
Hydroxamic Acids Containing a Bicyclic Pinane Backbone as Epigenetic and Metabolic Regulators: Synergizing Agents to Overcome Cisplatin Resistance

 , , , ,
, , , ,  ,
, 
Abstract
:Simple Summary
Abstract
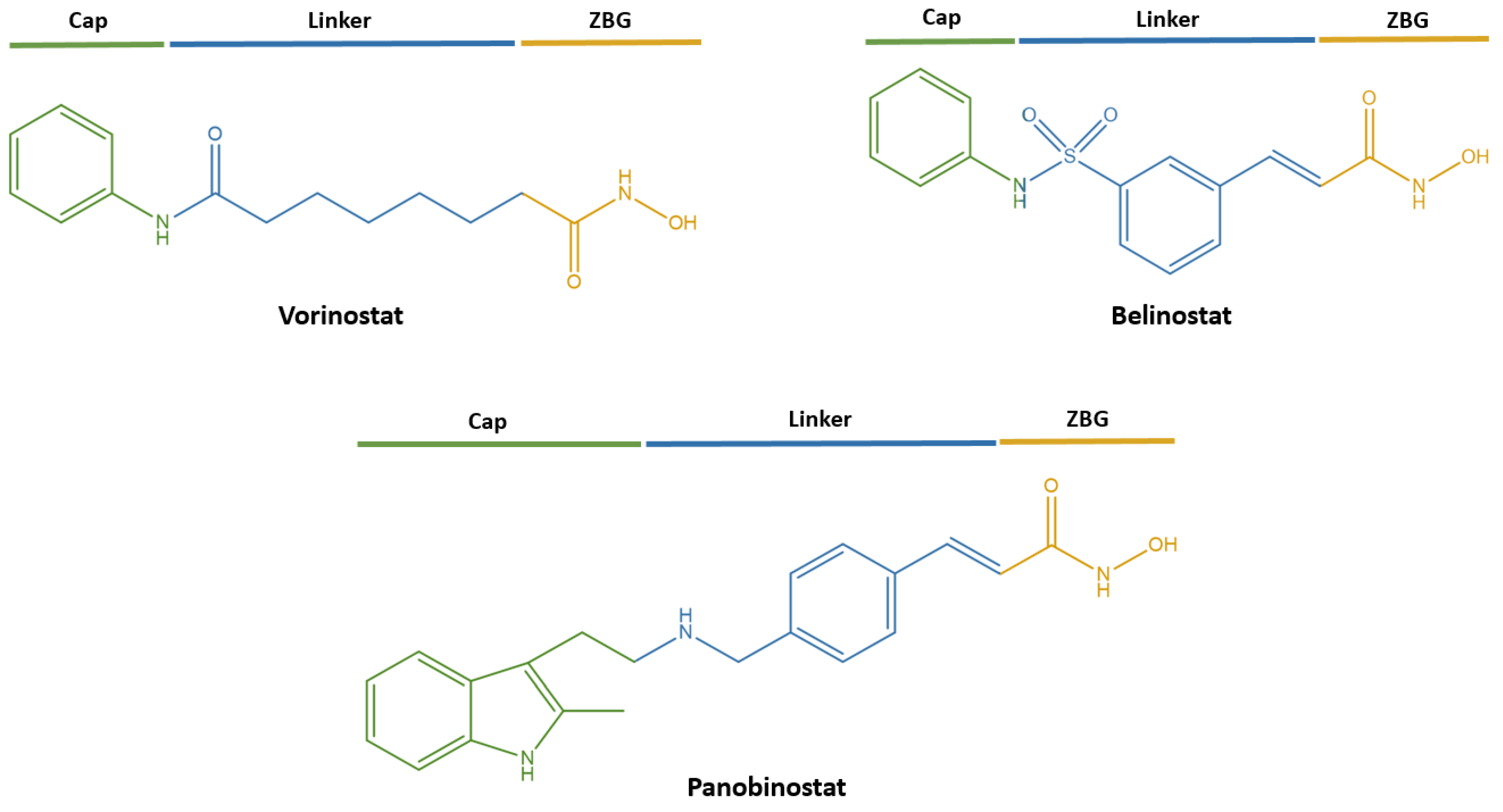
1. Introduction
2. Materials and Methods
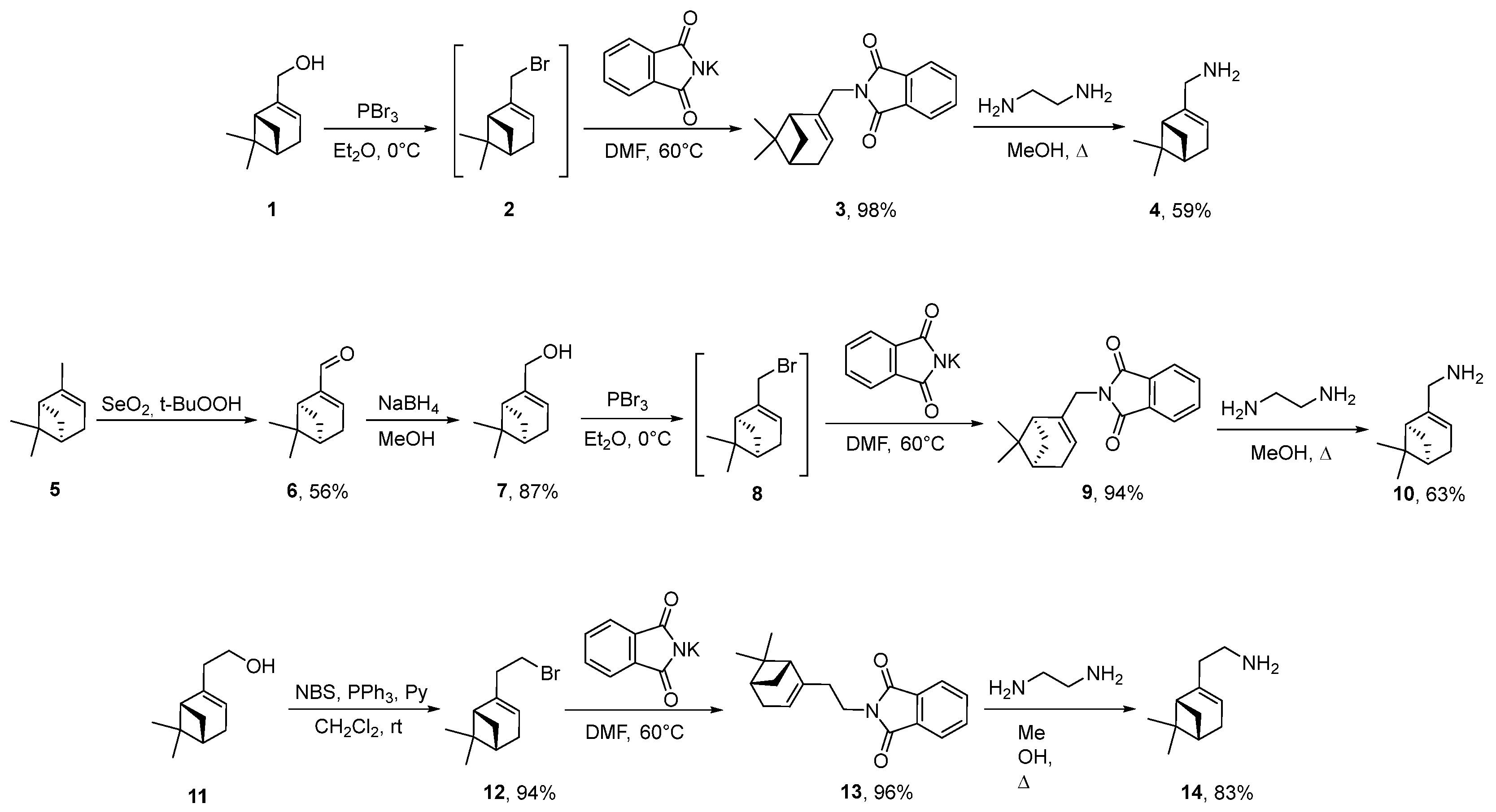
2.1. Chemistry
- Synthesis of (+)-myrtenol 7
- Synthesis of Bromides 2 and 8
- Synthesis of Bromide 12
- Synthesis of Phthalimides 3, 9 and 13
- 2-(((1R)-6,6-Dimethylbicyclo [3.1.1]hept-2-en-2-yl)methyl)isoindoline-1,3-dione 3
- Synthesis of Amines 4, 10 and 14
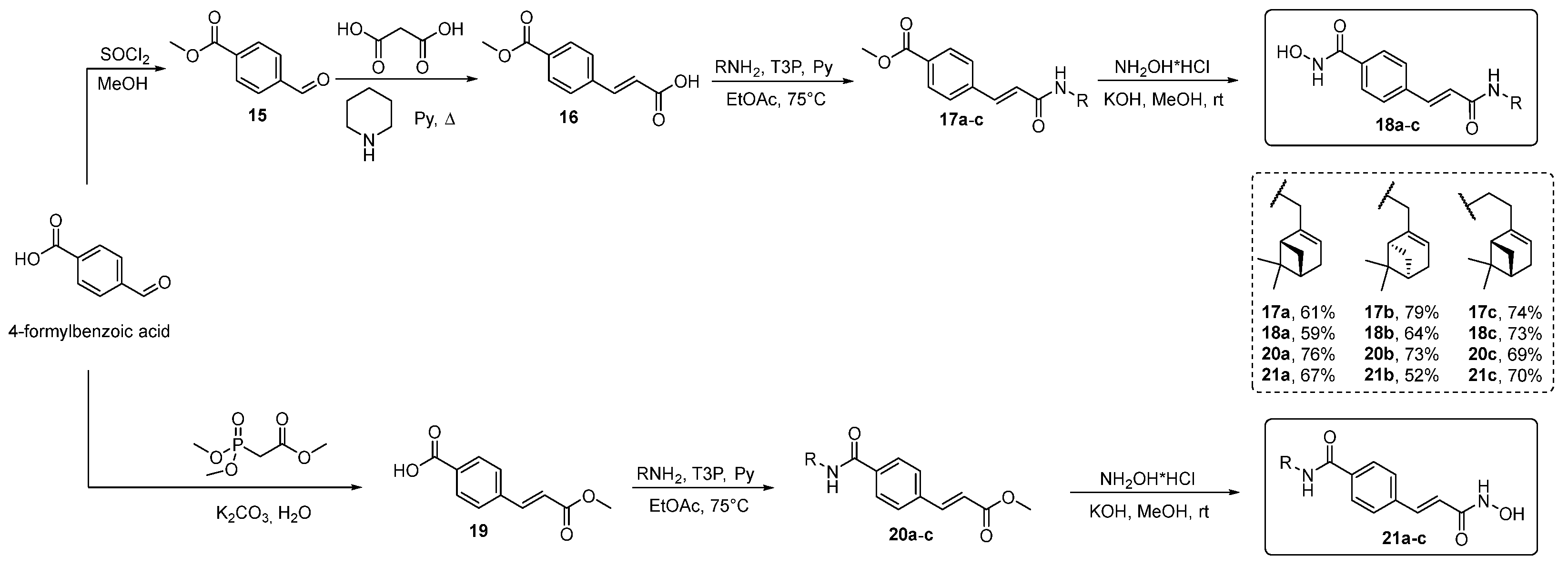
- Synthesis of methyl 4-formylbenzoate 15
- Synthesis of (E)-3-(4-methoxycarbonylphenyl)prop-2-enoic Acid 16
- Synthesis of (E)-4-(3-methoxy-3-oxoprop-1-en-1-yl)benzoic Acid 19
- Synthesis of Esters 17a–c and 20a–c
- Methyl 4-((E)-3-((((1R)-6,6-Dimethylbicyclo[3.1.1]hept-2-en-2-yl)methyl)amino)-3-oxoprop-1-en-1-yl)benzoate 17a
- White Solid, Yield 61%, m.p. 87.1–88.6 °C
- Methyl 4-((E)-3-((((1S)-6,6-Dimethylbicyclo[3.1.1]hept-2-en-2-yl)methyl)amino)-3-oxoprop-1-en-1-yl)benzoate 17b
- White Solid, Yield 79%
- Methyl 4-((E)-3-((2-((1R)-6,6-Dimethylbicyclo[3.1.1]hept-2-en-2-yl)ethyl)amino)-3-oxoprop-1-en-1-yl)benzoate 17c
- White Solid, Yield 74%, m.p. 104.0–105.2 °C
- Methyl (E)-3-(4-((((1R)-6,6-Dimethylbicyclo[3.1.1]hept-2-en-2-yl)methyl)carbamoyl)phenyl)acrylate 20a
- White Solid, Yield 76%, m.p. 100.5–101.7 °C
- Methyl (E)-3-(4-((((1S)-6,6-Dimethylbicyclo [3.1.1]hept-2-en-2-yl)methyl)carbamoyl)phenyl)acrylate 20b
- White Solid, Yield 73%
- Methyl (E)-3-(4-((2-((1R)-6,6-Dimethylbicyclo[3.1.1]hept-2-en-2-yl)ethyl)carbamoyl)phenyl)acrylate 20c
- White Solid, Yield 69%, m.p. 98.2–99.4 °C
- Synthesis of Hydroxamic Acids 18a–c and 21a–c
- 4-((E)-3-((((1R)-6,6-Dimethylbicyclo[3.1.1]hept-2-en-2-yl)methyl)amino)-3-oxoprop-1-en-1-yl)-N-hydroxybenzamide 18a
- White Solid, Yield 59%, m.p. 96.5 °C
- 4-((E)-3-((((1S)-6,6-Dimethylbicyclo[3.1.1]hept-2-en-2-yl)methyl)amino)-3-oxoprop-1-en-1-yl)-N-hydroxybenzamide 18b
- White Solid, Yield 64%
- 4-((E)-3-((2-((1R)-6,6-Dimethylbicyclo[3.1.1]hept-2-en-2-yl)ethyl)amino)-3-oxoprop-1-en-1-yl)-N-hydroxybenzamide 18c
- White Solid, Yield 73%, m.p. 189.7–191.1 °C
- N-(((1R)-6,6-Dimethylbicyclo[3.1.1]hept-2-en-2-yl)methyl)-4-((E)-3-(hydroxyamino)-3-oxoprop-1-en-1-yl)benzamide 21a
- White Solid, Yield 67%, m.p. 74.6 °C
- N-(((1S)-6,6-Dimethylbicyclo[3.1.1]hept-2-en-2-yl)methyl)-4-((E)-3-(hydroxyamino)-3-oxoprop-1-en-1-yl)benzamide 21b
- White Solid, Yield 52%
- N-(2-((1R)-6,6-Dimethylbicyclo [3.1.1]hept-2-en-2-yl)ethyl)-4-((E)-3-(hydroxyamino)-3-oxoprop-1-en-1-yl)benzamide 21c
- White Solid, Yield 70%, m.p. 132.5–132.6 °C
2.2. HDAC Inhibitory Activity Assay
2.3. Cell Culture
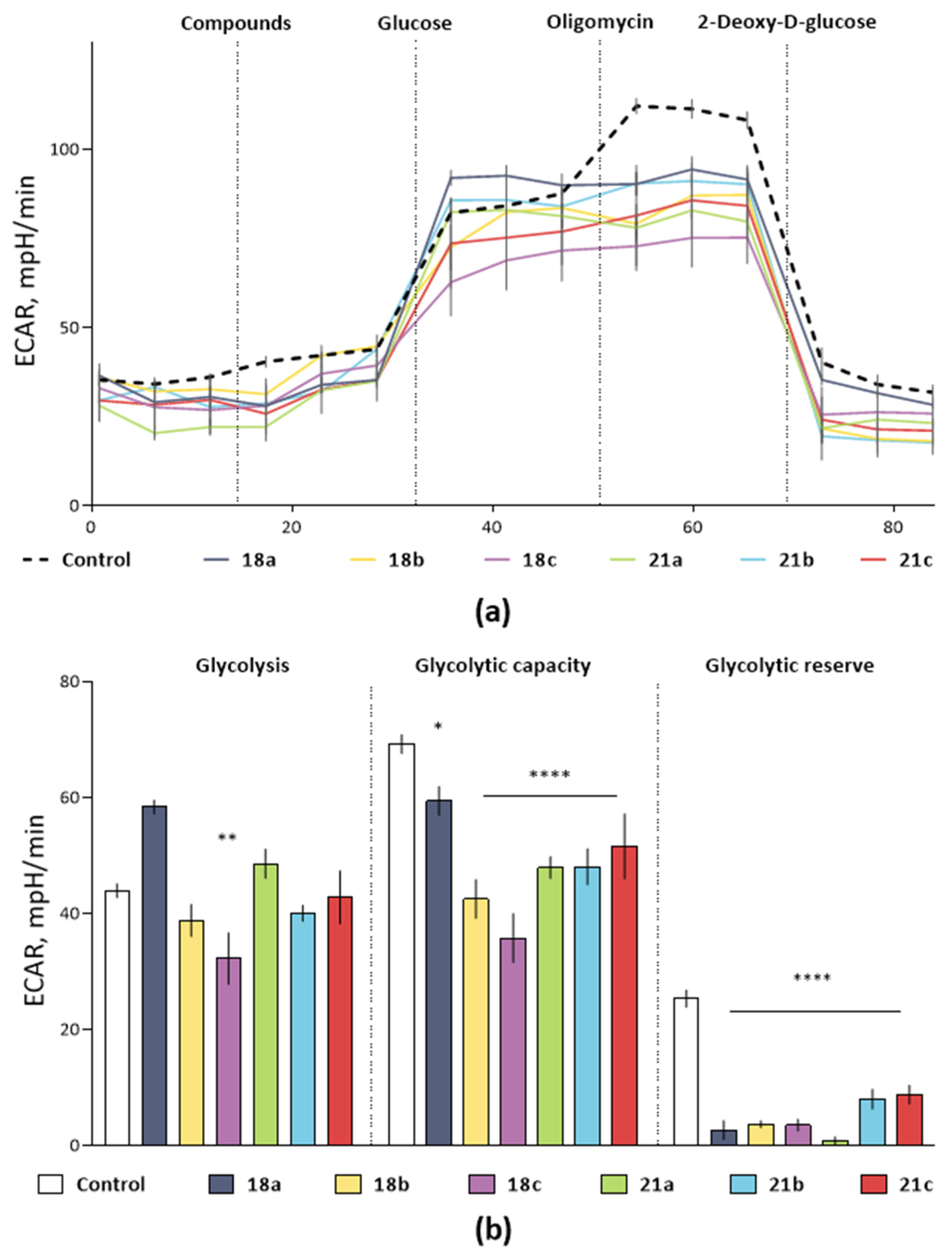
2.4. Glycolysis Flux Assay
2.5. Assay of Cell Death
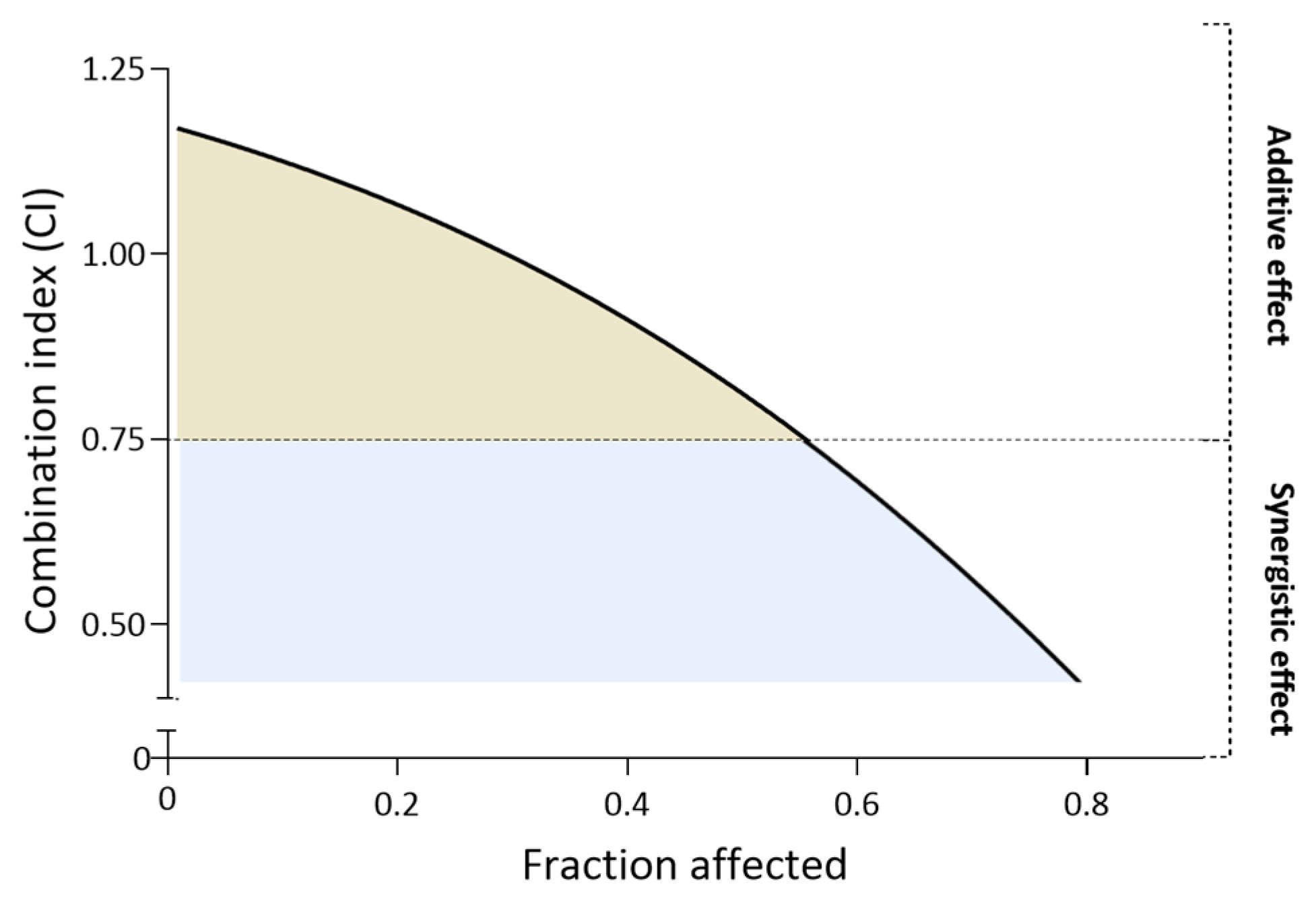
2.6. Drug Combination Analysis and Combination Index (CI) Calculation
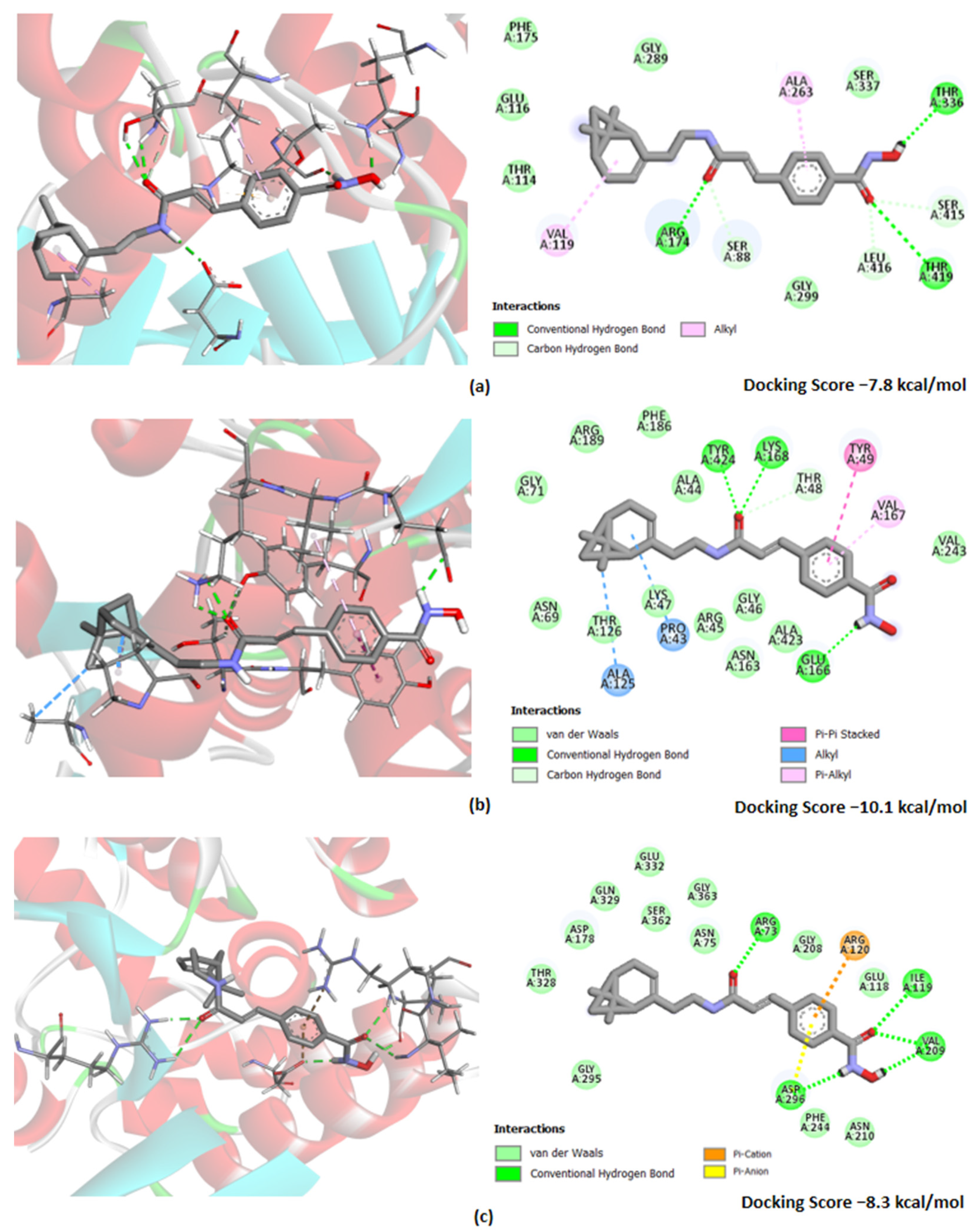
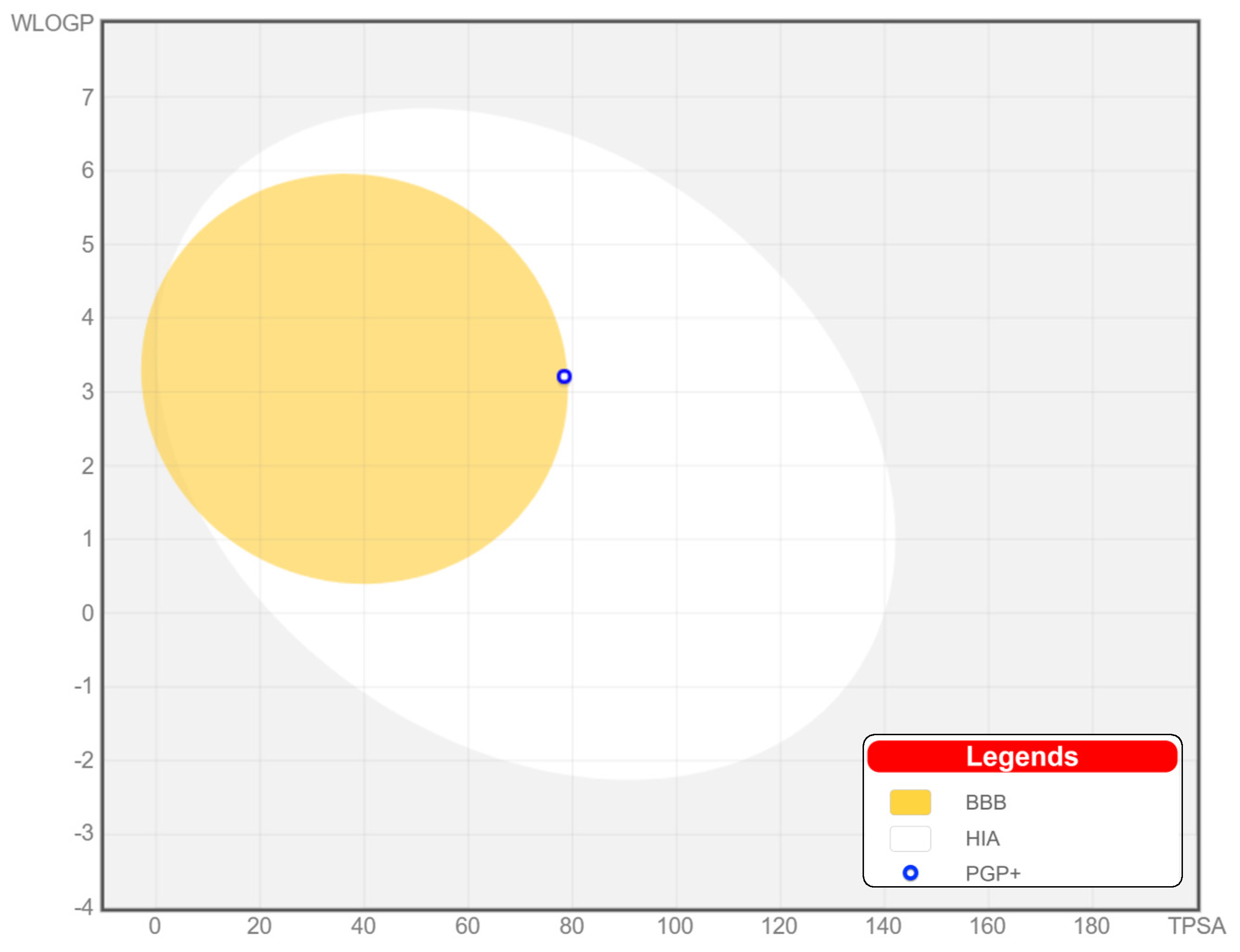
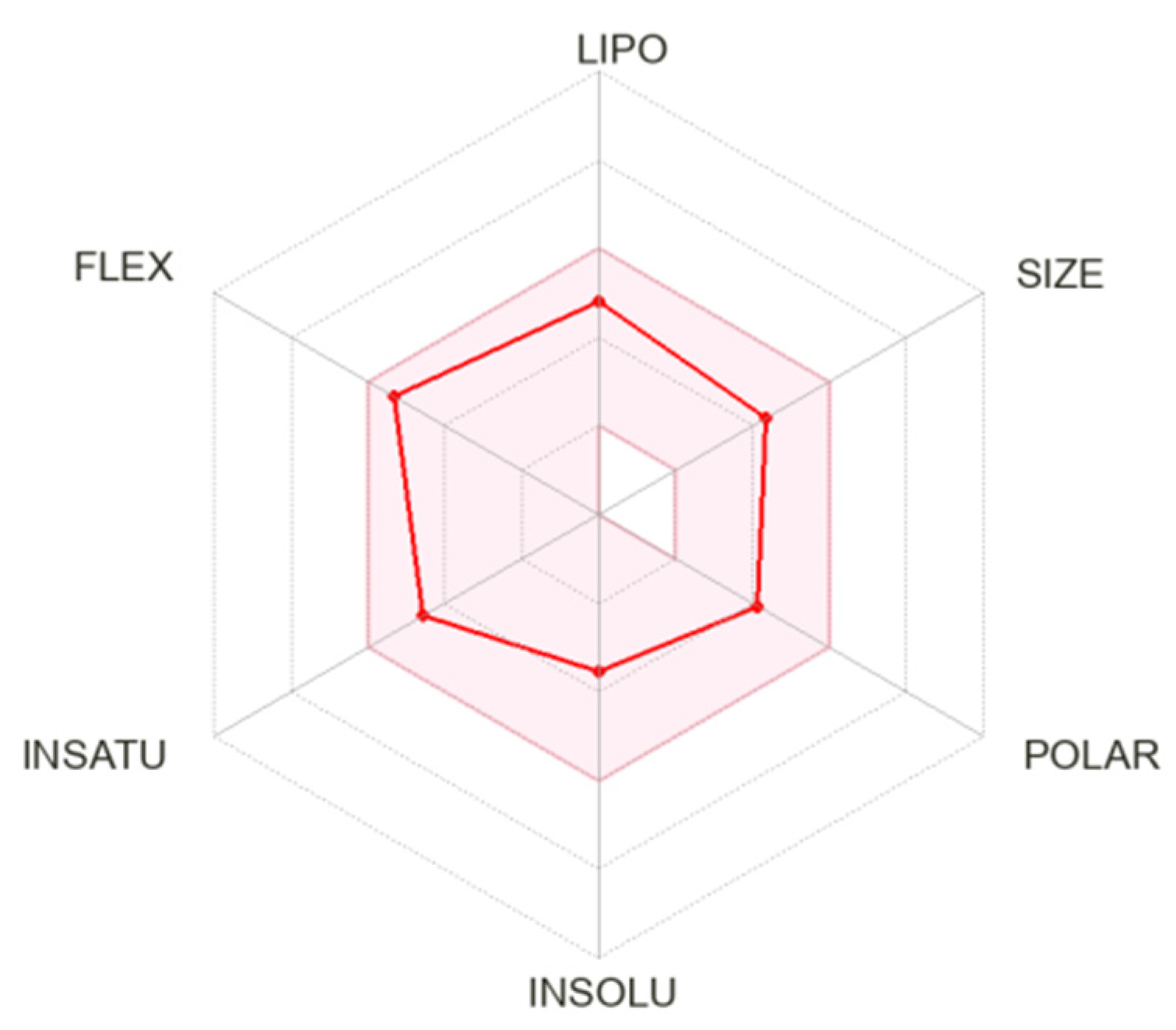
2.7. Molecular Docking and ADME Evaluation
2.8. Statistics
3. Results and Discussion
3.1. Chemistry
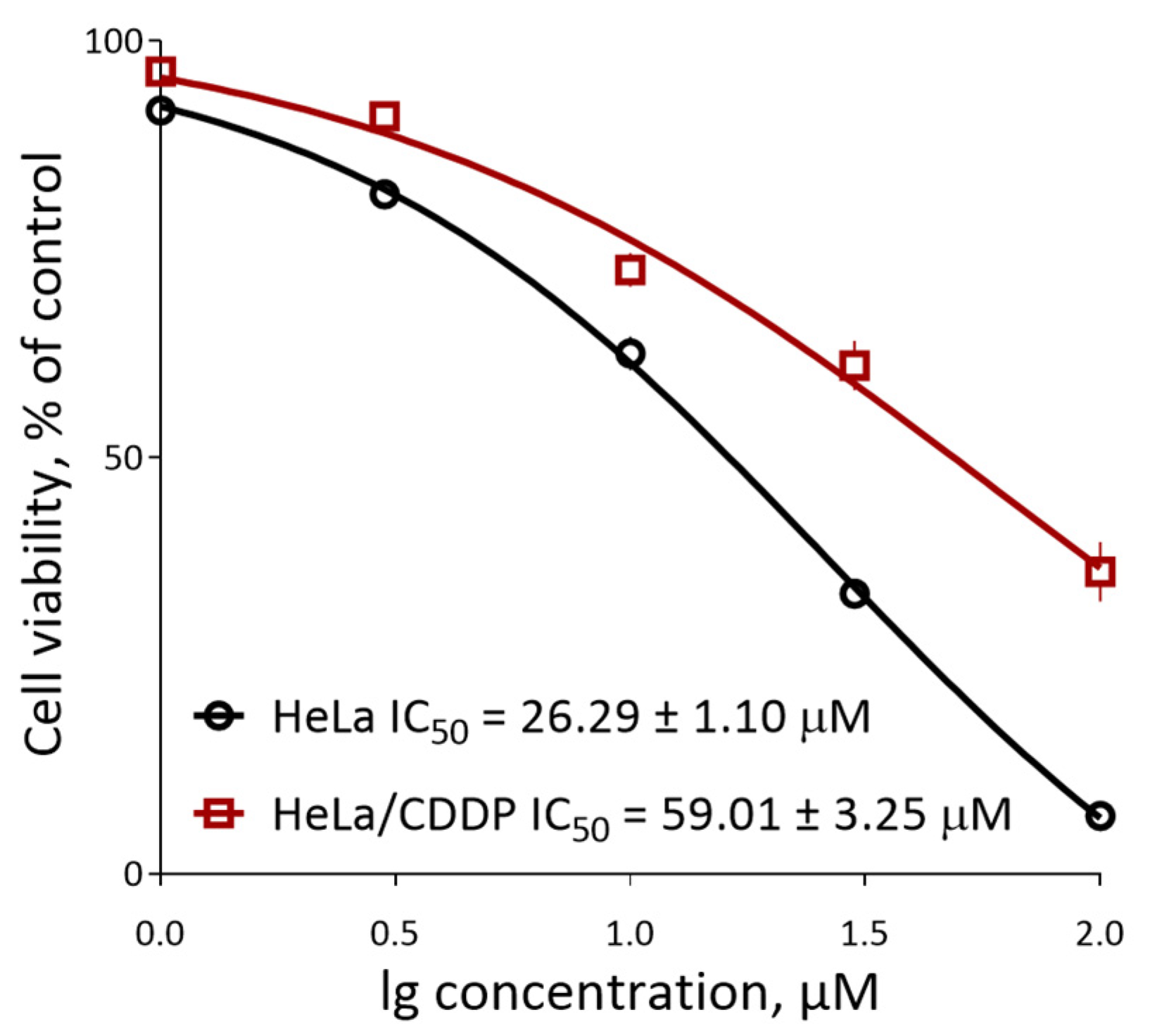
3.2. Biological Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Miller, K.D.; Nogueira, L.; Devasia, T.; Mariotto, A.B.; Yabroff, K.R.; Jemal, A.; Kramer, J.; Siegel, R.L. Cancer treatment and survivorship statistics, 2022. CA Cancer J. Clin. 2022, 72, 409–436. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Pomeroy, A.E.; Schmidt, E.V.; Sorger, P.K.; Palmer, A.C. Drug independence and the curability of cancer by combination chemotherapy. Trends Cancer 2022, 8, 915–929. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, F.U.; Sufiyan Chhipa, A.; Mishra, V.; Gupta, V.K.; Rawat, S.G.; Kumar, A.; Pathak, C. Molecular and cellular paradigms of multidrug resistance in cancer. Cancer Rep. 2022, 5, e1291. [Google Scholar] [CrossRef] [PubMed]
- Baguley, B.C. Multidrug resistance in cancer. Methods Mol. Biol. 2010, 596, 1–14. [Google Scholar] [PubMed]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Engle, K.; Kumar, G. Cancer multidrug-resistance reversal by ABCB1 inhibition: A recent update. Eur. J. Med. Chem. 2022, 239, 114542. [Google Scholar] [CrossRef]
- Hanna, C.; Kurian, K.M.; Williams, K.; Watts, C.; Jackson, A.; Carruthers, R.; Strathdee, K.; Cruickshank, G.; Dunn, L.; Erridge, S.; et al. Pharmacokinetics, safety, and tolerability of olaparib and temozolomide for recurrent glioblastoma: Results of the phase I OPARATIC trial. Neuro-Oncology 2020, 22, 1840–1850. [Google Scholar] [CrossRef]
- Li, W.; Zhang, H.; Assaraf, Y.G.; Zhao, K.; Xu, X.; Xie, J.; Yang, D.H.; Chen, Z.S. Overcoming ABC transporter-mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist. Updat. 2016, 27, 14–29. [Google Scholar] [CrossRef]
- Chari, A.; Martinez-Lopez, J.; Mateos, M.V.; Blade, J.; Benboubker, L.; Oriol, A.; Arnulf, B.; Rodriguez-Otero, P.; Pineiro, L.; Jakubowiak, A.; et al. Daratumumab plus carfilzomib and dexamethasone in patients with relapsed or refractory multiple myeloma. Blood 2019, 134, 421–431. [Google Scholar] [CrossRef]
- San-Miguel, J.; Dhakal, B.; Yong, K.; Spencer, A.; Anguille, S.; Mateos, M.V.; Fernandez de Larrea, C.; Martinez-Lopez, J.; Moreau, P.; Touzeau, C.; et al. Cilta-cel or Standard Care in Lenalidomide-Refractory Multiple Myeloma. N. Engl. J. Med. 2023, 389, 335–347. [Google Scholar] [CrossRef]
- Neganova, M.E.; Klochkov, S.G.; Aleksandrova, Y.R.; Aliev, G. The Hydroxamic Acids as Potential Anticancer and Neuroprotective Agents. Curr. Med. Chem. 2021, 28, 8139–8162. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liang, Y.; Si, X. Hydroxamic acid hybrids as the potential anticancer agents: An Overview. Eur. J. Med. Chem. 2020, 205, 112679. [Google Scholar] [CrossRef] [PubMed]
- Xue, K.; Gu, J.J.; Zhang, Q.; Mavis, C.; Hernandez-Ilizaliturri, F.J.; Czuczman, M.S.; Guo, Y. Vorinostat, a histone deacetylase (HDAC) inhibitor, promotes cell cycle arrest and re-sensitizes rituximab- and chemo-resistant lymphoma cells to chemotherapy agents. J. Cancer Res. Clin. Oncol. 2016, 142, 379–387. [Google Scholar] [CrossRef]
- El Omari, N.; Bakrim, S.; Khalid, A.; Albratty, M.; Abdalla, A.N.; Lee, L.H.; Goh, K.W.; Ming, L.C.; Bouyahya, A. Anticancer clinical efficiency and stochastic mechanisms of belinostat. Biomed. Pharmacother. 2023, 165, 115212. [Google Scholar] [CrossRef]
- Srinivas, N.R. Clinical pharmacokinetics of panobinostat, a novel histone deacetylase (HDAC) inhibitor: Review and perspectives. Xenobiotica 2017, 47, 354–368. [Google Scholar] [CrossRef] [PubMed]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schioth, H.B. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br. J. Clin. Pharmacol. 2021, 87, 4577–4597. [Google Scholar] [CrossRef]
- Posso, M.C.; Domingues, F.C.; Ferreira, S.; Silvestre, S. Development of Phenothiazine Hybrids with Potential Medicinal Interest: A Review. Molecules 2022, 27, 276. [Google Scholar] [CrossRef]
- Chugunova, E.A.; Burilov, A.R. Novel Structural Hybrids on the Base of Benzofuroxans and Furoxans. Mini-Review. Curr. Top. Med. Chem. 2017, 17, 986–1005. [Google Scholar] [CrossRef]
- Mohamed, M.F.A.; Abuo-Rahma, G.E.A. Molecular targets and anticancer activity of quinoline-chalcone hybrids: Literature review. RSC Adv. 2020, 10, 31139–31155. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Shang, C.; Wang, H.; Yun, J. Isatin-azole hybrids and their anticancer activities. Arch. Pharm. 2020, 353, e1900272. [Google Scholar] [CrossRef] [PubMed]
- Barreca, M.; Ingarra, A.M.; Raimondi, M.V.; Spano, V.; Piccionello, A.P.; De Franco, M.; Menilli, L.; Gandin, V.; Miolo, G.; Barraja, P.; et al. New tricyclic systems as photosensitizers towards triple negative breast cancer cells. Arch. Pharm. Res. 2022, 45, 806–821. [Google Scholar] [CrossRef]
- Li, G.; Lin, S.S.; Yu, Z.L.; Wu, X.H.; Liu, J.W.; Tu, G.H.; Liu, Q.Y.; Tang, Y.L.; Jiang, Q.N.; Xu, J.H.; et al. A PARP1 PROTAC as a novel strategy against PARP inhibitor resistance via promotion of ferroptosis in p53-positive breast cancer. Biochem. Pharmacol. 2022, 206, 115329. [Google Scholar] [CrossRef] [PubMed]
- Grillone, K.; Riillo, C.; Rocca, R.; Ascrizzi, S.; Spano, V.; Scionti, F.; Polera, N.; Maruca, A.; Barreca, M.; Juli, G.; et al. The New Microtubule-Targeting Agent SIX2G Induces Immunogenic Cell Death in Multiple Myeloma. Int. J. Mol. Sci. 2022, 23, 10222. [Google Scholar] [CrossRef]
- Soumoy, L.; Ghanem, G.E.; Saussez, S.; Journe, F. Bufalin for an innovative therapeutic approach against cancer. Pharmacol. Res. 2022, 184, 106442. [Google Scholar] [CrossRef]
- Zielinska-Blajet, M.; Pietrusiak, P.; Feder-Kubis, J. Selected Monocyclic Monoterpenes and Their Derivatives as Effective Anticancer Therapeutic Agents. Int. J. Mol. Sci. 2021, 22, 4763. [Google Scholar] [CrossRef]
- Silva, B.I.M.; Nascimento, E.A.; Silva, C.J.; Silva, T.G.; Aguiar, J.S. Anticancer activity of monoterpenes: A systematic review. Mol. Biol. Rep. 2021, 48, 5775–5785. [Google Scholar] [CrossRef]
- Vinothkumar, R.; Sudha, M.; Viswanathan, P.; Kabalimoorthy, J.; Balasubramanian, T.; Nalini, N. Modulating effect of d-carvone on 1,2-dimethylhydrazine-induced pre-neoplastic lesions, oxidative stress and biotransforming enzymes, in an experimental model of rat colon carcinogenesis. Cell Prolif. 2013, 46, 705–720. [Google Scholar] [CrossRef]
- Lima, L.T.F.; Ganzella, F.A.O.; Cardoso, G.C.; Pires, V.D.S.; Chequin, A.; Santos, G.L.; Braun-Prado, K.; Galindo, C.M.; Braz Junior, O.; Molento, M.B.; et al. l-carvone decreases breast cancer cells adhesion, migration, and invasion by suppressing FAK activation. Chem. Biol. Interact. 2023, 378, 110480. [Google Scholar] [CrossRef]
- Patel, P.B.; Thakkar, V.R. L-carvone induces p53, caspase 3 mediated apoptosis and inhibits the migration of breast cancer cell lines. Nutr. Cancer 2014, 66, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Yang, N.; Cao, Y.; Dang, J.; Cheng, L.; El-Sheikh, M.A.; Zhang, Y. d-Carvone inhibits the JAK/STAT3 signaling pathway and induced the apoptotic cell death in the human gastric cancer AGS cells. J. Biochem. Mol. Toxicol. 2021, 35, e22746. [Google Scholar] [CrossRef]
- Kang, W.; Choi, D.; Park, S.; Park, T. Carvone Decreases Melanin Content by Inhibiting Melanoma Cell Proliferation via the Cyclic Adenosine Monophosphate (cAMP) Pathway. Molecules 2020, 25, 5191. [Google Scholar] [CrossRef] [PubMed]
- Aydin, E.; Turkez, H.; Keles, M.S. Potential anticancer activity of carvone in N2a neuroblastoma cell line. Toxicol. Ind. Health 2015, 31, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Iyappan, P.; Bala, M.D.; Sureshkumar, M.; Veeraraghavan, V.P.; Palanisamy, A. D-carvone induced ROS mediated apoptotic cell death in human leukemic cell lines (Molt-4). Bioinformation 2021, 17, 171–180. [Google Scholar] [CrossRef]
- Booupathy, L.K.; Venkatachalam, S.; Natarajan, N.; Thamaraiselvan, R.; Arumugam, M.; Periyasamy, B.M. Chemopreventive effect of myrtenal on bacterial enzyme activity and the development of 1,2-dimethyl hydrazine-induced aberrant crypt foci in Wistar Rats. J. Food Drug Anal. 2016, 24, 206–213. [Google Scholar] [CrossRef]
- Farrag, M.A.; Ezz, M.K.; Ibrahim, N.K.; Ahmed, E.K. Chemopreventive Potential of Myrtenal against Nitrosamine-Initiated, Radiation-Promoted Rat Bladder Carcinogenesis. Nutr. Cancer 2022, 74, 288–298. [Google Scholar] [CrossRef]
- Anandakumar, P.; Kamaraj, S.; Vanitha, M.K. D-limonene: A multifunctional compound with potent therapeutic effects. J. Food Biochem. 2021, 45, e13566. [Google Scholar] [CrossRef]
- Mandal, D.; Sahu, B.R.; Parija, T. Combination of tamoxifen and D-limonene enhances therapeutic efficacy in breast cancer cells. Med. Oncol. 2023, 40, 216. [Google Scholar] [CrossRef]
- Kiesgen de Richter, R.; Bonato, M.; Follet, M.; Kamenka, J.M. The (+)- and (-)-[2-(1,3-dithianyl)]myrtanylborane. Solid and stable monoalkylboranes for asymmetric hydroboration. J. Org. Chem. 1990, 55, 2855–2860. [Google Scholar] [CrossRef]
- Liu, H.-X.; Tan, H.-B.; He, M.-T.; Ling, L.; Wang, Y.-H.; Long, C.-L. Isolation and synthesis of two hydroxychavicol heterodimers from Piper nudibaccatum. Tetrahedron 2015, 71, 2369–2375. [Google Scholar] [CrossRef]
- Akgun, B.; Hall, D.G. Fast and Tight Boronate Formation for Click Bioorthogonal Conjugation. Angew. Chem. Int. Ed. Engl. 2016, 55, 3909–3913. [Google Scholar] [CrossRef] [PubMed]
- Aleksandrova, Y.; Munkuev, A.; Mozhaitsev, E.; Suslov, E.; Tsypyshev, D.; Chaprov, K.; Begunov, R.; Volcho, K.; Salakhutdinov, N.; Neganova, M. Elaboration of the Effective Multi-Target Therapeutic Platform for the Treatment of Alzheimer’s Disease Based on Novel Monoterpene-Derived Hydroxamic Acids. Int. J. Mol. Sci. 2023, 24, 9743. [Google Scholar] [CrossRef] [PubMed]
- Karaj, E.; Dlamini, S.; Koranne, R.; Sindi, S.H.; Perera, L.; Taylor, W.R.; Viranga Tillekeratne, L.M. Pharmacophore optimization of imidazole chalcones to modulate microtubule dynamics. Bioorg Chem. 2022, 122, 105700. [Google Scholar] [CrossRef] [PubMed]
- Morera, L.; Roatsch, M.; Furst, M.C.; Hoffmann, I.; Senger, J.; Hau, M.; Franz, H.; Schule, R.; Heinrich, M.R.; Jung, M. 4-Biphenylalanine- and 3-Phenyltyrosine-Derived Hydroxamic Acids as Inhibitors of the JumonjiC-Domain-Containing Histone Demethylase KDM4A. ChemMedChem 2016, 11, 2063–2083. [Google Scholar] [CrossRef]
- Fang, K.; Dong, G.; Li, Y.; He, S.; Wu, Y.; Wu, S.; Wang, W.; Sheng, C. Discovery of Novel Indoleamine 2,3-Dioxygenase 1 (IDO1) and Histone Deacetylase (HDAC) Dual Inhibitors. ACS Med. Chem. Lett. 2018, 9, 312–317. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Q. Using Seahorse Machine to Measure OCR and ECAR in Cancer Cells. Methods Mol. Biol. 2019, 1928, 353–363. [Google Scholar]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the MTT Assay. Cold Spring Harb. Protoc. 2018, 2018. [Google Scholar] [CrossRef]
- Gao, Z.; Xu, J.; Fan, Y.; Qi, Y.; Wang, S.; Zhao, S.; Guo, X.; Xue, H.; Deng, L.; Zhao, R.; et al. PDIA3P1 promotes Temozolomide resistance in glioblastoma by inhibiting C/EBPbeta degradation to facilitate proneural-to-mesenchymal transition. J. Exp. Clin. Cancer Res. 2022, 41, 223. [Google Scholar] [CrossRef]
- Aleshin, A.E.; Malfois, M.; Liu, X.; Kim, C.S.; Fromm, H.J.; Honzatko, R.B.; Koch, M.H.; Svergun, D.I. Nonaggregating mutant of recombinant human hexokinase I exhibits wild-type kinetics and rod-like conformations in solution. Biochemistry 1999, 38, 8359–8366. [Google Scholar] [CrossRef]
- Lee, Y.H.; Li, Y.; Uyeda, K.; Hasemann, C.A. Tissue-specific structure/function differentiation of the liver isoform of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase. J. Biol. Chem. 2003, 278, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Nandi, S.; Dey, M. Biochemical and structural insights into how amino acids regulate pyruvate kinase muscle isoform 2. J. Biol. Chem. 2020, 295, 5390–5403. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Macpherson, J.A.; Theisen, A.; Masino, L.; Fets, L.; Driscoll, P.C.; Encheva, V.; Snijders, A.P.; Martin, S.R.; Kleinjung, J.; Barran, P.E.; et al. Functional cross-talk between allosteric effects of activating and inhibiting ligands underlies PKM2 regulation. Elife 2019, 8, e45068. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, S.; Wilson, J.E.; Mulichak, A.; Garavito, R.M. Allosteric regulation of type I hexokinase: A site-directed mutational study indicating location of the functional glucose 6-phosphate binding site in the N-terminal half of the enzyme. Arch. Biochem. Biophys. 1999, 362, 203–210. [Google Scholar] [CrossRef]
- Wang, Y.; Qu, C.; Liu, T.; Wang, C. PFKFB3 inhibitors as potential anticancer agents: Mechanisms of action, current developments, and structure-activity relationships. Eur. J. Med. Chem. 2020, 203, 112612. [Google Scholar] [CrossRef]
- Ureta, T.; Lazo, P.A.; Sols, A. Allosteric inhibition of brain hexokinase by glucose 6-phosphate in the reverse reaction. Arch. Biochem. Biophys. 1985, 239, 315–319. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Assaraf, Y.G.; Brozovic, A.; Goncalves, A.C.; Jurkovicova, D.; Line, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist. Updat. 2019, 46, 100645. [Google Scholar] [CrossRef]
- Yang, W.B.; Hsu, C.C.; Hsu, T.I.; Liou, J.P.; Chang, K.Y.; Chen, P.Y.; Liu, J.J.; Yang, S.T.; Wang, J.Y.; Yeh, S.H.; et al. Increased activation of HDAC1/2/6 and Sp1 underlies therapeutic resistance and tumor growth in glioblastoma. Neuro Oncol. 2020, 22, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Tang, Z.; Jiang, Y.; Luo, P.; Qing, B.; Wei, Y.; Zhang, S.; Tang, R. HDAC1 regulates the chemosensitivity of laryngeal carcinoma cells via modulation of interleukin-8 expression. Eur. J. Pharmacol. 2021, 896, 173923. [Google Scholar] [CrossRef] [PubMed]
- Fousek, K.; Horn, L.A.; Palena, C. Interleukin-8: A chemokine at the intersection of cancer plasticity, angiogenesis, and immune suppression. Pharmacol. Ther. 2021, 219, 107692. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Lin, Y.C.; Shih, J.Y.; Huang, W.J.; Chao, S.W.; Chang, Y.L.; Chen, C.C. DUSP1 expression induced by HDAC1 inhibition mediates gefitinib sensitivity in non-small cell lung cancers. Clin. Cancer Res. 2015, 21, 428–438. [Google Scholar] [CrossRef]
- Chen, S.H.; Chow, J.M.; Hsieh, Y.Y.; Lin, C.Y.; Hsu, K.W.; Hsieh, W.S.; Chi, W.M.; Shabangu, B.M.; Lee, C.H. HDAC1,2 Knock-Out and HDACi Induced Cell Apoptosis in Imatinib-Resistant K562 Cells. Int. J. Mol. Sci. 2019, 20, 2271. [Google Scholar] [CrossRef]
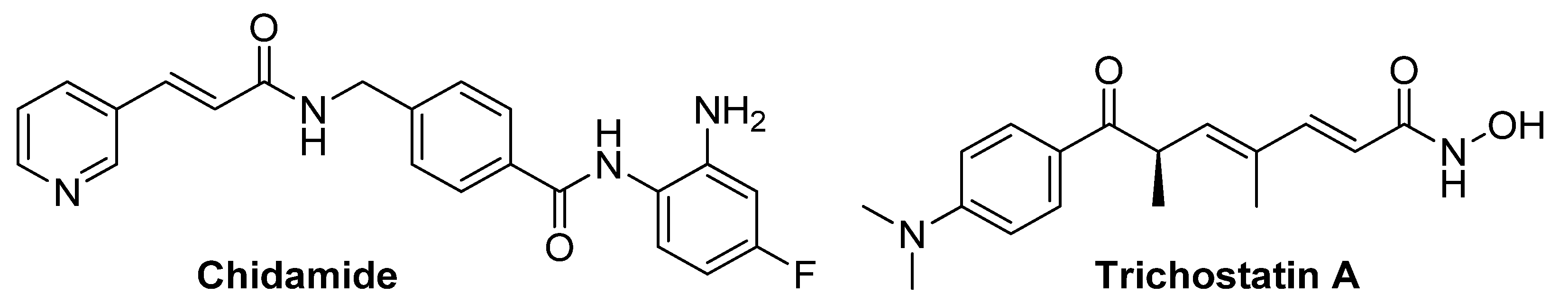
- Xue, K.; Wu, J.C.; Li, X.Y.; Li, R.; Zhang, Q.L.; Chang, J.J.; Liu, Y.Z.; Xu, C.H.; Zhang, J.Y.; Sun, X.J.; et al. Chidamide triggers BTG1-mediated autophagy and reverses the chemotherapy resistance in the relapsed/refractory B-cell lymphoma. Cell Death Dis. 2021, 12, 900. [Google Scholar] [CrossRef]
- Sato, H.; Uzu, M.; Kashiba, T.; Fujiwara, T.; Hatakeyama, H.; Ueno, K.; Hisaka, A. Trichostatin A modulates cellular metabolism in renal cell carcinoma to enhance sunitinib sensitivity. Eur. J. Pharmacol. 2019, 847, 143–157. [Google Scholar] [CrossRef]
- Andersen, C.L.; McMullin, M.F.; Ejerblad, E.; Zweegman, S.; Harrison, C.; Fernandes, S.; Bareford, D.; Knapper, S.; Samuelsson, J.; Lofvenberg, E.; et al. A phase II study of vorinostat (MK-0683) in patients with polycythaemia vera and essential thrombocythaemia. Br. J. Haematol. 2013, 162, 498–508. [Google Scholar] [CrossRef]
- Gimsing, P.; Hansen, M.; Knudsen, L.M.; Knoblauch, P.; Christensen, I.J.; Ooi, C.E.; Buhl-Jensen, P. A phase I clinical trial of the histone deacetylase inhibitor belinostat in patients with advanced hematological neoplasia. Eur. J. Haematol. 2008, 81, 170–176. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Wei, W.; Zhou, D.X. Histone Acetylation Enzymes Coordinate Metabolism and Gene Expression. Trends Plant Sci. 2015, 20, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef]
- Ganapathy-Kanniappan, S.; Geschwind, J.F. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer 2013, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W.; Cantley, L.C. Altered metabolism in cancer. BMC Biol. 2010, 8, 88. [Google Scholar] [CrossRef] [PubMed]
- Chelakkot, C.; Chelakkot, V.S.; Shin, Y.; Song, K. Modulating Glycolysis to Improve Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 2606. [Google Scholar] [CrossRef]
- Peng, J.; Cui, Y.; Xu, S.; Wu, X.; Huang, Y.; Zhou, W.; Wang, S.; Fu, Z.; Xie, H. Altered glycolysis results in drug-resistant in clinical tumor therapy. Oncol. Lett. 2021, 21, 369. [Google Scholar] [CrossRef]
- Wang, S.; Zhou, L.; Ji, N.; Sun, C.; Sun, L.; Sun, J.; Du, Y.; Zhang, N.; Li, Y.; Liu, W.; et al. Targeting ACYP1-mediated glycolysis reverses lenvatinib resistance and restricts hepatocellular carcinoma progression. Drug Resist. Updat. 2023, 69, 100976. [Google Scholar] [CrossRef]
- Liu, X.; Miao, W.; Huang, M.; Li, L.; Dai, X.; Wang, Y. Elevated Hexokinase II Expression Confers Acquired Resistance to 4-Hydroxytamoxifen in Breast Cancer Cells. Mol. Cell Proteom. 2019, 18, 2273–2284. [Google Scholar] [CrossRef]
- Liang, J.; Cao, R.; Wang, X.; Zhang, Y.; Wang, P.; Gao, H.; Li, C.; Yang, F.; Zeng, R.; Wei, P.; et al. Mitochondrial PKM2 regulates oxidative stress-induced apoptosis by stabilizing Bcl2. Cell Res. 2017, 27, 329–351. [Google Scholar] [CrossRef]
- Thomalla, D.; Beckmann, L.; Grimm, C.; Oliverio, M.; Meder, L.; Herling, C.D.; Nieper, P.; Feldmann, T.; Merkel, O.; Lorsy, E.; et al. Deregulation and epigenetic modification of BCL2-family genes cause resistance to venetoclax in hematologic malignancies. Blood 2022, 140, 2113–2126. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef]
- Du, D.; Wang-Kan, X.; Neuberger, A.; van Veen, H.W.; Pos, K.M.; Piddock, L.J.V.; Luisi, B.F. Multidrug efflux pumps: Structure, function and regulation. Nat. Rev. Microbiol. 2018, 16, 523–539. [Google Scholar] [CrossRef] [PubMed]
- Vodenkova, S.; Buchler, T.; Cervena, K.; Veskrnova, V.; Vodicka, P.; Vymetalkova, V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: Past, present and future. Pharmacol. Ther. 2020, 206, 107447. [Google Scholar] [CrossRef]
- Wu, H.; Du, J.; Li, C.; Li, H.; Guo, H.; Li, Z. Kaempferol Can Reverse the 5-Fu Resistance of Colorectal Cancer Cells by Inhibiting PKM2-Mediated Glycolysis. Int. J. Mol. Sci. 2022, 23, 3544. [Google Scholar] [CrossRef]
- Feng, J.; Dai, W.; Mao, Y.; Wu, L.; Li, J.; Chen, K.; Yu, Q.; Kong, R.; Li, S.; Zhang, J.; et al. Simvastatin re-sensitizes hepatocellular carcinoma cells to sorafenib by inhibiting HIF-1alpha/PPAR-gamma/PKM2-mediated glycolysis. J. Exp. Clin. Cancer Res. 2020, 39, 24. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Yang, X.F.; Tian, X.Q.; Tang, S.L.; Li, L.Q.; Zhao, S.; Zheng, H.C. The in vitro and vivo anti-tumor effects and molecular mechanisms of suberoylanilide hydroxamic acid (SAHA) and MG132 on the aggressive phenotypes of gastric cancer cells. Oncotarget 2016, 7, 56508–56525. [Google Scholar] [CrossRef] [PubMed]
- Neganova, M.E.; Klochkov, S.G.; Aleksandrova, Y.R.; Osipov, V.N.; Avdeev, D.V.; Pukhov, S.A.; Gromyko, A.V.; Aliev, G. New Spirocyclic Hydroxamic Acids as Effective Antiproliferative Agents. Anticancer Agents Med. Chem. 2021, 21, 597–610. [Google Scholar] [CrossRef]
- Caines, J.K.; Barnes, D.A.; Berry, M.D. The Use of Seahorse XF Assays to Interrogate Real-Time Energy Metabolism in Cancer Cell Lines. Methods Mol. Biol. 2022, 2508, 225–234. [Google Scholar]
- Anderson, A.S.; Roberts, P.C.; Frisard, M.I.; McMillan, R.P.; Brown, T.J.; Lawless, M.H.; Hulver, M.W.; Schmelz, E.M. Metabolic changes during ovarian cancer progression as targets for sphingosine treatment. Exp. Cell Res. 2013, 319, 1431–1442. [Google Scholar] [CrossRef]
- Dier, U.; Shin, D.H.; Hemachandra, L.P.; Uusitalo, L.M.; Hempel, N. Bioenergetic analysis of ovarian cancer cell lines: Profiling of histological subtypes and identification of a mitochondria-defective cell line. PLoS ONE 2014, 9, e98479. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhu, L.; Hao, B.; Gao, W.; Wang, Q.; Li, K.; Wang, M.; Huang, M.; Liu, Z.; Yang, Q.; et al. iNOS-derived nitric oxide promotes glycolysis by inducing pyruvate kinase M2 nuclear translocation in ovarian cancer. Oncotarget 2017, 8, 33047–33063. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yun, Y.; Wu, B.; Wen, L.; Wen, M.; Yang, H.; Zhao, L.; Liu, W.; Huang, S.; Wen, N.; et al. FOXM1 promotes reprogramming of glucose metabolism in epithelial ovarian cancer cells via activation of GLUT1 and HK2 transcription. Oncotarget 2016, 7, 47985–47997. [Google Scholar] [CrossRef] [PubMed]
- Beskow, L.M. Lessons from HeLa Cells: The Ethics and Policy of Biospecimens. Annu. Rev. Genomics Hum. Genet. 2016, 17, 395–417. [Google Scholar] [CrossRef]
- Amable, L. Cisplatin resistance and opportunities for precision medicine. Pharmacol. Res. 2016, 106, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Lugones, Y.; Loren, P.; Salazar, L.A. Cisplatin Resistance: Genetic and Epigenetic Factors Involved. Biomolecules 2022, 12, 1365. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, X.; Fu, J.; Xu, W.; Yuan, J. The Role of Tumour Metabolism in Cisplatin Resistance. Front. Mol. Biosci. 2021, 8, 691795. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.W.; Pouliot, L.M.; Hall, M.D.; Gottesman, M.M. Cisplatin resistance: A cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol. Rev. 2012, 64, 706–721. [Google Scholar] [CrossRef]
- Ramaiah, M.J.; Tangutur, A.D.; Manyam, R.R. Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy. Life Sci. 2021, 277, 119504. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | HDAC1-Inhibitory Ability (IC50, µM) | Compounds | HDAC1-Inhibitory Ability (IC50, µM) |
|---|---|---|---|
| Trichostatin A | 0.22 ± 0.01 | 18c | 4.45 ± 0.30 |
| 21a | 7.23 ± 0.46 | ||
| 18a | 6.90 ± 0.03 | 21b | 8.20 ± 0.49 |
| 18b | 7.96 ± 0.28 | 21c | 5.00 ± 0.12 |
| Compounds | HeLa | Hek-293 | Compounds | HeLa | Hek-293 |
|---|---|---|---|---|---|
| Trichostatin A | 0.38 ± 0.02 | 0.22 ± 0.01 | 18c | 33.81 ± 1.19 | 34.04 ± 3.04 |
| 21a | 18.01 ± 0.47 | 21.19 ± 1.66 | |||
| 18a | 29.25 ± 1.06 | 46.73 ± 2.93 | 21b | 42.78 ± 1.28 | 37.19 ± 1.89 |
| 18b | 43.59 ± 1.02 | 38.85 ± 1.24 | 21c | 19.10 ± 0.96 | 33.86 ± 2.59 |
| Compounds | IC50, µM | Compounds | IC50, µM |
|---|---|---|---|
| Cisplatin | 28.97 ± 2.80 | Cisplatin + 18c | 18.98 ± 1.16 * |
| Cisplatin + 21a | 27.04 ± 1.44 | ||
| Cisplatin + 18a | 29.10 ± 1.52 | Cisplatin + 21b | 32.69 ± 2.37 |
| Cisplatin + 18b | 30.00 ± 1.56 | Cisplatin + 21c | 25.73 ± 2.24 |
| Compounds | IC50, µM | Compounds | IC50, µM |
|---|---|---|---|
| Cisplatin | 59.01 ± 3.25 | Cisplatin + 18c | 29.24 ± 2.08 ** |
| Cisplatin + 21a | 59.56 ± 2.74 | ||
| Cisplatin + 18a | 60.24 ± 3.06 | Cisplatin + 21b | 61.55 ± 3.78 |
| Cisplatin + 18b | 60.69 ± 2.26 | Cisplatin + 21c | 56.07 ± 1.90 |
| Compound | MW (g/M) | Donors | Acceptors | TPSA | Log Po/v | BA |
|---|---|---|---|---|---|---|
| 18c | 354.44 | 3 | 3 | 78.43 Å2 | 2.72 | 0.55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aleksandrova, Y.; Munkuev, A.; Mozhaitsev, E.; Suslov, E.; Volcho, K.; Salakhutdinov, N.; Neganova, M. Hydroxamic Acids Containing a Bicyclic Pinane Backbone as Epigenetic and Metabolic Regulators: Synergizing Agents to Overcome Cisplatin Resistance. Cancers 2023, 15, 4985. https://doi.org/10.3390/cancers15204985
Aleksandrova Y, Munkuev A, Mozhaitsev E, Suslov E, Volcho K, Salakhutdinov N, Neganova M. Hydroxamic Acids Containing a Bicyclic Pinane Backbone as Epigenetic and Metabolic Regulators: Synergizing Agents to Overcome Cisplatin Resistance. Cancers. 2023; 15(20):4985. https://doi.org/10.3390/cancers15204985
Chicago/Turabian StyleAleksandrova, Yulia, Aldar Munkuev, Evgenii Mozhaitsev, Evgeniy Suslov, Konstantin Volcho, Nariman Salakhutdinov, and Margarita Neganova. 2023. "Hydroxamic Acids Containing a Bicyclic Pinane Backbone as Epigenetic and Metabolic Regulators: Synergizing Agents to Overcome Cisplatin Resistance" Cancers 15, no. 20: 4985. https://doi.org/10.3390/cancers15204985
APA StyleAleksandrova, Y., Munkuev, A., Mozhaitsev, E., Suslov, E., Volcho, K., Salakhutdinov, N., & Neganova, M. (2023). Hydroxamic Acids Containing a Bicyclic Pinane Backbone as Epigenetic and Metabolic Regulators: Synergizing Agents to Overcome Cisplatin Resistance. Cancers, 15(20), 4985. https://doi.org/10.3390/cancers15204985