
Two Faces of Glutaminase GLS2 in Carcinogenesis

Abstract
:Simple Summary
Abstract

1. Introduction
2. The GLS2 Gene and Its Products
3. GLS2 as a Promoter of Tumorigenesis
4. GLS2 as a Suppressor of Tumorigenesis
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to Clinic: Glutamine Metabolism to Cancer Therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef]
- Willems, L.; Jacque, N.; Jacquel, A.; Neveux, N.; Trovati Maciel, T.; Lambert, M.; Schmitt, A.; Poulain, L.; Green, A.S.; Uzunov, M.; et al. Inhibiting Glutamine Uptake Represents an Attractive New Strategy for Treating Acute Myeloid Leukemia. Blood 2013, 122, 3521–3532. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.-M.; Oh, M.-H.; Sun, I.-H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine Blockade Induces Divergent Metabolic Programs to Overcome Tumor Immune Evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Liang, Y.; Chen, Y.; Wang, L.; Li, D.; Liang, Z.; Sun, L.; Wang, Y.; Niu, H. Glutamine Affects T24 Bladder Cancer Cell Proliferation by Activating STAT3 through ROS and Glutaminolysis. Int. J. Mol. Med. 2019, 44, 2189–2200. [Google Scholar] [CrossRef]
- Jin, H.; Wang, S.; Zaal, E.A.; Wang, C.; Wu, H.; Bosma, A.; Jochems, F.; Isima, N.; Jin, G.; Lieftink, C.; et al. A Powerful Drug Combination Strategy Targeting Glutamine Addiction for the Treatment of Human Liver Cancer. eLife 2020, 9, 56749. [Google Scholar] [CrossRef]
- Yamashita, A.S.; da Costa Rosa, M.; Stumpo, V.; Rais, R.; Slusher, B.S.; Riggins, G.J. The Glutamine Antagonist Prodrug JHU-083 Slows Malignant Glioma Growth and Disrupts MTOR Signaling. Neurooncol. Adv. 2021, 3, 149. [Google Scholar] [CrossRef]
- Spada, M.; Piras, C.; Diana, G.; Leoni, V.P.; Frau, D.V.; Serreli, G.; Simbula, G.; Loi, R.; Noto, A.; Murgia, F.; et al. Glutamine Starvation Affects Cell Cycle, Oxidative Homeostasis and Metabolism in Colorectal Cancer Cells. Antioxidants 2023, 12, 683. [Google Scholar] [CrossRef]
- Aledo, J.C.; Gómez-Fabre, P.M.; Olalla, L.; Márquez, J. Identification of Two Human Glutaminase Loci and Tissue-Specific Expression of the Two Related Genes. Mamm. Genome 2000, 11, 1107–1110. [Google Scholar] [CrossRef]
- Elgadi, K.M.; Meguid, R.A.; Qian, M.; Souba, W.W.; Abcouwer, S.F. Cloning and Analysis of Unique Human Glutaminase Isoforms Generated by Tissue-Specific Alternative Splicing. Physiol. Genom. 1999, 1, 51–62. [Google Scholar] [CrossRef]
- Porter, L.D.; Ibrahim, H.; Taylor, L.; Curthoys, N.P. Complexity and Species Variation of the Kidney-Type Glutaminase Gene. Physiol. Genom. 2002, 9, 157–166. [Google Scholar] [CrossRef]
- Márquez, J.; Matés, J.M.; Campos-Sandoval, J.A. Glutaminases. In The Glutamate/GABA-Glutamine Cycle, Advances in Neurobiology 13, 1st ed.; Schousboe, A., Sonnewald, U., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 133–171. [Google Scholar] [CrossRef]
- Campos-Sandoval, J.A.; Martín-Rufián, M.; Cardona, C.; Lobo, C.; Peñalver, A.; Márquez, J. Glutaminases in Brain: Multiple Isoforms for Many Purposes. Neurochem. Int. 2015, 88, 1–5. [Google Scholar] [CrossRef]
- Cassago, A.; Ferreira, A.P.S.; Ferreira, I.M.; Fornezari, C.; Gomes, E.R.M.; Greene, K.S.; Pereira, H.M.; Garratt, R.C.; Dias, S.M.G.; Ambrosio, A.L.B. Mitochondrial Localization and Structure-Based Phosphate Activation Mechanism of Glutaminase C with Implications for Cancer Metabolism. Proc. Natl. Acad. Sci. USA 2012, 109, 1092–1097. [Google Scholar] [CrossRef]
- Szeliga, M.; Sidoryk, M.; Matyja, E.; Kowalczyk, P.; Albrecht, J. Lack of Expression of the Liver-Type Glutaminase (LGA) MRNA in Human Malignant Gliomas. Neurosci. Lett. 2005, 374, 171–173. [Google Scholar] [CrossRef]
- Pérez-Gómez, C.; Campos-Sandoval, J.A.; Alonso, F.J.; Segura, J.A.; Manzanares, E.; Riuz-Sánchez, P.; González, M.E.; Márquez, J.; Matés, J.M. Co-Expression of Glutaminase K and L Isoenzymes in Human Tumour Cells. Biochem. J. 2005, 386, 535–542. [Google Scholar] [CrossRef]
- Szeliga, M.; Matyja, E.; Obara, M.; Grajkowska, W.; Czernicki, T.; Albrecht, J. Relative Expression of MRNAS Coding for Glutaminase Isoforms in CNS Tissues and CNS Tumors. Neurochem. Res. 2008, 33, 808–813. [Google Scholar] [CrossRef]
- van den Heuvel, A.P.J.; Jing, J.; Wooster, R.F.; Bachman, K.E. Analysis of Glutamine Dependency in Non-Small Cell Lung Cancer. Cancer Biol. Ther. 2012, 13, 1185–1194. [Google Scholar] [CrossRef]
- Kim, S.; Jung, W.H.; Koo, J.S. The Expression of Glutamine-Metabolism-Related Proteins in Breast Phyllodes Tumors. Tumor Biol. 2013, 34, 2683–2689. [Google Scholar] [CrossRef]
- Huang, F.; Zhang, Q.; Ma, H.; Lv, Q.; Zhang, T. Expression of Glutaminase Is Upregulated in Colorectal Cancer and of Clinical Significance. Int. J. Clin Exp. Pathol. 2014, 7, 1093–1100. [Google Scholar] [PubMed]
- Pan, T.; Gao, L.; Wu, G.; Shen, G.; Xie, S.; Wen, H.; Yang, J.; Zhou, Y.; Tu, Z.; Qian, W. Elevated Expression of Glutaminase Confers Glucose Utilization via Glutaminolysis in Prostate Cancer. Biochem. Biophys. Res. Commun. 2015, 456, 452–458. [Google Scholar] [CrossRef]
- Yu, D.; Shi, X.; Meng, G.; Chen, J.; Yan, C.; Jiang, Y.; Wei, J.; Ding, Y. Kidney-Type Glutaminase (GLS1) Is a Biomarker for Pathologic Diagnosis and Prognosis of Hepatocellular Carcinoma. Oncotarget 2015, 6, 7619–7631. [Google Scholar] [CrossRef]
- Kim, H.M.; Lee, Y.K.; Koo, J.S. Expression of Glutamine Metabolism-Related Proteins in Thyroid Cancer. Oncotarget 2016, 7, 53628–53641. [Google Scholar] [CrossRef]
- Lobo, C.; Ruiz-Bellido, M.A.; Aledo, J.C.; Márquez, J.; Núñez De Castro, I.; Alonso, F.J. Inhibition of Glutaminase Expression by Antisense MRNA Decreases Growth and Tumourigenicity of Tumour Cells. Biochem. J. 2000, 348 Pt 2, 257–261. [Google Scholar] [CrossRef]
- Zhang, J.; Mao, S.; Guo, Y.; Wu, Y.; Yao, X.; Huang, Y. Inhibition of GLS Suppresses Proliferation and Promotes Apoptosis in Prostate Cancer. Biosci. Rep. 2019, 39, 1826. [Google Scholar] [CrossRef]
- Wang, J.-B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.B.; Dias, S.M.G.; Dang, C.V.; et al. Targeting Mitochondrial Glutaminase Activity Inhibits Oncogenic Transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef]
- Cheng, T.; Sudderth, J.; Yang, C.; Mullen, A.R.; Jin, E.S.; Matés, J.M.; DeBerardinis, R.J. Pyruvate Carboxylase Is Required for Glutamine-Independent Growth of Tumor Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8674–8679. [Google Scholar] [CrossRef]
- Martín-Rufián, M.; Nascimento-Gomes, R.; Higuero, A.; Crisma, A.R.; Campos-Sandoval, J.A.; Gómez-García, M.C.; Cardona, C.; Cheng, T.; Lobo, C.; Segura, J.A.; et al. Both GLS Silencing and GLS2 Overexpression Synergize with Oxidative Stress against Proliferation of Glioma Cells. J. Mol. Med. 2014, 92, 277–290. [Google Scholar] [CrossRef]
- Jacque, N.; Ronchetti, A.M.; Larrue, C.; Meunier, G.; Birsen, R.; Willems, L.; Saland, E.; Decroocq, J.; Maciel, T.T.; Lambert, M.; et al. Targeting Glutaminolysis Has Antileukemic Activity in Acute Myeloid Leukemia and Synergizes with BCL-2 Inhibition. Blood 2015, 126, 1346–1356. [Google Scholar] [CrossRef]
- Wang, D.; Li, X.; Gong, G.; Lu, Y.; Guo, Z.; Chen, R.; Huang, H.; Li, Z.; Bian, J. An Updated Patent Review of Glutaminase Inhibitors (2019–2022). Expert Opin. Ther. Pat. 2023, 33, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tan, L.; Gao, J.; Lin, C.; Wu, F.; Li, Y.; Zhang, J. Targeting Glutaminase 1 (GLS1) by Small Molecules for Anticancer Therapeutics. Eur. J. Med. Chem. 2023, 252, 115306. [Google Scholar] [CrossRef]
- Patel, M.; McGivan, J.D. Partial Purification and Properties of Rat Liver Glutaminase. Biochem. J. 1984, 220, 583–590. [Google Scholar] [CrossRef]
- Smith, E.M.; Watford, M. Rat Hepatic Glutaminase: Purification and Immunochemical Characterization. Arch. Biochem. Biophys. 1988, 260, 740–751. [Google Scholar] [CrossRef]
- Smith, E.M.; Watford, M. Molecular Cloning of a CDNA for Rat Hepatic Glutaminase. Sequence Similarity to Kidney-Type Glutaminase. J. Biol. Chem. 1990, 265, 10631–10636. [Google Scholar] [CrossRef]
- Pérez-Gómez, C.; Matés, J.M.; Gómez-Fabre, P.M.; Castillo-Olivares, A.D.; Alonso, F.J.; Márquez, J. Genomic Organization and Transcriptional Analysis of the Human L-Glutaminase Gene. Biochem. J. 2003, 370, 771–784. [Google Scholar] [CrossRef]
- Gómez-Fabre, P.M.; Aledo, J.C.; Del Castillo-Olivares, A.; Alonso, F.J.; Núñez De Castro, I.; Campos, J.A.; Márquez, J. Molecular Cloning, Sequencing and Expression Studies of the Human Breast Cancer Cell Glutaminase. Biochem. J. 2000, 345 Pt 2, 365–375. [Google Scholar] [CrossRef]
- de la Rosa, V.; Campos-Sandoval, J.A.; Martín-Rufián, M.; Cardona, C.; Matés, J.M.; Segura, J.A.; Alonso, F.J.; Márquez, J. A Novel Glutaminase Isoform in Mammalian Tissues. Neurochem. Int. 2009, 55, 76–84. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a Novel P53 Target Gene Regulating Energy Metabolism and Antioxidant Function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef]
- Martín-Rufián, M.; Tosina, M.; Campos-Sandoval, J.A.; Manzanares, E.; Lobo, C.; Segura, J.A.; Alonso, F.J.; Matés, J.M.; Márquez, J. Mammalian Glutaminase Gls2 Gene Encodes Two Functional Alternative Transcripts by a Surrogate Promoter Usage Mechanism. PLoS ONE 2012, 7, e38380. [Google Scholar] [CrossRef]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-Activated Glutaminase (GLS2), a P53-Inducible Regulator of Glutamine Metabolism and Reactive Oxygen Species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef]
- Arianna, G.; Bongiorno-Borbone, L.; Bernassola, F.; Terrinoni, A.; Markert, E.; Levine, A.J.; Fen, Z.; Agostini, M.; Zolla, L.; Finazzi Agro’, A.; et al. P63 Regulates Glutaminase 2 Expression. Cell Cycle 2013, 12, 1395–1405. [Google Scholar] [CrossRef]
- Xiao, D.; Ren, P.; Su, H.; Yue, M.; Xiu, R.; Hu, Y.; Liu, H.; Qing, G. Myc Promotes Glutaminolysis in Human Neuroblastoma through Direct Activation of Glutaminase 2. Oncotarget 2015, 6, 40655–40666. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, C.; Chen, M.; Cao, J.; Zhong, Y.; Chen, L.; Shen, H.-M.; Xia, D. Epigenetic Silencing of Glutaminase 2 in Human Liver and Colon Cancers. BMC Cancer 2013, 13, 601. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Lin, M.; Zhu, W.; Liang, Y.; Hong, X.; Zhao, Y.; Young, K.H.; Hu, W.; Feng, Z. Glutaminase 2 Negatively Regulates the PI3K/AKT Signaling and Shows Tumor Suppression Activity in Human Hepatocellular Carcinoma. Oncotarget 2014, 5, 2635–2647. [Google Scholar] [CrossRef]
- Szeliga, M.; Bogacińska-Karaś, M.; Kuźmicz, K.; Rola, R.; Albrecht, J. Downregulation of GLS2 in Glioblastoma Cells Is Related to DNA Hypermethylation but Not to the P53 Status. Mol. Carcinog. 2016, 55, 1309–1316. [Google Scholar] [CrossRef]
- Lukey, M.J.; Cluntun, A.A.; Katt, W.P.; Lin, M.J.; Druso, J.E.; Ramachandran, S.; Erickson, J.W.; Le, H.H.; Wang, Z.-E.; Blank, B.; et al. Liver-Type Glutaminase GLS2 Is a Druggable Metabolic Node in Luminal-Subtype Breast Cancer. Cell Rep. 2019, 29, 76–88.e7. [Google Scholar] [CrossRef]
- Zhang, M.; Song, J.; Yuan, W.; Zhang, W.; Sun, Z. Roles of RNA Methylation on Tumor Immunity and Clinical Implications. Front. Immunol. 2021, 12, 641507. [Google Scholar] [CrossRef]
- Chen, X.; Huang, L.; Yang, T.; Xu, J.; Zhang, C.; Deng, Z.; Yang, X.; Liu, N.; Chen, S.; Lin, S. METTL3 Promotes Esophageal Squamous Cell Carcinoma Metastasis Through Enhancing GLS2 Expression. Front. Oncol. 2021, 11, 667451. [Google Scholar] [CrossRef]
- Li, H.-J.; Li, X.; Pang, H.; Pan, J.-J.; Xie, X.-J.; Chen, W. Long Non-Coding RNA UCA1 Promotes Glutamine Metabolism by Targeting MiR-16 in Human Bladder Cancer. Jpn. J. Clin. Oncol. 2015, 45, 1055–1063. [Google Scholar] [CrossRef]
- Zhou, X.; Zhuo, M.; Zhang, Y.; Shi, E.; Ma, X.; Li, H. MiR-190a-5p Regulates Cardiomyocytes Response to Ferroptosis via Directly Targeting GLS2. Biochem. Biophys. Res. Commun. 2021, 566, 9–15. [Google Scholar] [CrossRef]
- Zhang, Q.; Song, C.; Zhang, M.; Liu, Y.; Wang, L.; Xie, Y.; Qi, H.; Ba, L.; Shi, P.; Cao, Y.; et al. Super-Enhancer-Driven LncRNA Snhg7 Aggravates Cardiac Hypertrophy via Tbx5/GLS2/Ferroptosis Axis. Eur. J. Pharmacol. 2023, 953, 175822. [Google Scholar] [CrossRef]
- Luo, J.; Bai, R.; Liu, Y.; Bi, H.; Shi, X.; Qu, C. Long Non-Coding RNA ATXN8OS Promotes Ferroptosis and Inhibits the Temozolomide-Resistance of Gliomas through the ADAR/GLS2 Pathway. Brain Res. Bull. 2022, 186, 27–37. [Google Scholar] [CrossRef]
- Sengupta, D.; Cassel, T.; Teng, K.; Aljuhani, M.; Chowdhary, V.K.; Hu, P.; Zhang, X.; Fan, T.W.-M.; Ghoshal, K. Regulation of Hepatic Glutamine Metabolism by MiR-122. Mol. Metab. 2020, 34, 174–186. [Google Scholar] [CrossRef]
- TURNER, A.; McGIVAN, J.D. Glutaminase Isoform Expression in Cell Lines Derived from Human Colorectal Adenomas and Carcinomas. Biochem. J. 2003, 370, 403–408. [Google Scholar] [CrossRef]
- Castell, L.; Vance, C.; Abbott, R.; Marquez, J.; Eggleton, P. Granule Localization of Glutaminase in Human Neutrophils and the Consequence of Glutamine Utilization for Neutrophil Activity. J. Biol. Chem. 2004, 279, 13305–13310. [Google Scholar] [CrossRef]
- Campos-Sandoval, J.A.; López de la Oliva, A.R.; Lobo, C.; Segura, J.A.; Matés, J.M.; Alonso, F.J.; Márquez, J. Expression of Functional Human Glutaminase in Baculovirus System: Affinity Purification, Kinetic and Molecular Characterization. Int. J. Biochem. Cell Biol. 2007, 39, 765–773. [Google Scholar] [CrossRef]
- López de la Oliva, A.R.; Campos-Sandoval, J.A.; Gómez-García, M.C.; Cardona, C.; Martín-Rufián, M.; Sialana, F.J.; Castilla, L.; Bae, N.; Lobo, C.; Peñalver, A.; et al. Nuclear Translocation of Glutaminase GLS2 in Human Cancer Cells Associates with Proliferation Arrest and Differentiation. Sci. Rep. 2020, 10, 2259. [Google Scholar] [CrossRef]
- Olalla, L.; Gutiérrez, A.; Campos, J.A.; Khan, Z.U.; Alonso, F.J.; Segura, J.A.; Márquez, J.; Aledo, J.C. Nuclear Localization of L-Type Glutaminase in Mammalian Brain. J. Biol. Chem. 2002, 277, 38939–38944. [Google Scholar] [CrossRef]
- Cardona, C.; Sánchez-Mejías, E.; Dávila, J.C.; Martín-Rufián, M.; Campos-Sandoval, J.A.; Vitorica, J.; Alonso, F.J.; Matés, J.M.; Segura, J.A.; Norenberg, M.D.; et al. Expression of Gls and Gls2 Glutaminase Isoforms in Astrocytes. Glia 2015, 63, 365–382. [Google Scholar] [CrossRef]
- Olalla, L.; Aledo, J.C.; Bannenberg, G.; Márquez, J. The C-terminus of Human Glutaminase L Mediates Association with PDZ Domain-containing Proteins. FEBS Lett. 2001, 488, 116–122. [Google Scholar] [CrossRef]
- Márquez, J.; López de la Oliva, A.R.; Matés, J.M.; Segura, J.A.; Alonso, F.J. Glutaminase: A Multifaceted Protein Not Only Involved in Generating Glutamate. Neurochem. Int. 2006, 48, 465–471. [Google Scholar] [CrossRef]
- Sfakianos, A.P.; Raven, R.M.; Willis, A.E. The Pleiotropic Roles of EIF5A in Cellular Life and Its Therapeutic Potential in Cancer. Biochem. Soc. Trans. 2022, 50, 1885–1895. [Google Scholar] [CrossRef]
- Aksu, M.; Trakhanov, S.; Görlich, D. Structure of the Exportin Xpo4 in Complex with RanGTP and the Hypusine-Containing Translation Factor EIF5A. Nat. Commun. 2016, 7, 11952. [Google Scholar] [CrossRef]
- Bhat, H.F.; Adams, M.E.; Khanday, F.A. Syntrophin Proteins as Santa Claus: Role(s) in Cell Signal Transduction. Cell. Mol. Life Sci. 2013, 70, 2533–2554. [Google Scholar] [CrossRef]
- Mohanty, S.; Ovee, M.; Banerjee, M. PDZ Domain Recognition: Insight from Human Tax-Interacting Protein 1 (TIP-1) Interaction with Target Proteins. Biology 2015, 4, 88–103. [Google Scholar] [CrossRef]
- Singh, N.; Bhalla, N. Moonlighting Proteins. Annu. Rev. Genet. 2020, 54, 265–285. [Google Scholar] [CrossRef]
- Ferreira, I.M.; Quesñay, J.E.N.; Bastos, A.C.; Rodrigues, C.T.; Vollmar, M.; Krojer, T.; Strain-Damerell, C.; Burgess-Brown, N.A.; von Delft, F.; Yue, W.W.; et al. Structure and Activation Mechanism of the Human Liver-Type Glutaminase GLS2. Biochimie 2021, 185, 96–104. [Google Scholar] [CrossRef]
- Nguyen, T.-T.T.; Katt, W.P.; Cerione, R.A. Alone and Together: Current Approaches to Targeting Glutaminase Enzymes as Part of Anti-Cancer Therapies. Future Drug Discov. 2022, 4, FDD79. [Google Scholar] [CrossRef]
- Saha, S.; Islam, S.M.; Abdullah-AL-Wadud, M.; Islam, S.; Ali, F.; Park, K. Multiomics Analysis Reveals That GLS and GLS2 Differentially Modulate the Clinical Outcomes of Cancer. J. Clin. Med. 2019, 8, 355. [Google Scholar] [CrossRef]
- Díaz-Serrano, A.; Gella, P.; Jiménez, E.; Zugazagoitia, J.; Paz-Ares Rodríguez, L. Targeting EGFR in Lung Cancer: Current Standards and Developments. Drugs 2018, 78, 893–911. [Google Scholar] [CrossRef]
- Kim, S.; Jeon, J.S.; Choi, Y.J.; Baek, G.H.; Kim, S.K.; Kang, K.W. Heterogeneity of Glutamine Metabolism in Acquired-EGFR-TKI-Resistant Lung Cancer. Life Sci. 2022, 291, 120274. [Google Scholar] [CrossRef]
- Ye, X.; Zhou, Q.; Matsumoto, Y.; Moriyama, M.; Kageyama, S.; Komatsu, M.; Satoh, S.; Tsuchida, M.; Saijo, Y. Inhibition of Glutaminolysis Inhibits Cell Growth via Down-Regulating Mtorc1 Signaling in Lung Squamous Cell Carcinoma. Anticancer Res. 2016, 36, 6021–6030. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Lee, Y.-Z.; Yang, C.-W.; Chang, H.-Y.; Hsu, H.-Y.; Chen, I.-S.; Chang, H.-S.; Lee, C.-H.; Lee, J.; Kumar, C.R.; Qiu, Y.-Q.; et al. Discovery of Selective Inhibitors of Glutaminase-2, Which Inhibit MTORC1, Activate Autophagy and Inhibit Proliferation in Cancer Cells. Oncotarget 2014, 5, 6087–6101. [Google Scholar] [CrossRef]
- Deng, Z.; Richardson, D.R. The Myc Family and the Metastasis Suppressor NDRG1: Targeting Key Molecular Interactions with Innovative Therapeutics. Pharmacol. Rev. 2023, 75, 1007–1035. [Google Scholar] [CrossRef]
- Xiang, L.; Xie, G.; Liu, C.; Zhou, J.; Chen, J.; Yu, S.; Li, J.; Pang, X.; Shi, H.; Liang, H. Knock-down of Glutaminase 2 Expression Decreases Glutathione, NADH, and Sensitizes Cervical Cancer to Ionizing Radiation. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2013, 1833, 2996–3005. [Google Scholar] [CrossRef]
- Kouros-Mehr, H.; Slorach, E.M.; Sternlicht, M.D.; Werb, Z. GATA-3 Maintains the Differentiation of the Luminal Cell Fate in the Mammary Gland. Cell 2006, 127, 1041–1055. [Google Scholar] [CrossRef]
- Dias, M.M.; Adamoski, D.; dos Reis, L.M.; Ascenção, C.F.R.; de Oliveira, K.R.S.; Mafra, A.C.P.; da Silva Bastos, A.C.; Quintero, M.; de, G. Cassago, C.; Ferreira, I.M.; et al. GLS2 Is Protumorigenic in Breast Cancers. Oncogene 2020, 39, 690–702. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Ramirez-Peña, E.; Arnold, J.; Shivakumar, V.; Joseph, R.; Vidhya Vijay, G.; den Hollander, P.; Bhangre, N.; Allegakoen, P.; Prasad, R.; Conley, Z.; et al. The Epithelial to Mesenchymal Transition Promotes Glutamine Independence by Suppressing GLS2 Expression. Cancers 2019, 11, 1610. [Google Scholar] [CrossRef]
- Yang, W.-H.; Qiu, Y.; Stamatatos, O.; Janowitz, T.; Lukey, M.J. Enhancing the Efficacy of Glutamine Metabolism Inhibitors in Cancer Therapy. Trends Cancer 2021, 7, 790–804. [Google Scholar] [CrossRef]
- Yu, C.-C.; Wu, P.-J.; Hsu, J.-L.; Ho, Y.-F.; Hsu, L.-C.; Chang, Y.-J.; Chang, H.-S.; Chen, I.-S.; Guh, J.-H. Ardisianone, a Natural Benzoquinone, Efficiently Induces Apoptosis in Human Hormone-Refractory Prostate Cancers through Mitochondrial Damage Stress and Survivin Downregulation. Prostate 2013, 73, 133–145. [Google Scholar] [CrossRef]
- Leu, W.-J.; Chang, H.-S.; Chen, I.-S.; Guh, J.-H.; Chan, S.-H. Antileukemic Natural Product Induced Both Apoptotic and Pyroptotic Programmed Cell Death and Differentiation Effect. Int. J. Mol. Sci. 2021, 22, 11239. [Google Scholar] [CrossRef]
- Robinson, M.M.; Mcbryant, S.J.; Tsukamoto, T.; Rojas, C.; Ferraris, D.V.; Hamilton, S.K.; Hansen, J.C.; Curthoys, N.P. Novel Mechanism of Inhibition of Rat Kidney-Type Glutaminase by Bis-2-(5-Phenylacetamido-1,2,4-Thiadiazol-2-Yl)Ethyl Sulfide (BPTES). Biochem. J. 2007, 406, 407–414. [Google Scholar] [CrossRef]
- Han, T.; Guo, M.; Zhang, T.; Gan, M.; Xie, C.; Wang, J.-B. A Novel Glutaminase Inhibitor-968 Inhibits the Migration and Proliferation of Non-Small Cell Lung Cancer Cells by Targeting EGFR/ERK Signaling Pathway. Oncotarget 2017, 8, 28063–28073. [Google Scholar] [CrossRef]
- Yuan, L.; Sheng, X.; Clark, L.H.; Zhang, L.; Guo, H.; Jones, H.M.; Willson, A.K.; Gehrig, P.A.; Zhou, C.; Bae-Jump, V.L. Glutaminase Inhibitor Compound 968 Inhibits Cell Proliferation and Sensitizes Paclitaxel in Ovarian Cancer. Am. J. Transl. Res. 2016, 8, 4265–4277. [Google Scholar]
- Guo, H.; Li, W.; Pan, G.; Wang, C.; Li, D.; Liu, N.; Sheng, X.; Yuan, L. The Glutaminase Inhibitor Compound 968 Exhibits Potent In Vitro and In Vivo Anti-Tumor Effects in Endometrial Cancer. Anticancer Agents Med. Chem. 2023, 23, 210–221. [Google Scholar] [CrossRef]
- Wang, D.; Meng, G.; Zheng, M.; Zhang, Y.; Chen, A.; Wu, J.; Wei, J. The Glutaminase-1 Inhibitor 968 Enhances Dihydroartemisinin-Mediated Antitumor Efficacy in Hepatocellular Carcinoma Cells. PLoS ONE 2016, 11, e0166423. [Google Scholar] [CrossRef]
- Yeh, T.-K.; Kuo, C.-C.; Lee, Y.-Z.; Ke, Y.-Y.; Chu, K.-F.; Hsu, H.-Y.; Chang, H.-Y.; Liu, Y.-W.; Song, J.-S.; Yang, C.-W.; et al. Design, Synthesis, and Evaluation of Thiazolidine-2,4-Dione Derivatives as a Novel Class of Glutaminase Inhibitors. J. Med. Chem. 2017, 60, 5599–5612. [Google Scholar] [CrossRef]
- Bode, B.P.; Souba, W.W. Modulation of Cellular Proliferation Alters Glutamine Transport and Metabolism in Human Hepatoma Cells. Ann. Surg. 1994, 220, 411–424. [Google Scholar] [CrossRef]
- Yuneva, M.O.; Fan, T.W.M.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Matés, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The Metabolic Profile of Tumors Depends on Both the Responsible Genetic Lesion and Tissue Type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef]
- Kuo, T.-C.; Chen, C.-K.; Hua, K.-T.; Yu, P.; Lee, W.-J.; Chen, M.-W.; Jeng, Y.-M.; Chien, M.-H.; Kuo, K.-T.; Hsiao, M.; et al. Glutaminase 2 Stabilizes Dicer to Repress Snail and Metastasis in Hepatocellular Carcinoma Cells. Cancer Lett. 2016, 383, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Krstic, J.; Galhuber, M.; Schulz, T.; Schupp, M.; Prokesch, A. P53 as a Dichotomous Regulator of Liver Disease: The Dose Makes the Medicine. Int. J. Mol. Sci. 2018, 19, 921. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Sui, X.; Weng, L.; Liu, Y. SNAIL1: Linking Tumor Metastasis to Immune Evasion. Front. Immunol. 2021, 12, 724200. [Google Scholar] [CrossRef] [PubMed]
- Vergani-Junior, C.A.; Tonon-da-Silva, G.; Inan, M.D.; Mori, M.A. DICER: Structure, Function, and Regulation. Biophys. Rev. 2021, 13, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Bid, H.K.; Roberts, R.D.; Manchanda, P.K.; Houghton, P.J. RAC1: An Emerging Therapeutic Option for Targeting Cancer Angiogenesis and Metastasis. Mol. Cancer Ther. 2013, 12, 1925–1934. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Zhao, Y.; Yue, X.; Zhu, Y.; Wang, X.; Wu, H.; Blanco, F.; Li, S.; Bhanot, G.; et al. Glutaminase 2 Is a Novel Negative Regulator of Small GTPase Rac1 and Mediates P53 Function in Suppressing Metastasis. eLife 2016, 5, e10727. [Google Scholar] [CrossRef]
- Suzuki, S.; Venkatesh, D.; Kanda, H.; Nakayama, A.; Hosokawa, H.; Lee, E.; Miki, T.; Stockwell, B.R.; Yokote, K.; Tanaka, T.; et al. GLS2 Is a Tumor Suppressor and a Regulator of Ferroptosis in Hepatocellular Carcinoma. Cancer Res. 2022, 82, 3209–3222. [Google Scholar] [CrossRef]
- Fujii, M.; Shibazaki, Y.; Wakamatsu, K.; Honda, Y.; Kawauchi, Y.; Suzuki, K.; Arumugam, S.; Watanabe, K.; Ichida, T.; Asakura, H.; et al. A Murine Model for Non-Alcoholic Steatohepatitis Showing Evidence of Association between Diabetes and Hepatocellular Carcinoma. Med. Mol. Morphol. 2013, 46, 141–152. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, L.; Wu, K.; Chen, Y.; Lee, D.; Gucek, M.; Sack, M.N. Mitochondrial General Control of Amino Acid Synthesis 5 Like 1 Regulates Glutaminolysis, Mammalian Target of Rapamycin Complex 1 Activity, and Murine Liver Regeneration. Hepatology 2020, 71, 643–657. [Google Scholar] [CrossRef]
- Zhang, T.; Cui, Y.; Wu, Y.; Meng, J.; Han, L.; Zhang, J.; Zhang, C.; Yang, C.; Chen, L.; Bai, X.; et al. Mitochondrial GCN5L1 Regulates Glutaminase Acetylation and Hepatocellular Carcinoma. Clin. Transl. Med. 2022, 12, e852. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhou, D.; Li, F.; Ji, L. Glutaminase 2 Functions as a Tumor Suppressor Gene in Gastric Cancer. Transl. Cancer Res. 2020, 9, 4906–4913. [Google Scholar] [CrossRef] [PubMed]
- WHO. Classification of Tumours Editorial Board. In Central Nervous System Tumours, 5th ed.; WHO Classification of Tumours series; International Agency for Research on Cancer: Lyon, France, 2021; Volume 6. [Google Scholar]
- Moreira Franco, Y.E.; Alves, M.J.; Uno, M.; Moretti, I.F.; Trombetta-Lima, M.; de Siqueira Santos, S.; dos Santos, A.F.; Arini, G.S.; Baptista, M.S.; Lerario, A.M.; et al. Glutaminolysis Dynamics during Astrocytoma Progression Correlates with Tumor Aggressiveness. Cancer Metab. 2021, 9, 18. [Google Scholar] [CrossRef]
- Szeliga, M.; Obara-Michlewska, M.; Matyja, E.; Łazarczyk, M.; Lobo, C.; Hilgier, W.; Alonso, F.J.; Márquez, J.; Albrecht, J. Transfection with Liver-type Glutaminase CDNA Alters Gene Expression and Reduces Survival, Migration and Proliferation of T98G Glioma Cells. Glia 2009, 57, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Majewska, E.; Márquez, J.; Albrecht, J.; Szeliga, M. Transfection with GLS2 Glutaminase (GAB) Sensitizes Human Glioblastoma Cell Lines to Oxidative Stress by a Common Mechanism Involving Suppression of the PI3K/AKT Pathway. Cancers 2019, 11, 115. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, A.; Hachem, L.D.; Mansouri, S.; Nassiri, F.; Laperriere, N.J.; Xia, D.; Lindeman, N.I.; Wen, P.Y.; Chakravarti, A.; Mehta, M.P.; et al. MGMT Promoter Methylation Status Testing to Guide Therapy for Glioblastoma: Refining the Approach Based on Emerging Evidence and Current Challenges. Neuro Oncol. 2019, 21, 167–178. [Google Scholar] [CrossRef]
- Szeliga, M.; Zgrzywa, A.; Obara-Michlewska, M.; Albrecht, J. Transfection of a Human Glioblastoma Cell Line with Liver-type Glutaminase (LGA) Down-regulates the Expression of DNA-repair Gene MGMT and Sensitizes the Cells to Alkylating Agents. J. Neurochem. 2012, 123, 428–436. [Google Scholar] [CrossRef]
- Chahal, M.; Abdulkarim, B.; Xu, Y.; Guiot, M.-C.; Easaw, J.C.; Stifani, N.; Sabri, S. O(6)-Methylguanine-DNA Methyltransferase Is a Novel Negative Effector of Invasion in Glioblastoma Multiforme. Mol. Cancer Ther. 2012, 11, 2440–2450. [Google Scholar] [CrossRef]
- Oliva, C.R.; Moellering, D.R.; Gillespie, G.Y.; Griguer, C.E. Acquisition of Chemoresistance in Gliomas Is Associated with Increased Mitochondrial Coupling and Decreased ROS Production. PLoS ONE 2011, 6, e24665. [Google Scholar] [CrossRef]
- de los Santos-Jiménez, J.; Campos-Sandoval, J.A.; Márquez-Torres, C.; Urbano-Polo, N.; Brøndegaard, D.; Martín-Rufián, M.; Lobo, C.; Peñalver, A.; Gómez-García, M.C.; Martín-Campos, J.; et al. Glutaminase Isoforms Expression Switches MicroRNA Levels and Oxidative Status in Glioblastoma Cells. J. Biomed. Sci. 2021, 28, 14. [Google Scholar] [CrossRef]
- Muir, A.; Danai, L.V.; Gui, D.Y.; Waingarten, C.Y.; Lewis, C.A.; Vander Heiden, M.G. Environmental Cystine Drives Glutamine Anaplerosis and Sensitizes Cancer Cells to Glutaminase Inhibition. eLife 2017, 6, e27713. [Google Scholar] [CrossRef]
- Kim, S.; Kim, J.E.; Kim, Y.H.; Hwang, T.; Kim, S.K.; Xu, W.J.; Shin, J.-Y.; Kim, J.-I.; Choi, H.; Kim, H.C.; et al. Glutaminase 2 Expression Is Associated with Regional Heterogeneity of 5-Aminolevulinic Acid Fluorescence in Glioblastoma. Sci. Rep. 2017, 7, 12221. [Google Scholar] [CrossRef]
- Hadjipanayis, C.G.; Widhalm, G.; Stummer, W. What Is the Surgical Benefit of Utilizing 5-Aminolevulinic Acid for Fluorescence-Guided Surgery of Malignant Gliomas? Neurosurgery 2015, 77, 663–673. [Google Scholar] [CrossRef]
- Goenka, A.; Tiek, D.; Song, X.; Huang, T.; Hu, B.; Cheng, S.-Y. The Many Facets of Therapy Resistance and Tumor Recurrence in Glioblastoma. Cells 2021, 10, 484. [Google Scholar] [CrossRef]
- Stummer, W.; Tonn, J.-C.; Goetz, C.; Ullrich, W.; Stepp, H.; Bink, A.; Pietsch, T.; Pichlmeier, U. 5-Aminolevulinic Acid-Derived Tumor Fluorescence. Neurosurgery 2014, 74, 310–320. [Google Scholar] [CrossRef]
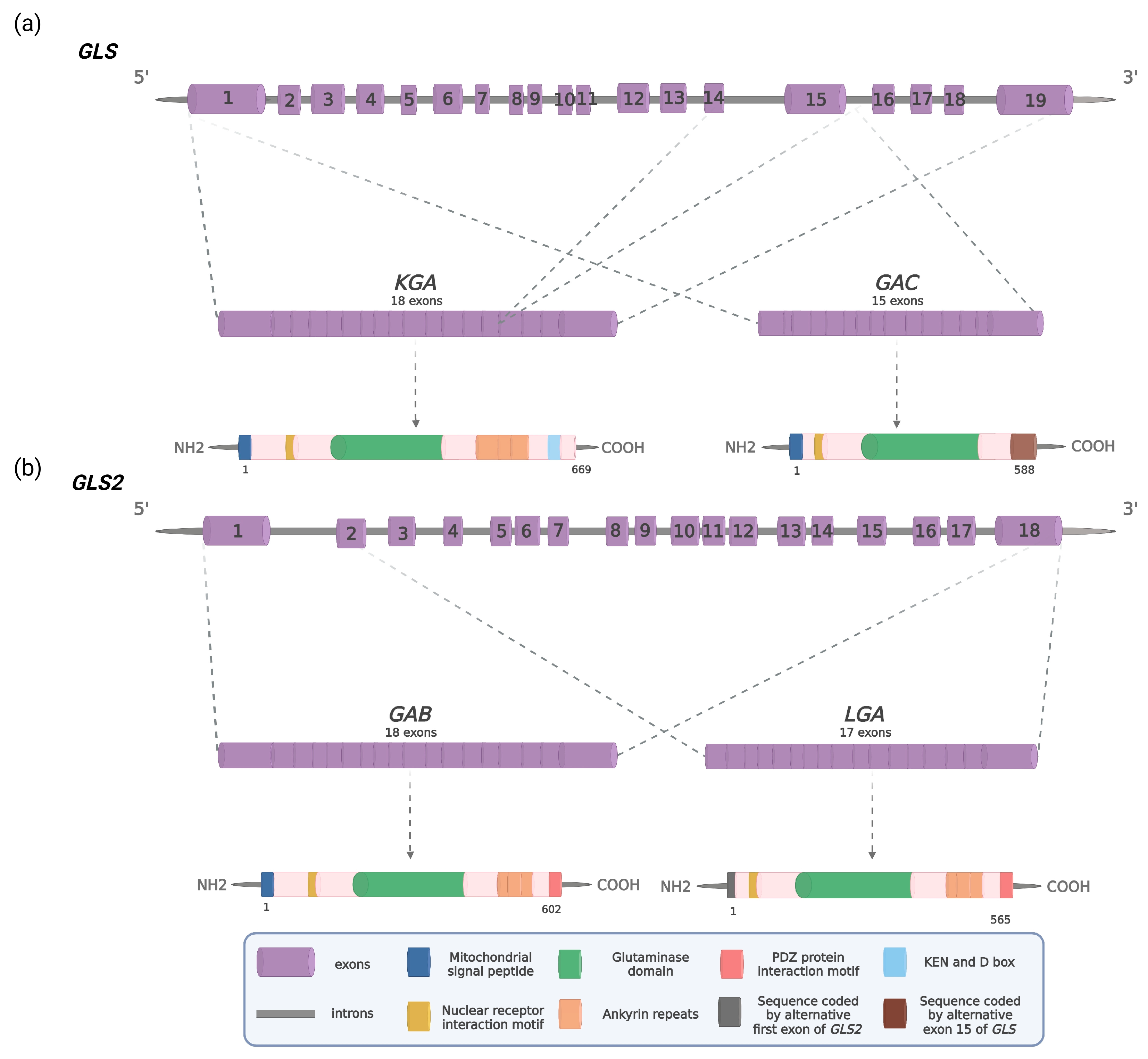
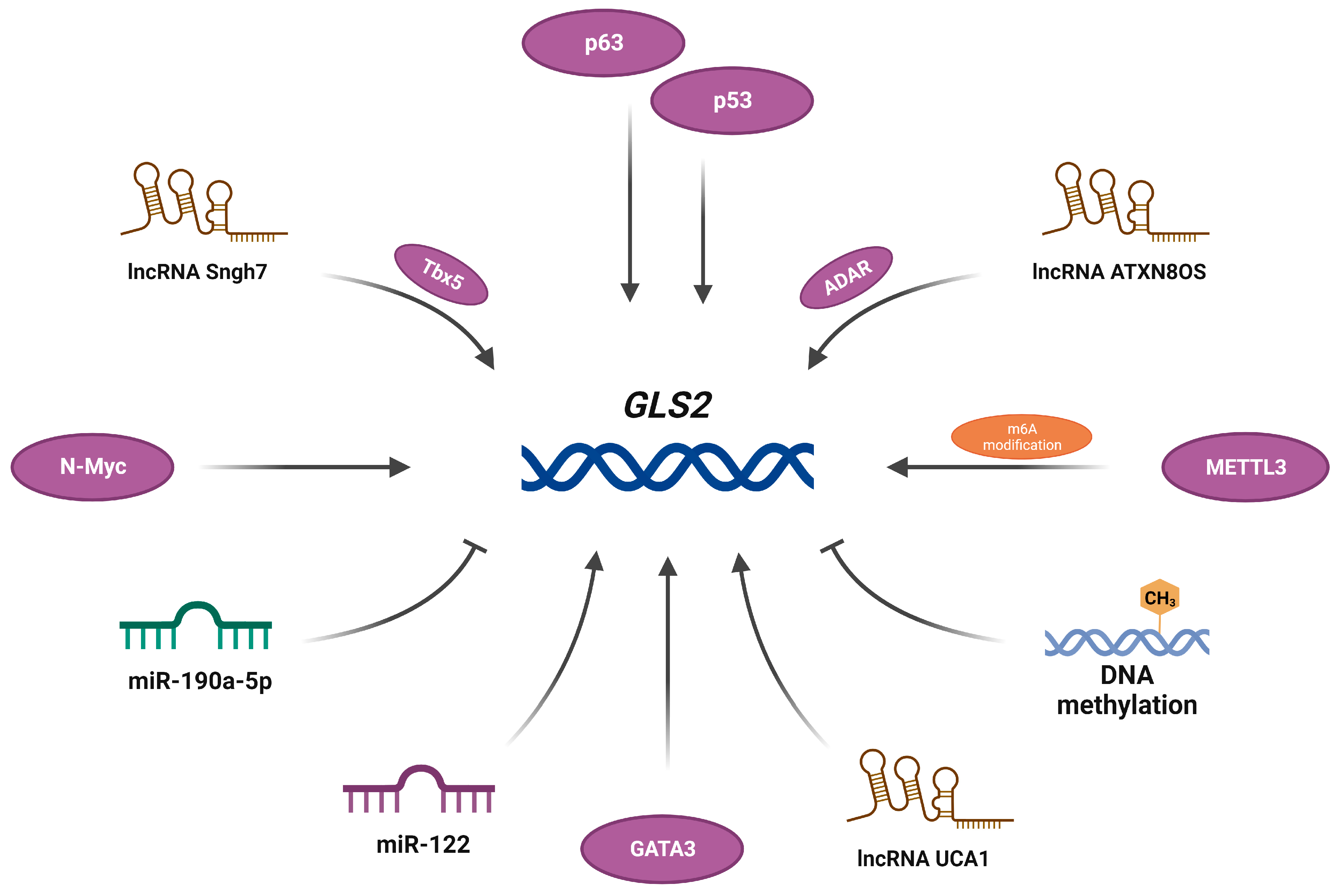

{kind=link}
{kind=link}
{kind=link}
| Cancer | Model | Outcome | Ref. |
|---|---|---|---|
| Effects of GLS2 Inhibition | |||
| Non-small cell lung cancer (NSCLC) | A549 cells with GLS2 knockdown | Decreased cell growth | [75] |
| Hepatocellular carcinoma (HCC) | HepG2 cells with GLS2 knockdown or AV-1 treatment | Decreased cell growth, inhibited anchorage-independent colony formation, autophagy induction, P13K/Akt inhibition, suppressed mTORC1 activity | |
| Lung squamous-cell carcinoma (SCC) | QG56 cells with GLS2 knockdown | Diminished viability, decreased mTORC1 activity | [73] |
| Neuroblastoma | MYCN-amplified Kelly and BE-2C cells with GLS2 knockdown | Reduced proliferation and clonogenic potential, attenuated tumors forming ability, decreased GSH, α-KG and ATP level, increased ROS level, inhibited Gln-dependent anaplerosis of TCA cycle and aerobic glycolysis | [43] |
| Cervical cancer | HeLa and HeLaR cells with GLS2 knockdown | Increased intracellular ROS levels, decreased production of NADH, NADPH, GSH, enhanced radiosensitivity | [77] |
| mouse model injected with HeLa, HeLaR and HeLaR cells with GLS2 knockdown | Enhanced radiosensitivity | ||
| Luminal-subtype breast cancer | MDA-MB-453 cells with GLS2 knockdown | Reduced proliferation, suppressed glutamine-mediated TCA cycle anaplerosis | [47] |
| mouse model injected with MDA-MB-453 cells | Reduced tumorigenesis | ||
| Triple-negative (TN) breast cancer | TN and non-TN cells with GLS2 knockdown | Diminished cell proliferation, increased oxidative stress | [79] |
| HMLE cells with GLS2 knockdown | Gln independence, reduced mitochondrial activity | [81] | |
| Effects of GLS2 Overexpression | |||
| Triple-negative (TN) breast cancer | TN and non-TN breast cancer cells overexpressing GLS2 | Elevated invasion capacity | [79] |
| mouse model injected with MDA-MB-231 cells | Pro-tumorigenic effect, decreased overall, disease-free and distant metastasis-free survival | ||
| SUM159 cells overexpressing GLS2 | Enhanced mitochondrial activity, glutamine independence, inhibited mammosphere formation, higher intracellular GSH and ATP level, higher basal respiration, reduced cells viability | [81] | |
| Compound | Class | Selectivity | Anticancer Activity |
|---|---|---|---|
| AV-1 (ardisianone) | alkyl benzoquinone | approximately tenfold selectivity against GLS2 over GLS [75] | human hormone-refractory prostate cancer cells PC-3 and DU-145145 [83], acute myeloid leukemia cells HL-60 [84] |
| 986 | benzophenanthridinone | threefold selectivity against GLS2 over GLS [47] | breast cancer cell lines DU4475 [85], MDA-MB-453, MDA-MB-453 xenografts [47], NSCLC cell lines A549, H23, H1299, and Spc-A1 [86], ovarian cell lines HEY, SKOV3, IGROV-1 [87], endometrial cancer cell lines Ishikawa and HEC-1B [88], HCC cell lines LM3, 7402 and HepG2 [89] |
| derivative 6 | thiazoli-dine- 2,4-dione | twofold selectivity against GLS over GLS2 [90] | human pancreas adenocarcinoma AsPC-1 cells AsPC-1 mice xenografts [90] |
| Cancer | Model | Outcome | Ref. |
|---|---|---|---|
| Effects of GLS2 Inhibition | |||
| Hepatocellular carcinoma (HCC) | HepG2 cell line | Increased ROS production, sensitization to H2O2-induced apoptosis, decreased oxygen consumption, α-ketoglutarate and ATP levels | [39] |
| PLC/PRF/5 cell line with GLS2 knockdown | Enhanced anchorage-independent cell growth and formation of tumors | [45] | |
| HepG2, HCC36 and Mahlavu cell lines with GLS2 knockdown | Enhanced cell migration and invasion, diminished level of Snail, reduction of miR-34a expression | [93] | |
| Mice with GLS2 knockout | Earlier development of larger tumors, resistance to ferroptosis, increased level of MDA and oxidative stress | [103] | |
| Effects of GLS2 Overexpression | |||
| Hepatocellular carcinoma (HCC) | HepG2 cells overexpressing GLS2 | Reduced cell colony formation | [39] |
| Huh1 and Huh7 cells overexpressing GLS2 | Reduced xenograft tumor growth, downregulation of PI3K/AKT signaling pathway | [45] | |
| PLC/PRF/5 cells with GLS2 knockdown | Downregulation of PI3K/AKT signaling pathway | ||
| Mahlavu and Huh7 cells overexpressing GLS2 | Decreased cell migration and invasion, stabilization of Dicer protein, induction of Dicer-dependent miR-34a maturation, repressed EMT, motility and metastasis | [93] | |
| Huh-1 cells co-transduced with GLS2 and RAC1 | Inhibited cell migration, invasion, metastasis and Rac1 activity | [98] | |
| HepG2, HepG3, SKHep1 cells overexpressing GLS2 | Enhanced ferroptosis through αKG-dependent increase of lipid ROS | [99] | |
| Mice model injected with SKHep1 cells overexpressing GLS2 | Reduced tumor formation, increased expression of ferroptosis markers | ||
| Colon cancer | HCT116 cells overexpressing GLS2 | Reduced viability and number of cell colonies, increased number of cells in G2/M phase, reduced levels of p21 and cyclin D1 | [44] |
| Gastric cancer | MGC-803 cells overexpressing GLS2 | Suppressed cell viability and ability to migration, induced apoptosis | [103] |
| Glioblastoma | T98G, U87MG and LN229 cells overexpressing GLS2 | Decreased cells viability, proliferation, ability to form colonies and migration, increased TMZ and H2O2-mediated oxidative stress sensitivity, downregulation of the PI3K/AKT pathway | [106,107] |
| T98G, SFxL and LN229 cells overexpressing GLS2 | Inhibition of cell migration, decreased apoptosis, antioxidant status and cellular motility, H2O2 and ATO sensitivity | [29] | |
| T98G and LN229 cells overexpressing GLS2 | Reduced CAT activity, increased total antioxidant capacity, decreased lipid peroxidation, miRNA-140-3p, miRNA-1246, miRNA-1260a, miRNA-21-5p and miRNA-146a-5p overexpression | [112] | |
| LN229 cells overexpressing GLS2 | Diminished SOD activity, miRNA-92a-3p upregulation | ||
| U251 and U251TR cells overexpressing GLS2; intracranial U251TR model | Diminished cell proliferation and migration, increased lipid ROS and E-cadherin level, decreased vimentin level, repressed EMT | [53] | |
| T98G cells overexpressing GLS2 | Decreased cell proliferation, increased level of p53 in nuclei, decreased level of c-Myc in cytoplasm | [58] | |
| Neuroblastoma | SH-SY5Y cells treated with PMA | Inhibited proliferation, increased p53 and p21 level, cell cycle arrested at G2/M stage | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buczkowska, J.; Szeliga, M. Two Faces of Glutaminase GLS2 in Carcinogenesis. Cancers 2023, 15, 5566. https://doi.org/10.3390/cancers15235566
Buczkowska J, Szeliga M. Two Faces of Glutaminase GLS2 in Carcinogenesis. Cancers. 2023; 15(23):5566. https://doi.org/10.3390/cancers15235566
Chicago/Turabian StyleBuczkowska, Joanna, and Monika Szeliga. 2023. "Two Faces of Glutaminase GLS2 in Carcinogenesis" Cancers 15, no. 23: 5566. https://doi.org/10.3390/cancers15235566
APA StyleBuczkowska, J., & Szeliga, M. (2023). Two Faces of Glutaminase GLS2 in Carcinogenesis. Cancers, 15(23), 5566. https://doi.org/10.3390/cancers15235566