

Targeting Proteasomes and the MHC Class I Antigen Presentation Machinery to Treat Cancer, Infections and Age-Related Diseases

{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
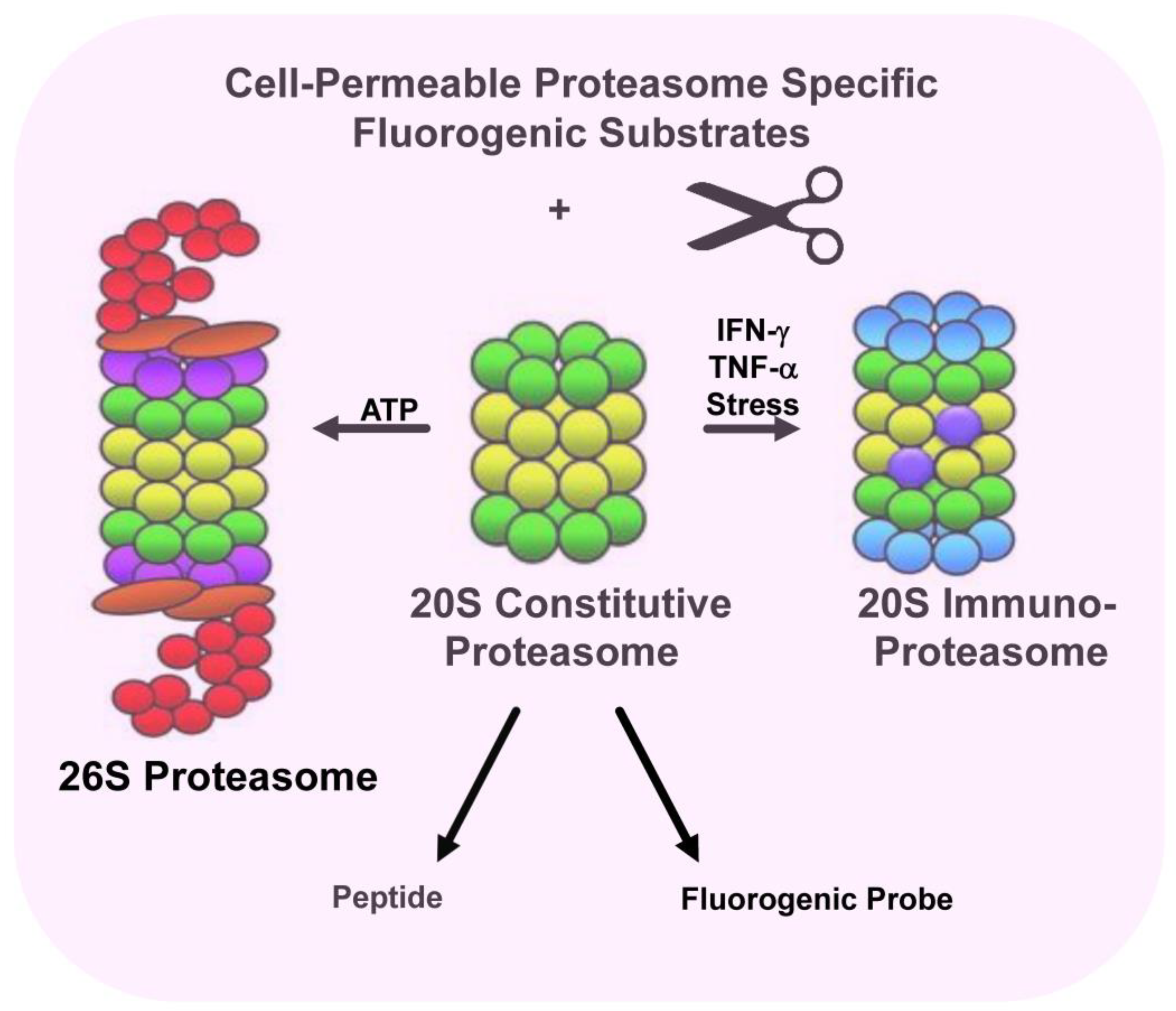
2. Immunoproteasomes
3. Targeting MHC Class I Antigen Presentation Machinery to Boost T-Cell Responses
4. Harnessing Proteasomes to Increase Neoantigens and Tumor-Associated Antigens
5. Targeting Proteasomes to Treat Infectious Diseases
6. Targeting Proteasomes to Treat Neurodegenerative Diseases
7. Proteasomes and Aging
8. Proteasomes and Autophagy
9. Modifiers of Proteasome Activators to Boost Immunotherapeutic Responses
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Park, S.H.; Hartl, F.U. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 2014, 24, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Schoenheimer, R. The Dynamic State of Body Constituents; Harvard University Press: Cambridge, MA, USA, 1942. [Google Scholar]
- Steinberg, D.; Vaughan, M.; Anfinsen, C.B. Kinetic aspects of assembly and degradation of proteins. Science 1956, 124, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Segal, H.L.; Matsuzawa, T.; Haider, M.; Abraham, G.J. What determines the half-life of proteins in vivo? Some experiences with alanine aminotransferase of rat tissues. Biochem. Biophys. Res. Commun. 1969, 36, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Schimke, R.T. Protein turnover and the control of enzyme levels in animal tissues. Acc. Chem. Res. 1970, 3, 113–120. [Google Scholar] [CrossRef]
- Hershko, A.; Tomkins, G.M. Studies on the degradation of tyrosine aminotransferase in hepatoma cells in culture: Influence of the composition of the medium and adenosine triphosphate dependence. J. Biol. Chem. 1971, 246, 710–714. [Google Scholar] [CrossRef]
- Ciechanover, A. Proteolysis: From the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell Biol. 2005, 6, 79–87. [Google Scholar] [CrossRef]
- Kleiger, G.; Mayor, T. Perilous journey: A tour of the ubiquitin-proteasome system. Trends Cell Biol. 2014, 24, 352–359. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Smith, D.M. A Practical Review of Proteasome Pharmacology. Pharmacol. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef]
- Schwartz, A.L.; Ciechanover, A. The ubiquitin-proteasome pathway and pathogenesis of human diseases. Annu. Rev. Med. 1999, 50, 57–74. [Google Scholar] [CrossRef]
- Shen, M.; Schmitt, S.; Buac, D.; Dou, Q.P. Targeting the ubiquitin-proteasome system for cancer therapy. Expert Opin. Ther. Targets 2013, 17, 1091–1108. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.F.; Gan, Z.Y.; Komander, D.; Dewson, G. Ubiquitin signaling in neurodegeneration: Mechanisms and therapeutic opportunities. Cell Death Differ. 2021, 28, 570–590. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Huang, T.; Zhang, L.; Zhou, Y.; Luo, H.; Xu, H.; Wang, X. Dysregulation of Ubiquitin-Proteasome System in Neurodegenerative Diseases. Front. Aging Neurosci. 2016, 8, 303. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, A.; Bertolotti, A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 697–712. [Google Scholar] [CrossRef] [PubMed]
- Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef]
- Livneh, I.; Cohen-Kaplan, V.; Cohen-Rosenzweig, C.; Avni, N.; Ciechanover, A. The life cycle of the 26S proteasome: From birth, through regulation and function, and onto its death. Cell Res. 2016, 26, 869–885. [Google Scholar] [CrossRef]
- Budenholzer, L.; Cheng, C.L.; Li, Y.; Hochstrasser, M. Proteasome Structure and Assembly. J. Mol. Biol. 2017, 429, 3500–3524. [Google Scholar] [CrossRef]
- Ignatz-Hoover, J.J.; Murphy, E.V.; Driscoll, J.J. Targeting Proteasomes in Cancer and Infectious Disease: A Parallel Strategy to Treat Malignancies and Microbes. Front. Cell. Infect. Microbiol. 2022, 12, 925804. [Google Scholar] [CrossRef]
- Driscoll, J.J.; Goldberg, A.L. The proteasome (multicatalytic protease) is a component of the 1500-kDa proteolytic complex which degrades ubiquitin-conjugated proteins. J. Biol. Chem. 1990, 286, 4789–4792. [Google Scholar] [CrossRef]
- Eytan, E.; Ganoth, D.; Armon, T.; Hershko, A. ATP-dependent incorporation of 20S protease into the 26S complex that degrades proteins conjugated to ubiquitin. Proc. Natl. Acad. Sci. USA 1990, 86, 7751–7755. [Google Scholar] [CrossRef]
- Tomko, R.J., Jr.; Hochstrasser, M. Molecular architecture and assembly of the eukaryotic proteasome. Annu. Rev. Biochem. 2013, 82, 415–445. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; DeMartino, G.N. Variably modulated gating of the 26S proteasome by ATP and polyubiquitin. Biochem. J. 2009, 421, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Kleijnen, M.F.; Roelofs, J.; Park, S.; Hathaway, N.A.; Glickman, M.; King, R.W.; Finley, D. Stability of the proteasome can be regulated allosterically through engagement of its proteolytic active sites. Nat. Struct. Mol. Biol. 2007, 14, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Bochtler, M.; Ditzel, L.; Groll, M.; Hartmann, C.; Huber, R. The proteasome. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 295–317. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12–36. [Google Scholar] [CrossRef]
- Bech-Otschir, D.; Helfrich, A.; Enenkel, C.; Consiglieri, G.; Seeger, M.; Holzhütter, H.G.; Dahlmann, B.; Kloetzel, P.M. Polyubiquitin substrates allosterically activate their own degradation by the 26S proteasome. Nat. Struct. Mol. Biol. 2009, 16, 219–225. [Google Scholar] [CrossRef]
- Schmidt, M.; Hanna, J.; Elsasser, S.; Finley, D. Proteasome-associated proteins: Regulation of a proteolytic machine. Biol. Chem. 2005, 386, 725–737. [Google Scholar] [CrossRef]
- Verma, R.; Oania, R.; Graumann, J.; Deshaies, R.J. Multiubiquitin chain receptors define a layer of substrate selectivity in the ubiquitin-proteasome system. Cell 2004, 118, 99–110. [Google Scholar] [CrossRef]
- Elsasser, S.; Chandler-Militello, D.; Müller, B.; Hanna, J.; Finley, D. Rad23 and Rpn10 serve as alternative ubiquitin receptors for the proteasome. J. Biol. Chem. 2004, 279, 26817–26822. [Google Scholar] [CrossRef]
- Madura, K. Rad23 and Rpn10: Perennial wallflowers join the melee. Trends Biochem. Sci. 2004, 29, 637–640. [Google Scholar] [CrossRef]
- Clarke, D.J.; Mondesert, G.; Segal, M.; Bertolaet, B.L.; Jensen, S.; Wolff, M.; Henze, M.; Reed, S.I. Dosage suppressors of pds1 implicate ubiquitin-associated domains in checkpoint control. Mol. Cell Biol. 2001, 21, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Gomez, T.A.; Kolawa, N.; Gee, M.; Sweredoski, M.J.; Deshaies, R.J. Identification of a functional docking site in the Rpn1 LRR domain for the UBA-UBL domain protein Ddi1. BMC Biol. 2011, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Hartmann-Petersen, R.; Gordon, C. Integral UBL domain proteins: A family of proteasome interacting proteins. Semin. Cell Dev. Biol. 2004, 15, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Hicke, L.; Schubert, H.L.; Hill, C.P. Ubiquitin-binding domains. Nat. Rev. Mol. Cell Biol. 2005, 6, 610–621. [Google Scholar] [CrossRef]
- Husnjak, K.; Elsasser, S.; Zhang, N.; Chen, X.; Randles, L.; Shi, Y.; Hofmann, K.; Walters, K.J.; Finley, D.; Dikic, I. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 2008, 453, 481–488. [Google Scholar] [CrossRef]
- Kaplun, L.; Tzirkin, R.; Bakhrat, A.; Shabek, N.; Ivantsiv, Y.; Raveh, D. The DNA damage-inducible UbL-UbA protein Ddi1 participates in Mec1-mediated degradation of HO endonuclease. Mol. Cell Biol. 2005, 25, 5355–5362. [Google Scholar] [CrossRef]
- Kleijnen, M.F.; Shih, A.H.; Zhou, P.; Kumar, S.; Soccio, R.E.; Kedersha, N.L.; Gill, G.; Howley, P.M. The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol. Cell 2000, 6, 409–419. [Google Scholar] [CrossRef]
- Lambertson, D.; Chen, L.; Madura, K. Pleiotropic defects caused by loss of the proteasome-interacting factors Rad23 and Rpn10 of Saccharomyces cerevisiae. Genetics 1999, 153, 69–79. [Google Scholar] [CrossRef]
- Saeki, Y.; Saitoh, A.; Toh-e, A.; Yokosawa, H. Ubiquitin-like proteins and Rpn10 play cooperative roles in ubiquitin-dependent proteolysis. Biochem. Biophys. Res. Commun. 2002, 293, 986–992. [Google Scholar] [CrossRef]
- Elsasser, S.; Finley, D. Delivery of ubiquitinated substrates to protein-unfolding machines. Nat. Cell Biol. 2005, 7, 742–749. [Google Scholar] [CrossRef]
- Saeki, Y. Ubiquitin recognition by the proteasome. J. Biochem. 2017, 161, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Stadtmueller, B.M.; Hill, C.P. Proteasome activators. Mol. Cell 2011, 41, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Caturegli, P.; Takahashi, M.; Suzuki, K. New insights into the function of the immunoproteasome in immune and nonimmune cells. J. Immunol. Res. 2015, 2015, 541984. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D.A.; Gregerson, D.S. Immunoproteasomes: Structure, function, and antigen presentation. Prog. Mol. Biol. Transl. Sci. 2012, 109, 75–112. [Google Scholar] [PubMed]
- Brown, M.G.; Driscoll, J.; Monaco, J.J. Structural and serological similarity of MHC-linked LMP and proteasome (multicatalytic proteinase) complexes. Nature 1991, 353, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Basler, M.; Kirk, C.J.; Groettrup, M. The immunoproteasome in antigen processing and other immunological functions. Curr. Opin. Immunol. 2013, 25, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Groettrup, M.; Kirk, C.; Basler, M. Proteasomes in immune cells: More than peptide producers? Nat. Rev. Immunol. 2010, 10, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, J.; Finley, D. A controlled breakdown: Antigen processing and the turnover of viral proteins. Cell 1992, 68, 823–825. [Google Scholar] [CrossRef]
- Driscoll, J.J.; Brown, M.G.; Finley, D.; Monaco, J.J. MHC-linked LMP gene products specifically alter peptidase activities of the proteasome. Nature 1993, 365, 262–264. [Google Scholar] [CrossRef]
- Groettrup, M.; Standera, S.; Stohwasser, R.; Kloetzel, P.M. The subunits MECL-1 and LMP2 are mutually required for incorporation into the 20S proteasome. Proc. Natl. Acad. Sci. USA 1997, 94, 8970–8975. [Google Scholar] [CrossRef]
- Groettrup, M.; Kraft, R.; Kostka, S.; Standera, S.; Stohwasser, R.; Kloetzel, P.M. A third interferon-gamma-induced subunit exchange in the 20S proteasome. Eur. J. Immunol. 1996, 26, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Trincado, J.L.; Gomez-Perosanz, M.; Reche, P.A. Fundamentals and methods for T-and B-cell epitope prediction. J. Immunol. Res. 2017, 2017, 2680160. [Google Scholar] [CrossRef] [PubMed]
- Diez-Rivero, C.M.; Lafuente, E.M.; Reche, P.A. Computational analysis and modeling of cleavage by the immunoproteasome and the constitutive proteasome. BMC Bioinform. 2010, 11, 479. [Google Scholar] [CrossRef] [PubMed]
- Kesmir, C.; Nussbaum, A.K.; Schild, H.; Detours, V.; Brunak, S. Prediction of proteasome cleavage motifs by neural networks. Protein Eng. 2002, 15, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Bhasin, M.; Raghava, G.P. Pcleavage: An SVM based method for prediction of constitutive proteasome and immunoproteasome cleavage sites in antigenic sequences. Nucleic Acids Res. 2005, 33, W202–W207. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.C.; Vedpathak, D.; Ostrin, E.J. The Functional and Mechanistic Roles of Immunoproteasome Subunits in Cancer. Cells 2021, 10, 3587. [Google Scholar] [CrossRef]
- Rammensee, H.G.; Falk, K.; Rötzschke, O. Peptides naturally presented by MHC class I molecules. Annu. Rev. Immunol. 1993, 11, 213–244. [Google Scholar] [CrossRef]
- Sijts, E.; Kloetzel, P.M. The role of the proteasome in the generation of MHC class I ligands and immune responses. Cell. Mol. Life Sci. 2011, 68, 1491–1502. [Google Scholar] [CrossRef]
- Seifert, U.; Bialy, L.P.; Ebstein, F.; Bech-Otschir, D.; Voigt, A.; Schröter, F.; Prozorovski, T.; Lange, N.; Steffen, J.; Rieger, M.; et al. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell 2010, 142, 613–624. [Google Scholar] [CrossRef]
- van Deventer, S.; Neefjes, J. The Immunoproteasome Cleans up after Inflammation. Cell 2010, 142, 517–518. [Google Scholar] [CrossRef][Green Version]
- Ebstein, F.; Kloetzel, P.M.; Krüger, E.; Seifert, U. Emerging roles of immunoproteasomes beyond MHC class I antigen processing. Cell. Mol. Life Sci. 2012, 69, 2543–2558. [Google Scholar] [CrossRef] [PubMed]
- Rivett, A.J.; Hearn, A.R. Proteasome function in antigen presentation: Immunoproteasome complexes, Peptide production, and interactions with viral proteins. Curr. Protein. Pept. Sci. 2004, 5, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Yewdell, J.W. DRiPs solidify: Progress in understanding endogenous MHC class I antigen processing. Trends Immunol. 2011, 32, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Yewdell, J.W.; Antón, L.C.; Bennink, J.R. Defective ribosomal products (DRiPs). A major source of antigenic peptides for MHC class I molecules? J. Immunol. 1996, 157, 1823–1826. [Google Scholar] [CrossRef] [PubMed]
- Blum, J.S.; Wearsch, P.A.; Cresswell, P. Pathways of antigen processing. Annu. Rev. Immunol. 2013, 31, 443–473. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Dougan, M.; Dranoff, G.; Dougan, S.K. Cancer Immunotherapy: Beyond Checkpoint Blockade. Annu. Rev. Cancer Biol. 2019, 3, 55–75. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Harel, M.; Ortenberg, R.; Varanasi, S.K.; Mangalhara, K.C.; Mardamshina, M.; Markovits, E.; Baruch, E.N.; Tripple, V.; Arama-Chayoth, M.; Greenberg, E.; et al. Proteomics of Melanoma Response to Immunotherapy Reveals Mitochondrial Dependence. Cell 2019, 179, 236–250.e18. [Google Scholar] [CrossRef]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e16. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.S.; Zhang, W.; Wang, X.; Jiang, P.; Traugh, N.; Li, Z.; Meyer, C.; Stewig, B.; Xie, Y.; Bu, X.; et al. Therapeutically Increasing MHC-I Expression Potentiates Immune Checkpoint Blockade. Cancer Discov. 2021, 11, 1524–1541. [Google Scholar] [CrossRef]
- Charles, A.; Bourne, C.M.; Korontsvit, T.; Aretz, Z.E.H.; Mun, S.S.; Dao, T.; Klatt, M.G.; Scheinberg, D.A. Low-dose CDK4/6 inhibitors induce presentation of pathway specific MHC ligands as potential targets for cancer immunotherapy. Oncoimmunology 2021, 10, 1916243. [Google Scholar] [CrossRef] [PubMed]
- Frederick, D.T.; Piris, A.; Cogdill, A.P.; Cooper, Z.A.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin. Cancer Res. 2013, 19, 1225–1231. [Google Scholar] [CrossRef] [PubMed]
- Brea, E.J.; Oh, C.Y.; Manchado, E.; Budhu, S.; Gejman, R.S.; Mo, G.; Mondello, P.; Han, J.E.; Jarvis, C.A.; Ulmert, D.; et al. Kinase Regulation of Human MHC Class I Molecule Expression on Cancer Cells. Cancer Immunol. Res. 2016, 4, 936–947. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, M.; Mezquita, L.; Guillebot De Nerville, G.; Tihy, I.; Malenica, I.; Chouaib, S.; Mami-Chouaib, F. Recent Advances in Lung Cancer Immunotherapy: Input of T-Cell Epitopes Associated With Impaired Peptide Processing. Front. Immunol. 2019, 10, 1505. [Google Scholar] [CrossRef]
- Hicklin, D.J.; Marincola, F.M.; Ferrone, S. HLA class I antigen downregulation in human cancers: T-cell immunotherapy revives an old story. Mol. Med. Today 1999, 5, 178–186. [Google Scholar] [CrossRef]
- Bubeník, J. Tumour MHC class I downregulation and immunotherapy. Oncol. Rep. 2003, 10, 2005–2008. [Google Scholar] [CrossRef]
- Durgeau, A.; El Hage, F.; Vergnon, I.; Validire, P.; de Montpréville, V.; Besse, B.; Soria, J.C.; van Hall, T.; Mami-Chouaib, F. Different expression levels of the TAP peptide transporter lead to recognition of different antigenic peptides by tumor-specific CTL. J. Immunol. 2011, 187, 5532–5539. [Google Scholar] [CrossRef]
- Einstein, M.H.; Leanza, S.; Chiu, L.G.; Schlecht, N.F.; Goldberg, G.L.; Steinberg, B.M.; Burk, R.D. Genetic variants in TAP are associated with high-grade cervical neoplasia. Clin. Cancer Res. 2009, 15, 1019–1023. [Google Scholar] [CrossRef]
- Leibowitz, M.S.; Andrade Filho, P.A.; Ferrone, S.; Ferris, R.L. Deficiency of activated STAT1 in head and neck cancer cells mediates TAP1-dependent escape from cytotoxic T lymphocytes. Cancer Immunol. Immunother. 2011, 60, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Marincola, F.M.; Jaffee, E.M.; Hicklin, D.J.; Ferrone, S. Escape of human solid tumors from T–cell recognition: Molecular mechanisms and functional significance. Adv. Immunol. 1999, 74, 181–273. [Google Scholar]
- Abele, R.; Tampé, R. Modulation of the antigen transport machinery TAP by friends and enemies. FEBS Lett. 2006, 580, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.Y.; Klatt, M.G.; Bourne, C.; Dao, T.; Dacek, M.M.; Brea, E.J.; Mun, S.S.; Chang, A.Y.; Korontsvit, T.; Scheinberg, D.A. ALK and RET inhibitors promote HLA class I antigen presentation and unmask new antigens within the tumor immunopeptidome. Cancer Immunol. Res. 2019, 7, 1984–1997. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Bastian, I.N.; Long, M.D.; Dow, M.; Li, W.; Liu, T.; Ngu, R.K.; Antonucci, L.; Huang, J.Y.; Phung, Q.T.; et al. Activation of NF-κB and p300/CBP potentiates cancer chemoimmunotherapy through induction of MHC-I antigen presentation. Proc. Natl. Acad. Sci. USA 2021, 118, e2025840118. [Google Scholar] [CrossRef]
- Arnold, D.; Driscoll, J.; Androlewicz, M.; Hughes, E.; Cresswell, P.; Spies, T. Proteasome subunits encoded in the MHC are not generally required for the processing of peptides bound by MHC class I molecules. Nature 1992, 360, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Glynne, R.; Powis, S.H.; Beck, S.; Kelly, A.; Kerr, L.; Trowsdale, J. A proteasome-related gene between the two ABC transporter loci in the class II region of the human MHC. Nature 1991, 353, 357–360. [Google Scholar] [CrossRef]
- Garbi, N.; Tiwari, N.; Momburg, F.; Hämmerling, G.J. A major role for tapasin as a stabilizer of the TAP peptide transporter and consequences for MHC class I expression. Eur. J. Immunol. 2003, 33, 264–273. [Google Scholar] [CrossRef]
- Diedrich, G.; Bangia, N.; Pan, M.; Cresswell, P. A role for calnexin in the assembly of the MHC class I loading complex in the endoplasmic reticulum. J. Immunol. 2001, 166, 1703–1709. [Google Scholar] [CrossRef]
- Schmidt, K.; Leisegang, M.; Kloetzel, P.M. ERAP2 supports TCR recognition of three immunotherapy targeted tumor epitopes. Mol. Immunol. 2023, 154, 61–68. [Google Scholar] [CrossRef]
- Xie, N.; Shen, G.; Gao, W.; Huang, Z.; Huang, C.; Fu, L. Neoantigens: Promising targets for cancer therapy. Signal Transduct. Target. Ther. 2023, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Borden, E.S.; Buetow, K.H.; Wilson, M.A.; Hastings, K.T. Cancer Neoantigens: Challenges and Future Directions for Prediction, Prioritization, and Validation. Front. Oncol. 2022, 12, 836821. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, A.H.; Hwang, M.S.; Konig, M.F.; Hsiue, E.H.; Douglass, J.; DiNapoli, S.R.; Mog, B.J.; Bettegowda, C.; Pardoll, D.M.; Gabelli, S.B.; et al. Targeting public neoantigens for cancer immunotherapy. Nat. Cancer 2021, 2, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Yewdell, J.W. MHC class I immunopeptidome: Past, present, and future. Mol. Cell. Proteom. 2022, 21, 100230. [Google Scholar] [CrossRef] [PubMed]
- Admon, A. The biogenesis of the immunopeptidome. Semin. Immunol. 2023, 67, 101766. [Google Scholar] [CrossRef] [PubMed]
- Rana, P.S.; Ignatz-Hoover, J.J.; Driscoll, J.J. Discovery of novel HDAC6 inhibitors that enhance proteasomal activity to boost antigen presentation and trigger anti-myeloma T-cell immunity. Cancer Res. 2023, 83, 6235. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple myeloma: 2022 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2022, 97, 1086–1107. [Google Scholar] [CrossRef]
- Cowan, A.J.; Green, D.J.; Kwok, M.; Lee, S.; Coffey, D.G.; Holmberg, L.A.; Tuazon, S.; Gopal, A.K.; Libby, E.N. Diagnosis and Management of Multiple Myeloma: A Review. JAMA 2022, 327, 464–477. [Google Scholar] [CrossRef]
- Driscoll, J.J.; Brailey, M. Emerging small molecule approaches to enhance the antimyeloma benefit of proteasome inhibitors. Cancer Metastasis Rev. 2017, 36, 585–598. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Kimbrough, E.O.; Lou, Y. Next generation of immune checkpoint inhibitors and beyond. J. Hematol. Oncol. 2021, 14, 45. [Google Scholar] [CrossRef]
- Cao, Y.; Zhu, H.; He, R.; Kong, L.; Shao, J.; Zhuang, R.; Xi, J.; Zhang, J. Proteasome, a Promising Therapeutic Target for Multiple Diseases Beyond Cancer. Drug Des. Dev. Ther. 2020, 14, 4327–4342. [Google Scholar] [CrossRef]
- Bibo-Verdugo, B.; Jiang, Z.; Caffrey, C.R.; O’Donoghue, A.J. Targeting proteasomes in infectious organisms to combat disease. FEBS J. 2017, 284, 1503–1517. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Tsu, C.; Dick, L.; Zhou, X.K.; Nathan, C. Distinct specificities of Mycobacterium. tuberculosis. and mammalian proteasomes for N-acetyl tripeptide substrates. J. Biol. Chem. 2008, 283, 34423–34431. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ponder, E.L.; Verdoes, M.; Asbjornsdottir, K.H.; Deu, E.; Edgington, L.E.; Lee, J.T.; Kirk, C.J.; Demo, S.D.; Williamson, K.C.; et al. Validation of the proteasome as a therapeutic target in plasmodium using an epoxyketone inhibitor with parasite-specific toxicity. Chem. Biol. 2012, 19, 1535–1545. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, K.; van Den Broek, M.; Kostka, S.; Kraft, R.; Soza, A.; Schmidtke, G.; Kloetzel, P.M.; Groettrup, M. Overexpression of the proteasome subunits LMP2, LMP7, and MECL-1, but not PA28 alpha/beta, enhances the presentation of an immunodominant lymphocytic choriomeningitis virus T cell epitope. J. Immunol. 2000, 165, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Nagle, A.S.; Biggart, A.; Lai, Y.H.; Liang, F.; Davis, L.C.; Barnes, S.W.; Mathison, C.J.; Myburgh, E.; Gao, M.Y.; et al. Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 2016, 537, 229–233. [Google Scholar] [CrossRef]
- Totaro, K.A.; Barthelme, D.; Simpson, P.T.; Jiang, X.; Lin, G.; Nathan, C.F.; Sauer, R.T.; Sello, J.K. Rational Design of Selective and Bioactive Inhibitors of the Mycobacterium tuberculosis Proteasome. ACS Infect. Dis. 2017, 3, 176–181. [Google Scholar] [CrossRef]
- Zhan, W.; Zhang, H.; Ginn, J.; Leung, A.; Liu, Y.J.; Michino, M.; Toita, A.; Okamoto, R.; Wong, T.T.; Imaeda, T.; et al. Development of a Highly Selective Plasmodium falciparum Proteasome Inhibitor with Anti-malaria Activity in Humanized Mice. Angew. Chem. Int. Ed. Engl. 2021, 60, 9279–9283. [Google Scholar] [CrossRef]
- LaMonte, G.M.; Almaliti, J.; Bibo-Verdugo, B.; Keller, L.; Zou, B.Y.; Yang, J.; Antonova-Koch, Y.; Orjuela-Sanchez, P.; Boyle, C.A.; Vigil, E.; et al. Development of a Potent Inhibitor of the Plasmodium Proteasome with Reduced Mammalian Toxicity. J. Med. Chem. 2017, 60, 6721–6732. [Google Scholar] [CrossRef]
- Augusto, D.G.; Hollenbach, J.A. HLA variation and antigen presentation in COVID-19 and SARS-CoV-2 infection. Curr. Opin. Immunol. 2022, 76, 102178. [Google Scholar] [CrossRef]
- Tola, F.S. The Role of Ubiquitin-Proteasome System in the Pathogenesis of Severe Acute Respiratory Syndrome Coronavirus-2 Disease. Int. J. Inflam. 2023, 2023, 6698069. [Google Scholar]
- Yewdell, J.W. Antigenic drift: Understanding COVID-19. Immunity 2021, 54, 2681–2687. [Google Scholar] [CrossRef] [PubMed]
- Wellington, D.; Yin, Z.; Yu, Z.; Heilig, R.; Davis, S.; Fischer, R.; Felce, S.L.; Hublitz, P.; Beveridge, R.; Dong, D.; et al. SARS-CoV-2 mutations affect proteasome processing to alter CD8 T cell responses. bioRxiv 2022. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein Aggregation and Neurodegenerative Disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Soto, C. Unfolding the Role of Protein Misfolding in Neurodegenerative Diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Sonntag, K.C.; Kim, W.; Cattaneo, E.; Isacson, O. Proteasome activator enhances survival of Huntington’s disease neuronal model cells. PLoS ONE 2007, 2, e238. [Google Scholar] [CrossRef]
- Hipp, M.S.; Patel, C.N.; Bersuker, K.; Riley, B.E.; Kaiser, S.E.; Shaler, T.A.; Brandeis, M.; Kopito, R.R. Indirect inhibition of 26S proteasome activity in a cellular model of Huntington’s disease. J. Cell Biol. 2012, 196, 573–587. [Google Scholar] [CrossRef]
- Grad, L.I.; Rouleau, G.A.; Ravits, J.; Cashman, N.R. Clinical Spectrum of Amyotrophic Lateral Sclerosis (ALS). Cold Spring Harb. Perspect. Med. 2017, 7, a024117. [Google Scholar] [CrossRef]
- Le Gall, L.; Anakor, E.; Connolly, O.; Vijayakumar, U.G.; Duddy, W.J.; Duguez, S. Molecular and Cellular Mechanisms Affected in ALS. J. Pers. Med. 2020, 10, 101. [Google Scholar] [CrossRef]
- Hirano, A. Familial Amyotrophic Lateral Sclerosis. Arch. Neurol. 1967, 16, 232. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Butti, Z.; Patten, S.A. RNA Dysregulation in Amyotrophic Lateral Sclerosis. Front. Genet. 2019, 9, 712. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA Processing Protein, Cause Familial Amyotrophic Lateral Sclerosis Type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Gill, C.; Phelan, J.P.; Hatzipetros, T.; Kidd, J.D.; Tassinari, V.R.; Levine, B.; Wang, M.Z.; Moreno, A.; Thompson, K.; Maier, M.; et al. SOD1-positive aggregate accumulation in the CNS predicts slower disease progression and increased longevity in a mutant SOD1 mouse model of ALS. Sci. Rep. 2019, 9, 6724. [Google Scholar] [CrossRef] [PubMed]
- Feneberg, E.; Gray, E.; Ansorge, O.; Talbot, K.; Turner, M.R. Towards a TDP-43-Based Biomarker for ALS and FTLD. Mol. Neurobiol. 2018, 55, 7789–7801. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.A.; Pickart, C.M.; Alban, A.; Landon, M.; Jamieson, C.; Ramage, R.; Mayer, R.J.; Layfield, R. Inhibition of the Ubiquitin-Proteasome System in Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2000, 97, 9902–9906. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.N.; Hanni, K.B.; Markesbery, W.R. Impaired Proteasome Function in Alzheimer’s Disease. J. Neurochem. 2000, 75, 436–439. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1097. [Google Scholar] [CrossRef]
- Lee, M.J.; Bhattarai, D.; Jang, H.; Baek, A.; Yeo, I.J.; Lee, S.; Miller, Z.; Lee, S.; Hong, J.T.; Kim, D.E.; et al. Macrocyclic Immunoproteasome Inhibitors as a Potential Therapy for Alzheimer’s Disease. J. Med. Chem. 2021, 64, 10934–10950. [Google Scholar] [CrossRef]
- Jones, C.L.; Njomen, E.; Sjögren, B.; Dexheimer, T.S.; Tepe, J.J. Small Molecule Enhancement of 20S Proteasome Activity Targets Intrinsically Disordered Proteins. ACS Chem. Biol. 2017, 12, 2240–2247. [Google Scholar] [CrossRef]
- Trader, D.J.; Simanski, S.; Dickson, P.; Kodadek, T. Establishment of a suite of assays that support the discovery of proteasome stimulators. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 892–899. [Google Scholar] [CrossRef]
- Vilchez, D.; Saez, I.; Dillin, A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 2014, 5, 5659. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Lee, C.K.; Klopp, R.G.; Weindruch, R.; Prolla, T.A. Gene expression profile of aging and its retardation by caloric restriction. Science 1999, 285, 1390–1393. [Google Scholar] [CrossRef]
- Ly, D.H.; Lockhart, D.J.; Lerner, R.A.; Schultz, P.G. Mitotic misregulation and human aging. Science 2000, 287, 2486–2492. [Google Scholar] [CrossRef]
- Tundo, G.R.; Sbardella, D.; Oddone, F.; Kudriaeva, A.A.; Lacal, P.M.; Belogurov, A.A., Jr.; Graziani, G.; Marini, S. At the cutting edge against cancer: A perspective on immunoproteasome and immune checkpoints modulation as a potential therapeutic intervention. Cancers 2021, 13, 4852. [Google Scholar] [CrossRef]
- Tundo, G.R.; Sbardella, D.; Santoro, A.M.; Coletta, A.; Oddone, F.; Grasso, G.; Milardi, D.; Lacal, P.M.; Marini, S.; Purrello, R.; et al. The proteasome as a druggable target with multiple therapeutic potentialities: Cutting and non-cutting edges. Pharmacol. Ther. 2020, 213, 107579. [Google Scholar] [CrossRef]
- Saez, I.; Vilchez, D. The Mechanistic Links Between Proteasome Activity, Aging and Age-related Diseases. Curr. Genom. 2014, 15, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D.A.; Husom, A.D.; Thompson, L.V. Altered proteasome structure, function, and oxidation in aged muscle. FASEB J. 2005, 19, 644–646. [Google Scholar] [CrossRef] [PubMed]
- Vernace, V.A.; Arnaud, L.; Schmidt-Glenewinkel, T.; Figueiredo-Pereira, M.E. Aging perturbs 26S proteasome assembly in Drosophila melanogaster. FASEB J. 2007, 21, 2672–2682. [Google Scholar] [CrossRef] [PubMed]
- Bulteau, A.L.; Petropoulos, I.; Friguet, B. Age-related alterations of proteasome structure and function in aging epidermis. Exp. Gerontol. 2000, 35, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Carrard, G.; Dieu, M.; Raes, M.; Toussaint, O.; Friguet, B. Impact of ageing on proteasome structure and function in human lymphocytes. Int. J. Biochem. Cell Biol. 2003, 35, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Hegde, A.N.; Duke, L.M.; Timm, L.E.; Nobles, H. The Proteasome and Ageing. Subcell. Biochem. 2023, 102, 99–112. [Google Scholar]
- Grune, T.; Jung, T.; Merker, K.; Davies, K.J. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ‘aggresomes’ during oxidative stress, aging, and disease. Int. J. Biochem. Cell Biol. 2004, 36, 2519–2530. [Google Scholar] [CrossRef]
- Andersson, V.; Hanzen, S.; Liu, B.; Molin, M.; Nystrom, T. Enhancing protein disaggregation restores proteasome activity in aged cells. Aging 2013, 5, 802–812. [Google Scholar] [CrossRef]
- De Duve, C. The lysosome. Sci. Am. 1963, 208, 64–72. [Google Scholar] [CrossRef]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Mansilla, A.; Menzies, F.M.; Rubinsztein, D.C. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell 2009, 33, 517–527. [Google Scholar] [CrossRef]
- Wang, X.J.; Yu, J.; Wong, S.H.; Cheng, A.S.; Chan, F.K.; Ng, S.S.; Cho, C.H.; Sung, J.J.; Wu, W.K. A novel crosstalk between two major protein degradation systems: Regulation of proteasomal activity by autophagy. Autophagy 2013, 9, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Myeku, N.; Figueiredo-Pereira, M.E. Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: Association with sequestosome 1/p62. J. Biol. Chem. 2011, 286, 22426–22440. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, S.; Vad, N.; Vallabhapurapu, S.; Vallabhapurapu, S.; Anderson, K.C.; Driscoll, J.J. MiR-29b replacement inhibits proteasomes and disrupts aggresome+autophagosome formation to enhance the antimyeloma benefit of bortezomib. Leukemia 2015, 29, 727–738. [Google Scholar] [CrossRef]
- Malek, E.; Abdel Malek, M.; Jagannathan, S.; Cottini, F.; Anderson, K.C.; Driscoll, J.J. Pharmacogenomics and chemical library screens reveal a novel SCFSKP2 inhibitor that overcomes bortezomib resistance in multiple myeloma. Leukemia 2016, 31, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Ignatz-Hoover, J.J.; Driscoll, J.J. Therapeutics to harness the immune microenvironment in multiple myeloma. Cancer Drug Resist. 2022, 5, 647–661. [Google Scholar] [CrossRef]
- Misund, K.; Baranowska, K.A.; Holien, T.; Starheim, K.; Johansson, I.; Buene, G.; Waage, A.; Bjørkøy, G.; Sundan, A. Chloroquine potentiates carfilzomib but not bortezomib effects on myeloma cells. Cancer Res. 2015, 75 (Suppl. S15), 1768. [Google Scholar] [CrossRef]
- Jagannathan, S.; Vad, N.; Latif, T.; Anderson, K.C.; Driscoll, J.J. Pharmacologic screens reveal metformin that suppresses GRP78-dependent autophagy to enhance the anti-myeloma effect to bortezomib. Leukemia 2015, 29, 2184–2191. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Lin, C.; Kuffour, E.O.; Fuchs, N.V.; Gertzen, C.G.W.; Kaiser, J.; Hirschenberger, M.; Tang, X.; Xu, H.C.; Michel, O.; Tao, R.; et al. Regulation of STING activity in DNA sensing by ISG15 modification. Cell Rep. 2023, 42, 113277. [Google Scholar] [CrossRef]
- Cho, W.; Won, S.; Choi, Y.; Yi, S.; Park, J.B.; Park, J.G.; Kim, C.E.; Narayana, C.; Kim, J.H.; Yim, J.; et al. Targeted Protein Upregulation of STING for Boosting the Efficacy of Immunotherapy. Angew. Chem. Int. Ed. Engl. 2023, 62, e202300978. [Google Scholar] [CrossRef]
- Gonugunta, V.K.; Sakai, T.; Pokatayev, V.; Yang, K.; Wu, J.; Dobbs, N.; Yan, N. Trafficking-Mediated STING Degradation Requires Sorting to Acidified Endolysosomes and Can Be Targeted to Enhance Anti-tumor Response. Cell Rep. 2017, 21, 3234–3242. [Google Scholar] [CrossRef]
- Liu, Z.; Hu, M.; Yang, Y.; Du, C.; Zhou, H.; Liu, C.; Chen, Y.; Fan, L.; Ma, H.; Gong, Y.; et al. An overview of PROTACs: A promising drug discovery paradigm. Mol. Biomed. 2022, 3, 46. [Google Scholar] [CrossRef]
- Cornel, A.M.; Mimpen, I.L.; Nierkens, S. MHC Class I downregulation in cancer: Underlying mechanisms and potential targets for cancer Immunotherapy. Cancers 2020, 12, 1760. [Google Scholar] [CrossRef]
- Desrichard, A.; Snyder, A.; Chan, T.A. Cancer Neoantigens and Applications for Immunotherapy. Clin. Cancer Res. 2016, 22, 807–812. [Google Scholar] [CrossRef]
- Kalaora, S.; Lee, J.S.; Barnea, E.; Levy, R.; Greenberg, P.; Alon, M.; Yagel, G.; Bar Eli, G.; Oren, R.; Peri, A.; et al. Immunoproteasome expression is associated with better prognosis and response to checkpoint therapies in melanoma. Nat. Commun. 2020, 11, 896. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rana, P.S.; Ignatz-Hoover, J.J.; Driscoll, J.J. Targeting Proteasomes and the MHC Class I Antigen Presentation Machinery to Treat Cancer, Infections and Age-Related Diseases. Cancers 2023, 15, 5632. https://doi.org/10.3390/cancers15235632
Rana PS, Ignatz-Hoover JJ, Driscoll JJ. Targeting Proteasomes and the MHC Class I Antigen Presentation Machinery to Treat Cancer, Infections and Age-Related Diseases. Cancers. 2023; 15(23):5632. https://doi.org/10.3390/cancers15235632
Chicago/Turabian StyleRana, Priyanka S., James J. Ignatz-Hoover, and James J. Driscoll. 2023. "Targeting Proteasomes and the MHC Class I Antigen Presentation Machinery to Treat Cancer, Infections and Age-Related Diseases" Cancers 15, no. 23: 5632. https://doi.org/10.3390/cancers15235632
APA StyleRana, P. S., Ignatz-Hoover, J. J., & Driscoll, J. J. (2023). Targeting Proteasomes and the MHC Class I Antigen Presentation Machinery to Treat Cancer, Infections and Age-Related Diseases. Cancers, 15(23), 5632. https://doi.org/10.3390/cancers15235632