
The Role of ZNF275/AKT Pathway in Carcinogenesis and Cisplatin Chemosensitivity of Cervical Cancer Using Patient-Derived Xenograft Models

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Tissue Samples
2.2. Cell lines and Culture
2.3. Compounds
2.4. Database Mining
2.5. Lentivirus and Transfection
2.6. Colony Formation Assay
2.7. Cell Viability Assay
2.8. Drug Combination Index
2.9. Transwell Migration and Invasion Assays
2.10. Flow Cytometric Analysis
2.11. Bromodeoxyuridine (BrdU) Proliferation Assay
2.12. Western Blotting Analysis
2.13. Establishment of Cervical Cancer PDX Models
2.14. Immunohistochemistry (IHC) Staining
2.15. In Vivo Drug Treatment Study
2.16. Statistical Analysis
3. Results
3.1. Expression Level of ZNF275 Protein in Cervical Cancer
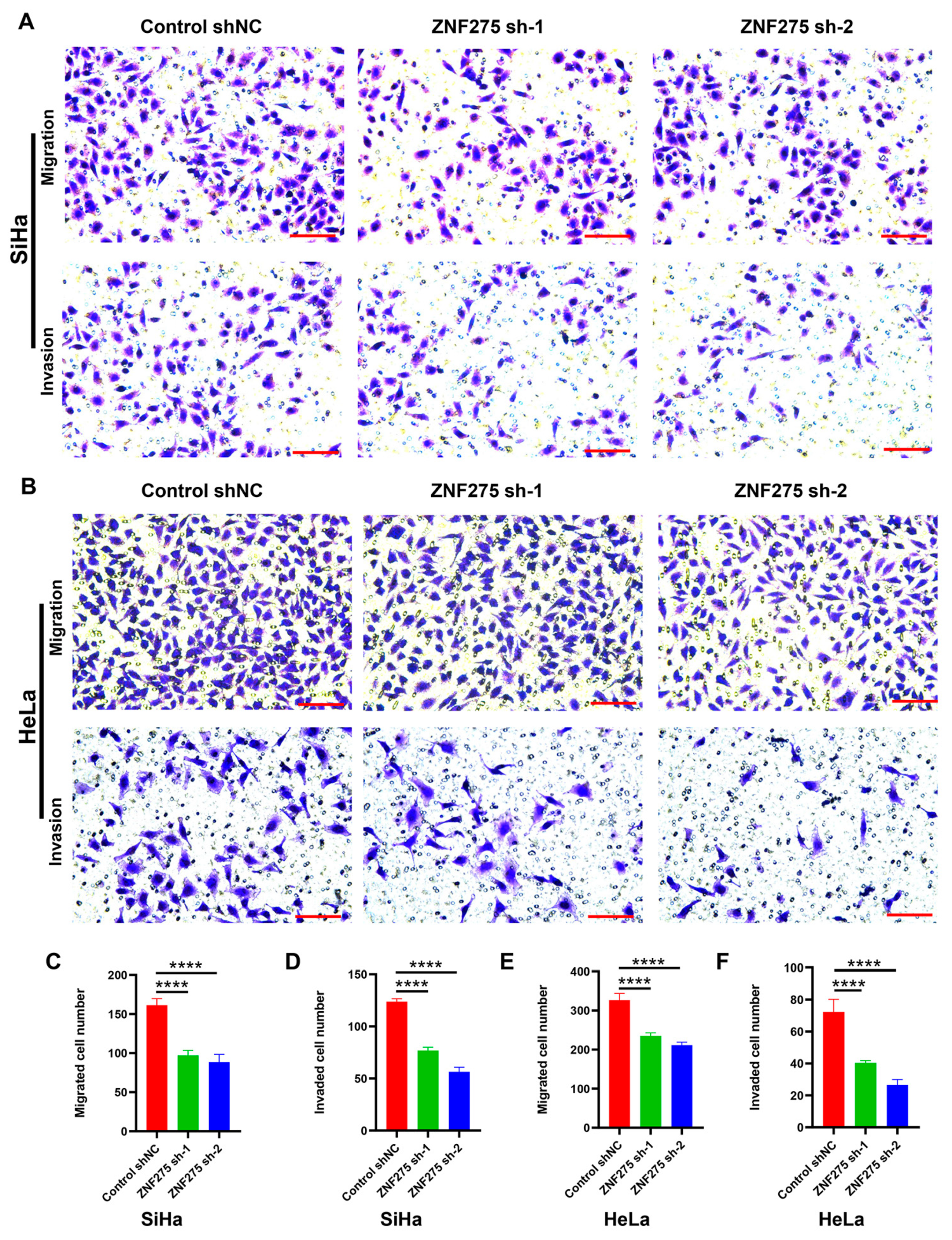
3.2. Effects of ZNF275 Downregulation on Migrative and Invasive Abilities of Cervical Cancer Cells
3.3. Effects of ZNF275 Downregulation on Apoptosis of Cervical Cancer Cells
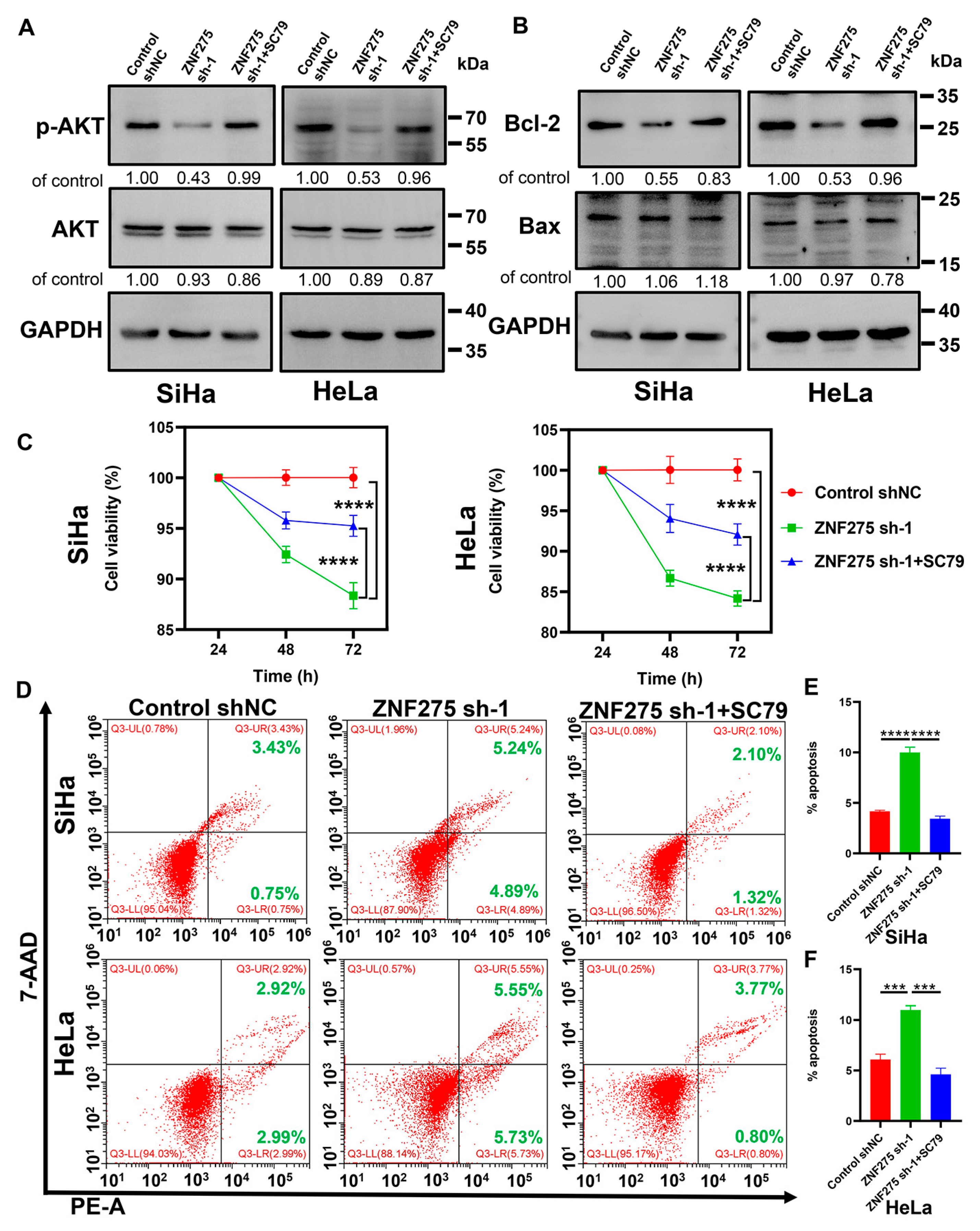
3.4. Activation of AKT/Bcl-2 Signaling Pathway Rescued ZNF275 Downregulation Mediated Effects on Cervical Cancer Cell Viability and Apoptosis
3.5. Therapeutically Targeting ZNF275-Induced AKT Pathway with Triciribine Influenced the Viabilities of SiHa and Caski Cells
3.6. Effects of Triciribine and Cisplatin on Biological Behaviors of SiHa Cells
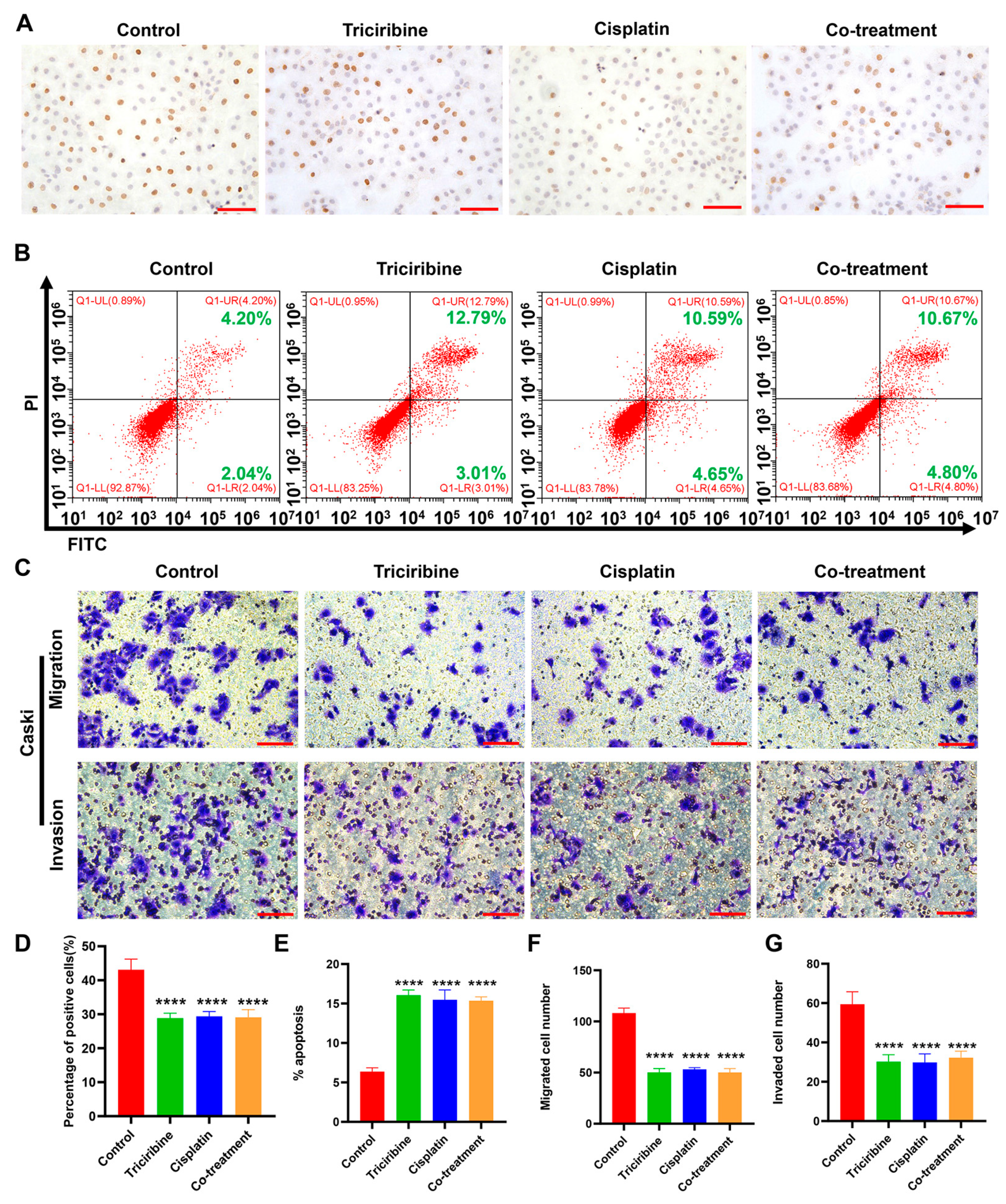
3.7. Effects of Triciribine and Cisplatin on Biological Behaviors of Caski Cells
3.8. Therapeutic Efficacies of Cisplatin, Triciribine, and the Co-Treatment in Cervical Cancer F3-PDX Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Small, W., Jr.; Bacon, M.A.; Bajaj, A.; Chuang, L.T.; Fisher, B.J.; Harkenrider, M.M.; Jhingran, A.; Kitchener, H.C.; Mileshkin, L.R.; Viswanathan, A.N.; et al. Cervical cancer: A global health crisis. Cancer 2017, 123, 2404–2412. [Google Scholar] [CrossRef] [PubMed]
- Masadah, R.; Rauf, S.; Pratama, M.Y.; Tiribelli, C.; Pascut, D. The Role of microRNAs in the Cisplatin- and Radio-Resistance of Cervical Cancer. Cancers 2021, 13, 1168. [Google Scholar] [CrossRef]
- Cohen, A.; Roane, B.; Leath, C. Novel Therapeutics for Recurrent Cervical Cancer: Moving Towards Personalized Therapy. Drugs 2020, 80, 217–227. [Google Scholar] [CrossRef]
- Tian, X.; Wang, X.; Cui, Z.; Liu, J.; Huang, X.; Shi, C.; Zhang, M.; Liu, T.; Du, X.; Li, R.; et al. A Fifteen-Gene Classifier to Predict Neoadjuvant Chemotherapy Responses in Patients with Stage IB to IIB Squamous Cervical Cancer. Adv. Sci. 2021, 8, 2001978. [Google Scholar] [CrossRef] [PubMed]
- Crowley, F.J.; O’Cearbhaill, R.E.; Collins, D.C. Exploiting somatic alterations as therapeutic targets in advanced and metastatic cervical cancer. Cancer Treat. Rev. 2021, 98, 102225. [Google Scholar] [CrossRef]
- Abdolahi, S.; Ghazvinian, Z.; Muhammadnejad, S.; Saleh, M.; Asadzadeh Aghdaei, H.; Baghaei, K. Patient-derived xenograft (PDX) models, applications and challenges in cancer research. J. Transl. Med. 2022, 20, 206. [Google Scholar] [CrossRef]
- Conte, N.; Mason, J.C.; Halmagyi, C.; Neuhauser, S.; Mosaku, A.; Yordanova, G.; Chatzipli, A.; Begley, D.A.; Krupke, D.M.; Parkinson, H.; et al. PDX Finder: A portal for patient-derived tumor xenograft model discovery. Nucleic Acids Res. 2019, 47, D1073–D1079. [Google Scholar] [CrossRef]
- Zou, S.; Ye, M.; Zhang, J.a.; Ji, H.; Chen, Y.; Zhu, X. Establishment and genetically characterization of patient-derived xenograft models of cervical cancer. BMC Med. Genom. 2022, 15, 191. [Google Scholar] [CrossRef]
- Bu, S.; Lv, Y.; Liu, Y.; Qiao, S.; Wang, H. Zinc Finger Proteins in Neuro-Related Diseases Progression. Front. Neurosci. 2021, 15, 760567. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xiao, Z.; Zhou, J.; Yang, M.; Feng, S.; Huang, Q.; Zou, J.; Zeng, T.; Li, Y.; Peng, L.; et al. ZNF382: A transcription inhibitor down-regulated in multiple tumors due to promoter methylation. Clin. Chim. Acta 2020, 500, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Cassandri, M.; Smirnov, A.; Novelli, F.; Pitolli, C.; Agostini, M.; Malewicz, M.; Melino, G.; Raschella, G. Zinc-finger proteins in health and disease. Cell Death Discov. 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Jen, J.; Wang, Y.C. Zinc finger proteins in cancer progression. J. Biomed. Sci. 2016, 23, 53. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, H.; Wang, Q.; Xu, S. ZNF217: The cerberus who fails to guard the gateway to lethal malignancy. Am. J. Cancer Res. 2021, 11, 3378–3405. [Google Scholar]
- Sayers, E.W.; Agarwala, R.; Bolton, E.E.; Brister, J.R.; Canese, K.; Clark, K.; Connor, R.; Fiorini, N.; Funk, K.; Hefferon, T.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2019, 47, D23–D28. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Som, A. Decoding molecular markers and transcriptional circuitry of naive and primed states of human pluripotency. Stem Cell Res. 2021, 53, 102334. [Google Scholar] [CrossRef]
- Halder, A.K.; Cordeiro, M. AKT Inhibitors: The Road Ahead to Computational Modeling-Guided Discovery. Int. J. Mol. Sci. 2021, 22, 3944. [Google Scholar] [CrossRef]
- Brown, J.S.; Banerji, U. Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol. Ther. 2017, 172, 101–115. [Google Scholar] [CrossRef]
- Berndt, N.; Yang, H.; Trinczek, B.; Betzi, S.; Zhang, Z.; Wu, B.; Lawrence, N.J.; Pellecchia, M.; Schonbrunn, E.; Cheng, J.Q.; et al. The Akt activation inhibitor TCN-P inhibits Akt phosphorylation by binding to the PH domain of Akt and blocking its recruitment to the plasma membrane. Cell Death Differ. 2010, 17, 1795–1804. [Google Scholar] [CrossRef]
- Toson, B.; Fortes, I.; Roesler, R.; Andrade, S. Targeting Akt/PKB in pediatric tumors: A review from preclinical to clinical trials. Pharmacol. Res. 2022, 183, 106403. [Google Scholar] [CrossRef]
- Liu, T.; Fang, Y.; Zhang, H.; Deng, M.; Gao, B.; Niu, N.; Yu, J.; Lee, S.; Kim, J.; Qin, B.; et al. HEATR1 Negatively Regulates Akt to Help Sensitize Pancreatic Cancer Cells to Chemotherapy. Cancer Res. 2016, 76, 572–581. [Google Scholar] [CrossRef]
- Northey, J.J.; Barrett, A.S.; Acerbi, I.; Hayward, M.K.; Talamantes, S.; Dean, I.S.; Mouw, J.K.; Ponik, S.M.; Lakins, J.N.; Huang, P.J.; et al. Stiff stroma increases breast cancer risk by inducing the oncogene ZNF217. J. Clin. Investig. 2020, 130, 5721–5737. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed]
- Kramer, D.; Stark, N.; Schulz-Heddergott, R.; Erytch, N.; Edmunds, S.; Rossmann, L.; Bastians, H.; Concin, N.; Moll, U.M.; Dobbelstein, M. Strong antitumor synergy between DNA crosslinking and HSP90 inhibition causes massive premitotic DNA fragmentation in ovarian cancer cells. Cell Death Differ. 2017, 24, 300–316. [Google Scholar] [CrossRef] [PubMed]
- Buglio, D.; Georgakis, G.; Hanabuchi, S.; Arima, K.; Khaskhely, N.; Liu, Y.; Younes, A. Vorinostat inhibits STAT6-mediated TH2 cytokine and TARC production and induces cell death in Hodgkin lymphoma cell lines. Blood 2008, 112, 1424–1433. [Google Scholar] [CrossRef]
- Deng, X.; Li, S.; Kong, F.; Ruan, H.; Xu, X.; Zhang, X.; Wu, Z.; Zhang, L.; Xu, Y.; Yuan, H.; et al. Long noncoding RNA PiHL regulates p53 protein stability through GRWD1/RPL11/MDM2 axis in colorectal cancer. Theranostics 2020, 10, 265–280. [Google Scholar] [CrossRef]
- Cohen, P.A.; Jhingran, A.; Oaknin, A.; Denny, L. Cervical cancer. Lancet 2019, 393, 169–182. [Google Scholar] [CrossRef]
- Zhou, X.; Lian, H.; Li, H.; Fan, M.; Xu, W.; Jin, Y. Nanotechnology in cervical cancer immunotherapy: Therapeutic vaccines and adoptive cell therapy. Front. Pharmacol. 2022, 13, 1065793. [Google Scholar] [CrossRef]
- Zhu, H.; Luo, H.; Zhang, W.; Shen, Z.; Hu, X.; Zhu, X. Molecular mechanisms of cisplatin resistance in cervical cancer. Drug Des. Dev. Ther. 2016, 10, 1885–1895. [Google Scholar] [CrossRef]
- Monk, B.J.; Tewari, K.S.; Dubot, C.; Caceres, M.V.; Hasegawa, K.; Shapira-Frommer, R.; Salman, P.; Yanez, E.; Gumus, M.; Hurtado de Mendoza, M.O.; et al. Health-related quality of life with pembrolizumab or placebo plus chemotherapy with or without bevacizumab for persistent, recurrent, or metastatic cervical cancer (KEYNOTE-826): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2023, 24, 392–402. [Google Scholar] [CrossRef]
- Hong, D.S.; Concin, N.; Vergote, I.; Bono, J.S.d.; Slomovitz, B.M.; Drew, Y.; Arkenau, H.-T.; Machiels, J.-P.; Spicer, J.F.; Jones, R.; et al. Tisotumab Vedotin in Previously Treated Recurrent or Metastatic Cervical Cancer. Clin. Cancer Res. 2020, 26, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, M.; Li, X.; Yin, S.; Wang, B. An Overview of Novel Agents for Cervical Cancer Treatment by Inducing Apoptosis: Emerging Drugs Ongoing Clinical Trials and Preclinical Studies. Front. Med. 2021, 8, 682366. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Qiu, B.; Li, L.; Xu, J.; Zhou, H.; Niu, T. An emerging prognosis prediction model for multiple myeloma: Hypoxia-immune related microenvironmental gene signature. Front. Oncol. 2022, 12, 992387. [Google Scholar] [CrossRef]
- Krig, S.R.; Miller, J.K.; Frietze, S.; Beckett, L.A.; Neve, R.M.; Farnham, P.J.; Yaswen, P.I.; Sweeney, C.A. ZNF217, a candidate breast cancer oncogene amplified at 20q13, regulates expression of the ErbB3 receptor tyrosine kinase in breast cancer cells. Oncogene 2010, 29, 5500–5510. [Google Scholar] [CrossRef]
- Xie, W.; Yu, J.; Yin, Y.; Zhang, X.; Zheng, X.; Wang, X. OCT4 induces EMT and promotes ovarian cancer progression by regulating the PI3K/AKT/mTOR pathway. Front. Oncol. 2022, 12, 876257. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Chen, H.; Wang, X.; Chen, T. Regorafenib induces Bim-mediated intrinsic apoptosis by blocking AKT-mediated FOXO3a nuclear export. Cell Death Discov. 2023, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Littlepage, L.E.; Adler, A.S.; Kouros-Mehr, H.; Huang, G.; Chou, J.; Krig, S.R.; Griffith, O.L.; Korkola, J.E.; Qu, K.; Lawson, D.A.; et al. The transcription factor ZNF217 is a prognostic biomarker and therapeutic target during breast cancer progression. Cancer Discov. 2012, 2, 638–651. [Google Scholar] [CrossRef]
- Gloesenkamp, C.R.; Nitzsche, B.; Ocker, M.; Fazio, P.D.; Quint, K.; Hoffmann, B.; Scherübl, H.; Höpfner, M. AKT inhibition by triciribine alone or as combination therapy for growth control of gastroenteropancreatic neuroendocrine tumors. Int. J. Oncol. 2012, 40, 876–888. [Google Scholar] [CrossRef]
- Balasis, M.E.; Forinash, K.D.; Chen, Y.A.; Fulp, W.J.; Coppola, D.; Hamilton, A.D.; Cheng, J.Q.; Sebti, S.M. Combination of farnesyltransferase and Akt inhibitors is synergistic in breast cancer cells and causes significant breast tumor regression in ErbB2 transgenic mice. Clin. Cancer Res. 2011, 17, 2852–2862. [Google Scholar] [CrossRef]
- Cho, S.Y.; Kang, W.; Han, J.Y.; Min, S.; Kang, J.; Lee, A.; Kwon, J.Y.; Lee, C.; Park, H. An Integrative Approach to Precision Cancer Medicine Using Patient-Derived Xenografts. Mol. Cells 2016, 39, 77–86. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, M.; Liu, T.; Miao, L.; Zou, S.; Ji, H.; Zhang, J.; Zhu, X. The Role of ZNF275/AKT Pathway in Carcinogenesis and Cisplatin Chemosensitivity of Cervical Cancer Using Patient-Derived Xenograft Models. Cancers 2023, 15, 5625. https://doi.org/10.3390/cancers15235625
Ye M, Liu T, Miao L, Zou S, Ji H, Zhang J, Zhu X. The Role of ZNF275/AKT Pathway in Carcinogenesis and Cisplatin Chemosensitivity of Cervical Cancer Using Patient-Derived Xenograft Models. Cancers. 2023; 15(23):5625. https://doi.org/10.3390/cancers15235625
Chicago/Turabian StyleYe, Miaomiao, Tingxian Liu, Liqing Miao, Shuangwei Zou, Huihui Ji, Jian’an Zhang, and Xueqiong Zhu. 2023. "The Role of ZNF275/AKT Pathway in Carcinogenesis and Cisplatin Chemosensitivity of Cervical Cancer Using Patient-Derived Xenograft Models" Cancers 15, no. 23: 5625. https://doi.org/10.3390/cancers15235625
APA StyleYe, M., Liu, T., Miao, L., Zou, S., Ji, H., Zhang, J., & Zhu, X. (2023). The Role of ZNF275/AKT Pathway in Carcinogenesis and Cisplatin Chemosensitivity of Cervical Cancer Using Patient-Derived Xenograft Models. Cancers, 15(23), 5625. https://doi.org/10.3390/cancers15235625