
Distinct Cellular Origins and Differentiation Process Account for Distinct Oncogenic and Clinical Behaviors of Leiomyosarcomas
 , ,
, ,  and
and  add
Show full author list
add
Show full author list
Abstract
Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Experimental Model and Subject Details
2.1.1. Human Samples
2.1.2. Cell Lines and Primary Culture
2.2. Method Details
2.2.1. Data Acquisition
Expression Microarray Data
Copy Number Data
Sequencing Data
2.2.2. Sequencing Data Analysis
RNA Sequencing (RNA-Seq)
miRNA Sequencing (miRNA-Seq)
Whole Genome Sequencing (WGS)
2.2.3. Detection of Single Nucleotide and Structural Variants
Single Nucleotide Variant (SNV)
Structural Variants
2.2.4. Experimental Validation
Fluorescent In-Situ Hybridization
Verification of Alterations
Sanger Sequencing
Immunohistochemistry
Immunofluorescence
Cytotoxicity Analysis
2.2.5. Quantification and Statistical Analysis
Normalization of Affymetrix and Agilent Micro-Arrays and Gene Selection
Gene Module Clustering
Sample Clustering and PCA Analysis
Patient Classification
Clinical Enrichment
Survival Analysis
Differential Expression Analysis
Functional Enrichment and Mapping
miRNA-mRNA Interaction Analysis
CNV Recurrence Analysis
Mutational Pattern Analysis
3. Results
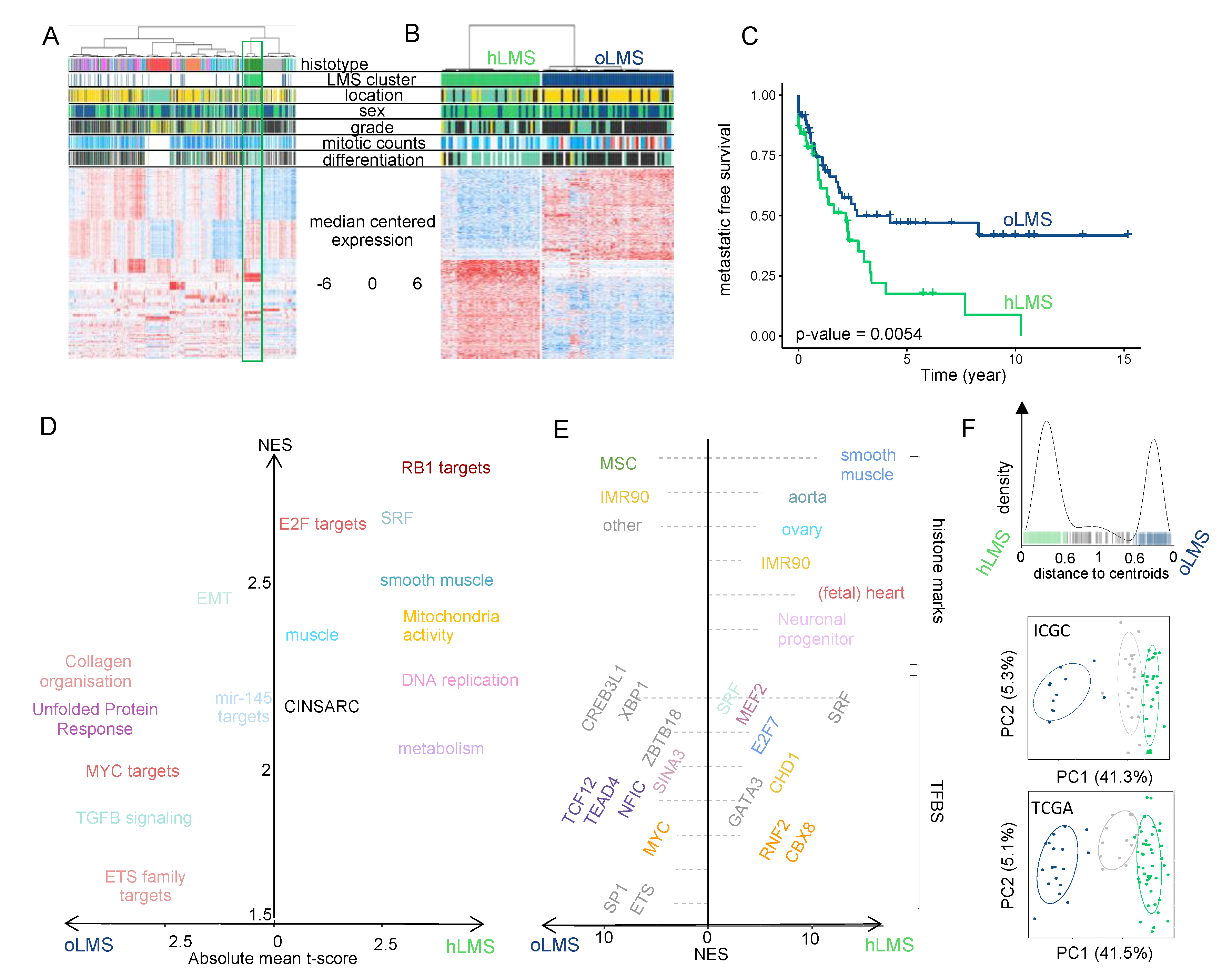
3.1. Identification of a Group of 42 LMS Behaving as Simple Genetic Sarcomas
3.2. hLMS Are Intra-Abdominal, Low-Grade, Metastatic LMS with Homogeneous Transcriptional Behavior
3.3. The Transcriptional Signature Highlights Cell Cycle and Differentiation Pathways Specific to LMS Subgroups
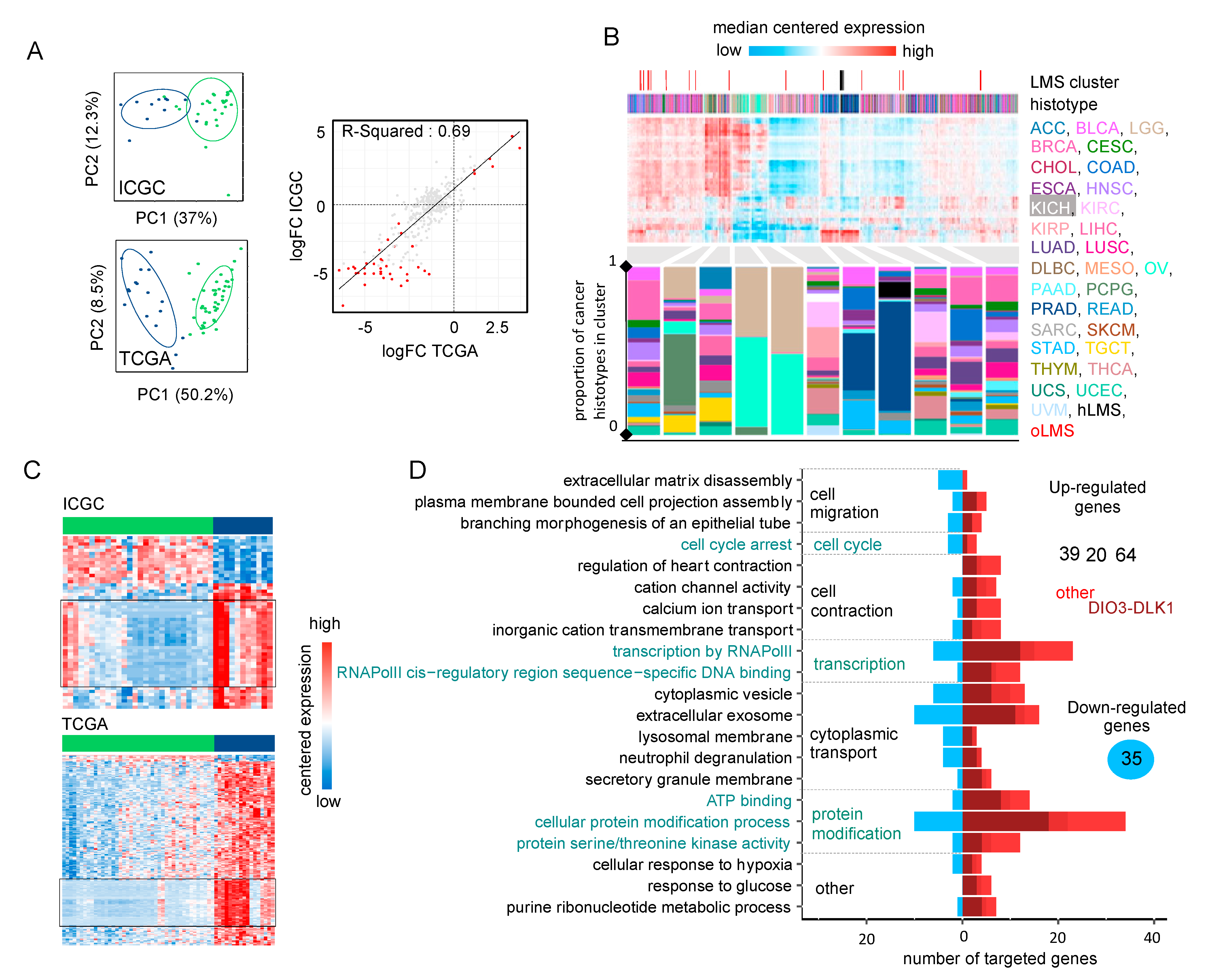
3.4. Gene Signature Identifies hLMS in Two Independent Cohorts
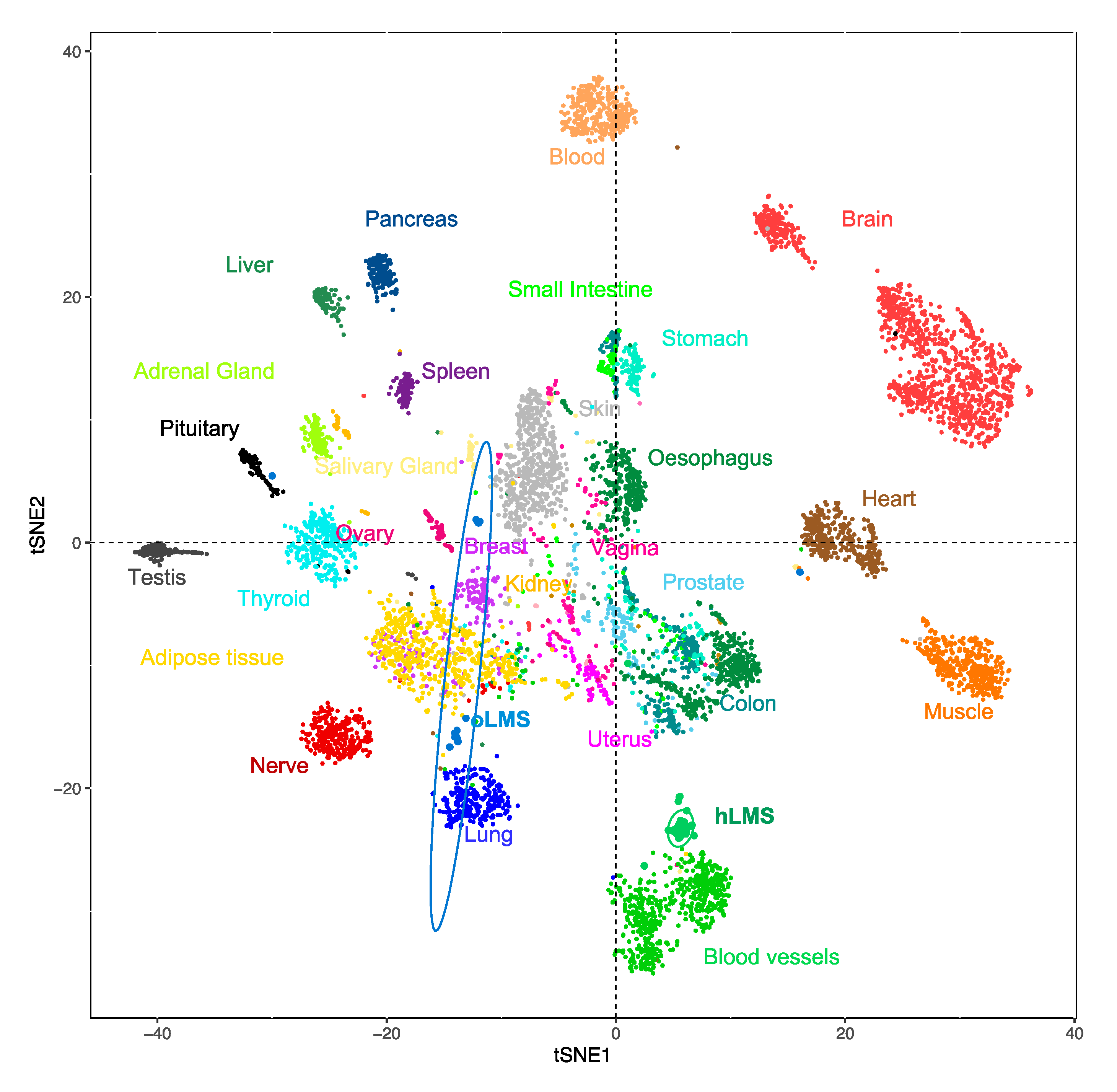
3.5. hLMS Originate from Vascular Smooth Muscle Cells
3.6. miRNAs Adopt Specific Behavior in hLMS
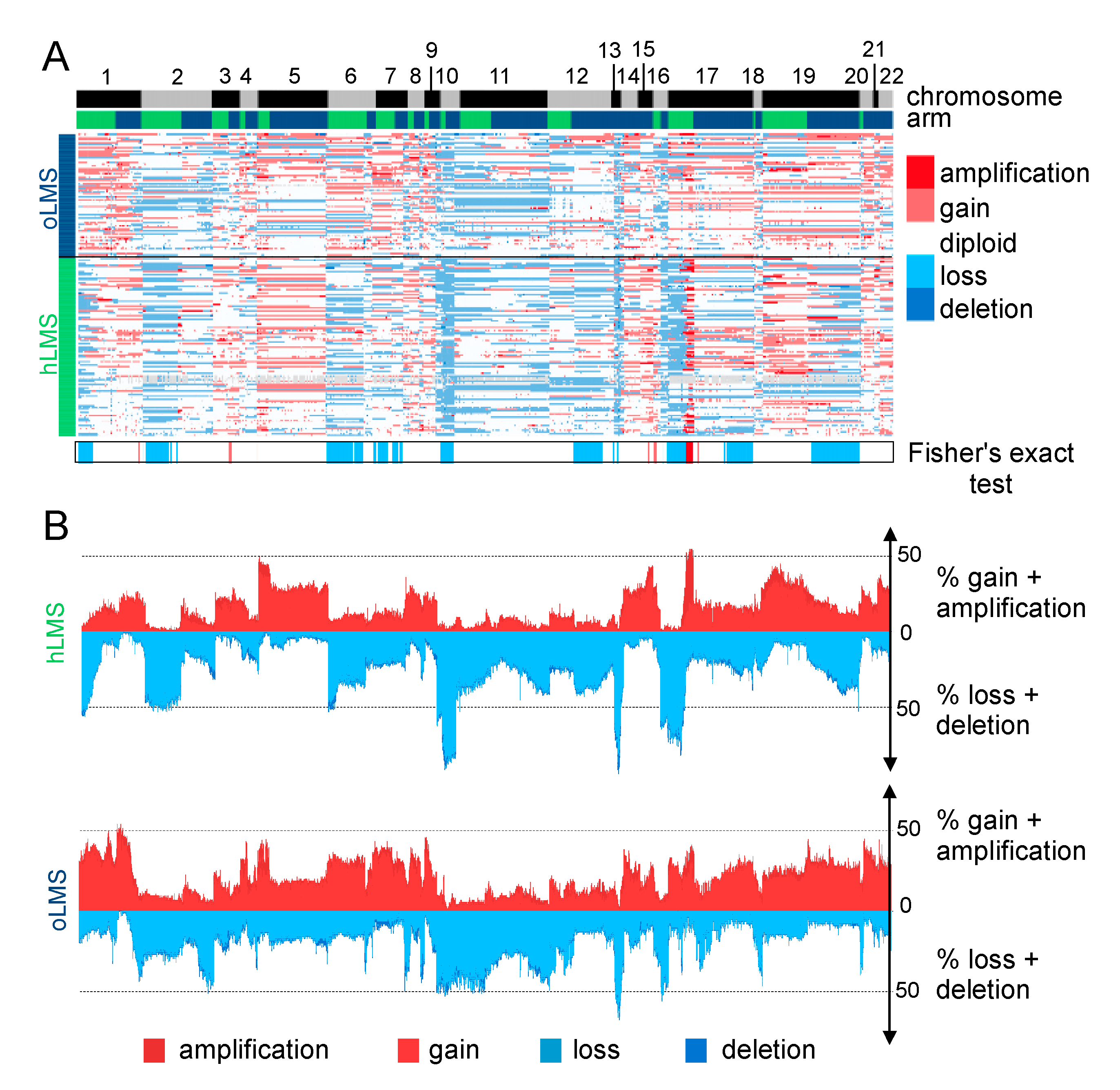
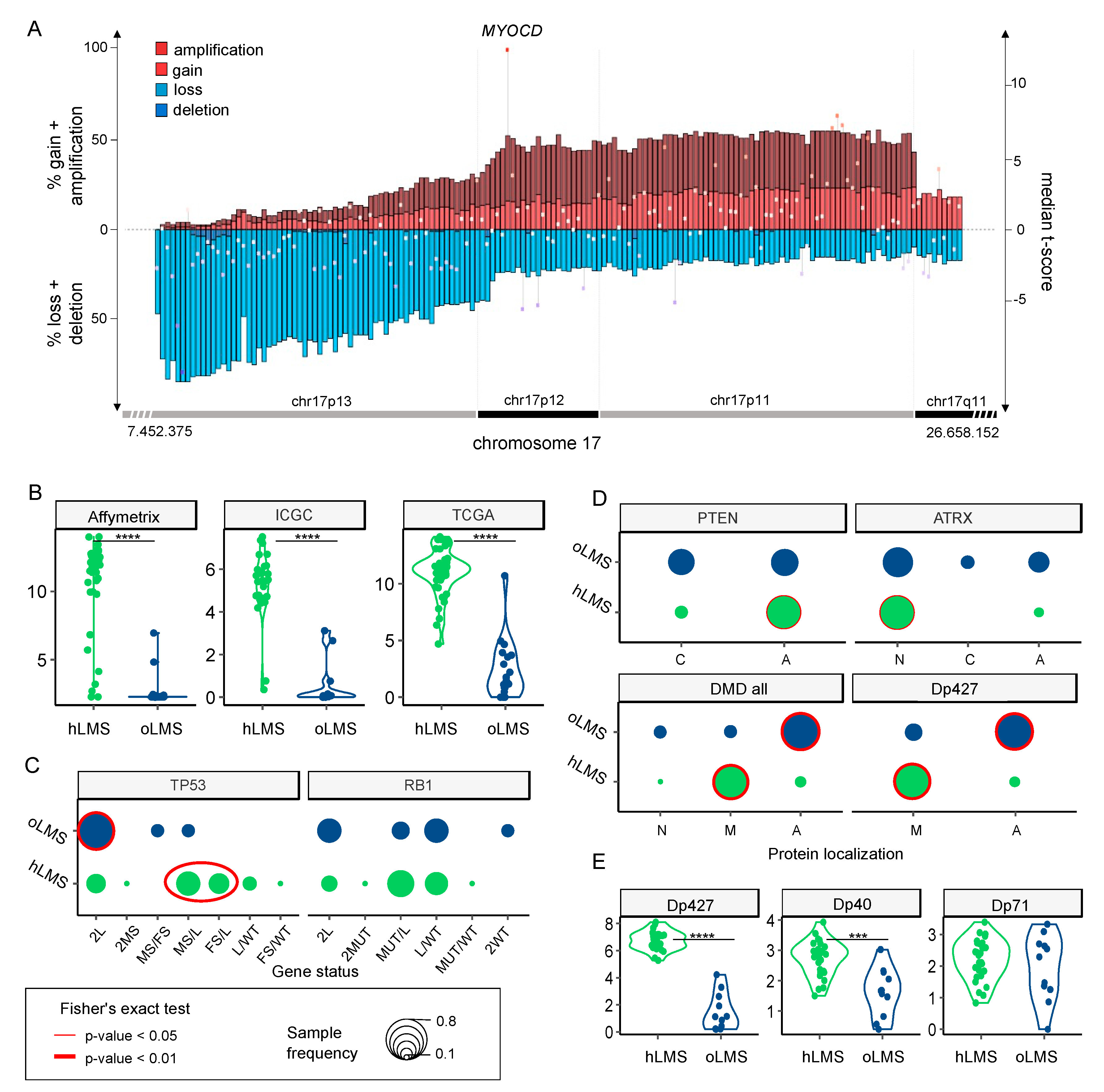
3.7. hLMS Show Recurrent and Specific Genomic Instability
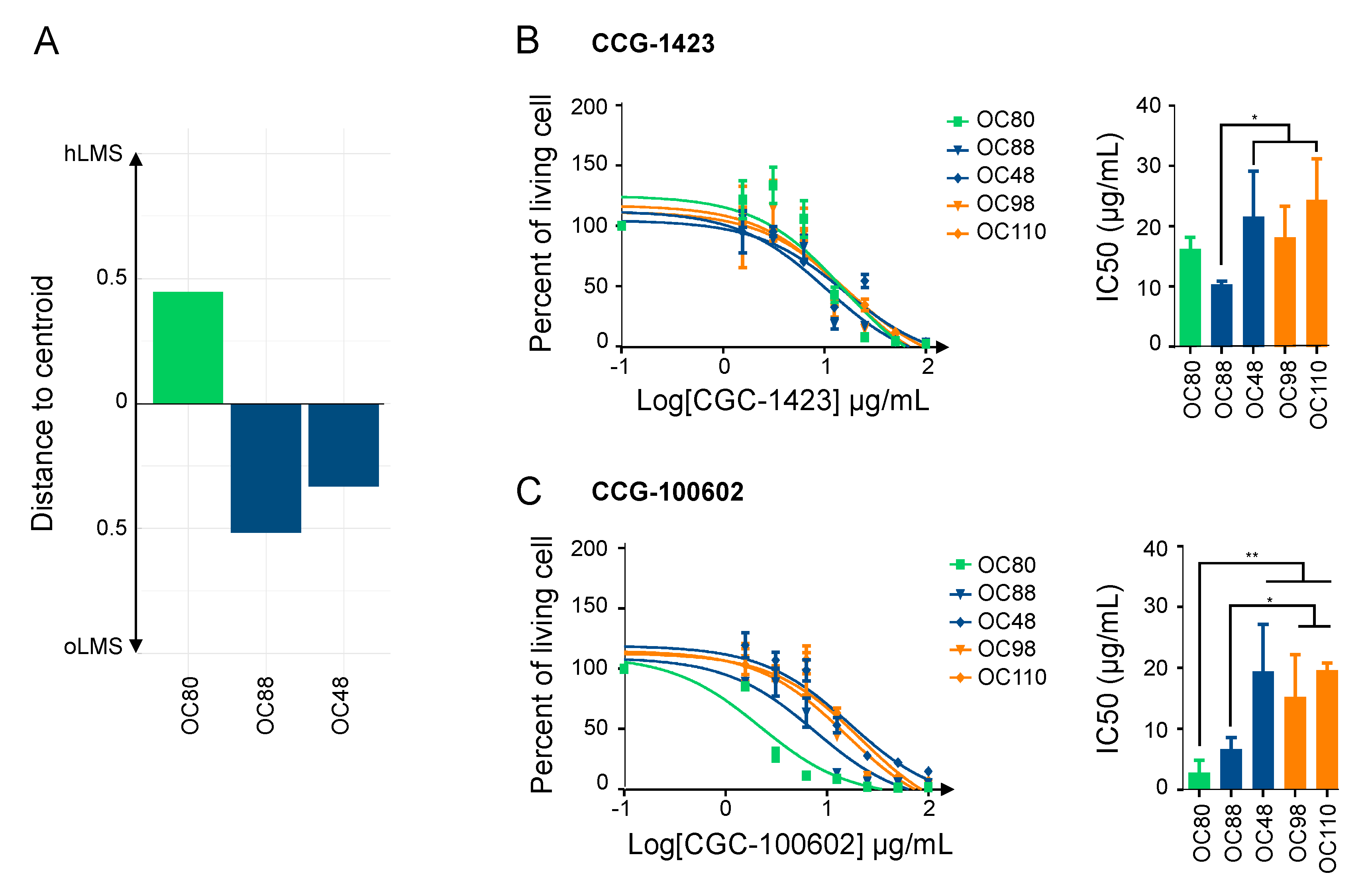
3.8. hLMS Can Be Targeted Specifically with an SRF/MYOCD Inhibitor
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| LMS | leiomyosacoma |
| hLMS | homogenous leiomyosarcoma |
| oLMS | other leiomyosarcoma |
| GIST | Gastrointestinal Stromal Tumor |
| PCC | Pearson’s Correlation coefficient |
| TF | transcription factor |
| ECM | extracellular matrix |
| DE | differentially expressed |
| vSMC | vascular smooth muscle cell |
| UPR | unfolded protein response |
References
- WHO Classification of Tumours Editorial Board. Soft Tissue and Bone Tumours, 5th ed.; WHO: Geneva, Switzerland, 2020; Volume 3, ISBN 978-92-832-4502-5. [Google Scholar]
- Judson, I.; Verweij, J.; Gelderblom, H.; Hartmann, J.T.; Schöffski, P.; Blay, J.-Y.; Kerst, J.M.; Sufliarsky, J.; Whelan, J.; Hohenberger, P.; et al. Doxorubicin Alone versus Intensified Doxorubicin plus Ifosfamide for First-Line Treatment of Advanced or Metastatic Soft-Tissue Sarcoma: A Randomised Controlled Phase 3 Trial. Lancet Oncol. 2014, 15, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Baird, K.; Davis, S.; Antonescu, C.R.; Harper, U.L.; Walker, R.L.; Chen, Y.; Glatfelter, A.A.; Duray, P.H.; Meltzer, P.S. Gene Expression Profiling of Human Sarcomas: Insights into Sarcoma Biology. Cancer Res. 2005, 65, 9226–9235. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.O.; West, R.B.; Linn, S.C.; Alter, O.; Knowling, M.A.; O’Connell, J.X.; Zhu, S.; Fero, M.; Sherlock, G.; Pollack, J.R.; et al. Molecular Characterisation of Soft Tissue Tumours: A Gene Expression Study. Lancet 2002, 359, 1301–1307. [Google Scholar] [CrossRef] [PubMed]
- Villacis, R.A.R.; Silveira, S.M.; Barros-Filho, M.C.; Marchi, F.A.; Domingues, M.A.C.; Scapulatempo-Neto, C.; Aguiar, S.; Lopes, A.; Cunha, I.W.; Rogatto, S.R. Gene Expression Profiling in Leiomyosarcomas and Undifferentiated Pleomorphic Sarcomas: SRC as a New Diagnostic Marker. PLoS ONE 2014, 9, e102281. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Jo, V.Y.; Mills, A.M.; Zhu, S.X.; Lee, C.-H.; Espinosa, I.; Nucci, M.R.; Varma, S.; Forgó, E.; Hastie, T.; et al. Clinically Relevant Molecular Subtypes in Leiomyosarcoma. Clin. Cancer Res. 2015, 21, 3501–3511. [Google Scholar] [CrossRef]
- Lee, Y.-F.; Roe, T.; Mangham, D.C.; Fisher, C.; Grimer, R.J.; Judson, I. Gene Expression Profiling Identifies Distinct Molecular Subgroups of Leiomyosarcoma with Clinical Relevance. Br. J. Cancer 2016, 115, 1000–1007. [Google Scholar] [CrossRef]
- Hemming, M.L.; Fan, C.; Raut, C.P.; Demetri, G.D.; Armstrong, S.A.; Sicinska, E.; George, S. Oncogenic Gene Expression Programs in Leiomyosarcoma and Characterization of Conventional, Inflammatory and Uterogenic Subtypes. Mol. Cancer Res. 2020, 18, 1302–1314. [Google Scholar] [CrossRef]
- Beck, A.H.; Lee, C.-H.; Witten, D.M.; Gleason, B.C.; Edris, B.; Espinosa, I.; Zhu, S.; Li, R.; Montgomery, K.D.; Marinelli, R.J.; et al. Discovery of Molecular Subtypes in Leiomyosarcoma through Integrative Molecular Profiling. Oncogene 2010, 29, 845–854. [Google Scholar] [CrossRef]
- Perot, G.; Derre, J.; Coindre, J.-M.; Tirode, F.; Lucchesi, C.; Mariani, O.; Gibault, L.; Guillou, L.; Terrier, P.; Aurias, A. Strong Smooth Muscle Differentiation Is Dependent on Myocardin Gene Amplification in Most Human Retroperitoneal Leiomyosarcomas. Cancer Res. 2009, 69, 2269–2278. [Google Scholar] [CrossRef]
- van der Graaf, W.T.; Blay, J.-Y.; Chawla, S.P.; Kim, D.-W.; Bui-Nguyen, B.; Casali, P.G.; Schöffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. Pazopanib for Metastatic Soft-Tissue Sarcoma (PALETTE): A Randomised, Double-Blind, Placebo-Controlled Phase 3 Trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Ben-Ami, E.; Barysauskas, C.M.; Solomon, S.; Tahlil, K.; Malley, R.; Hohos, M.; Polson, K.; Loucks, M.; Severgnini, M.; Patel, T.; et al. Immunotherapy with Single Agent Nivolumab for Advanced Leiomyosarcoma of the Uterus: Results of a Phase 2 Study: Nivolumab for Uterine Leiomyosarcoma. Cancer 2017, 123, 3285–3290. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network; Abeshouse, A.; Adebamowo, C.; Adebamowo, S.N.; Akbani, R.; Akeredolu, T.; Ally, A.; Anderson, M.L.; Anur, P.; Appelbaum, E.L.; et al. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017, 171, 950–965.e28. [Google Scholar] [CrossRef]
- Chudasama, P.; Mughal, S.S.; Sanders, M.A.; Hübschmann, D.; Chung, I.; Deeg, K.I.; Wong, S.-H.; Rabe, S.; Hlevnjak, M.; Zapatka, M.; et al. Integrative Genomic and Transcriptomic Analysis of Leiomyosarcoma. Nat. Commun. 2018, 9, 144. [Google Scholar] [CrossRef]
- Watson, S.; Perrin, V.; Guillemot, D.; Reynaud, S.; Coindre, J.-M.; Karanian, M.; Guinebretière, J.-M.; Freneaux, P.; Le Loarer, F.; Bouvet, M.; et al. Transcriptomic Definition of Molecular Subgroups of Small Round Cell Sarcomas: Molecular Classification of Sarcoma Subtypes. J. Pathol. 2018, 245, 29–40. [Google Scholar] [CrossRef]
- Chibon, F.; Lagarde, P.; Salas, S.; Pérot, G.; Brouste, V.; Tirode, F.; Lucchesi, C.; de Reynies, A.; Kauffmann, A.; Bui, B.; et al. Validated Prediction of Clinical Outcome in Sarcomas and Multiple Types of Cancer on the Basis of a Gene Expression Signature Related to Genome Complexity. Nat. Med. 2010, 16, 781–787. [Google Scholar] [CrossRef]
- Gibault, L.; Pérot, G.; Chibon, F.; Bonnin, S.; Lagarde, P.; Terrier, P.; Coindre, J.-M.; Aurias, A. New Insights in Sarcoma Oncogenesis: A Comprehensive Analysis of a Large Series of 160 Soft Tissue Sarcomas with Complex Genomics. J. Pathol. 2011, 223, 64–71. [Google Scholar] [CrossRef]
- Gibault, L.; Ferreira, C.; Pérot, G.; Audebourg, A.; Chibon, F.; Bonnin, S.; Lagarde, P.; Vacher-Lavenu, M.-C.; Terrier, P.; Coindre, J.-M.; et al. From PTEN Loss of Expression to RICTOR Role in Smooth Muscle Differentiation: Complex Involvement of the MTOR Pathway in Leiomyosarcomas and Pleomorphic Sarcomas. Mod. Pathol. 2012, 25, 197–211. [Google Scholar] [CrossRef]
- Lesluyes, T.; Pérot, G.; Largeau, M.R.; Brulard, C.; Lagarde, P.; Dapremont, V.; Lucchesi, C.; Neuville, A.; Terrier, P.; Vince-Ranchère, D.; et al. RNA Sequencing Validation of the Complexity INdex in SARComas Prognostic Signature. Eur. J. Cancer 2016, 57, 104–111. [Google Scholar] [CrossRef]
- Lagarde, P.; Perot, G.; Kauffmann, A.; Brulard, C.; Dapremont, V.; Hostein, I.; Neuville, A.; Wozniak, A.; Sciot, R.; Schoffski, P.; et al. Mitotic Checkpoints and Chromosome Instability Are Strong Predictors of Clinical Outcome in Gastrointestinal Stromal Tumors. Clin. Cancer Res. 2012, 18, 826–838. [Google Scholar] [CrossRef]
- Lagarde, P.; Przybyl, J.; Brulard, C.; Pérot, G.; Pierron, G.; Delattre, O.; Sciot, R.; Wozniak, A.; Schöffski, P.; Terrier, P.; et al. Chromosome Instability Accounts for Reverse Metastatic Outcomes of Pediatric and Adult Synovial Sarcomas. JCO 2013, 31, 608–615. [Google Scholar] [CrossRef]
- Fletcher, C.; Bridge, J.A.; Hogendoorn, P.; Mertens, F. WHO Classification of Tumours of Soft Tissue and Bone, 4th ed.; IARC Press: Lyon, France, 2013; Volume 5, ISBN 978-92-832-2434-1. [Google Scholar]
- Lagarde, P.; Brulard, C.; Pérot, G.; Mauduit, O.; Delespaul, L.; Neuville, A.; Stoeckle, E.; Le Guellec, S.; Rochaix, P.; Coindre, J.-M.; et al. Stable Instability of Sarcoma Cell Lines Genome Despite Intra-Tumoral Heterogeneity: A Genomic and Transcriptomic Study of Sarcoma Cell Lines. Austin J. Genet. Genom. Res. 2015, 2, 1014. [Google Scholar]
- McPherson, A.; Hormozdiari, F.; Zayed, A.; Giuliany, R.; Ha, G.; Sun, M.G.F.; Griffith, M.; Moussavi, A.H.; Senz, J.; Melnyk, N.; et al. DeFuse: An Algorithm for Gene Fusion Discovery in Tumor RNA-Seq Data. PLoS Comput. Biol. 2011, 7, e1001138. [Google Scholar] [CrossRef] [PubMed]
- Delespaul, L.; Lesluyes, T.; Pérot, G.; Brulard, C.; Lartigue, L.; Baud, J.; Lagarde, P.; Le Guellec, S.; Neuville, A.; Terrier, P.; et al. Recurrent TRIO Fusion in Nontranslocation-Related Sarcomas. Clin. Cancer Res. 2017, 23, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. MiRBase: Integrating MicroRNA Annotation and Deep-Sequencing Data. Nucleic Acids Res. 2011, 39, D152–D157. [Google Scholar] [CrossRef]
- Tam, S.; Tsao, M.-S.; McPherson, J.D. Optimization of MiRNA-Seq Data Preprocessing. Brief. Bioinform. 2015, 16, 950–963. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- García-Alcalde, F.; Okonechnikov, K.; Carbonell, J.; Cruz, L.M.; Götz, S.; Tarazona, S.; Dopazo, J.; Meyer, T.F.; Conesa, A. Qualimap: Evaluating next-Generation Sequencing Alignment Data. Bioinformatics 2012, 28, 2678–2679. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Joshi, N.; Fass, J. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files (Version 1.33); GitHub Repository. 2011. Available online: https://github.com/najoshi/sickle (accessed on 24 November 2022).
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Broad Institute. Picard Toolkit; Broad Institute, GitHub Repository: Cambridge, MA, USA, 2019. [Google Scholar]
- Li, H. A Statistical Framework for SNP Calling, Mutation Discovery, Association Mapping and Population Genetical Parameter Estimation from Sequencing Data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- The 1000 Genomes Project Consortium A Global Reference for Human Genetic Variation. Nature 2015, 526, 68–74. [CrossRef]
- Lesluyes, T.; Baud, J.; Pérot, G.; Charon-Barra, C.; You, A.; Valo, I.; Bazille, C.; Mishellany, F.; Leroux, A.; Renard-Oldrini, S.; et al. Genomic and Transcriptomic Comparison of Post-Radiation versus Sporadic Sarcomas. Mod. Pathol. 2019, 32, 1786–1794. [Google Scholar] [CrossRef]
- Klambauer, G.; Schwarzbauer, K.; Mayr, A.; Clevert, D.-A.; Mitterecker, A.; Bodenhofer, U.; Hochreiter, S.C. MOPS: Mixture of Poissons for Discovering Copy Number Variations in next-Generation Sequencing Data with a Low False Discovery Rate. Nucleic Acids Res. 2012, 40, e69. [Google Scholar] [CrossRef]
- Carlson, M. TxDb.Hsapiens.UCSC.Hg19.KnownGene: Annotation Package for TxDb Object(s); Bioconductor Repository. 2015. Available online: https://doi.org/10.18129/B9.bioc.TxDb.Hsapiens.UCSC.hg19.knownGene (accessed on 24 November 2022).
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New Capabilities and Interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Manie, E.; Vincent-Salomon, A.; Lehmann-Che, J.; Pierron, G.; Turpin, E.; Warcoin, M.; Gruel, N.; Lebigot, I.; Sastre-Garau, X.; Lidereau, R.; et al. High Frequency of TP53 Mutation in BRCA1 and Sporadic Basal-like Carcinomas but Not in BRCA1 Luminal Breast Tumors. Cancer Res. 2009, 69, 663–671. [Google Scholar] [CrossRef]
- Bolstad, B. PreprocessCore: A Collection of Pre-Processing Functions; Bioconductor Repository. 2019. Available online: https://doi.org/doi:10.18129/B9.bioc.preprocessCore (accessed on 24 November 2022).
- Csárdi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. InterJournal 2006, 1695, 1–9. [Google Scholar]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Kolde, R. Pheatmap: Pretty Heatmaps; CRAN Repository. 2019. Available online: https://CRAN.R-project.org/package=pheatmap (accessed on 24 November 2022).
- Vu, V.Q. Ggbiplot: A Ggplot2 Based Biplot; GitHub Repository. 2011; Available online: https://github.com/vqv/ggbiplot (accessed on 24 November 2022).
- Van der Maaten, L.; Hinton, G. Visualizing Non-Metric Similarities in Multiple Maps. Mach Learn 2012, 87, 33–55. [Google Scholar] [CrossRef]
- Marisa, L.; de Reyniès, A.; Duval, A.; Selves, J.; Gaub, M.P.; Vescovo, L.; Etienne-Grimaldi, M.-C.; Schiappa, R.; Guenot, D.; Ayadi, M.; et al. Gene Expression Classification of Colon Cancer into Molecular Subtypes: Characterization, Validation, and Prognostic Value. PLoS Med. 2013, 10, e1001453. [Google Scholar] [CrossRef] [PubMed]
- Scrucca, L.; Fop, M.; Murphy, T.B.; Raftery, A.E. Mclust 5: Clustering, Classification and Density Estimation Using Gaussian Finite Mixture Models. R J. 2016, 8, 289. [Google Scholar] [CrossRef] [PubMed]
- Therneau, T.M. A Package for Survival Analysis in R 2020; CRAN Repository. 2020. Available online: https://CRAN.R-project.org/package=survival (accessed on 24 November 2022).
- Kassambara, A.; Kosinski, M.; Biecek, P. Survminer: Drawing Survival Curves Using “Ggplot2”; CRAN Repository. 2019. Available online: https://CRAN.R-project.org/package=survminer (accessed on 24 November 2022).
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Vila-Casadesús, M.; Gironella, M.; Lozano, J.J. MiRComb: An R Package to Analyse MiRNA-MRNA Interactions. Examples across Five Digestive Cancers. PLoS ONE 2016, 11, e0151127. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-Responsive Genes Involved in Oxidative Phosphorylation Are Coordinately Downregulated in Human Diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Imrichová, H.; Hulselmans, G.; Kalender Atak, Z.; Potier, D.; Aerts, S. I-CisTarget 2015 Update: Generalized Cis-Regulatory Enrichment Analysis in Human, Mouse, and Fly. Nucleic Acids Res. 2015, 43, W57–W64. [Google Scholar] [CrossRef]
- Ayllon-Benitez, A.; Bourqui, R.; Thébault, P.; Mougin, F. GSAn: An Alternative to Enrichment Analysis for Annotating Gene Sets. NAR Genom. Bioinform. 2020, 2, lqaa017. [Google Scholar] [CrossRef]
- Xiao, F.; Zuo, Z.; Cai, G.; Kang, S.; Gao, X.; Li, T. MiRecords: An Integrated Resource for MicroRNA-Target Interactions. Nucleic Acids Res. 2009, 37, D105–D110. [Google Scholar] [CrossRef]
- Chou, C.-H.; Shrestha, S.; Yang, C.-D.; Chang, N.-W.; Lin, Y.-L.; Liao, K.-W.; Huang, W.-C.; Sun, T.-H.; Tu, S.-J.; Lee, W.-H.; et al. MiRTarBase Update 2018: A Resource for Experimentally Validated MicroRNA-Target Interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef]
- Blokzijl, F.; Janssen, R.; van Boxtel, R.; Cuppen, E. MutationalPatterns: Comprehensive Genome-Wide Analysis of Mutational Processes. Genome Med. 2018, 10, 33. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Darmusey, L.; Pérot, G.; Thébault, N.; Le Guellec, S.; Desplat, N.; Gaston, L.; Delespaul, L.; Lesluyes, T.; Darbo, E.; Gomez-Brouchet, A.; et al. ATRX Alteration Contributes to Tumor Growth and Immune Escape in Pleomorphic Sarcomas. Cancers 2021, 13, 2151. [Google Scholar] [CrossRef]
- Jiang, Y.; Yin, H.; Zheng, X.-L. MicroRNA-1 Inhibits Myocardin-Induced Contractility of Human Vascular Smooth Muscle Cells. J. Cell. Physiol. 2010, 225, 506–511. [Google Scholar] [CrossRef]
- Cordes, K.R.; Sheehy, N.T.; White, M.P.; Berry, E.C.; Morton, S.U.; Muth, A.N.; Lee, T.-H.; Miano, J.M.; Ivey, K.N.; Srivastava, D. MiR-145 and MiR-143 Regulate Smooth Muscle Cell Fate and Plasticity. Nature 2009, 460, 705–710. [Google Scholar] [CrossRef]
- Zhou, C.; Chen, Y.; Kang, W.; Lv, H.; Fang, Z.; Yan, F.; Li, L.; Zhang, W.; Shi, J. Mir-455-3p-1 Represses FGF7 Expression to Inhibit Pulmonary Arterial Hypertension through Inhibiting the RAS/ERK Signaling Pathway. J. Mol. Cell. Cardiol. 2019, 130, 23–35. [Google Scholar] [CrossRef]
- Wang, D.; Atanasov, A.G. The MicroRNAs Regulating Vascular Smooth Muscle Cell Proliferation: A Minireview. Int. J. Mol. Sci. 2019, 20, 324. [Google Scholar] [CrossRef]
- Xin, M.; Small, E.M.; Sutherland, L.B.; Qi, X.; McAnally, J.; Plato, C.F.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. MicroRNAs MiR-143 and MiR-145 Modulate Cytoskeletal Dynamics and Responsiveness of Smooth Muscle Cells to Injury. Genes Dev. 2009, 23, 2166–2178. [Google Scholar] [CrossRef]
- Horita, H.; Wysoczynski, C.L.; Walker, L.A.; Moulton, K.S.; Li, M.; Ostriker, A.; Tucker, R.; McKinsey, T.A.; Churchill, M.E.A.; Nemenoff, R.A.; et al. Nuclear PTEN Functions as an Essential Regulator of SRF-Dependent Transcription to Control Smooth Muscle Differentiation. Nat. Commun. 2016, 7, 10830. [Google Scholar] [CrossRef]
- Evelyn, C.R.; Wade, S.M.; Wang, Q.; Wu, M.; Iñiguez-Lluhí, J.A.; Merajver, S.D.; Neubig, R.R. CCG-1423: A Small-Molecule Inhibitor of RhoA Transcriptional Signaling. Mol. Cancer Ther. 2007, 6, 2249–2260. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Lee, J.-J.; Stoll, S.; Ma, B.; Costa, K.D.; Qiu, H. Rho Kinase Regulates Aortic Vascular Smooth Muscle Cell Stiffness Via Actin/SRF/Myocardin in Hypertension. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 44, 701. [Google Scholar] [CrossRef] [PubMed]
- Lartigue, L.; Merle, C.; Lagarde, P.; Delespaul, L.; Lesluyes, T.; Le Guellec, S.; Pérot, G.; Leroy, L.; Coindre, J.-M.; Chibon, F. Genome Remodeling upon Mesenchymal Tumor Cell Fusion Contributes to Tumor Progression and Metastatic Spread. Oncogene 2020, 39, 4198–4211. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.K.; Chae, S.-W.; Kim, H.-R.; Chae, H.J. Endoplasmic Reticulum Stress and Cancer. J. Cancer Prev. 2014, 19, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Miano, J. Serum Response Factor: Toggling between Disparate Programs of Gene Expression. J. Mol. Cell. Cardiol. 2003, 35, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Treisman, R. Identification of a Protein-Binding Site That Mediates Transcriptional Response of the c-Fos Gene to Serum Factors. Cell 1986, 46, 567–574. [Google Scholar] [CrossRef]
- Miano, J.M. Myocardin in Biology and Disease. J. Biomed. Res. 2015, 29, 3–19. [Google Scholar] [CrossRef]
- Turczyńska, K.M.; Swärd, K.; Hien, T.T.; Wohlfahrt, J.; Mattisson, I.Y.; Ekman, M.; Nilsson, J.; Sjögren, J.; Murugesan, V.; Hultgårdh-Nilsson, A.; et al. Regulation of Smooth Muscle Dystrophin and Synaptopodin 2 Expression by Actin Polymerization and Vascular Injury. Arter. Thromb. Vasc. Biol. 2015, 35, 1489–1497. [Google Scholar] [CrossRef]
- Vacante, F.; Denby, L.; Sluimer, J.C.; Baker, A.H. The Function of MiR-143, MiR-145 and the MiR-143 Host Gene in Cardiovascular Development and Disease. Vasc. Pharmacol. 2019, 112, 24–30. [Google Scholar] [CrossRef]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular Regulation of Vascular Smooth Muscle Cell Differentiation in Development and Disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef]
- Merlet, E.; Atassi, F.; Motiani, R.K.; Mougenot, N.; Jacquet, A.; Nadaud, S.; Capiod, T.; Trebak, M.; Lompré, A.-M.; Marchand, A. MiR-424/322 Regulates Vascular Smooth Muscle Cell Phenotype and Neointimal Formation in the Rat. Cardiovasc. Res. 2013, 98, 458–468. [Google Scholar] [CrossRef]
- Bi, R.; Ding, F.; He, Y.; Jiang, L.; Jiang, Z.; Mei, J.; Liu, H. MiR-503 Inhibits Platelet-Derived Growth Factor-Induced Human Aortic Vascular Smooth Muscle Cell Proliferation and Migration through Targeting the Insulin Receptor. Biomed. Pharmacother. 2016, 84, 1711–1716. [Google Scholar] [CrossRef]
- Geraldo, M.V.; Nakaya, H.I.; Kimura, E.T. Down-Regulation of 14q32-Encoded MiRNAs and Tumor Suppressor Role for MiR-654-3p in Papillary Thyroid Cancer. Oncotarget 2017, 8, 9597–9607. [Google Scholar] [CrossRef]
- Benetatos, L.; Hatzimichael, E.; Londin, E.; Vartholomatos, G.; Loher, P.; Rigoutsos, I.; Briasoulis, E. The MicroRNAs within the DLK1-DIO3 Genomic Region: Involvement in Disease Pathogenesis. Cell. Mol. Life Sci. 2013, 70, 795–814. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Affymetrix | ICGC + TCGA | ||||||
|---|---|---|---|---|---|---|---|
| Feature | Test | hLMS | oLMS | p-Value | hLMS | oLMS | p-Value |
| Differentiation (%) | Well (vs. Poor) | 88 | 24 | 3.87 × 10−9 (+) | 84 | 41 | 1.83 × 10−5 (+) |
| Grade (%) | Low (vs. High) | 58 | 24 | 6.51 × 10−4 (+) | |||
| Sex (%) | F (vs. M) | 76 | 48 | 0.003 (+) | 68 | 41 | 0.007 (+) |
| Location (%) | Internal trunk (vs. other) | 60 | 7 | 8.48 × 10−9 (+) | 82 | 27 | 1.53 × 10−7 (+) |
| Mitotic counts (median) | Ranks | 17 | 24.5 | 0.009 (−) | 11 | 35 | 0.0006 (−) |
| Gene expression variance (median) | Ranks | 0.7 | 1 | 2.90 × 10−13 (−) | ICGC | ||
| 0.25 | 0.36 | <2.2 × 10−16 (−) | |||||
| TCGA | |||||||
| 0.45 | 0.9 | <2.2 × 10−16 (−) | |||||
| Group | Alterations | TP53 | RB1 | PTEN | ATRX | DMD |
|---|---|---|---|---|---|---|
| hLMS | mutation | 60.7 (17) | 21.4 (6) | 0 (0) | 7.1 (2) | 3.6 (2) |
| oLMS | 18.2 (2) | 9 (1) | 0 (0) | 27.3 (3) | 9 (1) | |
| all | 48.7 (19) | 17.9 (7) | 0 (0) | 12.8 (5) | 5.1 (3) | |
| hLMS | SV | 25 (7) | 35.7 (10) | 3.6 (1) | 7.1 (2) | 14.3 (4) |
| oLMS | 36.4 (4) | 36.4 (4) | 0 (0) | 0(0) | 36.4 (4) | |
| all | 28.2 (11) | 35.9 (14) | 2.6 (1) | 5.1 (2) | 20.5 (8) | |
| hLMS | loss | 89.3 (25) | 92.9 (26) | 82.1 (23) | 7.1 (2) | 3.6 (1) |
| oLMS | 90.9 (10) | 81.8 (9) | 81.8 (9) | 18.1 (2) | 0 (0) | |
| all | 89.7 (35) | 89.7 (35) | 82 (32) | 10.2 (4) | 2.6 (1) | |
| hLMS | total | 100 (28) | 100 (28) | 82.1 (23) | 21.4 (6) | 17.8 (5) |
| oLMS | 100 (11) | 90.9 (10) | 81.8 (9) | 45.5 (5) | 45.5 (5) | |
| all | 100 (39) | 97.4 (38) | 82 (32) | 28.2 (11) | 25.6 (10) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Darbo, E.; Pérot, G.; Darmusey, L.; Le Guellec, S.; Leroy, L.; Gaston, L.; Desplat, N.; Thébault, N.; Merle, C.; Rochaix, P.; et al. Distinct Cellular Origins and Differentiation Process Account for Distinct Oncogenic and Clinical Behaviors of Leiomyosarcomas. Cancers 2023, 15, 534. https://doi.org/10.3390/cancers15020534
Darbo E, Pérot G, Darmusey L, Le Guellec S, Leroy L, Gaston L, Desplat N, Thébault N, Merle C, Rochaix P, et al. Distinct Cellular Origins and Differentiation Process Account for Distinct Oncogenic and Clinical Behaviors of Leiomyosarcomas. Cancers. 2023; 15(2):534. https://doi.org/10.3390/cancers15020534
Chicago/Turabian StyleDarbo, Elodie, Gaëlle Pérot, Lucie Darmusey, Sophie Le Guellec, Laura Leroy, Laëtitia Gaston, Nelly Desplat, Noémie Thébault, Candice Merle, Philippe Rochaix, and et al. 2023. "Distinct Cellular Origins and Differentiation Process Account for Distinct Oncogenic and Clinical Behaviors of Leiomyosarcomas" Cancers 15, no. 2: 534. https://doi.org/10.3390/cancers15020534
APA StyleDarbo, E., Pérot, G., Darmusey, L., Le Guellec, S., Leroy, L., Gaston, L., Desplat, N., Thébault, N., Merle, C., Rochaix, P., Valentin, T., Ferron, G., Chevreau, C., Bui, B., Stoeckle, E., Ranchere-Vince, D., Méeus, P., Terrier, P., Piperno-Neumann, S., ... Chibon, F. (2023). Distinct Cellular Origins and Differentiation Process Account for Distinct Oncogenic and Clinical Behaviors of Leiomyosarcomas. Cancers, 15(2), 534. https://doi.org/10.3390/cancers15020534