
Prevalence of Variants of Uncertain Significance in Patients Undergoing Genetic Testing for Hereditary Breast and Ovarian Cancer and Lynch Syndrome

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Study Population
2.3. Genetic Testing
2.4. Outcomes
2.5. Statistical Analysis
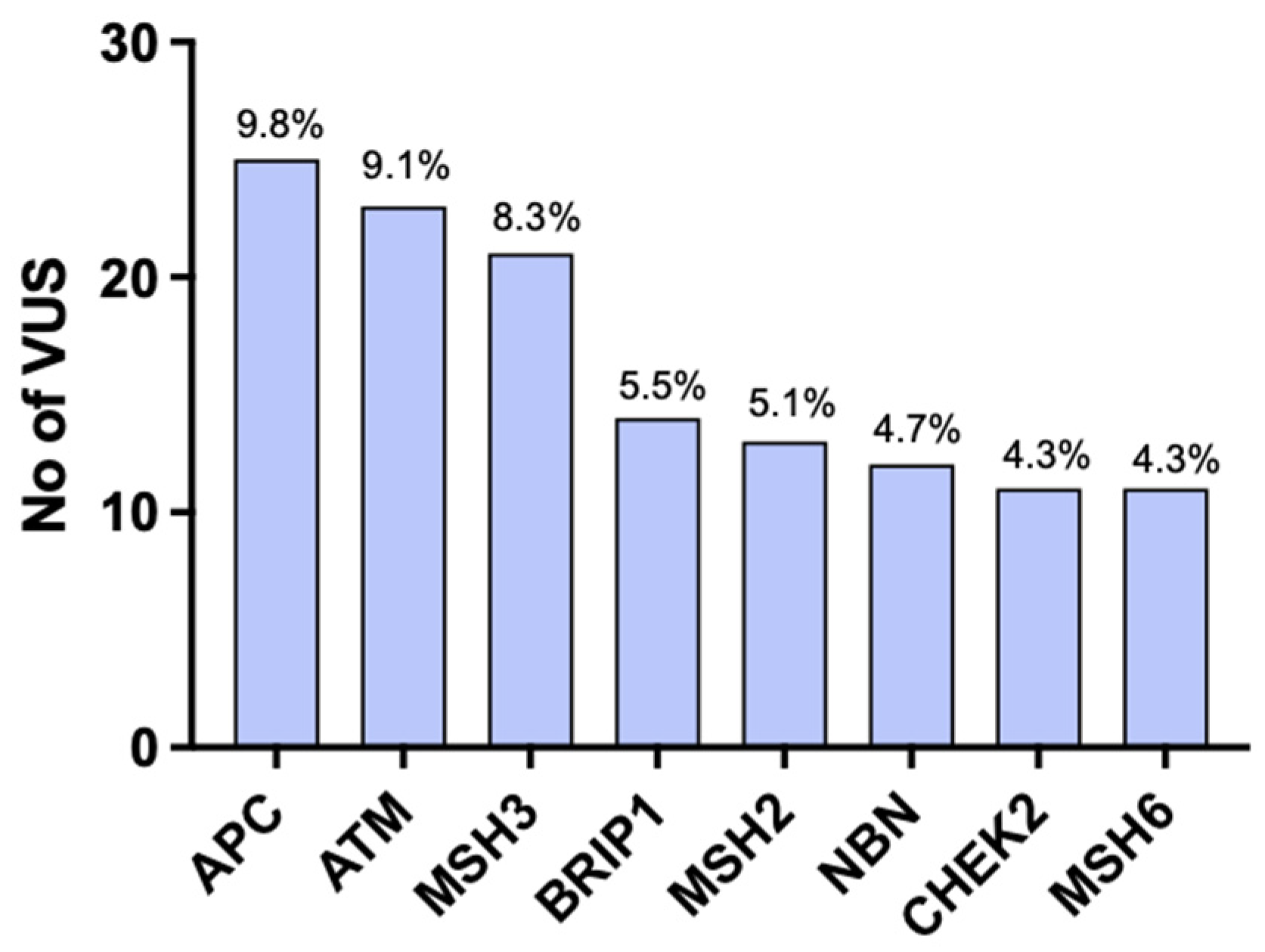
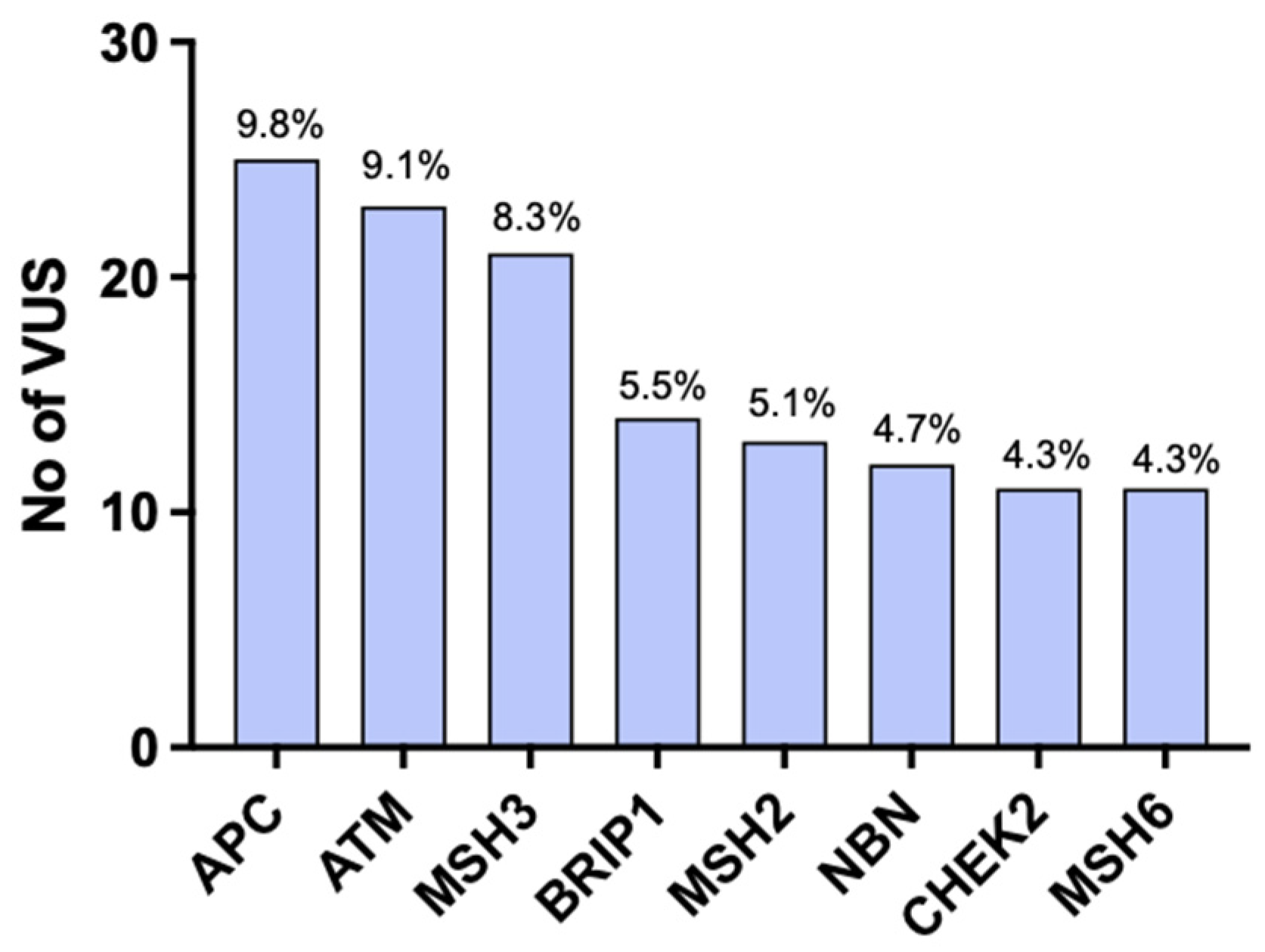
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Federici, G.; Soddu, S. Variants of uncertain significance in the era of high-throughput genome sequencing: A lesson from breast and ovary cancers. J. Exp. Clin. Cancer Res. 2020, 39, 46. [Google Scholar] [CrossRef] [PubMed]
- Yohe, S.; Thyagarajan, B. Review of Clinical Next-Generation Sequencing. Arch. Pathol. Lab. Med. 2017, 141, 1544–1557. [Google Scholar] [CrossRef] [PubMed]
- Burke, W.; Parens, E.; Chung, W.K.; Berger, S.M.; Appelbaum, P.S. The Challenge of Genetic Variants of Uncertain Clinical Significance: A Narrative Review. Ann. Intern. Med. 2022, 175, 994–1000. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Rahner, N.; Steinke, V. Hereditary cancer syndromes. Dtsch. Arztebl. Int. 2008, 105, 706–714. [Google Scholar] [CrossRef]
- Samadder, N.J.; Giridhar, K.V.; Baffy, N.; Riegert-Johnson, D.; Couch, F.J. Hereditary Cancer Syndromes—A Primer on Diagnosis and Management: Part 1: Breast-Ovarian Cancer Syndromes. Mayo Clin. Proc. 2019, 94, 1084–1098. [Google Scholar] [CrossRef]
- Samuel, D.; Diaz-Barbe, A.; Pinto, A.; Schlumbrecht, M.; George, S. Hereditary Ovarian Carcinoma: Cancer Pathogenesis Looking beyond BRCA1 and BRCA2. Cells 2022, 11, 539. [Google Scholar] [CrossRef]
- Giardiello, F.M.; Allen, J.I.; Axilbund, J.E.; Boland, C.R.; Burke, C.A.; Burt, R.W.; Church, J.M.; Dominitz, J.A.; Johnson, D.A.; Kaltenbach, T.; et al. Guidelines on genetic evaluation and management of Lynch syndrome: A consensus statement by the US Multi-Society Task Force on Colorectal Cancer. Dis. Colon. Rectum. 2014, 57, 1025–1048. [Google Scholar] [CrossRef]
- Sehgal, R.; Sheahan, K.; O’Connell, P.R.; Hanly, A.M.; Martin, S.T.; Winter, D.C. Lynch syndrome: An updated review. Genes 2014, 5, 497–507. [Google Scholar] [CrossRef]
- Saam, J.; Arnell, C.; Theisen, A.; Moyes, K.; Marino, I.; Roundy, K.M.; Wenstrup, R.J. Patients Tested at a Laboratory for Hereditary Cancer Syndromes Show an Overlap for Multiple Syndromes in Their Personal and Familial Cancer Histories. Oncology 2015, 89, 288–293. [Google Scholar] [CrossRef]
- Lynch, H.T.; Casey, M.J.; Snyder, C.L.; Bewtra, C.; Lynch, J.F.; Butts, M.; Godwin, A.K. Hereditary ovarian carcinoma: Heterogeneity, molecular genetics, pathology, and management. Mol. Oncol. 2009, 3, 97–137. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Ragone, A.; Lubinski, J.; Lynch, H.T.; Moller, P.; Ghadirian, P.; Foulkes, W.D.; Armel, S.; Eisen, A.; Neuhausen, S.L.; et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br. J. Cancer 2012, 107, 2005–2009. [Google Scholar] [CrossRef] [PubMed]
- Malander, S.; Rambech, E.; Kristoffersson, U.; Halvarsson, B.; Ridderheim, M.; Borg, A.; Nilbert, M. The contribution of the hereditary nonpolyposis colorectal cancer syndrome to the development of ovarian cancer. Gynecol. Oncol. 2006, 101, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Banno, K.; Yanokura, M.; Iida, M.; Adachi, M.; Masuda, K.; Ueki, A.; Kobayashi, Y.; Nomura, H.; Hirasawa, A.; et al. Features of ovarian cancer in Lynch syndrome (Review). Mol. Clin. Oncol. 2014, 2, 909–916. [Google Scholar] [CrossRef]
- Watson, P.; Vasen, H.F.A.; Mecklin, J.P.; Bernstein, I.; Aarnio, M.; Jarvinen, H.J.; Myrhøj, T.; Sunde, L.; Wijnen, J.T.; Lynch, H.T. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int. J. Cancer 2008, 123, 444–449. [Google Scholar] [CrossRef]
- Segev, Y.; Iqbal, J.; Lubinski, J.; Gronwald, J.; Lynch, H.T.; Moller, P.; Ghadirian, P.; Rosen, B.; Tung, N.; Kim-Sing, C.; et al. The incidence of endometrial cancer in women with BRCA1 and BRCA2 mutations: An international prospective cohort study. Gynecol. Oncol. 2013, 130, 127–131. [Google Scholar] [CrossRef]
- Sheehan, M.; Heald, B.; Yanda, C.; Kelly, E.D.; Grobmyer, S.; Eng, C.; Kalady, M.; Pederson, H. Investigating the Link between Lynch Syndrome and Breast Cancer. Eur. J. Breast Health 2020, 16, 106–109. [Google Scholar] [CrossRef]
- Buerki, N.; Gautier, L.; Kovac, M.; Marra, G.; Buser, M.; Mueller, H.; Heinimann, K. Evidence for breast cancer as an integral part of Lynch syndrome. Genes Chromosomes Cancer 2012, 51, 83–91. [Google Scholar] [CrossRef]
- Infante, M.; Arranz-Ledo, M.; Lastra, E.; Abella, L.E.; Ferreira, R.; Orozco, M.; Hernández, L.; Martínez, N.; Durán, M. Increased Co-Occurrence of Pathogenic Variants in Hereditary Breast and Ovarian Cancer and Lynch Syndromes: A Consequence of Multigene Panel Genetic Testing? Int. J. Mol. Sci. 2022, 23, 11499. [Google Scholar] [CrossRef]
- Clarke, J.E.; Magoon, S.; Forghani, I.; Alessandrino, F.; D’Amato, G.; Jonczak, E.; Subhawong, T.K. Radiologic screening and surveillance in hereditary cancers. Eur. J. Radiol. Open 2022, 9, 100422. [Google Scholar] [CrossRef]
- Hall, M.J.; Obeid, E.I.; Schwartz, S.C.; Mantia-Smaldone, G.; Forman, A.D.; Daly, M.B. Genetic testing for hereditary cancer predisposition: BRCA1/2, Lynch syndrome, and beyond. Gynecol. Oncol. 2016, 140, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, K.F.; Knerr, S.; Kauffman, T.L.; Lindberg, N.M.; Anderson, K.P.; Feigelson, H.S.; Gilmore, M.J.; Hunter, J.E.; Joseph, G.; Kraft, S.A.; et al. Systemic Barriers to Risk-Reducing Interventions for Hereditary Cancer Syndromes: Implications for Health Care Inequities. JCO Precis. Oncol. 2021, 5, 1709–1718. [Google Scholar] [CrossRef] [PubMed]
- Balmana, J.; Digiovanni, L.; Gaddam, P.; Walsh, M.F.; Joseph, V.; Stadler, Z.K.; Nathanson, K.L.; Garber, J.E.; Couch, F.J.; Offit, K.; et al. Conflicting Interpretation of Genetic Variants and Cancer Risk by Commercial Laboratories as Assessed by the Prospective Registry of Multiplex Testing. J. Clin. Oncol. 2016, 34, 4071. [Google Scholar] [CrossRef]
- Ready, K.; Gutierrez-Barrera, A.M.; Amos, C.; Meric-Bernstam, F.; Lu, K.; Hortobagyi, G.; Arun, B. Cancer risk management decisions of women with BRCA1 or BRCA2 variants of uncertain significance. Breast J. 2011, 17, 210–212. [Google Scholar] [CrossRef] [PubMed]
- Kurian, A.W.; Ward, K.C.; Abrahamse, P.; Bondarenko, I.; Hamilton, A.S.; Deapen, D.; Morrow, M.; Berek, J.S.; Hofer, T.P.; Katz, S.J. Time Trends in Receipt of Germline Genetic Testing and Results for Women Diagnosed with Breast Cancer or Ovarian Cancer, 2012–2019. J. Clin. Oncol. 2021, 39, 1631–1640. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) Version 2.2024—27 September 2023; National Comprehensive Cancer Network: Plymouth Meeting, PA, USA, 2023.
- Vasen, H.F.; Watson, P.; Mecklin, J.P.; Lynch, H.T. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999, 116, 1453–1456. [Google Scholar] [CrossRef] [PubMed]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Ruschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; Van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef]
- Stolarova, L.; Kleiblova, P.; Janatova, M.; Soukupova, J.; Zemankova, P.; Macurek, L.; Kleibl, Z. CHEK2 Germline Variants in Cancer Predisposition: Stalemate Rather than Checkmate. Cells 2020, 9, 2675. [Google Scholar] [CrossRef]
- Weiss, J.M.; Gupta, S.; Burke, C.A.; Axell, L.; Chen, L.M.; Chung, D.C.; Clayback, K.M.; Dallas, S.; Felder, S.; Gbolahan, O.; et al. NCCN Guidelines(R) Insights: Genetic/Familial High-Risk Assessment: Colorectal, Version 1.2021. J. Natl. Compr. Cancer Netw. 2021, 19, 1122–1132. [Google Scholar]
- Guindalini, R.S.C.; Viana, D.V.; Kitajima, J.; Rocha, V.M.; Lopez, R.V.M.; Zheng, Y.; Freitas, É.; Monteiro, F.P.M.; Valim, A.; Schlesinger, D.; et al. Detection of germline variants in Brazilian breast cancer patients using multigene panel testing. Sci. Rep. 2022, 12, 4190. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Razeq, H.; Tamimi, F.; Abujamous, L.; Abdel-Razeq, R.; Abunasser, M.; Edaily, S.; Abdulelah, H.; Khashabeh, R.A.; Bater, R. Rates of Variants of Uncertain Significance Among Patients with Breast Cancer Undergoing Genetic Testing: Regional Perspectives. Front. Oncol. 2022, 12, 673094. [Google Scholar] [CrossRef] [PubMed]
- Caswell-Jin, J.L.; Gupta, T.; Hall, E.; Petrovchich, I.M.; Mills, M.A.; Kingham, K.E.; Koff, R.; Chun, N.M.; Levonian, P.; Lebensohn, A.P.; et al. Racial/ethnic differences in multiple-gene sequencing results for hereditary cancer risk. Genet. Med. 2018, 20, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Ndugga-Kabuye, M.K.; Issaka, R.B. Inequities in multi-gene hereditary cancer testing: Lower diagnostic yield and higher VUS rate in individuals who identify as Hispanic, African or Asian and Pacific Islander as compared to European. Fam. Cancer 2019, 18, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Makhnoon, S.; Shirts, B.H.; Bowen, D.J. Patients’ perspectives of variants of uncertain significance and strategies for uncertainty management. J. Genet. Couns. 2019, 28, 313–325. [Google Scholar] [CrossRef]
- Monteiro, A.N.; Bouwman, P.; Kousholt, A.N.; Eccles, D.M.; Millot, G.A.; Masson, J.Y.; Schmidt, M.K.; Sharan, S.K.; Scully, R.; Wiesmüller, L.; et al. Variants of uncertain clinical significance in hereditary breast and ovarian cancer genes: Best practices in functional analysis for clinical annotation. J. Med. Genet. 2020, 57, 509–518. [Google Scholar] [CrossRef]
- Woods, N.T.; Baskin, R.; Golubeva, V.; Jhuraney, A.; De-Gregoriis, G.; Vaclova, T.; Goldgar, D.E.; Couch, F.J.; Carvalho, M.A.; Iversen, E.S.; et al. Functional assays provide a robust tool for the clinical annotation of genetic variants of uncertain significance. NPJ Genom. Med. 2016, 1, 16001. [Google Scholar] [CrossRef]
- Tricarico, R.; Kasela, M.; Mareni, C.; Thompson, B.A.; Drouet, A.; Staderini, L.; Gorelli, G.; Crucianelli, F.; Ingrosso, V.; Kantelinen, J.; et al. Assessment of the InSiGHT Interpretation Criteria for the Clinical Classification of 24 MLH1 and MSH2 Gene Variants. Hum. Mutat. 2017, 38, 64–77. [Google Scholar] [CrossRef]
- Khandakji, M.; Habish, H.H.A.; Abdulla, N.B.S.; Kusasi, S.A.A.; Abdou, N.M.G.; Al-Mulla, H.; Al Sulaiman, R.J.A.; Bu Jassoum, S.M.; Mifsud, B. BRCA1-specific machine learning model predicts variant pathogenicity with high accuracy. Physiol. Genom. 2023, 55, 315–323. [Google Scholar] [CrossRef]
- Stolarova, L.; Kleiblova, P.; Zemankova, P.; Stastna, B.; Janatova, M.; Soukupova, J.; Achatz, M.I.; Ambrosone, C.; Apostolou, P.; Arun, B.K.; et al. ENIGMA CHEK2gether Project: A Comprehensive Study Identifies Functionally Impaired CHEK2 Germline Missense Variants Associated with Increased Breast Cancer Risk. Clin. Cancer Res. 2023, 29, 3037–3050. [Google Scholar] [CrossRef]
- Hoffman-Andrews, L. The known unknown: The challenges of genetic variants of uncertain significance in clinical practice. J. Law Biosci. 2017, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| (a) | |
| Personal history of cancer | Breast cancer diagnosed at ≤50 y.o. |
| Triple-negative breast cancer | |
| ≥2 primary breast cancers | |
| Lobular breast cancer with concurrent history of diffuse gastric cancer | |
| Epithelial ovarian cancer, fallopian cancer, or primary peritoneal cancer | |
| Male breast cancer | |
| Ashkenazi Jewish ancestry | |
| High-risk family history: | |
| |
| |
| |
| No personal history of cancer | Close relative meeting any of the above criteria |
| Individual with >5% of BRCA1/2 pathogenic variant in risk calculators (i.e., Tyrer-Cuzick, BRCAPro, CanRisk) | |
| (b) | |
| Amsterdam II | Three relatives with Lynch syndrome–related cancers with all the following criteria met: |
| |
| |
| |
| |
| |
| Bethesda Criteria | Test if any of the following: |
| |
| |
| |
| |
| |
| Number of Total (%) | ||
|---|---|---|
| Age (mean ± SD) | 50 ± 15 | |
| Gender | ||
| Males | 63 (9.50%) | |
| Females | 597 (90.04%) | |
| Transgender | 3 (0.45%) | |
| Ethnicity | ||
| Hispanic | 35 (5.27%) | F: 32/35 |
| Non-Hispanic | 378 (57.01%) | F: 338/378 |
| Unknown | 250 (37.70%) | F: 227/250 |
| Race * | ||
| White | 333 (50.20%) | F: 298/333 |
| African American | 20 (3.01%) | F:17/20 |
| Asian | 34 (5.12%) | F: 30/34 |
| Native American | 10 (1.50%) | F: 10/10 |
| Unknown | 280 (42.23%) | F: 254/280 |
| Ashkenazi Jews | 95 (14.3%) | |
| Personal history of breast cancer | 162 (24.43%) | |
| Personal history of colon cancer | 20 (3.01%) | |
| Family history of cancer in 1st-degree relative | 351 (52.94%) | |
| Breast cancer in 1st-degree relative | 116 (17.49%) | |
| Ovarian cancer in 1st-degree relative | 23 (3.46%) | |
| Prostate cancer in 1st-degree relative | 31 (4.67%) | |
| Colon cancer in 1st-degree relative | 21 (3.16%) | |
| Family history of cancer in 2nd-degree relative | 396 (59.7%) |
| Age at Testing | Syndrome Tested for | VUS | Reference Sequence (RS) |
|---|---|---|---|
| 51 | HBOC | c.1190A>G (p.Gln397Arg) in AXIN2 c.3650C>G (p.Ser1217Cys) in BRCA1 c.1027-1G>T (splice acceptor) in RAD51C | rs774887154 rs398122676 rs1567818502 |
| 52 | HBOC | c.8246A>T (p.Lys2749Ile) in ATM c.556A>G (p.Ser186Gly) in BARD1 c.743G>A (p.Arg248Gln) in MSH6 | rs779145081 rs16852741 rs764870249 |
| 71 | HBOC | c.803C>T (p.Pro268Leu) in RECQL4 c.899A>G (p.Gln300Arg) in SMARCE1 | rs760340046 rs766568737 |
| 37 | HBOC | c.4002-8dup in MSH6 c.210A>G (p.Ser70Ser) in PALB2 | rs267608139 rs786202650 |
| 24 | HBOC | c.6179G>A (p.Arg2060His) in ATM c.470T>C (p.Ile157Thr) in CHEK2 | rs376521407 rs17879961 |
| 79 | HBOC | c.3440dup (p.Asn1147fs) in BRIP1 c.470T>C (p.Ile157Thr) in CHEK2 | rs753683450 rs17879961 |
| 35 | HBOC | c.2075A>C (p.His692Pro) in BRCA1 c.730A>G (p.Ile244Val) in RAD51C | rs2053831947 rs199886026 |
| 42 | HBOC | c.1348A>C (p.Asn450His) in PALB2 c.3362G>A (p.Gly1121Asp) in PALB2 | rs62625274 rs62625282 |
| 62 | HBOC | c.6865A>G (p.Thr2289Ala) in APC c.1655T>C (p.Ile552Thr) in BRIP1 | rs1554087807 rs369340666 |
| 50 | HBOC | c.7415A>C (p.Lys2472Thr) in BRCA2 c.5C>T (p.Ser2Phe) in MSH3 | rs80358963 rs768844493 |
| 82 | HBOC | c.722C>T (p.Ala241Val) in STK11 | rs2080777192 |
| 50 | HBOC | c.970G>A (p.Glu324Lys) in SDHA | rs147014102 |
| 35 | HBOC | c.26G>C (p.Cys9Ser) in RAD51D | rs140825795 |
| 46 | HBOC | c.961G>A (p.Gly321Ser) in POLD1 | rs41554817 |
| 70 | HBOC | c.1168C>T (p.Pro390Ser) in POLD1 | rs2038747136 |
| 73 | HBOC | c.1425G>T(p.Glu475Asp) in PDGFRA | rs200309940 |
| 42 | HBOC | c.1186G>T (p.Ala396Ser) in PDGFRA | rs1327567130 |
| 35 | HBOC | c.2317A>G (p.Met773Val) in PDGFRA | rs191808397 |
| 65 | HBOC | c.3014T>C (p.Phe1005Ser) in PALB2 | rs879254268 |
| 60 | HBOC | c.3025C>G (p.Pro1009Ala) in PALB2 | rs764669864 |
| 64 | HBOC | c.821T>C (p.Leu274Pro) in NTHL1 | Variation ID: 2517325 |
| 69 | HBOC | c.458G>A (p.Arg153Gln) in NTHL1 | Variation ID: 2518789 |
| 60 | HBOC | c.1720T>A (p.Leu574Ile) in NBN | rs142334798 |
| 74 | HBOC | c.643C>T (p.Arg215Trp) in NBN | rs34767364 |
| 31 | HBOC | VUS in NBN (Missing specific sequence) | NA |
| 81 | HBOC | VUS in NBN (Missing specific sequence) | NA |
| 63 | HBOC | c.2060A>C (p.Lys687Thr) in NBN | rs186371605 |
| 67 | HBOC | c.1447A>G (p.Thr483Ala) in MUTYH | Not previously reported |
| 60 | HBOC | c.1306C>G (p.Leu43Val) in MUTYH | Not previously reported |
| 68 | HBOC | c.1778G>A (p.Arg593Gln) in MSH3 | rs764832633 |
| 37 | HBOC | c.1764-9_1764-8del in MSH3 | rs41559616 |
| 79 | HBOC | VUS in MSH3 (Missing specific sequence) | NA |
| 41 | HBOC | c.2732T>G (p.Leu911Trp) in MSH3 | rs41545019 |
| 71 | HBOC | c.1028-6T>C in MSH3 | rs769258876 |
| 48 | HBOC | VUS in MSH2 (Missing specific sequence) | NA |
| 49 | HBOC | VUS in MLH1 (Missing specific sequence) | NA |
| 67 | HBOC | c.359G>C (p.Arg120Pro) in GALNT12 | rs202137559 |
| 64 | HBOC | c.907G>A (p.Asp303Asn) in GALNT12 | rs145236923 |
| 75 | HBOC | c.4010A>C (p.Asp1337Ala) in DICER1 | rs1891070773 |
| 50 | HBOC | c.248A>G (p.Tyr83Cys) in DICER1 | rs373646414 |
| 61 | HBOC | VUS in CTNNA1 (Missing specific sequence) | NA |
| 44 | HBOC | c.1111C>T (p.His371Tyr) in CHEK2 c.2357T>C (p.Val786Ala) in MSH3 | rs531398630 |
| 32 | HBOC | c.1111C>T (p.His371Tyr) in CHEK2 c.719C>T (p.Pro240Leu) in GALNT12 | rs531398630 rs59362219 |
| 58 | HBOC | c.580A>T (p.Ser194Cys) in CHEK2 | rs786203042 |
| 53 | HBOC | c.593-3_593-2insSVA in CHEK2 | Not previously reported |
| 46 | HBOC | c.369T>A (p.His123Gln) in CHEK2 | Not previously reported |
| 55 | HBOC | c.-2G>A in CDKN2A | rs191394143 |
| 69 | HBOC | c.310C>A (p.Leu104Met) in CDK4 | rs759535768 |
| 61 | HBOC | c.1897A>C (p.Ile663Leu) in BRIP1 | rs765314472 |
| 52 | HBOC | c.2469G>T (p.Arg823Ser) in BRIP1 | rs587780239 |
| 31 | HBOC | c.415T>G (p.Ser139Ala) in BRIP1 | rs202072866 |
| 47 | HBOC | c.226G>A (p.Val76Ile) in BRIP1 | rs769573395 |
| 40 | HBOC | c.2423G>T (p.Arg808Ile) in BRIP1 | rs781153382 |
| 75 | HBOC | c.9816T>G (p.Asp3272Glu) in BRCA2 | rs56111359 |
| 32 | HBOC | c.2779A>G (p.Met927Val) in BRCA2 | rs786201837 |
| 36 | HBOC | R2784Q (8579G>A) in BRCA2 | rs80359076 |
| 33 | HBOC | c.3318C>G (p.Ser1106Arg) in BRCA2 | rs1298550035 |
| 48 | HBOC | c.1360C>G (p.Pro454Ala) in BARD1 | rs730881408 |
| 76 | HBOC | c.2284T>C (p.Trp762Arg) in BARD1 | rs878854008 |
| 78 | HBOC | VUS in AXIN2 (Missing specific sequence) | NA |
| 32 | HBOC | c.1235A>G (p.Asn412Ser) in AXIN2 | rs115931022 |
| 67 | HBOC | c.1985T>C (p.Leu662Pro) in AXIN2 | rs142476324 |
| 79 | HBOC | c.6019C>T (p.Leu762Arg) in ATM | Not previously reported |
| 43 | HBOC | c.7871G>C (p.Cys2624Ser) in ATM | rs759392666 |
| 51 | HBOC | c.2698A>G (p.Met900Val) in ATM | rs138468963 |
| 35 | HBOC | VUS in ATM (Missing specific sequence) | NA |
| 48 | HBOC | c.6919C>T (p.Leu2307Phe) in ATM | rs56009889 |
| 39 | HBOC | c.8155C>T (p.Arg2719Cys) in ATM | rs138526014 |
| 74 | HBOC | VUS in ATM (Missing specific sequence) | NA |
| 39 | HBOC | VUS in ATM (Missing specific sequence) | NA |
| 48 | HBOC | c.2011A>G (p.Ile671Val) in ATM | rs730881344 |
| 57 | HBOC | c.6248T>C (p.Ile2083Thr) in APC | rs758715972 |
| 48 | HBOC | VUS (Missing specific sequence) | NA |
| 57 | HBOC | c.1169C>T (p.Thr390Met) in TSC2 | rs1596303442 |
| 33 | HBOC | c.758A>G (p.His253Arg) in SMARCA4 | rs2086063382 |
| 47 | HBOC | c.367C>T (p.Pro123Ser) in SDHC c.1307C>T (p.Ala436Val) in TERT | rs773039986 rs986886145 |
| 33 | HBOC | c.172A>T (p.Thr58Ser) in RNF43 | rs142864107 |
| 57 | HBOC | c.1720A>C (p.Lys574Gln) in RAD50 | rs779597467 |
| 40 | HBOC | c.6767G>C (p.Gly2256Ala) in POLE | rs749707316 |
| 60 | HBOC | c.101G>T (p.Arg34Leu) in POLE | rs747005851 |
| 38 | HBOC | c.1016A>T (p.Asp339Val) in POLE | rs1060500865 |
| 38 | HBOC | c.75del (p.Asp25Glufs*16) in POLD1 | rs772855121 |
| 57 | HBOC | c.1510G>C (p.Glu504Gln) in PMS2 | rs368516768 |
| 33 | HBOC | c.97A>T (p.Asn33Tyr) in PDGFRA | rs200979664 |
| 32 | HBOC | c.2317A>G (p.Met773Val) in PDGFRA | rs191808397 |
| 72 | HBOC | c.1730C>T (p.Pro577Leu) in PDGFRA | rs778015444 |
| 63 | HBOC | c.1651C>A (p.Gln551Lys) in PDGFRA | rs770950644 |
| 53 | HBOC | c.155T>C (p.Val52Ala) in PALB2 | rs373970237 |
| 41 | HBOC | c.208G>A (p.Gly70Ser) in NTHL1 | Variation ID: 2096931 |
| 41 | HBOC | c.5360C>T (p.Thr1787Met) in NF1 | rs760649828 |
| 27 | HBOC | c.4009C>T (p.Arg1337Trp) in NF1 | rs146306756 |
| 49 | HBOC | c.3883A>G (p.Thr1295Ala) in NF1 | rs143836226 |
| 27 | HBOC | c.3315A>G (Silent) in NF1 | rs1555614915 |
| 51 | HBOC | c.169G>A (p.Gly57Ser) in NF1 | rs779727341 |
| 48 | HBOC | c.595C>T (p.Pro199Ser) in NBN | rs587780097 |
| 61 | HBOC | c.536A>T (p.Glu179Val) in NBN | rs864622578 |
| 47 | HBOC | c.430A>G (p.Thr144Ala) in NBN | rs1812023859 |
| 56 | HBOC | c.2056A>G (p.Lys686Glu) in NBN | rs786203920 |
| 44 | HBOC | c.1343A>T (p.Gln448Leu) in NBN c.1120A>G (p.Ser374Gly) in TSC1 | rs146403088 Variation ID: 1063383 |
| 43 | HBOC | c.100G>C (p.Glu34Gln) in MUTYH | rs1557492431 |
| 42 | HBOC | c.458G>C (p.Gly153Ala) in MSH6 c.3290C>T (p.Pro1097Leu) in PALB2 | rs1251899870 rs587781308 |
| 39 | HBOC | c.1720C>T (p.Arg574Trp) in MSH3 c.3762A>T (p.Glu1254Asp) in MSH6 | rs771054581 rs375459388 |
| 42 | HBOC | c.909G>C (p.Lys303Asn) in MSH3 | rs757164724 |
| 82 | HBOC | c.582C>G (p.Asp194Glu) in MSH3 | rs749446559 |
| 37 | HBOC | c.350G>A (p.Gly117Asp) in MSH3 | rs1456712758 |
| 31 | HBOC | c.2732T>G (p.Leu911Trp) in MSH3 | rs41545019 |
| 56 | HBOC | c.2185C>G (p.His729Asp) in MSH3 | rs145353158 |
| 71 | HBOC | c.2173G>A (p.Glu725Lys) in MSH3 c.845C>T (p.Thr282Ile) in MSH3 c.1255G>A (p.Ala419Thr) in MUTYH | rs200612739 rs202184623 rs58778044 |
| 52 | HBOC | c.1568+5G>A (Intronic) in MSH3 | rs778804919 |
| 61 | HBOC | c.1019T>C (p.Ile340Thr) in MSH3 | rs1228031532 |
| 64 | HBOC | c.1172C>A (p.Ala391Asp) in MSH2 | rs864622674 |
| 59 | HBOC | c.1064G>A (p.Arg355Lys) in MSH2 | Variation ID: 1781308 |
| 66 | HBOC | c.1856A>G (p.Tyr619Lys) in MSH2 | rs63749982 |
| 28 | HBOC | c.1172C>A (p.Ala391Asp) in MSH2 | rs864622674 |
| 84 | HBOC | c.2606C>A (p.Ala869Glu) in MSH2 c.257C>T (p.Ala86Val) in POLD1 | rs730881772 rs148040399 |
| 73 | HBOC | c.123C>G (p.Asp41Glu) in MSH2 | rs761960690 |
| 43 | HBOC | c.2156T>C (p.Ile719Thr) in MLH1 | rs757603534 |
| 39 | HBOC | c.2045T>C (p.Met682Thr) in MLH1 c.1408A>G (p.Thr470Ala) in PALB2 | rs1060500693 rs150636811 |
| 76 | HBOC | c.941G>A (p.Arg314Gln) in MEN1 | rs771645621 |
| 44 | HBOC | c.1553C>T (p.Pro518Leu) in KIT c.334A>G (p.Asn112Asp) in MSH6 | rs569408054 rs864622397 |
| 77 | HBOC | c.429C>G (p.Phe143Leu) in FLCN c.2377G>A (p.Gly793Ser) in PALB2 | Variation ID: 388510 rs878855109 |
| 67 | HBOC | c.4352G>C (p.Arg1451Thr) in DICER1 | Variation ID: 1056894 |
| 48 | HBOC | c.1143+5T>C (Intronic) in CTNNA1 | rs766106863 |
| 25 | HBOC | c.539G>T (p.Arg180His) in CHEK2 | rs137853009 |
| 51 | HBOC | c.772A>G (p.Ile258Val) in CHEK2 c.-2A>C in RAD51D | rs876658690 rs2091800072 |
| 61 | HBOC | c.663C>G (p.Ile221Met) in CHEK2 | rs200451612 |
| 36 | HBOC | c.331G>T (p.Asp111Tyr) in CHEK2 c.1019T>C (p.Phe340Ser) in MSH6 c.6674G>A (p.Arg2225His) in POLE | rs1569159072 rs61753793 rs538875477 |
| 39 | HBOC | c.1567C>G (p.Arg523Gly) in CHEK2 c.4859C>T (p.Ser1620Phe) in SMARCA4 | rs149501505 rs1600649021 |
| 41 | HBOC | c.949T>C (p.Phe317Leu) in CDH1 c.2085A>G (Silent) in MSH3 | rs1555515643 rs777245977 |
| 35 | HBOC | c.1784C>G (p.Pro595Arg) CDH1 | rs1555516843 |
| 39 | HBOC | c.436A>G (p.Ile146Val) in BRIP1 | rs1567868598 |
| 25 | HBOC | c.3533A>T (p.Glu1178Val) in BRIP1 c.3379A>G (p.Asn1127Asp) in MSH3 c.1009G>A (p.Val337Ile) in PDGFRA | rs752850661 |
| 64 | HBOC | c.337A>C (p.Thr113Pro) in BRIP1 c.728G>A (p.Arg243Gln) in MSH2 | rs1555617812 rs63751455 |
| 53 | HBOC | c.2233G>A (p.Ala745Thr) in BRIP1 | rs587780235 |
| 63 | HBOC | c.1660C>G (p.Gln554Glu) in BRIP1 | rs777217004 |
| 56 | HBOC | c.891_902del (p.Glu297_Val300del) in BRCA2 | rs2072399471 |
| 36 | HBOC | c.6703A>T (p.Met2235Leu) in BRCA2 | Variation ID: 1056020 |
| 83 | HBOC | c.343A>G (p.Lys115Glu) in BRCA2 c.1075C>A (p.Pro359Thr) in MUTYH | rs56242644 Not previously reported |
| 33 | HBOC | c.4339C>A (p.Gln1447Lys) in BRCA1 | rs1567868598 |
| 50 | HBOC | c.1022G>T (p.Gly341Val) in BMPR1A | rs1564724250 |
| 51 | HBOC | c.80C>A in BARD1 | NA |
| 36 | HBOC | c.748T>C (p.Ser250Pro) in BARD1 c.1618G>A (p.Asp540Asn) in MUTYH | rs570022823 Not previously reported |
| 38 | HBOC | c.617A>G (p.Gln206Arg) in BARD1 | rs760718143 |
| 54 | HBOC | c.1835A>T (p.Asp612Val) in BARD1 | rs201140528 |
| 55 | HBOC | c.2770C>T (p.Arg924Trp) in ATM | rs55723361 |
| 62 | HBOC | c.8968G>A (p.Glu2990Lys) in ATM | rs1800558 |
| 37 | HBOC | c.8187A>C (p.Gln2729His) in ATM c.821G>T (p.Gly274Val) in CDH1 | rs587781946 rs876660861 |
| 51 | HBOC | c.7743C>A (p.Ser2581Arg) in ATM | rs2086306575 |
| 54 | HBOC | c.670A>G (p.Lys224Glu) in ATM c.1004A>G (p.Asn335Ser) in PMS2 | rs145053092 rs200513014 |
| 58 | HBOC | c.4375G>A (p.Gly1459Arg) in ATM | rs145667735 |
| 26 | HBOC | c.4349T>C (p.Leu1450Pro) in ATM c.1192C>T (p.Arg398Cys) in GALNT12 | rs750306932 rs747755624 |
| 39 | HBOC | c.238C>T (p.Pro80Ser) in ATM | rs750597831 |
| 37 | HBOC | c.133C>T (p.Arg45Trp) in ATM c.7457C>T (p.Thr2486Ile) in NF1 | rs3218684 Not previously reported |
| 36 | HBOC | c.4088A>G (p.Lys1363Arg) in APC c.5392A>G (p.Asn1798Asp) in APC c.1489A>G (p.Ile497Val) in MSH2 c.157G>T (p.Ala53Ser) in MSH2 | rs373607243 rs200794097 rs755501968 rs755931648 |
| 52 | HBOC | c.6944A>G (p.Gln2315Arg) in APC c.1037C>T (p.Ser346Phe) in MSH6 c.503C>G (p.Ala168Gly) in MSH6 | rs1060503273 rs567785169 rs774162322 |
| 79 | HBOC | c.7903A>G (p.Thr2635Ala) in ATM | rs886059799 |
| 42 | HBOC | c.2438A>G (p.Asn813Ser) in ATM | Not previously reported |
| 38 | HBOC | c.8462A>G (p.Asp2821Gly) in APC | rs780049836 |
| 68 | HBOC | c.8276G>A (p.Arg2759His) in APC c.582C>G (p.Asp194Glu) in MSH3 | rs538289470 rs749446559 |
| 41 | HBOC | c.7399C>A (p.Pro2467Thr) in APC c.5026A>G (p.Arg1676Gly) in APC | rs372305287 rs370560998 |
| 29 | HBOC | c.688C>T (p.Arg230Cys) in APC | rs587779805 |
| 45 | HBOC | c.6724A>G (p.Ser2242Gly) in APC c.511A>G (p.Ile171Val) in NBN | rs201375478 rs61754966 |
| 53 | HBOC | c.6520A>G (p.Ser2174Gly) in APC c.1117G>A (p.Gly373Arg) in MLH1 | rs754536901 rs587776934 |
| 48 | HBOC | c.6338G>C (p.Ser2113Thr) in APC | rs1766189874 |
| 28 | HBOC | c.5240T>C (p.Met1747Thr) in APC c.1094G>A (p.Arg365Gln) in RAD50 | rs864622751 rs146370443 |
| 23 | HBOC | c.5216A>G (p.Lys1739Arg) in APC | rs769558291 |
| 66 | HBOC | c.5026_5028del (p.Arg1676del) in APC c.3715A>G (p.Ile1239Val) in MSH6 c.668T>C (p.Ile223Thr) in RAD50 | rs768369050 rs1469961964 rs1750475890 |
| 40 | HBOC | c.4372C>T (p.Pro1458Ser) in APC | rs143796828 |
| 34 | HBOC | c.2222A>G (p.Asn741Ser) in APC | rs150209825 |
| 62 | HBOC | c.-30369A>G (Non-coding) in APC | NA |
| 59 | HBOC | c.203G>A, p.Arg68Gln in AXIN2 | rs138056036 |
| 26 | HBOC | c.1267C>T (p.Leu423Phe) in AXIN2 c.1567G>A (p.Glu523Lys) in MSH3 | rs376630432 rs34058399 |
| 55 | HBOC | c.111G>T (p.Gln37His) in AXIN2 c.1643G>A (p.Gly548Asp) in MSH2 c.1361G>A (p.Arg454Gln) in MSH3 | Variation ID: 1494944 rs1573553753 rs144798521 |
| 79 | HBOC | c.3352A>G (p.Asn1118Asp) in APC c.1660C>T (p.Arg554Trp) in RNF43 | rs140493115 Variation ID: 1140674 |
| 47 | HBOC | c.797C>G (p.Thr266Ser) in BMPR1A c.3762A>T (p.Glu1254Asp) in MSH6 | rs1554890797 rs375459388 |
| 35 | Lynch Syndrome | c.6363_6365dupTG (p.Ala2122dup) in APC c.2804C>T (p.Thr935Met) in ATM | rs587780602 rs3218708 |
| 48 | HBOC | c.dup exon 2 (p14ARF) in CDKN2A c.dup entire (p16INK4a) in CDKN2A | Not previously Not previously reported |
| 60 | HBOC | c.5026A>G (p.Arg1676Gly) in APC c.7399C>A (p.Pro2467Thr) in APC | rs200794097 rs372305287 |
| 52 | HBOC | c.626A>G (p.Ile2076Val) in ATM c.317G>C (p.Arg106Thr) in MSH2 | Not previously reported rs41295286 |
| 47 | HBOC | c.1007A>G (p.Asn336Ser) in POLE c.667C>T (p.Arg223Cys) in RNF43 | rs5744760 rs755478993 |
| 73 | HBOC | c.1353A>G (p.Asn118Ser) in BARD1 c.2081C>G (p.Pro694Arg) in NBN | rs142864491 rs746090959 |
| 50 | HBOC | VUS in KIT and MSH6 (Missing specific sequence) | NA |
| 70 | HBOC | c.1655G>A (p.Arg552Lys) in GALNT12 c.527T>C (p.Ile176Thr) in NHTL1 | rs1285871027 rs1805378 |
| 35 | HBOC | c.3444C>A (p.Asp1148Glu) in BRIP1 c.-2G>A in CDKN2A | rs28997573 rs191394143 |
| 36 | HBOC | VUS in ATM and RAD51D (Missing specific sequence) | NA |
| 50 | Lynch Syndrome | c.1883A>G (p.Asn628Ser) in DICER1 | rs756051157 |
| 45 | HBOC | c.536A>G (p.Tyr170Cys) in NHTL1 | Not previously reported |
| 43 | HBOC | c.118G>A (p.Gly40Ser) in MSH2 | rs63751260 |
| Gene | VUS |
|---|---|
| ATM | c.6019C>T (p.Leu762Arg) |
| ATM | c.626A>G (p.Ile2076Val) |
| ATM | c.2438A>G (p.Asn813Ser) |
| CHEK2 | c.593-3_593-2insSVA |
| CHEK2 | c.369T>A (p.His123Gln) |
| MUTYH | c.1447A>G (p.Thr483Ala) |
| MUTYH | c.1306C>G (p.Leu43Val) |
| MUTYH | c.1075C>A (p.Pro359Thr) |
| MUTYH | c.1618G>A (p.Asp540Asn) |
| NF1 | c.7457C>T (p.Thr2486Ile) |
| NHTL1 | c.536A>G (p.Tyr170Cys) |
| Gene | VUS | Reference Sequence | Uncertain Significance | Likely Benign |
|---|---|---|---|---|
| APC | c.5392A>G (p.Asn1798Asp) | rs200794097 | yes | x |
| APC | c.8462A>G (p.Asp2821Gly) | rs780049836 | x | x |
| APC | c.7399C>A (p.Pro2467Thr) | rs372305287 | x | x |
| APC | c.5026A>G (p.Arg1676Gly) | rs370560998 | x | x |
| ATM | c.8246A>T (p.Lys2749Ile) | rs779145081 | x | x |
| ATM | c.6179G>A (p.Arg2060His) | rs376521407 | x | x |
| ATM | c.6919C>T (p.Leu2307Phe) | rs56009889 | x | x |
| ATM | c.670A>G (p.Lys224Glu) | rs145053092 | x | x |
| ATM | c.7903A>G (p.Thr2635Ala) | rs886059799 | x | x |
| AXIN2 | c.1985T>C (p.Leu662Pro) | rs142476324 | x | x |
| AXIN2 | c.203G>A (p.Arg68Gln) | rs138056036 | x | x |
| BARD1 | c.556A>G (p.Ser186Gly) | rs16852741 | x | x |
| BARD1 | c.1360C>G (p.Pro454Ala) | rs730881408 | x | x |
| BARD1 | c.748T>C (p.Ser250Pro) | rs570022823 | x | x |
| BARD1 | c.1835A>T (p.Asp612Val) | rs201140528 | x | x |
| BRCA1 | c.3650C>G (p.Ser1217Cys) | rs398122676 | x | x |
| BRCA1 | c.2075A>C (p.His692Pro) | rs2053831947 | x | x |
| BRCA1 | c.4339C>A (p.Gln1447Lys) | rs1567868598 | x | x |
| BRCA2 | c.3318C>G (p.Ser1106Arg) | rs1298550035 | x | x |
| BRCA2 | c.343A>G (p.Lys115Glu) | rs56242644 | x | x |
| BRIP1 | c.226G>A (p.Val76Ile) | rs769573395 | x | x |
| BRIP1 | c.436A>G (p.Ile146Val) | rs1567868598 | x | x |
| CHEK2 | c.1111C>T (p.His371Tyr) | rs531398630 | x | x |
| CHEK2 | c.580A>T (p.Ser194Cys) | rs786203042 | x | x |
| CHEK2 | c.539G>T (p.Arg180His) | rs137853009 | x | x |
| CHEK2 | c.663C>G (p.Ile221Met) | rs200451612 | x | x |
| DICER1 | c.248A>G (p.Tyr83Cys) | rs373646414 | x | x |
| GALNT12 | c.907G>A (p.Asp303Asn) | rs145236923 | x | x |
| GALNT12 | c.1655G>A (p.Arg552Lys) | rs1285871027 | x | x |
| MEN1 | c.941G>A (p.Arg314Gln) | rs771645621 | x | x |
| MSH2 | c.1856A>G (p.Tyr619Lys) | rs63749982 | x | x |
| MSH2 | c.2606C>A (p.Ala869Glu) | rs730881772 | x | x |
| MSH2 | c.123C>G (p.Asp41Glu) | rs761960690 | x | x |
| MSH2 | c.118G>A (p.Gly40Ser) | rs63751260 | x | x |
| MSH2 | c.1489A>G (p.Ile497Val) | rs755501968 | x | x |
| MSH2 | c.157G>T (p.Ala53Ser) | rs755931648 | x | x |
| MSH3 | c.2732T>G (p.Leu911Trp) | rs41545019 | x | x |
| MSH3 | c.909G>C (p.Lys303Asn) | rs757164724 | x | x |
| MSH3 | c.582C>G (p.Asp194Glu) | rs749446559 | x | x |
| MSH3 | c.2732T>G (p.Leu911Trp) | rs41545019 | x | x |
| MSH3 | c.2173G>A (p.Glu725Lys) | rs200612739 | x | x |
| MSH3 | c.1361G>A (p.Arg454Gln) | rs144798521 | x | x |
| MSH6 | c.743G>A (p.Arg248Gln) | rs764870249 | x | x |
| MSH6 | c.4002-8dup | rs267608139 | x | x |
| MSH6 | c.3762A>T (p.Glu1254Asp) | rs375459388 | x | x |
| MSH6 | c.3762A>T (p.Glu1254Asp) | rs375459388 | x | x |
| MUTYH | c.1255G>A (p.Ala419Thr) | rs587780744 | x | x |
| NBN | c.1720T>A (p.Leu574Ile) | rs142334798 | x | x |
| NBN | c.643C>T (p.Arg215Trp) | rs34767364 | x | x |
| NBN | c.595C>T (p.Pro199Ser) | rs587780097 | x | x |
| NBN | c.1343A>T (p.Gln448Leu) | rs146403088 | x | x |
| NBN | c.511A>G (p.Ile171Val) | rs61754966 | x | x |
| NF1 | c.3883A>G (p.Thr1295Ala) | rs143836226 | x | x |
| NF1 | c.3315A>G (Silent) | rs1555614915 | x | x |
| NF1 | c.169G>A (p.Gly57Ser) | rs779727341 | x | x |
| NHTL1 | c.527T>C (p.Ile176Thr) | rs1805378 | x | x |
| PALB2 | 210A>G (p.Ser70Ser) | rs786202650 | x | x |
| PGDFRA | c.1425G>T (p.Glu475Asp) | rs200309940 | x | x |
| PMS2 | c.1510G>C (p.Glu504Gln) | rs368516768 | x | x |
| PMS2 | c.1004A>G (p.Asn335Ser) | rs200513014 | x | x |
| POLD1 | c.961G>A (p.Gly321Ser) | rs41554817 | x | x |
| RAD50 | c.1720A>C (p.Lys574Gln) | rs779597467 | x | x |
| RAD50 | c.1094G>A (p.Arg365Gln) | rs146370443 | x | x |
| RAD51D | c.26G>C (p.Cys9Ser) | rs140825795 | x | x |
| RNF43 | c.1660C>T (p.Arg554Trp) | Variation ID: 1140674 | x | x |
| All Patients N (%) * | Positive for VUS N (%) ** | p-Value | ||
|---|---|---|---|---|
| Age at time of testing | ≤40 | 204 (30.7%) | 56 (27.4%) | 0.730 |
| >40 | 459 (69.2%) | 132 (28.7%) | ||
| Ethnicity | Hispanic | 35 (5.27%) | 12 (34.2%) | 0.374 |
| Non-Hispanic | 378 (57.01%) | 103 (27.2%) | ||
| Race | White | 333 (50.20%) | 81 (24.3%) | 0.019 |
| African-American | 20 (3.01%) | 8 (40%) | ||
| Asian | 34 (5.12%) | 15 (44.1%) | ||
| Personal history of breast cancer | Yes | 176 (26.5%) | 62 (35.2%) | 0.018 |
| No | 487 (73.4%) | 126 (25.8%) | ||
| Family history of cancer in 1st-degree relative | Yes | 351 (52.9%) | 107 (30.4%) | 0.253 |
| No | 299 (45.1%) | 79 (26.4%) | ||
| Family history of breast cancer in 1st-degree relative | Yes | 116 (17.4%) | 44 (37.9%) | 0.018 |
| No | 394 (59.4%) | 105 (26.6%) | ||
| Family history of ovarian cancer in 1st-degree relative | Yes | 23 (3.4%) | 11 (47.8%) | 0.047 |
| No | 490 (73.9%) | 140 (28.5%) | ||
| Family history of prostate cancer in 1st-degree relative | Yes | 31 (4.89%) | 6 (19.3%) | 0.203 |
| No | 482 (72.6%) | 145 (30.0%) | ||
| Family history of colon cancer in 1st-degree relative | Yes | 21 (3.1%) | 4 (19.0%) | 0.286 |
| No | 492 (74.2%) | 147 (29.8%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chrysafi, P.; Jani, C.T.; Lotz, M.; Al Omari, O.; Singh, H.; Stafford, K.; Agarwal, L.; Rupal, A.; Dar, A.Q.; Dangelo, A.; et al. Prevalence of Variants of Uncertain Significance in Patients Undergoing Genetic Testing for Hereditary Breast and Ovarian Cancer and Lynch Syndrome. Cancers 2023, 15, 5762. https://doi.org/10.3390/cancers15245762
Chrysafi P, Jani CT, Lotz M, Al Omari O, Singh H, Stafford K, Agarwal L, Rupal A, Dar AQ, Dangelo A, et al. Prevalence of Variants of Uncertain Significance in Patients Undergoing Genetic Testing for Hereditary Breast and Ovarian Cancer and Lynch Syndrome. Cancers. 2023; 15(24):5762. https://doi.org/10.3390/cancers15245762
Chicago/Turabian StyleChrysafi, Pavlina, Chinmay T. Jani, Margaret Lotz, Omar Al Omari, Harpreet Singh, Katherine Stafford, Lipisha Agarwal, Arashdeep Rupal, Abdul Qadir Dar, Abby Dangelo, and et al. 2023. "Prevalence of Variants of Uncertain Significance in Patients Undergoing Genetic Testing for Hereditary Breast and Ovarian Cancer and Lynch Syndrome" Cancers 15, no. 24: 5762. https://doi.org/10.3390/cancers15245762
APA StyleChrysafi, P., Jani, C. T., Lotz, M., Al Omari, O., Singh, H., Stafford, K., Agarwal, L., Rupal, A., Dar, A. Q., Dangelo, A., & Lam, P. (2023). Prevalence of Variants of Uncertain Significance in Patients Undergoing Genetic Testing for Hereditary Breast and Ovarian Cancer and Lynch Syndrome. Cancers, 15(24), 5762. https://doi.org/10.3390/cancers15245762