


Nuclear Glycoprotein A Repetitions Predominant (GARP) Is a Common Trait of Glioblastoma Stem-like Cells and Correlates with Poor Survival in Glioblastoma Patients


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Western Blot
2.3. Flow Cytometry
2.4. Confocal Microscopy
2.5. Animal Experiments
2.6. GARP Immunohistochemistry and Immunofluorescence
2.7. Cell Sorting
2.8. Extreme Limiting Dilution Assay
2.9. Bioinformatic Pipeline
2.10. Statistics
3. Results
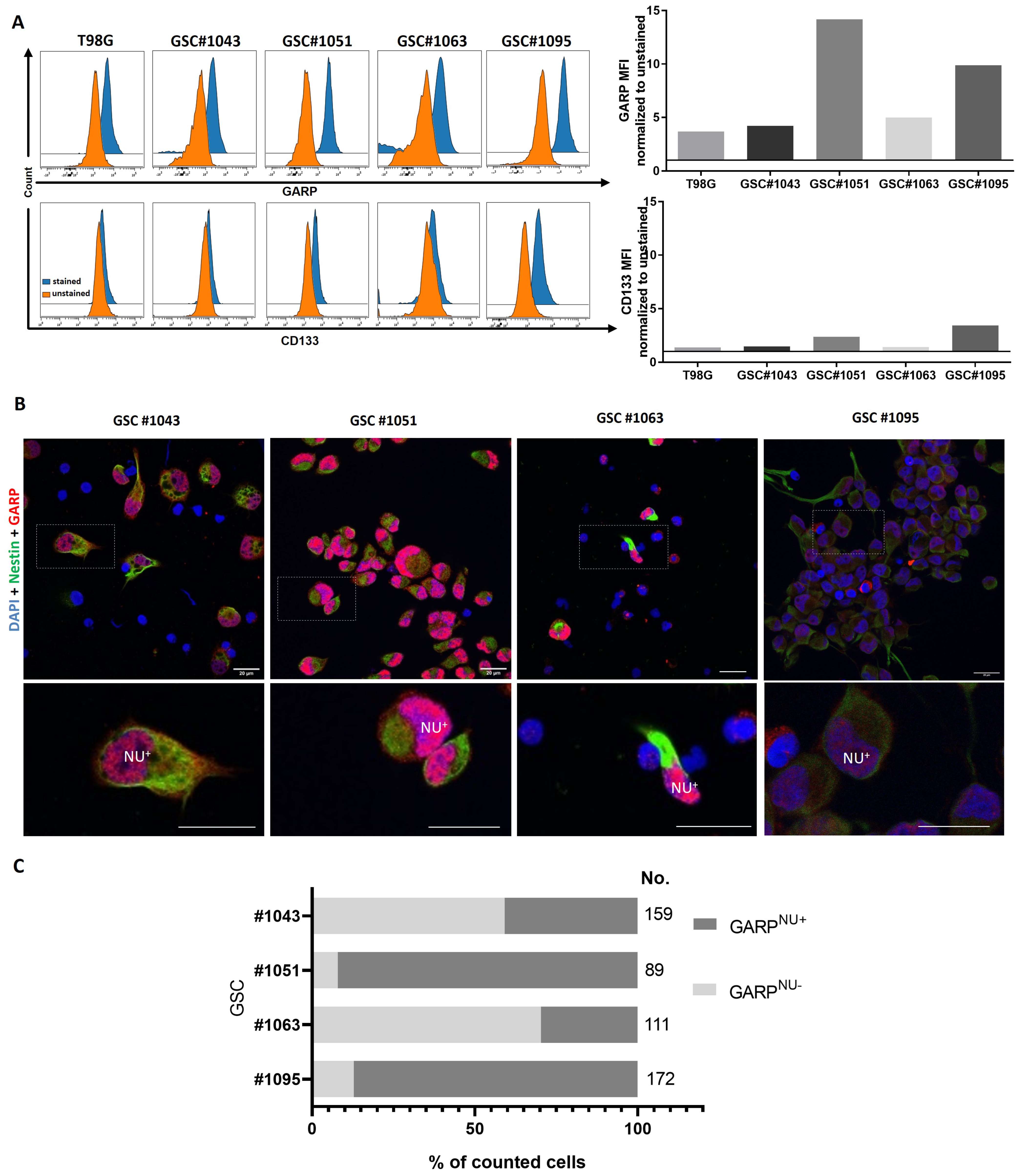
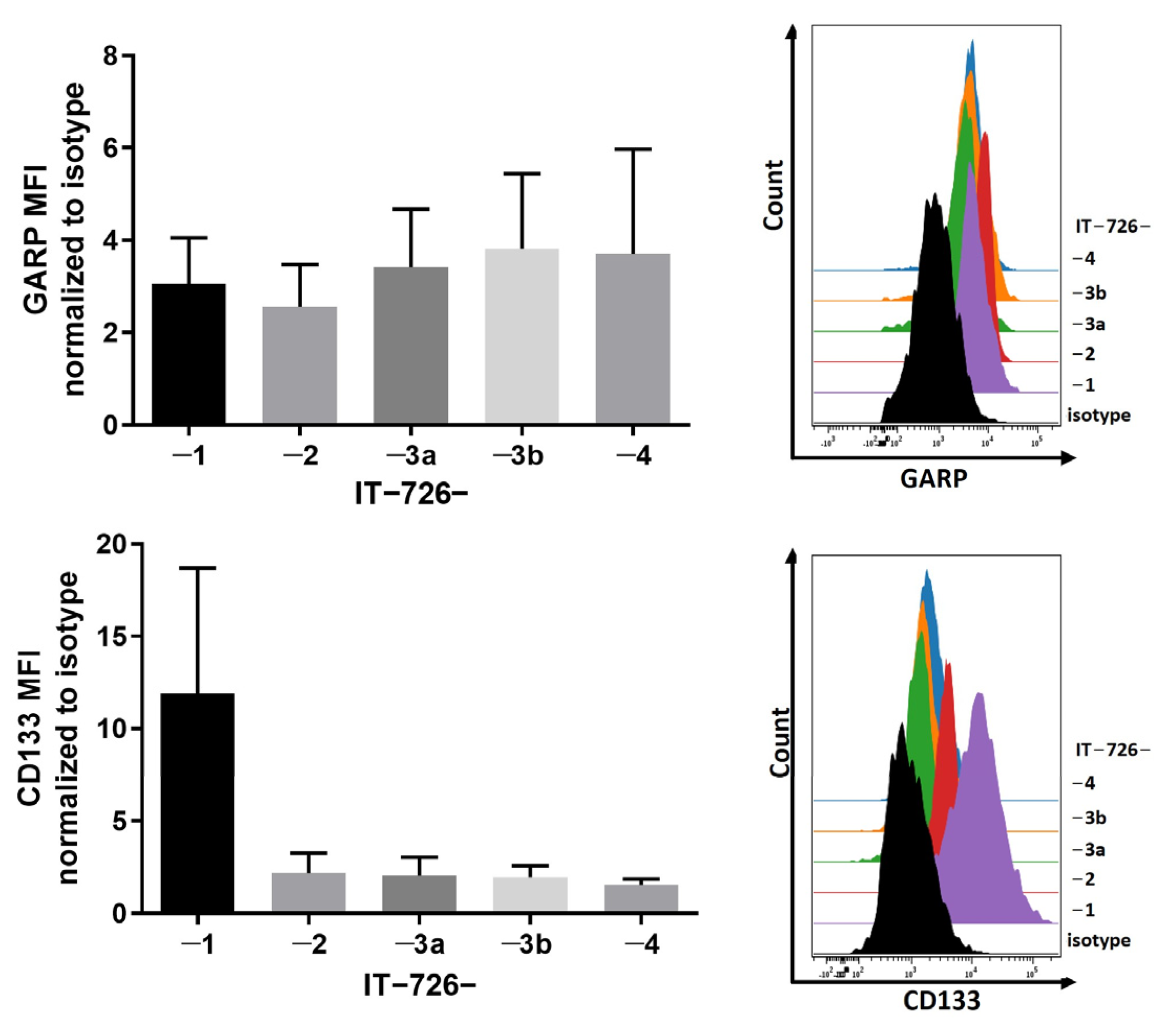
3.1. GARP Expression Is Conserved across Different Types of GSCs In Vitro and In Vivo
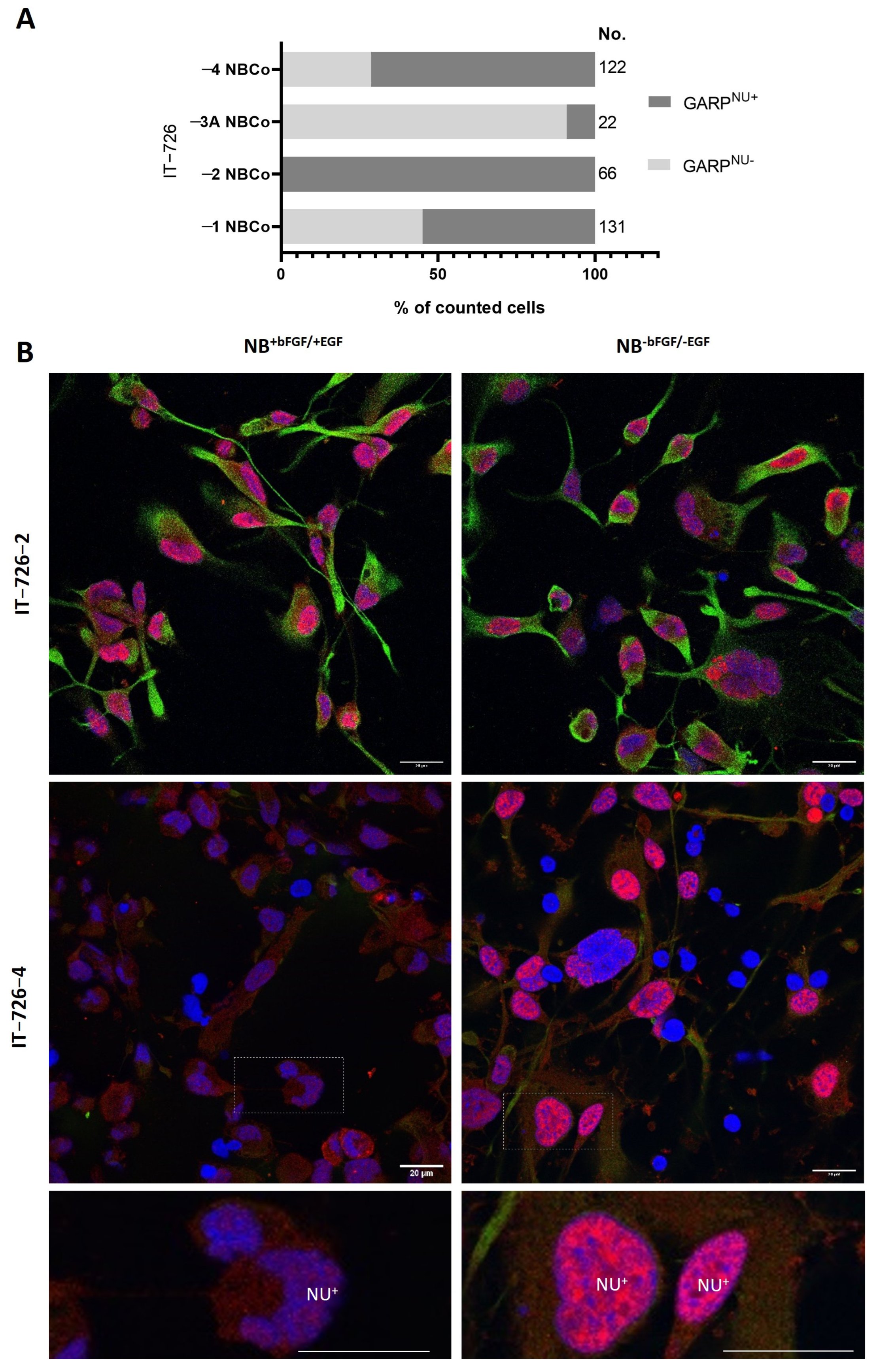
3.2. Intratumoral Heterogeneity of Subcellular Distribution Patterns of GARP
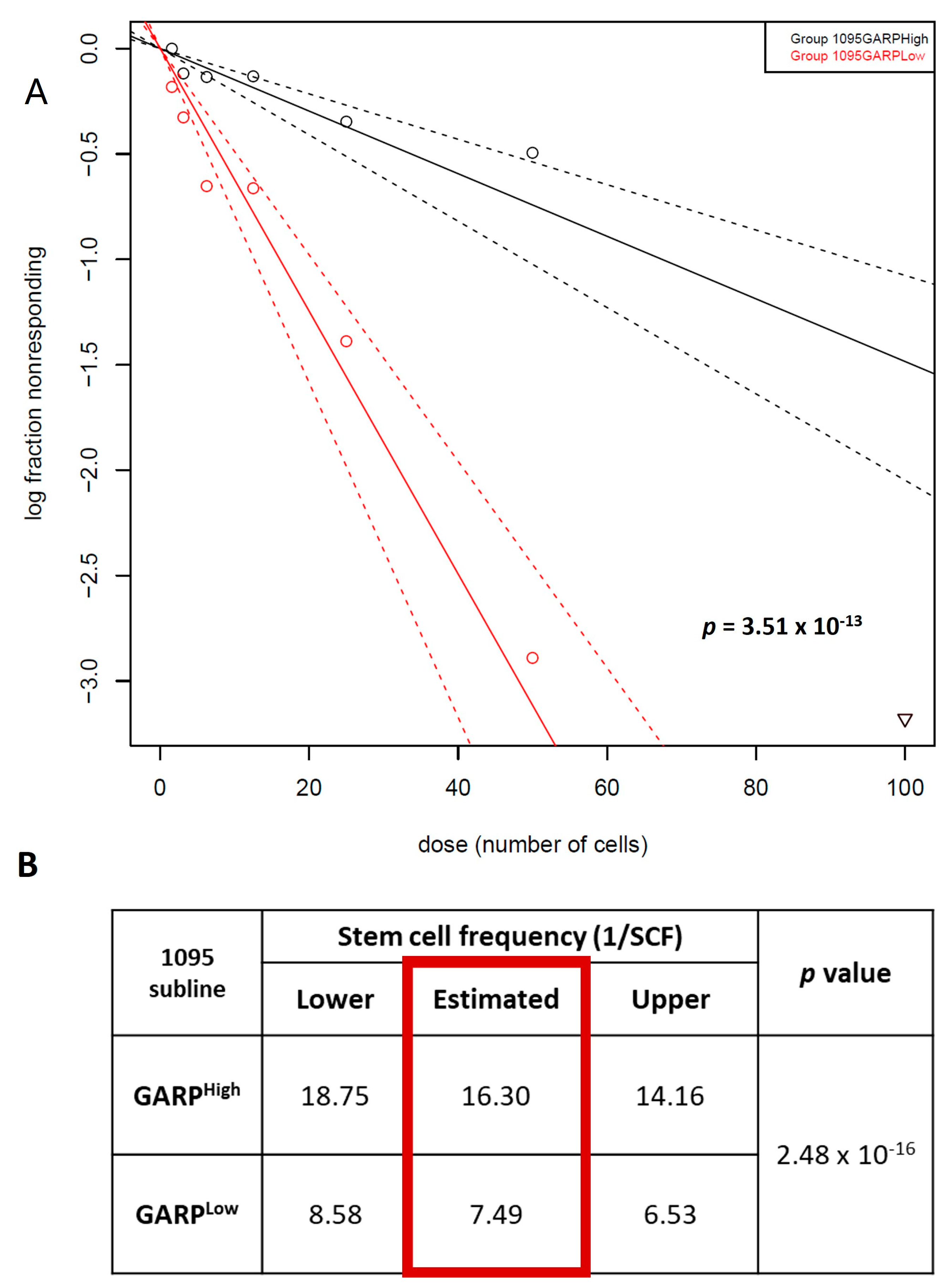
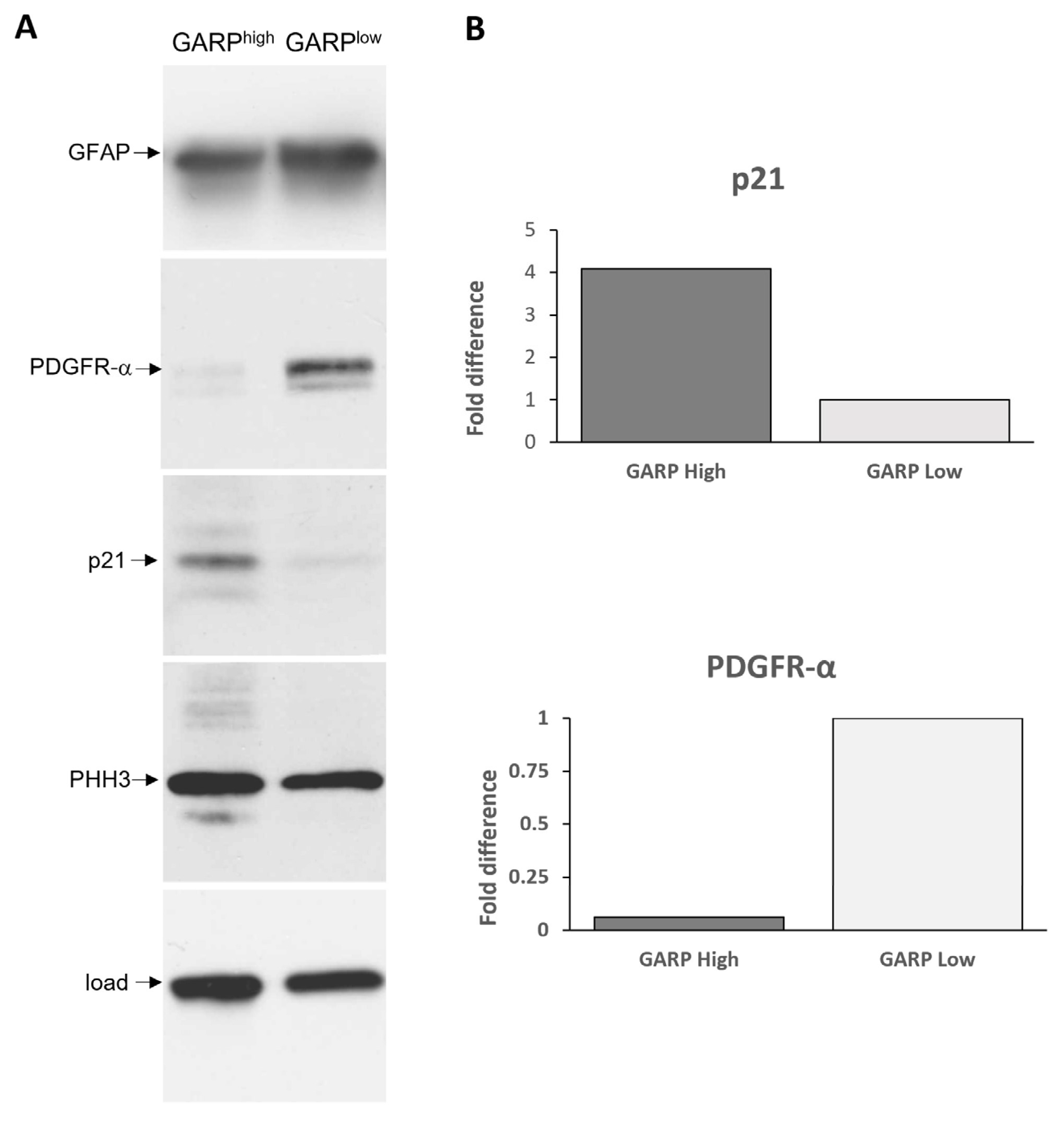
3.3. Relationship between GARP Expression and GSCs Stemness
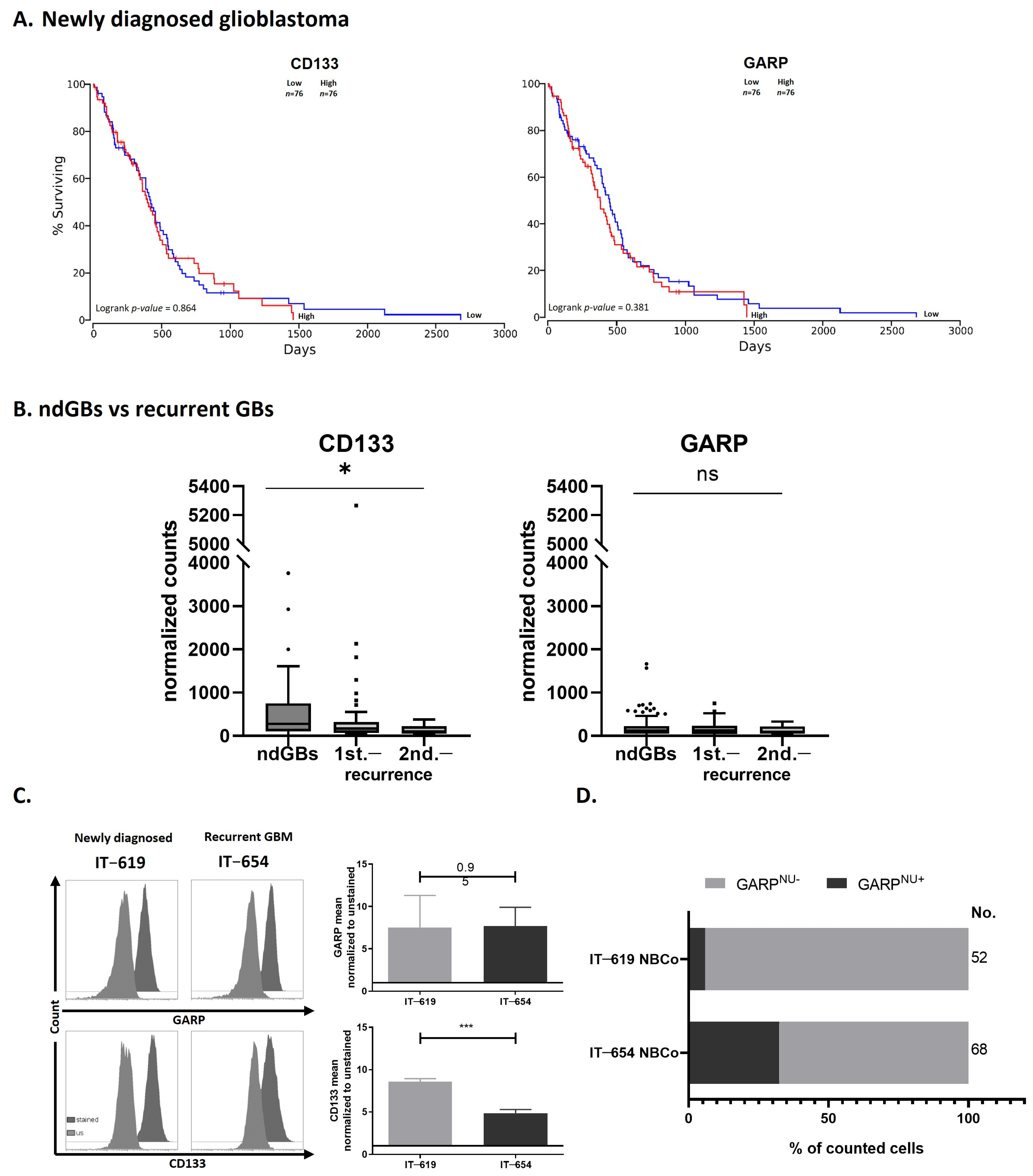
3.4. GARP mRNA and Surface Protein Levels Do Not Predict GB Patient Survival
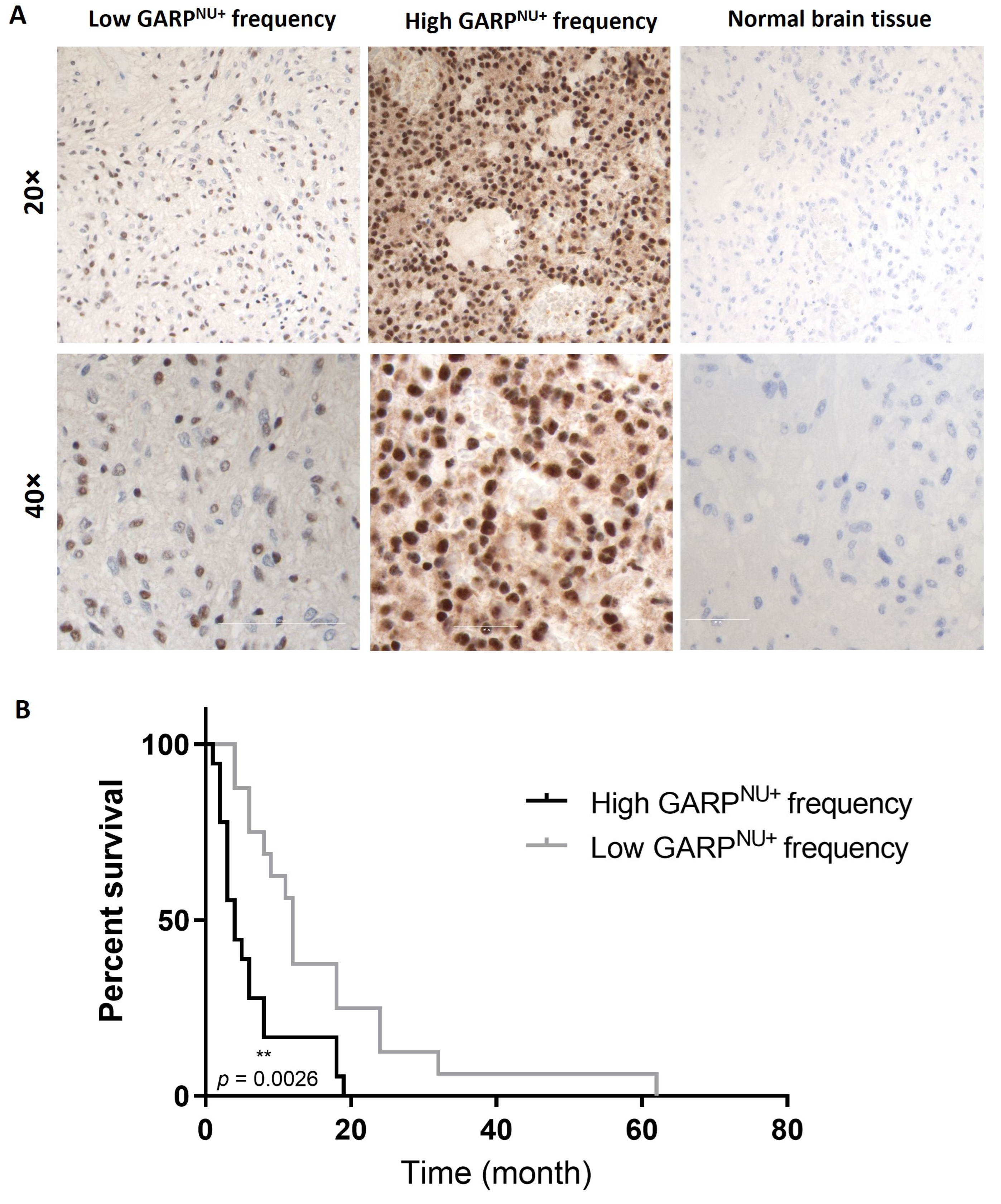
3.5. Nuclear Localization of GARP Correlates with Poor Survival in Patients with GB
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020, 22, 1073–1113. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Campos, B.; Olsen, L.R.; Urup, T.; Poulsen, H.S. A comprehensive profile of recurrent glioblastoma. Oncogene 2016, 35, 5819–5825. [Google Scholar] [CrossRef] [PubMed]
- Gallego, O. Nonsurgical treatment of recurrent glioblastoma. Curr. Oncol. 2015, 22, e273–e281. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, N.; Kim, E.; Sprang, B.; Leukel, P.; Khafaji, F.; Ringel, F.; Sommer, C.; Tuettenberg, J.; Tuettenberg, A. GARP as an Immune Regulatory Molecule in the Tumor Microenvironment of Glioblastoma Multiforme. Int. J. Mol. Sci. 2019, 20, 3676. [Google Scholar] [CrossRef]
- Cai, X.; Sughrue, M.E. Glioblastoma: New therapeutic strategies to address cellular and genomic complexity. Oncotarget 2018, 9, 9540–9554. [Google Scholar] [CrossRef]
- Ghosh, D.; Nandi, S.; Bhattacharjee, S. Combination therapy to checkmate Glioblastoma: Clinical challenges and advances. Clin. Transl. Med. 2018, 7, 33. [Google Scholar] [CrossRef]
- Bonavia, R.; Inda, M.-M.; Cavenee, W.K.; Furnari, F.B. Heterogeneity maintenance in glioblastoma: A social network. Cancer Res. 2011, 71, 4055–4060. [Google Scholar] [CrossRef]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef] [PubMed]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef] [PubMed]
- Kolenda, J.; Jensen, S.S.; Aaberg-Jessen, C.; Christensen, K.; Andersen, C.; Brünner, N.; Kristensen, B.W. Effects of hypoxia on expression of a panel of stem cell and chemoresistance markers in glioblastoma-derived spheroids. J. Neurooncol. 2011, 103, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, D.; Mellai, M.; Annovazzi, L.; Caldera, V.; Piazzi, A.; Denysenko, T.; Melcarne, A. Stem cell niches in glioblastoma: A neuropathological view. Biomed Res. Int. 2014, 2014, 725921. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, V.N.; Hei, T.K. Induction of apoptotic death and retardation of neuronal differentiation of human neural stem cells by sodium arsenite treatment. Exp. Cell Res. 2013, 319, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Natsume, A.; Ito, M.; Katsushima, K.; Ohka, F.; Hatanaka, A.; Shinjo, K.; Sato, S.; Takahashi, S.; Ishikawa, Y.; Takeuchi, I.; et al. Chromatin regulator PRC2 is a key regulator of epigenetic plasticity in glioblastoma. Cancer Res. 2013, 73, 4559–4570. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Günther, H.S.; Schmidt, N.O.; Phillips, H.S.; Kemming, D.; Kharbanda, S.; Soriano, R.; Modrusan, Z.; Meissner, H.; Westphal, M.; Lamszus, K. Glioblastoma-derived stem cell-enriched cultures form distinct subgroups according to molecular and phenotypic criteria. Oncogene 2008, 27, 2897–2909. [Google Scholar] [CrossRef]
- Chen, R.; Nishimura, M.C.; Bumbaca, S.M.; Kharbanda, S.; Forrest, W.F.; Kasman, I.M.; Greve, J.M.; Soriano, R.H.; Gilmour, L.L.; Rivers, C.S.; et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell 2010, 17, 362–375. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Son, M.J.; Woolard, K.; Nam, D.-H.; Lee, J.; Fine, H.A. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell 2009, 4, 440–452. [Google Scholar] [CrossRef]
- Ludwig, K.; Kornblum, H.I. Molecular markers in glioma. J. Neurooncol. 2017, 134, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Beier, D.; Hau, P.; Proescholdt, M.; Lohmeier, A.; Wischhusen, J.; Oefner, P.J.; Aigner, L.; Brawanski, A.; Bogdahn, U.; Beier, C.P. CD133(+) and CD133(-) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007, 67, 4010–4015. [Google Scholar] [CrossRef] [PubMed]
- Ogden, A.T.; Waziri, A.E.; Lochhead, R.A.; Fusco, D.; Lopez, K.; Ellis, J.A.; Kang, J.; Assanah, M.; McKhann, G.M.; Sisti, M.B.; et al. Identification of A2B5+CD133- tumor-initiating cells in adult human gliomas. Neurosurgery 2008, 62, 505–514. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Tang, X.; Zuo, C.; Fang, P.; Liu, G.; Qiu, Y.; Huang, Y.; Tang, R. Targeting Glioblastoma Stem Cells: A Review on Biomarkers, Signal Pathways and Targeted Therapy. Front. Oncol. 2021, 11, 701291. [Google Scholar] [CrossRef]
- Barrantes-Freer, A.; Renovanz, M.; Eich, M.; Braukmann, A.; Sprang, B.; Spirin, P.; Pardo, L.A.; Giese, A.; Kim, E.L. CD133 Expression Is Not Synonymous to Immunoreactivity for AC133 and Fluctuates throughout the Cell Cycle in Glioma Stem-Like Cells. PLoS ONE 2015, 10, e0130519. [Google Scholar] [CrossRef]
- Bidlingmaier, S.; Zhu, X.; Liu, B. The utility and limitations of glycosylated human CD133 epitopes in defining cancer stem cells. J. Mol. Med. 2008, 86, 1025–1032. [Google Scholar] [CrossRef]
- Zimmer, N.; Trzeciak, E.R.; Graefen, B.; Satoh, K.; Tuettenberg, A. GARP as a Therapeutic Target for the Modulation of Regulatory T Cells in Cancer and Autoimmunity. Front. Immunol. 2022, 13, 928450. [Google Scholar] [CrossRef]
- Fauß, J.; Sprang, B.; Leukel, P.; Sommer, C.; Nikolova, T.; Ringel, F.; Kim, E.L. ALDH1A3 Segregated Expression and Nucleus-Associated Proteasomal Degradation Are Common Traits of Glioblastoma Stem Cells. Biomedicines 2021, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.L.; Sorokin, M.; Kantelhardt, S.R.; Kalasauskas, D.; Sprang, B.; Fauss, J.; Ringel, F.; Garazha, A.; Albert, E.; Gaifullin, N.; et al. Intratumoral Heterogeneity and Longitudinal Changes in Gene Expression Predict Differential Drug Sensitivity in Newly Diagnosed and Recurrent Glioblastoma. Cancers 2020, 12, 520. [Google Scholar] [CrossRef] [PubMed]
- Barrantes-Freer, A.; Kim, E.; Bielanska, J.; Giese, A.; Mortensen, L.S.; Schulz-Schaeffer, W.J.; Stadelmann, C.; Brück, W.; Pardo, L.A. Human glioma-initiating cells show a distinct immature phenotype resembling but not identical to NG2 glia. J. Neuropathol. Exp. Neurol. 2013, 72, 307–324. [Google Scholar] [CrossRef] [PubMed]
- Kalasauskas, D.; Sorokin, M.; Sprang, B.; Elmasri, A.; Viehweg, S.; Salinas, G.; Opitz, L.; Rave-Fraenk, M.; Schulz-Schaeffer, W.; Kantelhardt, S.R.; et al. Diversity of Clinically Relevant Outcomes Resulting from Hypofractionated Radiation in Human Glioma Stem Cells Mirrors Distinct Patterns of Transcriptomic Changes. Cancers 2020, 12, 570. [Google Scholar] [CrossRef]
- Müller, A.; Weyerhäuser, P.; Berte, N.; Jonin, F.; Lyubarskyy, B.; Sprang, B.; Kantelhardt, S.R.; Salinas, G.; Opitz, L.; Schulz-Schaeffer, W.; et al. Concurrent Activation of Both Survival-Promoting and Death-Inducing Signaling by Chloroquine in Glioblastoma Stem Cells: Implications for Potential Risks and Benefits of Using Chloroquine as Radiosensitizer. Cells 2023, 12, 1290. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, N.; Krutzik, P.O.; Irish, J.M. Web-based analysis and publication of flow cytometry experiments. Curr. Protoc. Cytom. 2010, 53, 10–17. [Google Scholar] [CrossRef]
- Kim, E.L.; Wüstenberg, R.; Rübsam, A.; Schmitz-Salue, C.; Warnecke, G.; Bücker, E.-M.; Pettkus, N.; Speidel, D.; Rohde, V.; Schulz-Schaeffer, W.; et al. Chloroquine activates the p53 pathway and induces apoptosis in human glioma cells. Neuro Oncol. 2010, 12, 389–400. [Google Scholar] [CrossRef]
- Hahn, S.A.; Neuhoff, A.; Landsberg, J.; Schupp, J.; Eberts, D.; Leukel, P.; Bros, M.; Weilbaecher, M.; Schuppan, D.; Grabbe, S.; et al. A key role of GARP in the immune suppressive tumor microenvironment. Oncotarget 2016, 7, 42996–43009. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Hu, Y.; Smyth, G.K. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 2009, 347, 70–78. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Anaya, J. OncoLnc: Linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Comput. Sci. 2016, 2, e67. [Google Scholar] [CrossRef]
- Brat, D.J.; Verhaak, R.G.W.; Aldape, K.D.; Yung, W.K.A.; Salama, S.R.; Cooper, L.A.D.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; Morozova, O.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, A.; Wickremsekera, A.; Tan, S.T.; Peng, L.; Davis, P.F.; Itinteang, T. Cancer Stem Cell Hierarchy in Glioblastoma Multiforme. Front. Surg. 2016, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Jörg, D.J.; Cavalli, F.M.G.; Richards, L.M.; Nguyen, L.V.; Vanner, R.J.; Guilhamon, P.; Lee, L.; Kushida, M.M.; Pellacani, D.; et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 2017, 549, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Silver, A.; Feier, D.; Ghosh, T.; Rahman, M.; Huang, J.; Sarkisian, M.R.; Deleyrolle, L.P. Heterogeneity of glioblastoma stem cells in the context of the immune microenvironment and geospatial organization. Front. Oncol. 2022, 12, 1022716. [Google Scholar] [CrossRef]
- Miyashita, S.; Hoshino, M. Transit Amplifying Progenitors in the Cerebellum: Similarities to and Differences from Transit Amplifying Cells in Other Brain Regions and between Species. Cells 2022, 11, 726. [Google Scholar] [CrossRef]
- Barrett, L.E.; Granot, Z.; Coker, C.; Iavarone, A.; Hambardzumyan, D.; Holland, E.C.; Nam, H.; Benezra, R. Self-renewal does not predict tumor growth potential in mouse models of high-grade glioma. Cancer Cell 2012, 21, 11–24. [Google Scholar] [CrossRef]
- Jin, X.; Kuang, Y.; Li, L.; Li, H.; Zhao, T.; He, Y.; Di, C.; Kang, J.; Yuan, L.; Yu, B.; et al. A positive feedback circuit comprising p21 and HIF-1α aggravates hypoxia-induced radioresistance of glioblastoma by promoting Glut1/LDHA-mediated glycolysis. FASEB J. 2022, 36, e22229. [Google Scholar] [CrossRef] [PubMed]
- Marqués-Torrejón, M.Á.; Porlan, E.; Banito, A.; Gómez-Ibarlucea, E.; Lopez-Contreras, A.J.; Fernández-Capetillo, O.; Vidal, A.; Gil, J.; Torres, J.; Fariñas, I. Cyclin-dependent kinase inhibitor p21 controls adult neural stem cell expansion by regulating Sox2 gene expression. Cell Stem Cell 2013, 12, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Abdoli Shadbad, M.; Hosseinkhani, N.; Asadzadeh, Z.; Brunetti, O.; Silvestris, N.; Baradaran, B. The Prognostic Value of CD133 in Predicting the Relapse and Recurrence Pattern of High-Grade Gliomas on MRI: A Meta-Analysis. Front. Oncol. 2021, 11, 722833. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.; Meziani, S.; Shah, S.; Kaci, R.; Pimpie, C.; Pocard, M.; Mirshahi, M. Differentiation of cancer cells upregulates HLA-G and PD-L1. Oncol. Rep. 2020, 43, 1797–1804. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.Y.; Sui, H.X.; Liu, Y.; Ke, S.Z.; Wang, Y.N.; Gao, F.G. TGF-β of lung cancer microenvironment upregulates B7H1 and GITRL expression in dendritic cells and is associated with regulatory T cell generation. Oncol. Rep. 2012, 28, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Yuan, P.; Wu, H.; Chen, J.; Fu, J.; Li, P.; Lu, J.; Wei, W. Dendritic cells with an increased PD-L1 by TGF-β induce T cell anergy for the cytotoxicity of hepatocellular carcinoma cells. Int. Immunopharmacol. 2014, 20, 117–123. [Google Scholar] [CrossRef] [PubMed]
- De Streel, G.; Bertrand, C.; Chalon, N.; Liénart, S.; Bricard, O.; Lecomte, S.; Devreux, J.; Gaignage, M.; de Boeck, G.; Mariën, L.; et al. Selective inhibition of TGF-β1 produced by GARP-expressing Tregs overcomes resistance to PD-1/PD-L1 blockade in cancer. Nat. Commun. 2020, 11, 4545. [Google Scholar] [CrossRef]
- Bertrand, C.; van Meerbeeck, P.; de Streel, G.; Vaherto-Bleeckx, N.; Benhaddi, F.; Rouaud, L.; Noël, A.; Coulie, P.G.; van Baren, N.; Lucas, S. Combined Blockade of GARP:TGF-β1 and PD-1 Increases Infiltration of T Cells and Density of Pericyte-Covered GARP+ Blood Vessels in Mouse MC38 Tumors. Front. Immunol. 2021, 12, 704050. [Google Scholar] [CrossRef]
- Hahn, S.A.; Stahl, H.F.; Becker, C.; Correll, A.; Schneider, F.-J.; Tuettenberg, A.; Jonuleit, H. Soluble GARP has potent antiinflammatory and immunomodulatory impact on human CD4⁺ T cells. Blood 2013, 122, 1182–1191. [Google Scholar] [CrossRef]
- Zimmer, N.; Krebs, F.K.; Zimmer, S.; Mitzel-Rink, H.; Kumm, E.J.; Jurk, K.; Grabbe, S.; Loquai, C.; Tuettenberg, A. Platelet-Derived GARP Induces Peripheral Regulatory T Cells-Potential Impact on T Cell Suppression in Patients with Melanoma-Associated Thrombocytosis. Cancers 2020, 12, 3653. [Google Scholar] [CrossRef]
- Xing, H.; Liang, C.; Xu, X.; Sun, H.; Ma, X.; Jiang, Z. Mesenchymal stroma/stem-like cells of GARP knockdown inhibits cell proliferation and invasion of mouse colon cancer cells (MC38) through exosomes. J. Cell. Mol. Med. 2020, 24, 13984–13990. [Google Scholar] [CrossRef]
- Asp, M.; Bergenstråhle, J.; Lundeberg, J. Spatially Resolved Transcriptomes-Next Generation Tools for Tissue Exploration. Bioessays 2020, 42, e1900221. [Google Scholar] [CrossRef] [PubMed]
- Ghebeh, H.; Lehe, C.; Barhoush, E.; Al-Romaih, K.; Tulbah, A.; Al-Alwan, M.; Hendrayani, S.-F.; Manogaran, P.; Alaiya, A.; Al-Tweigeri, T.; et al. Doxorubicin downregulates cell surface B7-H1 expression and upregulates its nuclear expression in breast cancer cells: Role of B7-H1 as an anti-apoptotic molecule. Breast Cancer Res. 2010, 12, R48. [Google Scholar] [CrossRef]
- Urciuoli, E.; Coletta, I.; Rizzuto, E.; de Vito, R.; Petrini, S.; D’Oria, V.; Pezzullo, M.; Milano, G.M.; Cozza, R.; Locatelli, F.; et al. Src nuclear localization and its prognostic relevance in human osteosarcoma. J. Cell. Physiol. 2018, 233, 1658–1670. [Google Scholar] [CrossRef] [PubMed]
- Satelli, A.; Batth, I.S.; Brownlee, Z.; Rojas, C.; Meng, Q.H.; Kopetz, S.; Li, S. Potential role of nuclear PD-L1 expression in cell-surface vimentin positive circulating tumor cells as a prognostic marker in cancer patients. Sci. Rep. 2016, 6, 28910. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Src protein-tyrosine kinase structure and regulation. Biochem. Biophys. Res. Commun. 2004, 324, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Alvarez-Breckenridge, C.A.; Wang, Q.-E.; Yu, J. TGF-β signaling and its targeting for glioma treatment. Am. J. Cancer Res. 2015, 5, 945–955. [Google Scholar] [PubMed]
- Ikushima, H.; Todo, T.; Ino, Y.; Takahashi, M.; Miyazawa, K.; Miyazono, K. Autocrine TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell 2009, 5, 504–514. [Google Scholar] [CrossRef]
- Peñuelas, S.; Anido, J.; Prieto-Sánchez, R.M.; Folch, G.; Barba, I.; Cuartas, I.; García-Dorado, D.; Poca, M.A.; Sahuquillo, J.; Baselga, J.; et al. TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell 2009, 15, 315–327. [Google Scholar] [CrossRef]
- Shaim, H.; Shanley, M.; Basar, R.; Daher, M.; Gumin, J.; Zamler, D.B.; Uprety, N.; Wang, F.; Huang, Y.; Gabrusiewicz, K.; et al. Targeting the αv integrin/TGF-β axis improves natural killer cell function against glioblastoma stem cells. J. Clin. Investig. 2021, 131, e142116. [Google Scholar] [CrossRef]
- Golestaneh, N.; Mishra, B. TGF-beta, neuronal stem cells and glioblastoma. Oncogene 2005, 24, 5722–5730. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zimmer, N.; Trzeciak, E.R.; Müller, A.; Licht, P.; Sprang, B.; Leukel, P.; Mailänder, V.; Sommer, C.; Ringel, F.; Tuettenberg, J.; et al. Nuclear Glycoprotein A Repetitions Predominant (GARP) Is a Common Trait of Glioblastoma Stem-like Cells and Correlates with Poor Survival in Glioblastoma Patients. Cancers 2023, 15, 5711. https://doi.org/10.3390/cancers15245711
Zimmer N, Trzeciak ER, Müller A, Licht P, Sprang B, Leukel P, Mailänder V, Sommer C, Ringel F, Tuettenberg J, et al. Nuclear Glycoprotein A Repetitions Predominant (GARP) Is a Common Trait of Glioblastoma Stem-like Cells and Correlates with Poor Survival in Glioblastoma Patients. Cancers. 2023; 15(24):5711. https://doi.org/10.3390/cancers15245711
Chicago/Turabian StyleZimmer, Niklas, Emily R. Trzeciak, Andreas Müller, Philipp Licht, Bettina Sprang, Petra Leukel, Volker Mailänder, Clemens Sommer, Florian Ringel, Jochen Tuettenberg, and et al. 2023. "Nuclear Glycoprotein A Repetitions Predominant (GARP) Is a Common Trait of Glioblastoma Stem-like Cells and Correlates with Poor Survival in Glioblastoma Patients" Cancers 15, no. 24: 5711. https://doi.org/10.3390/cancers15245711
APA StyleZimmer, N., Trzeciak, E. R., Müller, A., Licht, P., Sprang, B., Leukel, P., Mailänder, V., Sommer, C., Ringel, F., Tuettenberg, J., Kim, E., & Tuettenberg, A. (2023). Nuclear Glycoprotein A Repetitions Predominant (GARP) Is a Common Trait of Glioblastoma Stem-like Cells and Correlates with Poor Survival in Glioblastoma Patients. Cancers, 15(24), 5711. https://doi.org/10.3390/cancers15245711