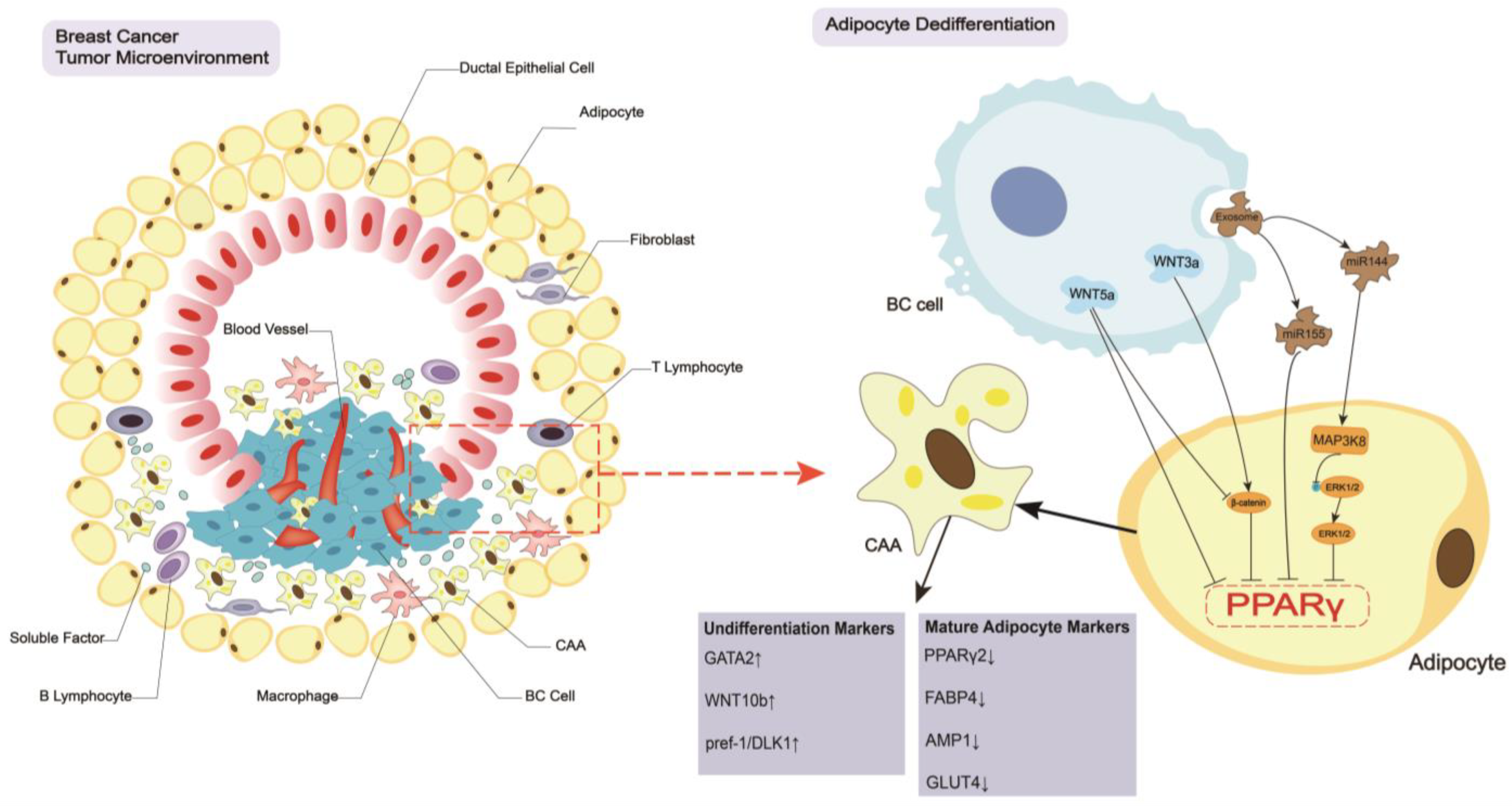
2. Breast Cancer Regulates the CAA Formation and Biology
2.1. Dedifferentiation of Adipocytes
2.2. Biological Changes in CAAs
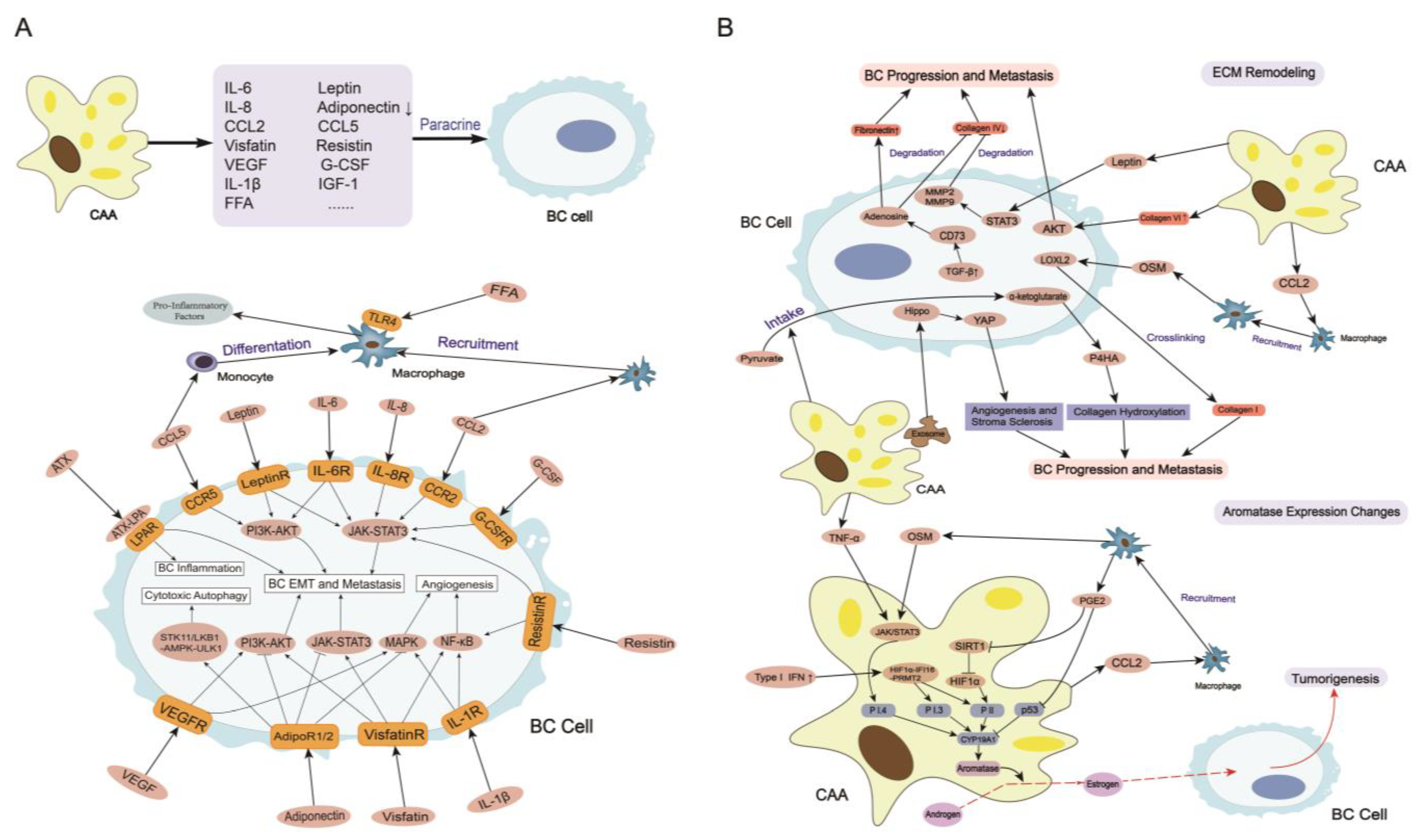
3. CAAs Induce Breast Cancer Tumorigenesis, Progression, and Metastasis
3.1. Secretome in CAAs
3.1.1. Leptin
3.1.2. Adiponectin
3.1.3. IL-6
3.1.4. CCL2
3.1.5. CCL5
3.1.6. IL-1β
3.1.7. Other Adipokines and Cytokines
3.2. ECM Remodeling
3.3. Aromatase Expression Changes
3.4. Metabolism Remodeling
3.4.1. Glucose Metabolism
3.4.2. Fatty Acid Metabolism
3.4.3. Amino Acid Metabolism
3.5. Immune System Changes
3.5.1. Tumor-Associated Macrophages
3.5.2. Other Immune Cells
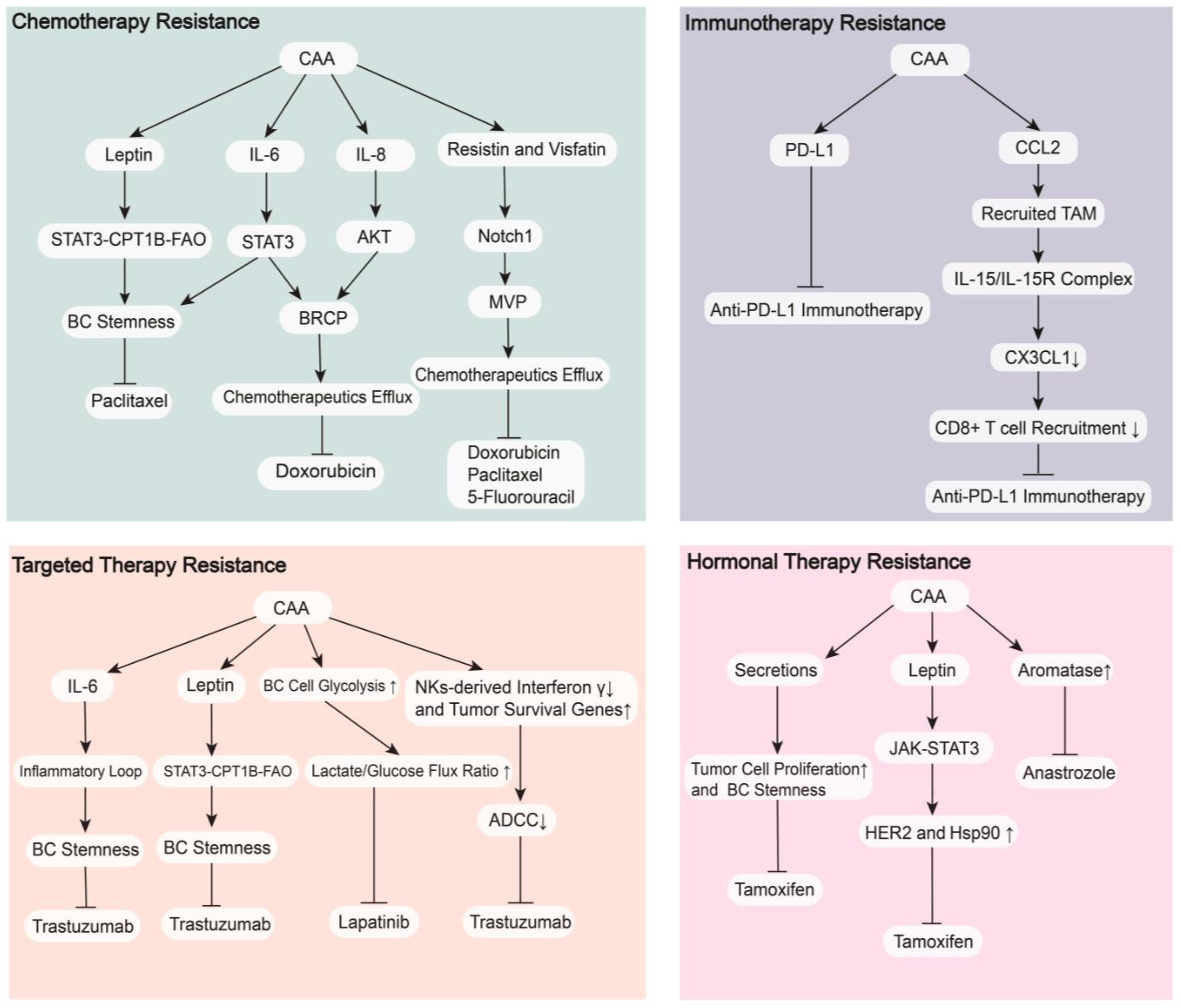
4. CAAs in Breast Cancer Therapy
4.1. Therapeutic Resistance
4.1.1. Chemotherapy Resistance
4.1.2. Targeted Therapy Resistance
4.1.3. Hormonal Therapy Resistance
4.1.4. Immunotherapy Resistance
4.2. Targeting CAAs Strategies
4.2.1. Drugs Targeting the Formation of CAAs
4.2.2. Drugs Targeting Immune and Metabolism Changes in CAAs
4.2.3. Drugs Targeting Cytokines and Adipokines Secreted by CAAs
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer. J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Danenberg, E.; Bardwell, H.; Zanotelli, V.R.T.; Provenzano, E.; Chin, S.; Rueda, O.M.; Green, A.; Rakha, E.; Aparicio, S.; Ellis, I.O.; et al. Breast tumor microenvironment structures are associated with genomic features and clinical outcome. Nat. Genet. 2022, 54, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Singleton, D.C.; Macann, A.; Wilson, W.R. Therapeutic targeting of the hypoxic tumour microenvironment. Nat. Rev. Clin. Oncol. 2021, 18, 751–772. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, W.; Zhou, Q.; Chen, C.; Yuan, W.; Liu, J.; Li, X.; Sun, Z. Roles of circRNAs in the tumour microenvironment. Mol. Cancer 2020, 19, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, M.; Zhang, Y.; Su, Q.; Xie, Z.; Chen, X.; Yan, R.; Li, P.; Li, T.; Qin, X.; et al. Functions and clinical significance of mechanical tumor microenvironment: Cancer cell sensing, mechanobiology and metastasis. Cancer Commun. 2022, 42, 374–400. [Google Scholar] [CrossRef]
- Bejarano, L.; Jordāo, M.J.C.; Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef]
- Wu, Q.; Li, B.; Li, Z.; Li, J.; Sun, S.; Sun, S. Cancer-associated adipocytes: Key players in breast cancer progression. J. Hematol. Oncol. 2019, 12, 95. [Google Scholar] [CrossRef]
- Fraser, J.K.; Wulur, I.; Alfonso, Z.; Hedrick, M.H. Fat tissue: An underappreciated source of stem cells for biotechnology. Trends Biotechnol. 2006, 24, 150–154. [Google Scholar] [CrossRef]
- Gregoire, F.M.; Smas, C.M.; Sul, H.S. Understanding Adipocyte Differentiation. Physiol. Rev. 1998, 78, 783–809. [Google Scholar] [CrossRef]
- Zhao, C.; Wu, M.; Zeng, N.; Xiong, M.; Hu, W.; Lv, W.; Yi, Y.; Zhang, Q.; Wu, Y. Cancer-associated adipocytes: Emerging supporters in breast cancer. J. Exp. Clin. Cancer Res. 2020, 39, 156. [Google Scholar] [CrossRef]
- Rybinska, I.; Mangano, N.; Tagliabue, E.; Triulzi, T. Cancer-Associated Adipocytes in Breast Cancer: Causes and Consequences. Int. J. Mol. Sci. 2021, 22, 3775. [Google Scholar] [CrossRef] [PubMed]
- Protani, M.; Coory, M.; Martin, J.H. Effect of obesity on survival of women with breast cancer: Systematic review and meta-analysis. Breast Cancer Res. Treat. 2010, 123, 627–635. [Google Scholar] [CrossRef]
- Namazi, N.; Irandoost, P.; Heshmati, J.; Larijani, B.; Azadbakht, L. The association between fat mass and the risk of breast cancer: A systematic review and meta-analysis. Clin. Nutr. 2019, 38, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Rybinska, I.; Agresti, R.; Trapani, A.; Tagliabue, E.; Triulzi, T. Adipocytes in Breast Cancer, the Thick and the Thin. Cells 2020, 9, 560. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.S.; Almeida, C.R.; Helguero, L.A.; Duarte, I.F. Metabolic crosstalk in the breast cancer microenvironment. Eur. J. Cancer 2019, 121, 154–171. [Google Scholar] [CrossRef]
- Tang, Q.Q.; Lane, M.D. Adipogenesis: From stem cell to adipocyte. Annu. Rev. Biochem. 2012, 81, 715–736. [Google Scholar] [CrossRef]
- Hall, J.A.; Ramachandran, D.; Roh, H.C.; DiSpirito, J.R.; Belchior, T.; Zushin, P.H.; Palmer, C.; Hong, S.; Mina, A.I.; Liu, B.; et al. Obesity-Linked PPARγ S273 Phosphorylation Promotes Insulin Resistance through Growth Differentiation Factor 3. Cell Metab. 2020, 32, 665–675. [Google Scholar] [CrossRef]
- Madsen, M.S.; Siersbæk, R.; Boergesen, M.; Nielsen, R.; Mandrup, S. Peroxisome Proliferator-Activated Receptor γ and C/EBPα Synergistically Activate Key Metabolic Adipocyte Genes by Assisted Loading. Mol. Cell. Biol. 2014, 34, 939–954. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, M.; Xu, F.; Jiang, S. Wnt signaling in breast cancer: Biological mechanisms, challenges and opportunities. Mol. Cancer 2020, 19, 165. [Google Scholar] [CrossRef]
- Ross, S.E.; Hemati, N.; Longo, K.A.; Bennett, C.N.; Lucas, P.C.; Erickson, R.L.; Macdougald, O.A. Inhibition of Adipogenesis by Wnt Signaling. Science 2000, 289, 950–953. [Google Scholar] [CrossRef]
- Gustafson, B.; Smith, U. Activation of Canonical Wingless-type MMTV Integration Site Family (Wnt) Signaling in Mature Adipocytes Increases β-Catenin Levels and Leads to Cell Dedifferentiation and Insulin Resistance. J. Biol. Chem. 2010, 285, 14031–14041. [Google Scholar] [CrossRef] [PubMed]
- Zoico, E.; Darra, E.; Rizzatti, V.; Budui, S.; Franceschetti, G.; Mazzali, G.; Rossi, A.P.; Fantin, F.; Menegazzi, M.; Cinti, S.; et al. Adipocytes WNT5a mediated dedifferentiation: A possible target in pancreatic cancer microenvironment. Oncotarget 2016, 7, 20223–20235. [Google Scholar] [CrossRef]
- Topol, L.; Jiang, X.; Choi, H.; Garrett-Beal, L.; Carolan, P.J.; Yang, Y. Wnt-5a inhibits the canonical Wnt pathway by promoting GSK-3–independent β-catenin degradation. J. Cell Biol. 2003, 162, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Li, J.; Li, Z.; Sun, S.; Zhu, S.; Wang, L.; Wu, J.; Yuan, J.; Zhang, Y.; Sun, S.; et al. Exosomes from the tumour-adipocyte interplay stimulate beige/brown differentiation and reprogram metabolism in stromal adipocytes to promote tumour progression. J. Exp. Clin. Cancer Res. 2019, 38, 223. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Sun, S.; Li, Z.; Yang, Q.; Li, B.; Zhu, S.; Wang, L.; Wu, J.; Yuan, J.; Yang, C.; et al. Tumour-originated exosomal miR-155 triggers cancer-associated cachexia to promote tumour progression. Mol. Cancer 2018, 17, 155. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Su, X.; Xu, M.; Xiao, X.; Li, X.; Li, H.; Keating, A.; Zhao, R.C. Exosomes secreted by mesenchymal stromal/stem cell-derived adipocytes promote breast cancer cell growth via activation of Hippo signaling pathway. Stem Cell Res. Ther. 2019, 10, 117. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, J.; Lv, W.; Zhao, C.; Xia, Y.; Wu, Y.; Zhang, Q. The pleiotropic roles of adipocyte secretome in remodeling breast cancer. J. Exp. Clin. Cancer Res. 2022, 41, 203. [Google Scholar] [CrossRef]
- Mubtasim, N.; Moustaid-Moussa, N.; Gollahon, L. The Complex Biology of the Obesity-Induced, Metastasis-Promoting Tumor Microenvironment in Breast Cancer. Int. J. Mol. Sci. 2022, 23, 2480. [Google Scholar] [CrossRef]
- Lyu, X.; Zhang, Q.; Fares, H.M.; Wang, Y.; Han, Y.; Sun, L. Contribution of adipocytes in the tumor microenvironment to breast cancer metabolism. Cancer Lett. 2022, 534, 215616. [Google Scholar] [CrossRef]
- Zahid, H.; Simpson, E.R.; Brown, K.A. Inflammation, dysregulated metabolism and aromatase in obesity and breast cancer. Curr. Opin. Pharm. 2016, 31, 90–96. [Google Scholar] [CrossRef]
- García-Estevez, L.; González-Martínez, S.; Moreno-Bueno, G. The Leptin Axis and Its Association with the Adaptive Immune System in Breast Cancer. Front. Immunol. 2021, 12, 784823. [Google Scholar] [CrossRef]
- Sánchez-Jiménez, F.; Pérez-Pérez, A.; de la Cruz-Merino, L.; Sánchez-Margalet, V. Obesity and Breast Cancer: Role of Leptin. Front. Oncol. 2019, 9, 596. [Google Scholar] [CrossRef] [PubMed]
- Catalano, S.; Giordano, C.; Rizza, P.; Gu, G.; Barone, I.; Bonofiglio, D.; Giordano, F.; Malivindi, R.; Gaccione, D.; Lanzino, M.; et al. Evidence that leptin through STAT and CREB signaling enhances cyclin D1 expression and promotes human endometrial cancer proliferation. J. Cell Physiol. 2009, 218, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Wang, B.; Li, M.; Cui, C.; Liu, F.; Gao, Y. Celastrol inhibits the proliferation and migration of MCF-7 cells through the leptin-triggered PI3K/AKT pathway. Comput. Struct. Biotechnol. J. 2022, 20, 3173–3181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yue, C.; Herrmann, A.; Song, J.; Egelston, C.; Wang, T.; Zhang, Z.; Li, W.; Lee, H.; Aftabizadeh, M.; et al. STAT3 Activation-Induced Fatty Acid Oxidation in CD8+ T Effector Cells Is Critical for Obesity-Promoted Breast Tumor Growth. Cell Metab. 2020, 31, 148–161. [Google Scholar] [CrossRef]
- Li, S.; Wei, X.; Zhan, X.; He, J.; Zeng, Y.; Tian, X.; Yuan, S.; Sun, L. Adipocyte-Derived Leptin Promotes PAI-1-Mediated Breast Cancer Metastasis in a STAT3/miR-34a Dependent Manner. Cancers 2020, 12, 3864. [Google Scholar] [CrossRef]
- Gonzalez, R.R.; Cherfils, S.; Escobar, M.; Yoo, J.H.; Carino, C.; Styer, A.K.; Sullivan, B.T.; Sakamoto, H.; Olawaiye, A.; Serikawa, T.; et al. Leptin Signaling Promotes the Growth of Mammary Tumors and Increases the Expression of Vascular Endothelial Growth Factor (VEGF) and Its Receptor Type Two (VEGF-R2). J. Biol. Chem. 2006, 281, 26320–26328. [Google Scholar] [CrossRef]
- Kim, H.G.; Jin, S.W.; Kim, Y.A.; Khanal, T.; Lee, G.H.; Kim, S.J.; Rhee, S.D.; Chung, Y.C.; Hwang, Y.J.; Jeong, T.C.; et al. Leptin induces CREB-dependent aromatase activation through COX-2 expression in breast cancer cells. Food Chem. Toxicol. 2017, 106, 232–241. [Google Scholar] [CrossRef]
- Luo, Y.; Li, H.; Zhang, Y.; Wu, Y.; Shen, D.; Che, Y. Combination of Endogenous Estradiol and Adipokine Leptin in Breast Cancer Risk and Prognosis Assessment in Postmenopausal Chinese Women. Front. Endocrinol. 2021, 12, 766463. [Google Scholar] [CrossRef]
- Van Gemert, W.A.; May, A.M.; Schuit, A.J.; Oosterhof, B.Y.M.; Peeters, P.H.; Monninkhof, E.M. Effect of Weight Loss with or without Exercise on Inflammatory Markers and Adipokines in Postmenopausal Women: The SHAPE-2 Trial, A Randomized Controlled Trial. Cancer Epidemiol. Biomark. Prev. 2016, 25, 799–806. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Arthur, R.; Manson, J.E.; Chlebowski, R.T.; Kroenke, C.H.; Peterson, L.; Cheng, T.D.; Feliciano, E.C.; Lane, D.; Luo, J.; et al. Association of Body Fat and Risk of Breast Cancer in Postmenopausal Women with Normal Body Mass Index. JAMA Oncol. 2019, 5, 155. [Google Scholar] [CrossRef]
- Sturgeon, K.; Digiovanni, L.; Good, J.; Salvatore, D.; Fenderson, D.; Domchek, S.; Stopfer, J.; Galantino, M.L.; Bryan, C.; Hwang, W.; et al. Exercise-Induced Dose-Response Alterations in Adiponectin and Leptin Levels Are Dependent on Body Fat Changes in Women at Risk for Breast Cancer. Cancer. Epidemiol. Biomark. Prev. 2016, 25, 1195–1200. [Google Scholar] [CrossRef]
- Choi, J.; Cha, Y.J.; Koo, J.S. Adipocyte biology in breast cancer: From silent bystander to active facilitator. Prog. Lipid Res. 2018, 69, 11–20. [Google Scholar] [CrossRef]
- Shahril, M.R.; Zakarai, N.S.; Appannah, G.; Nurnazahiah, A.; Mohamed, H.J.J.; Ahmad, A.; Lua, P.L.; Fenech, M. ‘Energy-Dense, High-SFA and Low-Fiber’ Dietary Pattern Lowered Adiponectin but Not Leptin Concentration of Breast Cancer Survivors. Nutrients 2021, 13, 3339. [Google Scholar] [CrossRef] [PubMed]
- Andò, S.; Naimo, G.D.; Gelsomino, L.; Catalano, S.; Mauro, L. Novel insights into adiponectin action in breast cancer: Evidence of its mechanistic effects mediated by ERα expression. Obes. Rev. 2020, 21, e13004. [Google Scholar] [CrossRef] [PubMed]
- Pham, D.V.; Park, P.H. Adiponectin triggers breast cancer cell death via fatty acid metabolic reprogramming. J. Exp. Clin. Cancer Res. 2022, 41, 9. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.J.; Nagaraju, G.P.; Nagalingam, A.; Muniraj, N.; Kuppusamy, P.; Walker, A.; Woo, J.; Gyorffy, B.; Gabrielson, E.; Saxena, N.K.; et al. ADIPOQ/adiponectin induces cytotoxic autophagy in breast cancer cells through STK11/LKB1-mediated activation of the AMPK-ULK1 axis. Autophagy 2017, 13, 1386–1403. [Google Scholar] [CrossRef]
- Macis, D.; Guerrieri-Gonzaga, A.; Gandini, S. Circulating adiponectin and breast cancer risk: A systematic review and meta-analysis. Int. J. Epidemiol. 2014, 43, 1226–1236. [Google Scholar] [CrossRef]
- Yoon, Y.S.; Kwon, A.R.; Lee, Y.K.; Oh, S.W. Circulating adipokines and risk of obesity related cancers: A systematic review and meta-analysis. Obes. Res. Clin. Pract. 2019, 13, 329–339. [Google Scholar] [CrossRef]
- Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 2021, 33, 127–148. [Google Scholar] [CrossRef]
- Kim, H.S.; Jung, M.; Choi, S.K.; Woo, J.; Piao, Y.J.; Hwang, E.H.; Kim, H.; Kim, S.J.; Moon, W.K. IL-6-mediated cross-talk between human preadipocytes and ductal carcinoma in situ in breast cancer progression. J. Exp. Clin. Cancer Res. 2018, 37, 200. [Google Scholar] [CrossRef]
- He, J.; Wei, X.; Li, S.; Liu, Y.; Hu, H.; Li, Z.; Kuang, X.; Wang, L.; Shi, X.; Yuan, S.; et al. Adipocyte-derived IL-6 and leptin promote breast Cancer metastasis via upregulation of Lysyl Hydroxylase-2 expression. Cell Commun. Signal. 2018, 16, 100. [Google Scholar] [CrossRef]
- Weng, Y.; Tseng, H.; Chen, Y.; Shen, P.; Al Haq, A.T.; Chen, L.; Tung, Y.; Hsu, H. MCT-1/miR-34a/IL-6/IL-6R signaling axis promotes EMT progression, cancer stemness and M2 macrophage polarization in triple-negative breast cancer. Mol. Cancer 2019, 18, 42. [Google Scholar] [CrossRef]
- Siersbæk, R.; Scabia, V.; Nagarajan, S.; Chernukhin, I.; Papachristou, E.K.; Broome, R.; Johnston, S.J.; Joosten, S.E.P.; Green, A.R.; Kumar, S.; et al. IL6/STAT3 Signaling Hijacks Estrogen Receptor α Enhancers to Drive Breast Cancer Metastasis. Cancer Cell 2020, 38, 412–423. [Google Scholar] [CrossRef]
- Benoy, I.; Salgado, R.; Colpaert, C.; Weytjens, R.; Vermeulen, P.B.; Dirix, L.Y. Serum Interleukin 6, Plasma VEGF, Serum VEGF, and VEGF Platelet Load in Breast Cancer Patients. Clin. Breast Cancer 2002, 2, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Salgado, R.; Junius, S.; Benoy, I.; Van Dam, P.; Vermeulen, P.; Van Marck, E.; Huget, P.; Dirix, L.Y. Circulating interleukin-6 predicts survival in patients with metastatic breast cancer. Int. J. Cancer 2003, 103, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Goswami, B.; Mittal, P. Effect of standard anthracycline based neoadjuvant chemotherapy on circulating levels of serum IL-6 in patients of locally advanced carcinoma breast—A prospective study. Int. J. Surg. 2012, 10, 638–640. [Google Scholar] [CrossRef]
- Midavaine, É.; Côté, J.; Sarret, P. The multifaceted roles of the chemokines CCL2 and CXCL12 in osteophilic metastatic cancers. Cancer Metast. Rev. 2021, 40, 427–445. [Google Scholar] [CrossRef]
- Arendt, L.M.; McCready, J.; Keller, P.J.; Baker, D.D.; Naber, S.P.; Seewaldt, V.; Kuperwasser, C. Obesity Promotes Breast Cancer by CCL2-Mediated Macrophage Recruitment and Angiogenesis. Cancer Res. 2013, 73, 6080–6093. [Google Scholar] [CrossRef] [PubMed]
- Bonapace, L.; Coissieux, M.; Wyckoff, J.; Mertz, K.D.; Varga, Z.; Junt, T.; Bentires-Alj, M. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 2014, 515, 130–133. [Google Scholar] [CrossRef]
- Müller, A.K.; Köhler, U.A.; Trzebanski, S.; Vinik, Y.; Raj, H.M.; Girault, J.A.; Ben Chetrit, N.; Maraver, A.; Jung, S.; Lev, S. Mouse Modeling Dissecting Macrophage–Breast Cancer Communication Uncovered Roles of PYK2 in Macrophage Recruitment and Breast Tumorigenesis. Adv. Sci. 2022, 9, 2105696. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Cao, W.; Qiu, Y.; Zhou, Y.; Guo, Q.; Gao, Y.; Lu, N. Oroxylin A suppresses ACTN1 expression to inactivate cancer-associated fibroblasts and restrain breast cancer metastasis. Pharm. Res. 2020, 159, 104981. [Google Scholar] [CrossRef]
- Rogic, A.; Pant, I.; Grumolato, L.; Fernandez-Rodriguez, R.; Edwards, A.; Das, S.; Sun, A.; Yao, S.; Qiao, R.; Jaffer, S.; et al. High endogenous CCL2 expression promotes the aggressive phenotype of human inflammatory breast cancer. Nat. Commun. 2021, 12, 6889. [Google Scholar] [CrossRef]
- Velasco-Velázquez, M.; Xolalpa, W.; Pestell, R.G. The potential to target CCL5/CCR5 in breast cancer. Expert. Opin. Ther. Targets 2014, 18, 1265–1275. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Song, X.; Zhou, X.; Qin, Y.; Yang, J.; Wang, Y.; Sun, Z.; Yu, K.; Zhang, S.; Liu, S. Emodin inhibits epithelial-mesenchymal transition and metastasis of triple negative breast cancer via antagonism of CC-chemokine ligand 5 secreted from adipocytes. Int. J. Mol. Med. 2018, 42, 579–588. [Google Scholar]
- An, G.; Wu, F.; Huang, S.; Feng, L.; Bai, J.; Gu, S.; Zhao, X. Effects of CCL5 on the biological behavior of breast cancer and the mechanisms of its interaction with tumor-associated macrophages. Oncol. Rep. 2019, 42, 2499–2511. [Google Scholar] [CrossRef] [PubMed]
- Ban, Y.; Mai, J.; Li, X.; Mitchell-Flack, M.; Zhang, T.; Zhang, L.; Chouchane, L.; Ferrari, M.; Shen, H.; Ma, X. Targeting Autocrine CCL5–CCR5 Axis Reprograms Immunosuppressive Myeloid Cells and Reinvigorates Antitumor Immunity. Cancer Res. 2017, 77, 2857–2868. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Qin, J.; Zhong, L.; Gong, L.; Zhang, B.; Zhang, Y.; Gao, W. CCL5-Mediated Th2 Immune Polarization Promotes Metastasis in Luminal Breast Cancer. Cancer Res. 2015, 75, 4312–4321. [Google Scholar] [CrossRef] [PubMed]
- Sax, M.J.; Gasch, C.; Athota, V.R.; Freeman, R.; Rasighaemi, P.; Westcott, D.E.; Day, C.J.; Nikolic, I.; Elsworth, B.; Wei, M.; et al. Cancer cell CCL5 mediates bone marrow independent angiogenesis in breast cancer. Oncotarget 2016, 7, 85437–85449. [Google Scholar] [CrossRef]
- Zazo, S.; González-Alonso, P.; Martín-Aparicio, E.; Chamizo, C.; Luque, M.; Sanz-Álvarez, M.; Mínguez, P.; Gómez-López, G.; Cristóbal, I.; Caramés, C.; et al. Autocrine CCL5 Effect Mediates Trastuzumab Resistance by ERK Pathway Activation in HER2-Positive Breast Cancer. Mol. Cancer Ther. 2020, 19, 1696–1707. [Google Scholar] [CrossRef]
- Zhou, J.; Tulotta, C.; Ottewell, P.D. IL-1β in breast cancer bone metastasis. J. Expert. Rev. Mol. Med. 2022, 24, e11. [Google Scholar] [CrossRef]
- Perrier, S.; Caldefie-Chézet, F.; Vasson, M. IL-1 family in breast cancer: Potential interplay with leptin and other adipocytokines. FEBS Lett. 2009, 583, 259–265. [Google Scholar] [CrossRef]
- Pein, M.; Insua-Rodríguez, J.; Hongu, T.; Riedel, A.; Meier, J.; Wiedmann, L.; Decker, K.; Essers, M.A.G.; Sinn, H.; Spaich, S.; et al. Metastasis-initiating cells induce and exploit a fibroblast niche to fuel malignant colonization of the lungs. Nat. Commun. 2020, 11, 1494. [Google Scholar] [CrossRef]
- Pradhan, A.K.; Maji, S.; Bhoopathi, P.; Talukdar, S.; Mannangatti, P.; Guo, C.; Wang, X.; Cartagena, L.C.; Idowu, M.; Landry, J.W.; et al. Pharmacological inhibition of MDA-9/Syntenin blocks breast cancer metastasis through suppression of IL-1β. Proc. Natl. Acad. Sci. USA 2021, 118, e2103180118. [Google Scholar] [CrossRef] [PubMed]
- Kolb, R.; Kluz, P.; Tan, Z.W.; Borcherding, N.; Bormann, N.; Vishwakarma, A.; Balcziak, L.; Zhu, P.; Davies, B.S.; Gourronc, F.; et al. Obesity-associated inflammation promotes angiogenesis and breast cancer via angiopoietin-like 4. Oncogene 2019, 38, 2351–2363. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.A.; Fredrikson, G.N.; Ericson, U.; Gullberg, B.; Hedblad, B.; Engström, G.; Borgquist, S.; Nilsson, J.; Wirfält, E. Low-Grade Inflammation, Oxidative Stress and Risk of Invasive Post-Menopausal Breast Cancer—A Nested Case-Control Study from the Malmö Diet and Cancer Cohort. PLoS ONE 2016, 11, e158959. [Google Scholar] [CrossRef]
- Vilsmaier, T.; Rack, B.; König, A.; Friese, K.; Janni, W.; Jeschke, U.; Acher, T.W. Influence of Circulating Tumour Cells on Production of IL-1α, IL-1β and IL-12 in Sera of Patients with Primary Diagnosis of Breast Cancer Before Treatment. Anticancer Res. 2016, 36, 5227–5236. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hung, A.C.; Lo, S.; Yuan, S.F. Adipocytokines visfatin and resistin in breast cancer: Clinical relevance, biological mechanisms, and therapeutic potential. Cancer Lett. 2021, 498, 229–239. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, X.; He, Q.; Gimple, R.C.; Liao, Y.; Wang, L.; Wu, R.; Xie, Q.; Rich, J.N.; Shen, K.; et al. Adipocytes promote breast tumorigenesis through TAZ-dependent secretion of Resistin. Proc. Natl. Acad. Sci. USA 2020, 117, 33295–33304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, M.; Yin, N.; Zhang, J. The Expression Regulation and Biological Function of Autotaxin. Cells 2021, 10, 939. [Google Scholar] [CrossRef]
- Zeng, J.; Hu, J.; Lian, Y.; Jiang, Y.; Chen, B. SFRP5 is a target gene transcriptionally regulated by PPARgamma in 3T3-L1 adipocytes. Gene 2018, 641, 190–195. [Google Scholar] [CrossRef]
- Zhou, W.; Ye, C.; Li, L.; Liu, L.; Wang, F.; Yu, L.; Zhou, F.; Xiang, Y.; Wang, Y.; Yin, G.; et al. Adipocyte-derived SFRP5 inhibits breast cancer cells migration and invasion through Wnt and epithelial-mesenchymal transition signaling pathways. Chin. J. Cancer Res. 2020, 32, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Li, B.; Li, J.; Sun, S.; Yuan, J.; Sun, S. Cancer-associated adipocytes as immunomodulators in cancer. Biomark. Res. 2021, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Queen, M.M.; Ryan, R.E.; Holzer, R.G.; Keller-Peck, C.R.; Jorcyk, C.L. Breast Cancer Cells Stimulate Neutrophils to Produce Oncostatin M: Potential Implications for Tumor Progression. Cancer Res. 2005, 65, 8896–8904. [Google Scholar] [CrossRef] [PubMed]
- Bell, L.N.; Cai, L.; Johnstone, B.H.; Traktuev, D.O.; March, K.L.; Considine, R.V. A central role for hepatocyte growth factor in adipose tissue angiogenesis. Am. J. Physiol.-Endoc. Metab. 2008, 294, E336–E344. [Google Scholar] [CrossRef]
- Sam, M.R.; Elliott, B.E.; Mueller, C.R. A novel activating role of SRC and STAT3 on HGF transcription in human breast cancer cells. Mol. Cancer 2007, 6, 69. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, J.; Ogawa, M.; Yamashita, S.; Nomura, K.; Kuramoto, M.; Saishoji, T.; Shin, S. Immunoreactive hepatocyte growth factor is a strong and independent predictor of recurrence and survival in human breast cancer. Cancer Res. 1994, 54, 1630–1633. [Google Scholar]
- Liu, L.; Wu, Y.; Zhang, C.; Zhou, C.; Li, Y.; Zeng, Y.; Zhang, C.; Li, R.; Luo, D.; Wang, L.; et al. Cancer-associated adipocyte-derived G-CSF promotes breast cancer malignancy via Stat3 signaling. J. Mol. Cell. Biol. 2020, 12, 723–737. [Google Scholar] [CrossRef]
- Su, X.; Xu, Y.; Fox, G.C.; Xiang, J.; Kwakwa, K.A.; Davis, J.L.; Belle, J.I.; Lee, W.; Wong, W.H.; Fontana, F.; et al. Breast cancer–derived GM-CSF regulates arginase 1 in myeloid cells to promote an immunosuppressive microenvironment. J. Clin. Investig. 2021, 131, e145296. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Foekens, J.A.; Peters, H.A.; Grebenchtchikov, N.; Look, M.P.; Meijer-van, G.M.; Geurts-Moespot, A.; van der Kwast, T.H.; Sweep, C.G.; Klijn, J.G. High tumor levels of vascular endothelial growth factor predict poor response to systemic therapy in advanced breast cancer. Cancer Res. 2001, 61, 5407–5414. [Google Scholar]
- D’Esposito, V.; Passaretti, F.; Hammarstedt, A.; Liguoro, D.; Terracciano, D.; Molea, G.; Canta, L.; Miele, C.; Smith, U.; Beguinot, F.; et al. Adipocyte-released insulin-like growth factor-1 is regulated by glucose and fatty acids and controls breast cancer cell growth in vitro. Diabetologia 2012, 55, 2811–2822. [Google Scholar] [CrossRef]
- Zeng, L.; Biernacka, K.M.; Holly, J.M.P.; Jarrett, C.; Morrison, A.A.; Morgan, A.; Winters, Z.E.; Foulstone, E.J.; Shield, J.P.; Perks, C.M. Hyperglycaemia confers resistance to chemotherapy on breast cancer cells: The role of fatty acid synthase. Endocr.-Relat. Cancer 2010, 17, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; He, X.; Tong, C.; Li, H.; Xie, C.; Wu, Y.; Wang, L.; Yan, X.; Luo, D.; Tang, Y.; et al. Cancer-associated adipocytes promote the invasion and metastasis in breast cancer through LIF/CXCLs positive feedback loop. Int. J. Biol. Sci. 2022, 18, 1363–1380. [Google Scholar] [CrossRef]
- Cox, T.R. The matrix in cancer. Nat. Rev. Cancer 2021, 21, 217–238. [Google Scholar] [CrossRef] [PubMed]
- Dinca, S.C.; Greiner, D.; Weidenfeld, K.; Bond, L.; Barkan, D.; Jorcyk, C.L. Novel mechanism for OSM-promoted extracellular matrix remodeling in breast cancer: LOXL2 upregulation and subsequent ECM alignment. Breast Cancer Res. 2021, 23, 56. [Google Scholar] [CrossRef] [PubMed]
- Juárez-Cruz, J.C.; Zuñiga-Eulogio, M.D.; Olea-Flores, M.; Castañeda-Saucedo, E.; Mendoza-Catalán, M.Á.; Ortuño-Pineda, C.; Moreno-Godínez, M.E.; Villegas-Comonfort, S.; Padilla-Benavides, T.; Navarro-Tito, N. Leptin induces cell migration and invasion in a FAK-Src-dependent manner in breast cancer cells. Endocr. Connect. 2019, 8, 1539–1552. [Google Scholar] [CrossRef]
- Iyengar, P.; Espina, V.; Williams, T.W.; Lin, Y.; Berry, D.; Jelicks, L.A.; Lee, H.; Temple, K.; Graves, R.; Pollard, J.; et al. Adipocyte-derived collagen VI affects early mammary tumor progression in vivo, demonstrating a critical interaction in the tumor/stroma microenvironment. J. Clin. Investig. 2005, 115, 1163–1176. [Google Scholar] [CrossRef]
- Vasiukov, G.; Novitskaya, T.; Zijlstra, A.; Owens, P.; Ye, F.; Zhao, Z.; Moses, H.L.; Blackwell, T.; Feoktistov, I.; Novitskiy, S.V. Myeloid Cell–Derived TGFβ Signaling Regulates ECM Deposition in Mammary Carcinoma via Adenosine-Dependent Mechanisms. Cancer Res. 2020, 80, 2628–2638. [Google Scholar] [CrossRef]
- Elia, I.; Rossi, M.; Stegen, S.; Broekaert, D.; Doglioni, G.; van Gorsel, M.; Boon, R.; Escalona-Noguero, C.; Torrekens, S.; Verfaillie, C.; et al. Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature 2019, 568, 117–121. [Google Scholar] [CrossRef]
- Germain, D. Estrogen Carcinogenesis in Breast Cancer. Endocrin. Metab. Clin. 2011, 40, 473–484. [Google Scholar] [CrossRef]
- Van Landeghem, A.A.; Poortman, J.; Nabuurs, M.; Thijssen, J.H. Endogenous concentration and subcellular distribution of androgens in normal and malignant human breast tissue. Cancer Res. 1985, 45, 2907–2912. [Google Scholar]
- Miller, W.R. Aromatase activity in breast tissue. J. Steroid Biochem. Mol. Biol. 1991, 39, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, P.; Au, C.C.; Benito-Martin, A.; Ladumor, H.; Oshchepkova, S.; Moges, R.; Brown, K.A. Estrogens and breast cancer: Mechanisms involved in obesity-related development, growth and progression. J. Steroid Biochem. Mol. Biol. 2019, 189, 161–170. [Google Scholar] [CrossRef]
- Wang, X.; Simpson, E.R.; Brown, K.A. Aromatase overexpression in dysfunctional adipose tissue links obesity to postmenopausal breast cancer. J. Steroid Biochem. Mol. Biol. 2015, 153, 35–44. [Google Scholar] [CrossRef]
- Ka, N.L.; Lim, G.Y.; Kim, S.S.; Hwang, S.; Han, J.; Lee, Y.H.; Lee, M.O. Type I IFN stimulates IFI16-mediated aromatase expression in adipocytes that promotes E2-dependent growth of ER-positive breast cancer. Cell. Mol. Life Sci. 2022, 79, 306. [Google Scholar] [CrossRef] [PubMed]
- Provance, O.K.; Lewis-Wambi, J. Deciphering the role of interferon alpha signaling and microenvironment crosstalk in inflammatory breast cancer. Breast Cancer Res. 2019, 21, 59. [Google Scholar] [CrossRef]
- Pouyssegur, J.; Marchiq, I.; Parks, S.K.; Durivault, J.; Zdralevic, M.; Vucetic, M. ‘Warburg effect’ controls tumor growth, bacterial, viral infections and immunity—Genetic deconstruction and therapeutic perspectives. Semin. Cancer Biol. 2022, 86, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.A. Metabolic pathways in obesity-related breast cancer. Nat. Rev. Endocrinol. 2021, 17, 350–363. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Docanto, M.M.; Sasano, H.; Lo, C.; Simpson, E.R.; Brown, K.A. Prostaglandin E2 Inhibits p53 in Human Breast Adipose Stromal Cells: A Novel Mechanism for the Regulation of Aromatase in Obesity and Breast Cancer. Cancer Res. 2015, 75, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C. Metabolism and cancer: The future is now. Br. J. Cancer 2020, 122, 133–135. [Google Scholar] [CrossRef] [PubMed]
- Felmlee, M.A.; Jones, R.S.; Rodriguez-Cruz, V.; Follman, K.E.; Morris, M.E. Monocarboxylate Transporters (SLC16): Function, Regulation, and Role in Health and Disease. Pharm. Rev. 2020, 72, 466–485. [Google Scholar] [CrossRef]
- Sotgia, F.; Martinez-Outschoorn, U.E.; Howell, A.; Pestell, R.G.; Pavlides, S.; Lisanti, M.P. Caveolin-1 and cancer metabolism in the tumor microenvironment: Markers, models, and mechanisms. Annu. Rev. Pathol. 2012, 7, 423–467. [Google Scholar] [CrossRef]
- Sotgia, F.; Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Pestell, R.G.; Lisanti, M.P. Understanding the Warburg effect and the prognostic value of stromal caveolin-1 as a marker of a lethal tumor microenvironment. Breast Cancer Res. 2011, 13, 213. [Google Scholar] [CrossRef]
- Singh, R.; Parveen, M.; Basgen, J.M.; Fazel, S.; Meshesha, M.F.; Thames, E.C.; Moore, B.; Martinez, L.; Howard, C.B.; Vergnes, L.; et al. Increased Expression of Beige/Brown Adipose Markers from Host and Breast Cancer Cells Influence Xenograft Formation in Mice. Mol. Cancer Res. 2016, 14, 78–92. [Google Scholar] [CrossRef]
- Grabner, G.F.; Xie, H.; Schweiger, M.; Zechner, R. Lipolysis: Cellular mechanisms for lipid mobilization from fat stores. Nat. Metab. 2021, 3, 1445–1465. [Google Scholar] [CrossRef]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V. Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef]
- Glatz, J.F.; Luiken, J.J. From fat to FAT (CD36/SR-B2): Understanding the regulation of cellular fatty acid uptake. Biochimie 2017, 136, 21–26. [Google Scholar] [CrossRef]
- Guaita-Esteruelas, S.; Saavedra-García, P.; Bosquet, A.; Borràs, J.; Girona, J.; Amiliano, K.; Rodríguez-Balada, M.; Heras, M.; Masana, L.; Gumà, J. Adipose-Derived Fatty Acid-Binding Proteins Plasma Concentrations Are Increased in Breast Cancer Patients. Oncologist 2017, 22, 1309–1315. [Google Scholar] [CrossRef]
- Yang, D.; Li, Y.; Xing, L.; Tan, Y.; Sun, J.; Zeng, B.; Xiang, T.; Tan, J.; Ren, G.; Wang, Y. Utilization of adipocyte-derived lipids and enhanced intracellular trafficking of fatty acids contribute to breast cancer progression. Cell Commun. Signal. 2018, 16, 1–12. [Google Scholar] [CrossRef]
- Gyamfi, J.; Yeo, J.H.; Kwon, D.; Min, B.S.; Cha, Y.J.; Koo, J.S.; Jeong, J.; Lee, J.; Choi, J. Interaction between CD36 and FABP4 modulates adipocyte-induced fatty acid import and metabolism in breast cancer. NPJ Breast Cancer 2021, 7, 129. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Attané, C.; Milhas, D.; Dirat, B.; Dauvillier, S.; Guerard, A.; Gilhodes, J.; Lazar, I.; Alet, N.; Laurent, V.; et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2017, 2, e87489. [Google Scholar] [CrossRef] [PubMed]
- Puig, T.; Aguilar, H.; Cufi, S.; Oliveras, G.; Turrado, C.; Ortega-Gutierrez, S.; Benhamu, B.; Lopez-Rodriguez, M.L.; Urruticoechea, A.; Colomer, R. A novel inhibitor of fatty acid synthase shows activity against HER2+ breast cancer xenografts and is active in anti-HER2 drug-resistant cell lines. Breast Cancer Res. 2011, 13, R131. [Google Scholar] [CrossRef]
- Simińska, E.; Koba, M. Amino acid profiling as a method of discovering biomarkers for early diagnosis of cancer. Amino Acids 2016, 48, 1339–1345. [Google Scholar] [CrossRef]
- Munteanu, R.; Onaciu, A.; Moldovan, C.; Zimta, A.; Gulei, D.; Paradiso, A.; Lazar, V.; Berindan-Neagoe, I. Adipocyte-Based Cell Therapy in Oncology: The Role of Cancer-Associated Adipocytes and Their Reinterpretation as Delivery Platforms. Pharmaceutics 2020, 12, 402. [Google Scholar] [CrossRef]
- Huang, J.; Diaz-Meco, M.T.; Moscat, J. The macroenviromental control of cancer metabolism by p62. Cell Cycle 2018, 17, 2110–2121. [Google Scholar] [CrossRef] [PubMed]
- Balaban, S.; Shearer, R.F.; Lee, L.S.; van Geldermalsen, M.; Schreuder, M.; Shtein, H.C.; Cairns, R.; Thomas, K.C.; Fazakerley, D.J.; Grewal, T.; et al. Adipocyte lipolysis links obesity to breast cancer growth: Adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 2017, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Salimian Rizi, B.; Caneba, C.; Nowicka, A.; Nabiyar, A.W.; Liu, X.; Chen, K.; Klopp, A.; Nagrath, D. Nitric Oxide Mediates Metabolic Coupling of Omentum-Derived Adipose Stroma to Ovarian and Endometrial Cancer Cells. Cancer Res. 2015, 75, 456–471. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Marabelle, A.; Eggermont, A.; Soria, J.C.; Kroemer, G.; Zitvogel, L. Targeting the tumor microenvironment: Removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. 2016, 27, 1482–1492. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.B.; Yeh, E.S.; Soloff, A.C. Tumor-associated macrophages: Unwitting accomplices in breast cancer malignancy. NPJ Breast Cancer 2016, 2, 15025. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Maller, O.; Drain, A.P.; Barrett, A.S.; Borgquist, S.; Ruffell, B.; Zakharevich, I.; Pham, T.T.; Gruosso, T.; Kuasne, H.; Lakins, J.N.; et al. Tumour-associated macrophages drive stromal cell-dependent collagen crosslinking and stiffening to promote breast cancer aggression. Nat. Mater. 2021, 20, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Sossey-Alaoui, K.; Pluskota, E.; Bialkowska, K.; Szpak, D.; Parker, Y.; Morrison, C.D.; Lindner, D.J.; Schiemann, W.P.; Plow, E.F. Kindlin-2 Regulates the Growth of Breast Cancer Tumors by Activating CSF-1–Mediated Macrophage Infiltration. Cancer Res. 2017, 77, 5129–5141. [Google Scholar] [CrossRef]
- Corrêa, L.H.; Corrêa, R.; Farinasso, C.M.; de Sant Ana Dourado, L.P.; Magalhães, K.G. Adipocytes and Macrophages Interplay in the Orchestration of Tumor Microenvironment: New Implications in Cancer Progression. Front. Immunol. 2017, 8, 1129. [Google Scholar] [CrossRef]
- Mu, X.; Shi, W.; Xu, Y.; Xu, C.; Zhao, T.; Geng, B.; Yang, J.; Pan, J.; Hu, S.; Zhang, C.; et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle 2018, 17, 428–438. [Google Scholar] [CrossRef]
- Engin, A.B.; Engin, A.; Gonul, I.I. The effect of adipocyte–macrophage crosstalk in obesity-related breast cancer. J. Mol. Endocrinol. 2019, 62, R201–R222. [Google Scholar] [CrossRef]
- Vazquez Rodriguez, G.; Abrahamsson, A.; Jensen, L.D.E.; Dabrosin, C. Adipocytes Promote Early Steps of Breast Cancer Cell Dissemination via Interleukin-8. Front. Immunol. 2018, 9, 1767. [Google Scholar] [CrossRef]
- Michelet, X.; Dyck, L.; Hogan, A.; Loftus, R.M.; Duquette, D.; Wei, K.; Beyaz, S.; Tavakkoli, A.; Foley, C.; Donnelly, R.; et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat. Immunol. 2018, 19, 1330–1340. [Google Scholar] [CrossRef]
- Wu, B.; Sun, X.; Gupta, H.B.; Yuan, B.; Li, J.; Ge, F.; Chiang, H.C.; Zhang, X.; Zhang, C.; Zhang, D.; et al. Adipose PD-L1 Modulates PD-1/PD-L1 Checkpoint Blockade Immunotherapy Efficacy in Breast Cancer. Oncoimmunology 2018, 7, e1500107. [Google Scholar] [CrossRef] [PubMed]
- Hillers-Ziemer, L.E.; Kuziel, G.; Williams, A.E.; Moore, B.N.; Arendt, L.M. Breast cancer microenvironment and obesity: Challenges for therapy. Cancer. Metastasis Rev. 2022, 41, 627–647. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Fahrmann, J.F.; Lee, H.; Li, Y.; Tripathi, S.C.; Yue, C.; Zhang, C.; Lifshitz, V.; Song, J.; Yuan, Y.; et al. JAK/STAT3-Regulated Fatty Acid β-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab. 2018, 27, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.S.; Zeng, D.; Liang, Y.K.; Wu, Y.; Li, M.F.; Qi, Y.Z.; Wei, X.L.; Huang, W.H.; Chen, M.; Zhang, G.J. Major vault protein is a direct target of Notch1 signaling and contributes to chemoresistance in triple-negative breast cancer cells. Cancer Lett. 2019, 440–441, 156–167. [Google Scholar] [CrossRef]
- Lehuédé, C.; Li, X.; Dauvillier, S.; Vaysse, C.; Franchet, C.; Clement, E.; Esteve, D.; Longué, M.; Chaltiel, L.; Le Gonidec, S.; et al. Adipocytes promote breast cancer resistance to chemotherapy, a process amplified by obesity: Role of the major vault protein (MVP). Breast Cancer Res. 2019, 21, 7. [Google Scholar] [CrossRef] [PubMed]
- Plava, J.; Cihova, M.; Burikova, M.; Matuskova, M.; Kucerova, L.; Miklikova, S. Recent advances in understanding tumor stroma-mediated chemoresistance in breast cancer. Mol. Cancer 2019, 18, 67. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Kim, G.I.; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; D’Angelo, R.; Paulson, A.K.; et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol. Cell 2012, 47, 570–584. [Google Scholar] [CrossRef]
- Han, J.; Qu, H.; Han, M.; Ding, Y.; Xie, M.; Hu, J.; Chen, Y.; Dong, H. MSC-induced lncRNA AGAP2-AS1 promotes stemness and trastuzumab resistance through regulating CPT1 expression and fatty acid oxidation in breast cancer. Oncogene 2021, 40, 833–847. [Google Scholar] [CrossRef]
- Duong, M.N.; Cleret, A.; Matera, E.; Chettab, K.; Mathé, D.; Valsesia-Wittmann, S.; Clémenceau, B.; Dumontet, C. Adipose cells promote resistance of breast cancer cells to trastuzumab-mediated antibody-dependent cellular cytotoxicity. Breast Cancer Res. 2015, 17, 57. [Google Scholar] [CrossRef]
- Komurov, K.; Tseng, J.T.; Muller, M.; Seviour, E.G.; Moss, T.J.; Yang, L.; Nagrath, D.; Ram, P.T. The glucose-deprivation network counteracts lapatinib-induced toxicity in resistant ErbB2-positive breast cancer cells. Mol. Syst. Biol. 2012, 8, 596. [Google Scholar] [CrossRef]
- Morgan, M.M.; Arendt, L.M.; Alarid, E.T.; Beebe, D.J.; Johnson, B.P. Mammary adipose stromal cells derived from obese women reduce sensitivity to the aromatase inhibitor anastrazole in an organotypic breast model. FASEB J. 2019, 33, 8623–8633. [Google Scholar] [CrossRef]
- Giordano, C.; Vizza, D.; Panza, S.; Barone, I.; Bonofiglio, D.; Lanzino, M.; Sisci, D.; De Amicis, F.; Fuqua, S.A.W.; Catalano, S.; et al. Leptin increases HER2 protein levels through a STAT3-mediated up-regulation of Hsp90 in breast cancer cells. Mol. Oncol. 2013, 7, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Delort, L.; Bougaret, L.; Cholet, J.; Vermerie, M.; Billard, H.; Decombat, C.; Bourgne, C.; Berger, M.; Dumontet, C.; Caldefie-Chezet, F. Hormonal Therapy Resistance and Breast Cancer: Involvement of Adipocytes and Leptin. Nutrients. 2019, 11, 2839. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, Q.; Yang, N.; Shi, Q.; Su, H.; Lin, T.; He, Z.; Wang, W.; Guo, H.; Shen, P. Crosstalk between IL-15Rα+ tumor-associated macrophages and breast cancer cells reduces CD8+ T cell recruitment. Cancer Commun. 2022, 42, 536–557. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.J.; Koo, J.S. Adipokines as therapeutic targets in breast cancer treatment. Expert Opin. Ther. Targets 2018, 22, 941–953. [Google Scholar] [CrossRef]
- Teufelsbauer, M.; Rath, B.; Plangger, A.; Staud, C.; Nanobashvili, J.; Huk, I.; Neumayer, C.; Hamilton, G.; Radtke, C. Effects of metformin on adipose-derived stromal cell (ADSC) – Breast cancer cell lines interaction. Life Sci. 2020, 261, 118371. [Google Scholar] [CrossRef] [PubMed]
- Rasha, F.; Ramalingam, L.; Menikdiwela, K.; Hernandez, A.; Moussa, H.; Gollahon, L.; Layeequr Rahman, R.; Moustaid-Moussa, N. Renin angiotensin system inhibition attenuates adipocyte-breast cancer cell interactions. Exp. Cell Res. 2020, 394, 112114. [Google Scholar] [CrossRef]
- Gonzalez, S.N.; Rodriguez, T.S.; Ouanouki, A.; El, C.L.; Annabi, B. EGCG Inhibits Adipose-Derived Mesenchymal Stem Cells Differentiation into Adipocytes and Prevents a STAT3-Mediated Paracrine Oncogenic Control of Triple-Negative Breast Cancer Cell Invasive Phenotype. Molecules 2021, 26, 1506. [Google Scholar] [CrossRef]
- Hsieh, C.; Chiu, H.; Wang, C.; Kuo, C. Aspirin Modifies Inflammatory Mediators and Metabolomic Profiles and Contributes to the Suppression of Obesity-Associated Breast Cancer Cell Growth. Int. J. Mol. Sci. 2020, 21, 4652. [Google Scholar] [CrossRef]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef]
- Otvos, L.J.; Haspinger, E.; La Russa, F.; Maspero, F.; Graziano, P.; Kovalszky, I.; Lovas, S.; Nama, K.; Hoffmann, R.; Knappe, D.; et al. Design and development of a peptide-based adiponectin receptor agonist for cancer treatment. BMC Biotechnol. 2011, 11, 90. [Google Scholar] [CrossRef]
- Taliaferro-Smith, L.; Nagalingam, A.; Knight, B.B.; Oberlick, E.; Saxena, N.K.; Sharma, D. Integral Role of PTP1B in Adiponectin-Mediated Inhibition of Oncogenic Actions of Leptin in Breast Carcinogenesis. Neoplasia 2013, 15, 11–23. [Google Scholar] [CrossRef]
- Rene Gonzalez, R.; Watters, A.; Xu, Y.; Singh, U.P.; Mann, D.R.; Rueda, B.R.; Penichet, M.L. Leptin-signaling inhibition results in efficient anti-tumor activity in estrogen receptor positive or negative breast cancer. Breast Cancer Res. 2009, 11, R36. [Google Scholar] [CrossRef]
- Hong, C.; Schubert, M.; Tijhuis, A.E.; Requesens, M.; Roorda, M.; van den Brink, A.; Ruiz, L.A.; Bakker, P.L.; van der Sluis, T.; Pieters, W.; et al. cGAS–STING drives the IL-6-dependent survival of chromosomally instable cancers. Nature 2022, 607, 366–373. [Google Scholar] [CrossRef]
- Wolfsberger, J.; Sakil, H.A.M.; Zhou, L.; van Bree, N.; Baldisseri, E.; de Souza Ferreira, S.; Zubillaga, V.; Stantic, M.; Fritz, N.; Hartman, J.; et al. TAp73 represses NF-κB–mediated recruitment of tumor-associated macrophages in breast cancer. Proc. Natl. Acad. Sci. USA 2021, 118, e2017089118. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tiruthani, K.; Wang, M.; Zhou, X.; Qiu, N.; Xiong, Y.; Pecot, C.V.; Liu, R.; Huang, L. Tumor-targeted gene therapy with lipid nanoparticles inhibits tumor-associated adipocytes and remodels the immunosuppressive tumor microenvironment in triple-negative breast cancer. Nanoscale Horiz. 2021, 6, 319–329. [Google Scholar] [CrossRef]
- Nie, Y.; Huang, H.; Guo, M.; Chen, J.; Wu, W.; Li, W.; Xu, X.; Lin, X.; Fu, W.; Yao, Y.; et al. Breast Phyllodes Tumors Recruit and Repolarize Tumor-Associated Macrophages via Secreting CCL5 to Promote Malignant Progression, Which Can Be Inhibited by CCR5 Inhibition Therapy. Clin. Cancer Res. 2019, 25, 3873–3886. [Google Scholar] [CrossRef] [PubMed]
- Tulotta, C.; Lefley, D.V.; Freeman, K.; Gregory, W.M.; Hanby, A.M.; Heath, P.R.; Nutter, F.; Wilkinson, J.M.; Spicer-Hadlington, A.R.; Liu, X.; et al. Endogenous Production of IL1B by Breast Cancer Cells Drives Metastasis and Colonization of the Bone Microenvironment. Clin. Cancer Res. 2019, 25, 2769–2782. [Google Scholar] [CrossRef] [PubMed]
- Reggiani, F.; Labanca, V.; Mancuso, P.; Rabascio, C.; Talarico, G.; Orecchioni, S.; Manconi, A.; Bertolini, F. Adipose Progenitor Cell Secretion of GM-CSF and MMP9 Promotes a Stromal and Immunological Microenvironment That Supports Breast Cancer Progression. Cancer Res. 2017, 77, 5169–5182. [Google Scholar] [CrossRef]
- Al-Jawadi, A.; Rasha, F.; Ramalingam, L.; Alhaj, S.; Moussa, H.; Gollahon, L.; Dharmawardhane, S.; Moustaid-Moussa, N. Protective effects of eicosapentaenoic acid in adipocyte-breast cancer cell cross talk. J. Nutr. Biochem. 2020, 75, 108244. [Google Scholar] [CrossRef]
- Adinew, G.M.; Taka, E.; Mochona, B.; Badisa, R.B.; Mazzio, E.A.; Elhag, R.; Soliman, K.F.A. Therapeutic Potential of Thymoquinone in Triple-Negative Breast Cancer Prevention and Progression through the Modulation of the Tumor Microenvironment. Nutrients 2022, 14, 79. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Suarez, N.; Fernandez-Marrero, Y.; Torabidastgerdooei, S.; Annabi, B. EGCG Prevents the Onset of an Inflammatory and Cancer-Associated Adipocyte-like Phenotype in Adipose-Derived Mesenchymal Stem/Stromal Cells in Response to the Triple-Negative Breast Cancer Secretome. Nutrients 2022, 14, 1099. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.; Dong, S.; Huang, R.; Chen, X. Cancer-Associated Adipocytes and Breast Cancer: Intertwining in the Tumor Microenvironment and Challenges for Cancer Therapy. Cancers 2023, 15, 726. https://doi.org/10.3390/cancers15030726
Wu C, Dong S, Huang R, Chen X. Cancer-Associated Adipocytes and Breast Cancer: Intertwining in the Tumor Microenvironment and Challenges for Cancer Therapy. Cancers. 2023; 15(3):726. https://doi.org/10.3390/cancers15030726
Chicago/Turabian StyleWu, Chenghui, Shuwen Dong, Renhong Huang, and Xiaosong Chen. 2023. "Cancer-Associated Adipocytes and Breast Cancer: Intertwining in the Tumor Microenvironment and Challenges for Cancer Therapy" Cancers 15, no. 3: 726. https://doi.org/10.3390/cancers15030726
APA StyleWu, C., Dong, S., Huang, R., & Chen, X. (2023). Cancer-Associated Adipocytes and Breast Cancer: Intertwining in the Tumor Microenvironment and Challenges for Cancer Therapy. Cancers, 15(3), 726. https://doi.org/10.3390/cancers15030726