


Deletion of Glycogen Synthase Kinase 3 Beta Reprograms NK Cell Metabolism

 , , , and
, , , and 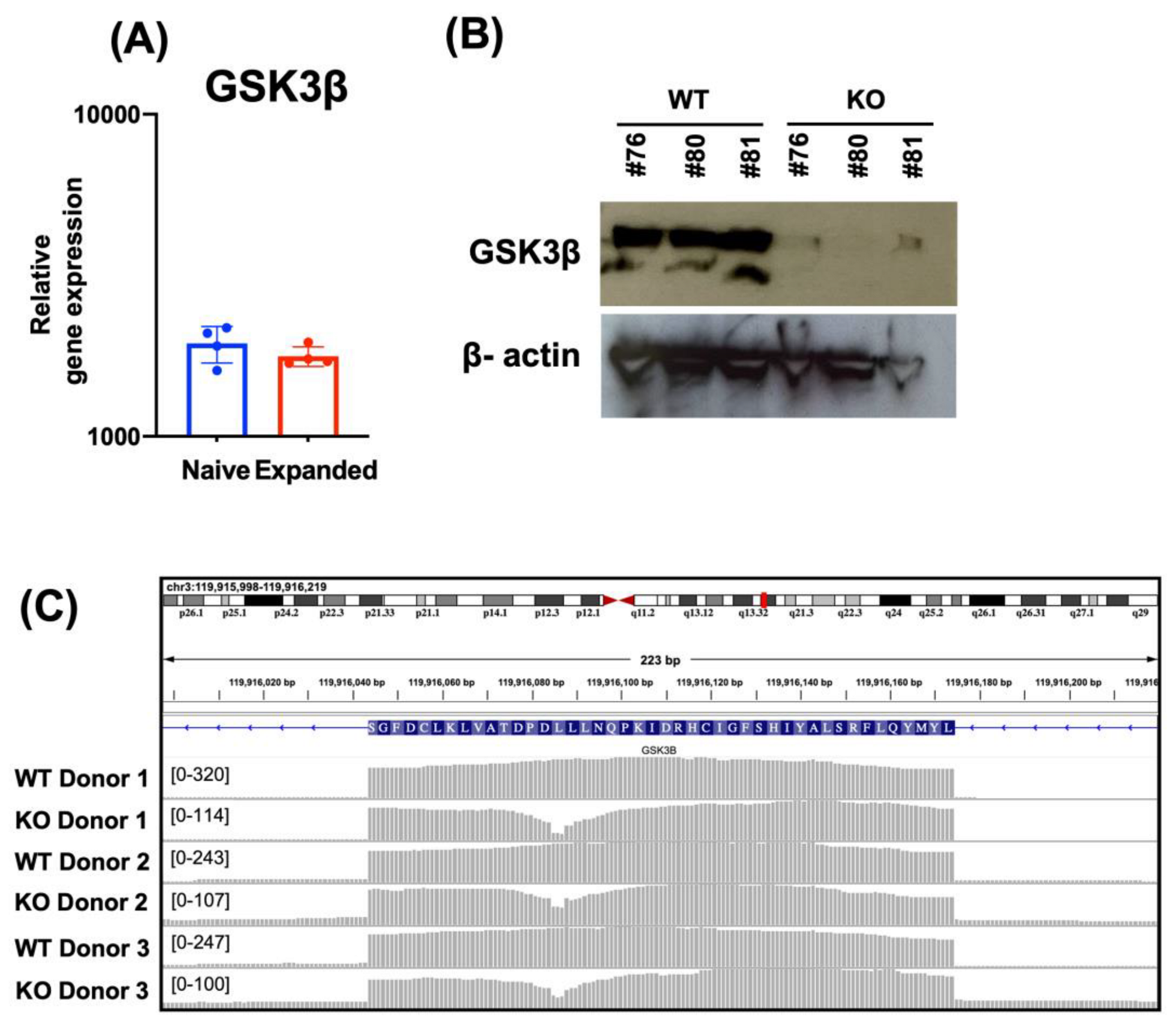{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Source of Primary Human NK Cells and Regulatory Approvals
2.2. Purification and Expansion of NK Cells
2.3. Tumor Cell Lines
2.4. Generation of CRISPR-Edited NK Cells
2.5. NK Cell Functional Assays
2.6. Metabolic Assays
2.7. Antibodies
2.8. Cytokine Secretion
2.9. RNA-Sequencing on Non-Expanded Healthy and AML-NK
2.10. RNA-Sequencing on Expanded WT and GSK3B-KO NK Cells
3. Results
3.1. Deletion of GSK3B in FC-21 Expanded NK Cells
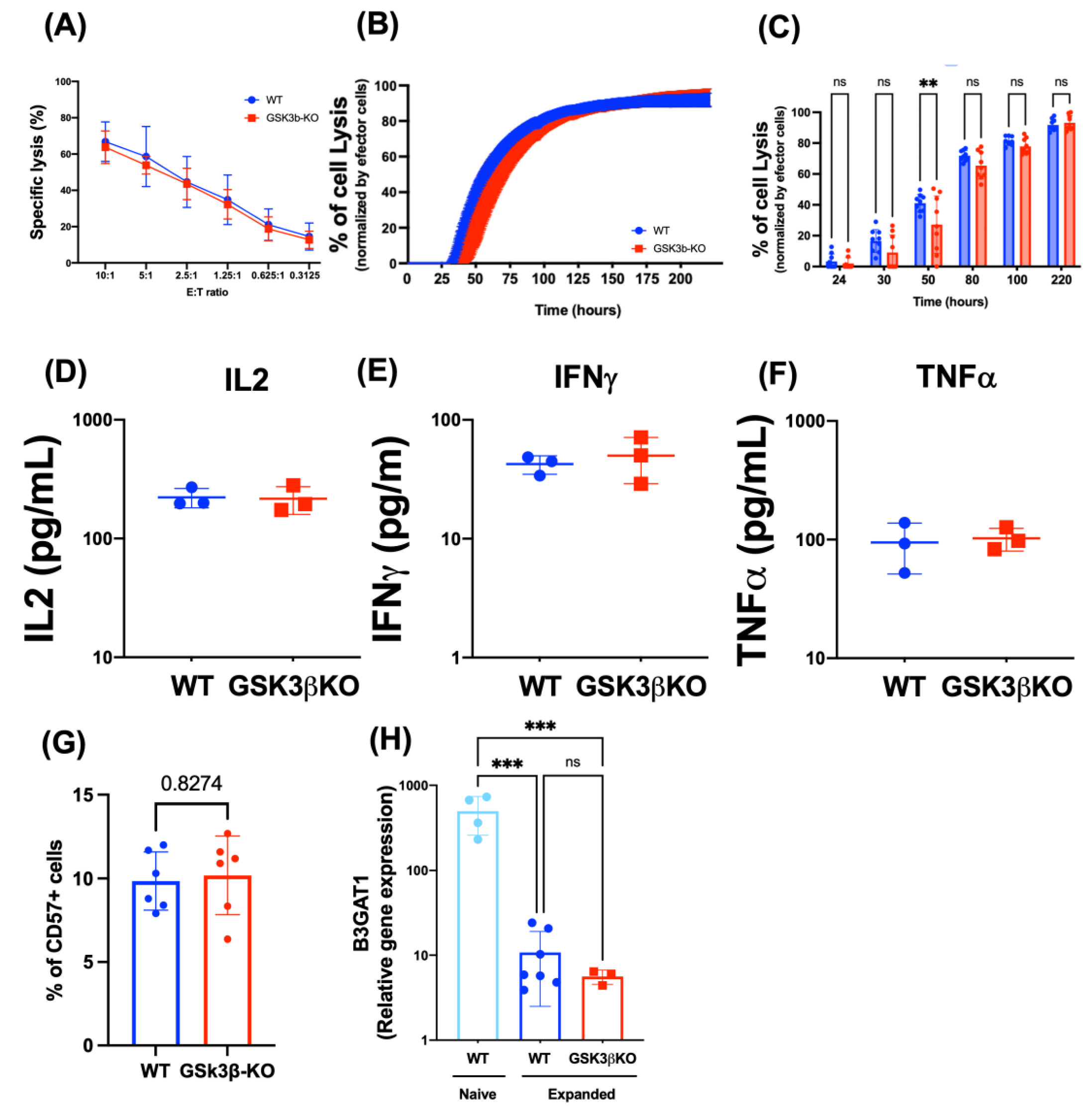
3.2. Deletion of GSK3B Does Not Alter Proliferation, Killing Potency, Cytokine Secretion, or Maturation of FC-21 Expanded NK Cells
3.3. Transcriptional Changes in GSK3B-KO NK Cells
3.4. Deletion of GSK3B Leads to Metabolic Reprogramming of NK Cells
3.5. GSK3β Is Highly Expressed in NK Cells at Stage 5 and 6 of Maturation from AML Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, D.A. Cellular therapy: Adoptive immunotherapy with expanded natural killer cells. Immunol. Rev. 2019, 290, 85–99. [Google Scholar] [CrossRef]
- Loftus, R.M.; Finlay, D.K. Immunometabolism: Cellular Metabolism Turns Immune Regulator. J. Biol. Chem. 2016, 291, 1–10. [Google Scholar] [CrossRef]
- Poznanski, S.M.; Singh, K.; Ritchie, T.M.; Aguiar, J.A.; Fan, I.Y.; Portillo, A.L.; Rojas, E.A.; Vahedi, F.; El-Sayes, A.; Xing, S.; et al. Metabolic flexibility determines human NK cell functional fate in the tumor microenvironment. Cell Metab. 2021, 33, 1205–1220.e5. [Google Scholar] [CrossRef] [PubMed]
- Naeimi Kararoudi, M.; Tullius, B.P.; Chakravarti, N.; Pomeroy, E.J.; Moriarity, B.S.; Beland, K.; Colamartino, A.B.L.; Haddad, E.; Chu, Y.; Cairo, M.S.; et al. Genetic and epigenetic modification of human primary NK cells for enhanced antitumor activity. Semin Hematol. 2020, 57, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Cichocki, F.; Valamehr, B.; Bjordahl, R.; Zhang, B.; Rezner, B.; Rogers, P.; Gaidarova, S.; Moreno, S.; Tuininga, K.; Dougherty, P.; et al. GSK3 Inhibition Drives Maturation of NK Cells and Enhances Their Antitumor Activity. Cancer Res. 2017, 77, 5664–5675. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.K.; Miller, J.S. Current strategies exploiting NK-cell therapy to treat haematologic malignancies. Int. J. Immunogenet. 2018, 45, 237–246. [Google Scholar] [CrossRef]
- Parameswaran, R.; Ramakrishnan, P.; Moreton, S.A.; Xia, Z.; Hou, Y.; Lee, D.A.; Gupta, K.; deLima, M.; Beck, R.C.; Wald, D.N. Repression of GSK3 restores NK cell cytotoxicity in AML patients. Nat. Commun. 2016, 7, 11154. [Google Scholar] [CrossRef] [PubMed]
- Chretien, A.S.; Fauriat, C.; Orlanducci, F.; Galseran, C.; Rey, J.; Bouvier Borg, G.; Gautherot, E.; Granjeaud, S.; Hamel-Broza, J.F.; Demerle, C.; et al. Natural Killer Defective Maturation Is Associated with Adverse Clinical Outcome in Patients with Acute Myeloid Leukemia. Front. Immunol. 2017, 8, 573. [Google Scholar] [CrossRef]
- Chretien, A.S.; Devillier, R.; Granjeaud, S.; Cordier, C.; Demerle, C.; Salem, N.; Wlosik, J.; Orlanducci, F.; Gorvel, L.; Fattori, S.; et al. High-dimensional mass cytometry analysis of NK cell alterations in AML identifies a subgroup with adverse clinical outcome. Proc. Natl. Acad. Sci. USA 2021, 118, e2020459118. [Google Scholar] [CrossRef]
- Mundy-Bosse, B.L.; Weigel, C.; Wu, Y.Z.; Abdelbaky, S.; Youssef, Y.; Casas, S.B.; Polley, N.; Ernst, G.; Young, K.A.; McConnell, K.K.; et al. Identification and Targeting of the Developmental Blockade in Extranodal Natural Killer/T-cell Lymphoma. Blood Cancer Discov. 2022, 3, 154–169. [Google Scholar] [CrossRef]
- Freud, A.G.; Becknell, B.; Roychowdhury, S.; Mao, H.C.; Ferketich, A.K.; Nuovo, G.J.; Hughes, T.L.; Marburger, T.B.; Sung, J.; Baiocchi, R.A.; et al. A human CD34(+) subset resides in lymph nodes and differentiates into CD56bright natural killer cells. Immunity 2005, 22, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Freud, A.G.; Yokohama, A.; Becknell, B.; Lee, M.T.; Mao, H.C.; Ferketich, A.K.; Caligiuri, M.A. Evidence for discrete stages of human natural killer cell differentiation in vivo. J. Exp. Med. 2006, 203, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Youssef, Y.; Robinson, C.; Ernst, G.F.; Carson, M.Y.; Young, K.A.; Scoville, S.D.; Zhang, X.; Harris, R.; Sekhri, P.; et al. CD56 Expression Marks Human Group 2 Innate Lymphoid Cell Divergence from a Shared NK Cell and Group 3 Innate Lymphoid Cell Developmental Pathway. Immunity 2018, 49, 464–476.e464. [Google Scholar] [CrossRef] [PubMed]
- Scoville, S.D.; Mundy-Bosse, B.L.; Zhang, M.H.; Chen, L.; Zhang, X.; Keller, K.A.; Hughes, T.; Chen, L.; Cheng, S.; Bergin, S.M.; et al. A Progenitor Cell Expressing Transcription Factor RORgammat Generates All Human Innate Lymphoid Cell Subsets. Immunity 2016, 44, 1140–1150. [Google Scholar] [CrossRef]
- Freud, A.G.; Keller, K.A.; Scoville, S.D.; Mundy-Bosse, B.L.; Cheng, S.; Youssef, Y.; Hughes, T.; Zhang, X.; Mo, X.; Porcu, P.; et al. NKp80 Defines a Critical Step during Human Natural Killer Cell Development. Cell Rep. 2016, 16, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Moseman, J.E.; Foltz, J.A.; Sorathia, K.; Heipertz, E.L.; Lee, D.A. Evaluation of serum-free media formulations in feeder cell-stimulated expansion of natural killer cells. Cytotherapy 2020, 22, 322–328. [Google Scholar] [CrossRef]
- Naeimi Kararoudi, M.; Nagai, Y.; Elmas, E.; de Souza Fernandes Pereira, M.; Ali, S.A.; Imus, P.H.; Wethington, D.; Borrello, I.M.; Lee, D.A.; Ghiaur, G. CD38 deletion of human primary NK cells eliminates daratumumab-induced fratricide and boosts their effector activity. Blood 2020, 136, 2416–2427. [Google Scholar] [CrossRef]
- Naeimi Kararoudi, M.; Dolatshad, H.; Trikha, P.; Hussain, S.A.; Elmas, E.; Foltz, J.A.; Moseman, J.E.; Thakkar, A.; Nakkula, R.J.; Lamb, M.; et al. Generation of Knock-out Primary and Expanded Human NK Cells Using Cas9 Ribonucleoproteins. J. Vis. Exp. 2018, e58237. [Google Scholar] [CrossRef]
- Kararoudi, M.N.; Likhite, S.; Elmas, E.; Yamamoto, K.; Schwartz, M.; Sorathia, K.; Pereira, M.D.S.F.; Sezgin, Y.; Devine, R.D.; Lyberger, J.M.; et al. Optimization and validation of CAR transduction into human primary NK cells using CRISPR and AAV. Cell Rep. Methods 2022, 2, 100236. [Google Scholar] [CrossRef]
- Somanchi, S.S.; McCulley, K.J.; Somanchi, A.; Chan, L.L.; Lee, D.A. A Novel Method for Assessment of Natural Killer Cell Cytotoxicity Using Image Cytometry. PLoS ONE 2015, 10, e0141074. [Google Scholar] [CrossRef]
- Cerignoli, F.; Abassi, Y.A.; Lamarche, B.J.; Guenther, G.; Santa Ana, D.; Guimet, D.; Zhang, W.; Zhang, J.; Xi, B. In vitro immunotherapy potency assays using real-time cell analysis. PLoS ONE 2018, 13, e0193498. [Google Scholar] [CrossRef]
- Naeimi Kararoudi, M.; Likhite, S.; Elmas, E.; Schwartz, M.; Sorathia, K.; Yamamoto, K.; Chakravarti, N.; Moriarity, B.S.; Meyer, K.; Lee, D.A. CD33 Targeting Primary CAR-NK Cells Generated By CRISPR Mediated Gene Insertion Show Enhanced Anti-AML Activity. Blood 2020, 136, 3. [Google Scholar] [CrossRef]
- Foltz, J.A.; Moseman, J.E.; Thakkar, A.; Chakravarti, N.; Lee, D.A. TGFbeta Imprinting During Activation Promotes Natural Killer Cell Cytokine Hypersecretion. Cancers 2018, 10, 423. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Su, S.; Law, C.W.; Ah-Cann, C.; Asselin-Labat, M.L.; Blewitt, M.E.; Ritchie, M.E. Glimma: Interactive graphics for gene expression analysis. Bioinformatics 2017, 33, 2050–2052. [Google Scholar] [CrossRef]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.H.; Jones, R.G. Fueling immunity: Insights into metabolism and lymphocyte function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Blum, R.H.; Bernareggi, D.; Ask, E.H.; Wu, Z.; Hoel, H.J.; Meng, Z.; Wu, C.; Guan, K.L.; Malmberg, K.J.; et al. Metabolic Reprograming via Deletion of CISH in Human iPSC-Derived NK Cells Promotes In Vivo Persistence and Enhances Anti-tumor Activity. Cell Stem Cell 2020, 27, 224–237.e226. [Google Scholar] [CrossRef]
- Lichtenegger, F.S.; Lorenz, R.; Gellhaus, K.; Hiddemann, W.; Beck, B.; Subklewe, M. Impaired NK cells and increased T regulatory cell numbers during cytotoxic maintenance therapy in AML. Leuk. Res. 2014, 38, 964–969. [Google Scholar] [CrossRef] [PubMed]
- Khaznadar, Z.; Henry, G.; Setterblad, N.; Agaugue, S.; Raffoux, E.; Boissel, N.; Dombret, H.; Toubert, A.; Dulphy, N. Acute myeloid leukemia impairs natural killer cells through the formation of a deficient cytotoxic immunological synapse. Eur. J. Immunol. 2014, 44, 3068–3080. [Google Scholar] [CrossRef] [PubMed]
- Costello, R.T.; Sivori, S.; Marcenaro, E.; Lafage-Pochitaloff, M.; Mozziconacci, M.J.; Reviron, D.; Gastaut, J.A.; Pende, D.; Olive, D.; Moretta, A. Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukemia. Blood 2002, 99, 3661–3667. [Google Scholar] [CrossRef]
- Ohnishi, T. NADH-quinone oxidoreductase, the most complex complex. J. Bioenerg. Biomembr. 1993, 25, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Ugalde, C.; Hinttala, R.; Timal, S.; Smeets, R.; Rodenburg, R.J.; Uusimaa, J.; van Heuvel, L.P.; Nijtmans, L.G.; Majamaa, K.; Smeitink, J.A. Mutated ND2 impairs mitochondrial complex I assembly and leads to Leigh syndrome. Mol. Genet. Metab. 2007, 90, 10–14. [Google Scholar] [CrossRef]
- Masoud, R.; Reyes-Castellanos, G.; Lac, S.; Garcia, J.; Dou, S.; Shintu, L.; Abdel Hadi, N.; Gicquel, T.; El Kaoutari, A.; Dieme, B.; et al. Targeting Mitochondrial Complex I Overcomes Chemoresistance in High OXPHOS Pancreatic Cancer. Cell Rep. Med. 2020, 1, 100143. [Google Scholar] [CrossRef]
- Bou-Tayeh, B.; Laletin, V.; Salem, N.; Just-Landi, S.; Fares, J.; Leblanc, R.; Balzano, M.; Kerdiles, Y.M.; Bidaut, G.; Herault, O.; et al. Chronic IL-15 Stimulation and Impaired mTOR Signaling and Metabolism in Natural Killer Cells During Acute Myeloid Leukemia. Front. Immunol. 2021, 12, 730970. [Google Scholar] [CrossRef]
- Nielsen, C.M.; White, M.J.; Goodier, M.R.; Riley, E.M. Functional Significance of CD57 Expression on Human NK Cells and Relevance to Disease. Front. Immunol. 2013, 4, 422. [Google Scholar] [CrossRef]
- Cichocki, F.; Cooley, S.; Davis, Z.; DeFor, T.E.; Schlums, H.; Zhang, B.; Brunstein, C.G.; Blazar, B.R.; Wagner, J.; Diamond, D.J.; et al. CD56dimCD57+NKG2C+ NK cell expansion is associated with reduced leukemia relapse after reduced intensity HCT. Leukemia 2016, 30, 456–463. [Google Scholar] [CrossRef]
- Gorska, A.; Gruchala-Niedoszytko, M.; Niedoszytko, M.; Maciejewska, A.; Chelminska, M.; Skrzypski, M.; Wasag, B.; Kaczkan, M.; Lange, M.; Nedoszytko, B.; et al. The Role of TRAF4 and B3GAT1 Gene Expression in the Food Hypersensitivity and Insect Venom Allergy in Mastocytosis. Arch. Immunol. Ther. Exp. 2016, 64, 497–503. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, M.S.F.; Sorathia, K.; Sezgin, Y.; Thakkar, A.; Maguire, C.; Collins, P.L.; Mundy-Bosse, B.L.; Lee, D.A.; Naeimi Kararoudi, M. Deletion of Glycogen Synthase Kinase 3 Beta Reprograms NK Cell Metabolism. Cancers 2023, 15, 705. https://doi.org/10.3390/cancers15030705
Pereira MSF, Sorathia K, Sezgin Y, Thakkar A, Maguire C, Collins PL, Mundy-Bosse BL, Lee DA, Naeimi Kararoudi M. Deletion of Glycogen Synthase Kinase 3 Beta Reprograms NK Cell Metabolism. Cancers. 2023; 15(3):705. https://doi.org/10.3390/cancers15030705
Chicago/Turabian StylePereira, Marcelo S. F., Kinnari Sorathia, Yasemin Sezgin, Aarohi Thakkar, Colin Maguire, Patrick L. Collins, Bethany L. Mundy-Bosse, Dean A. Lee, and Meisam Naeimi Kararoudi. 2023. "Deletion of Glycogen Synthase Kinase 3 Beta Reprograms NK Cell Metabolism" Cancers 15, no. 3: 705. https://doi.org/10.3390/cancers15030705
APA StylePereira, M. S. F., Sorathia, K., Sezgin, Y., Thakkar, A., Maguire, C., Collins, P. L., Mundy-Bosse, B. L., Lee, D. A., & Naeimi Kararoudi, M. (2023). Deletion of Glycogen Synthase Kinase 3 Beta Reprograms NK Cell Metabolism. Cancers, 15(3), 705. https://doi.org/10.3390/cancers15030705