
A Narrative Review on CD44’s Role in Glioblastoma Invasion, Proliferation, and Tumor Recurrence


 ,
,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
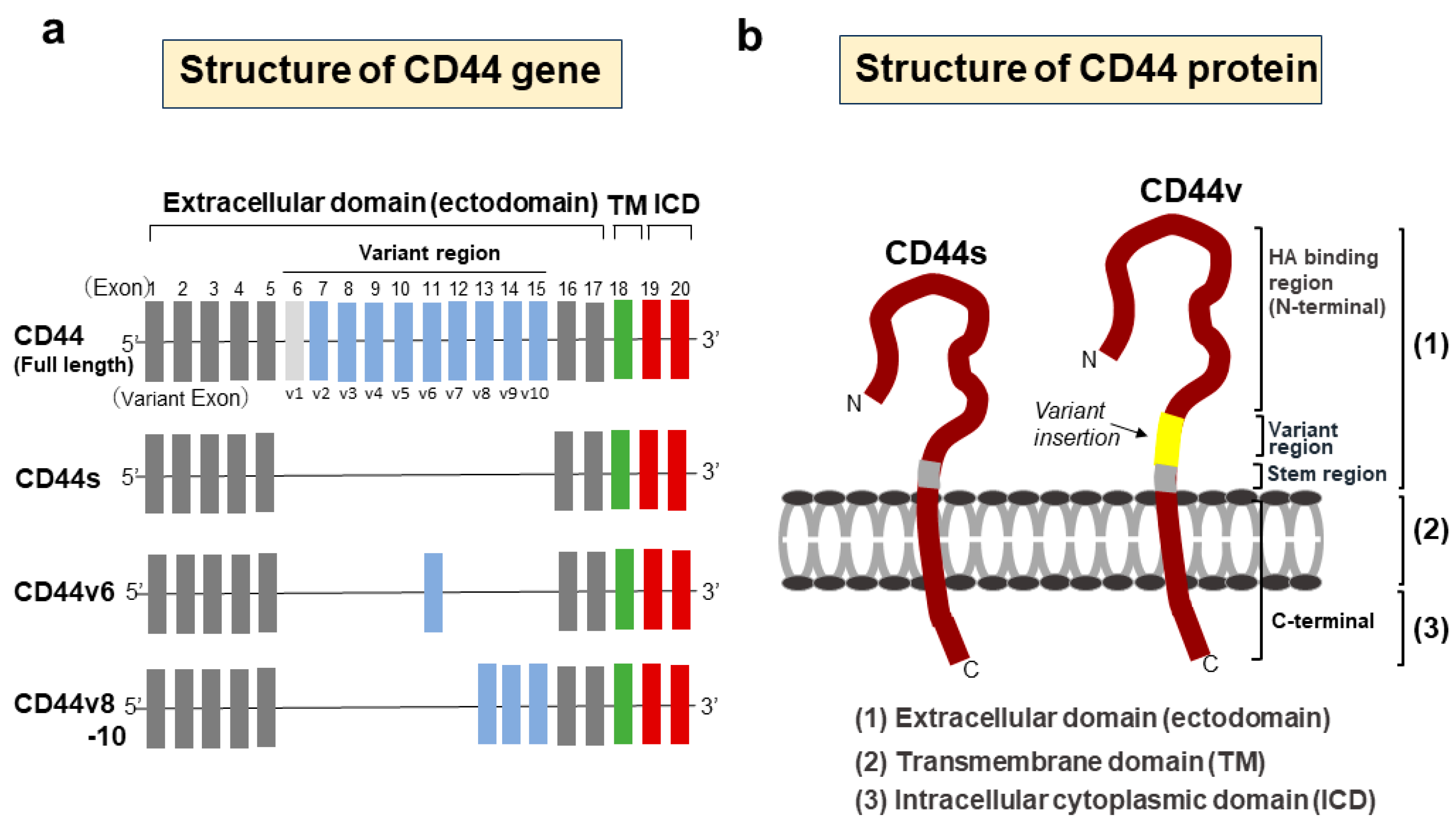
2. Structure and Function of CD44
3. Expression of CD44 in GBM
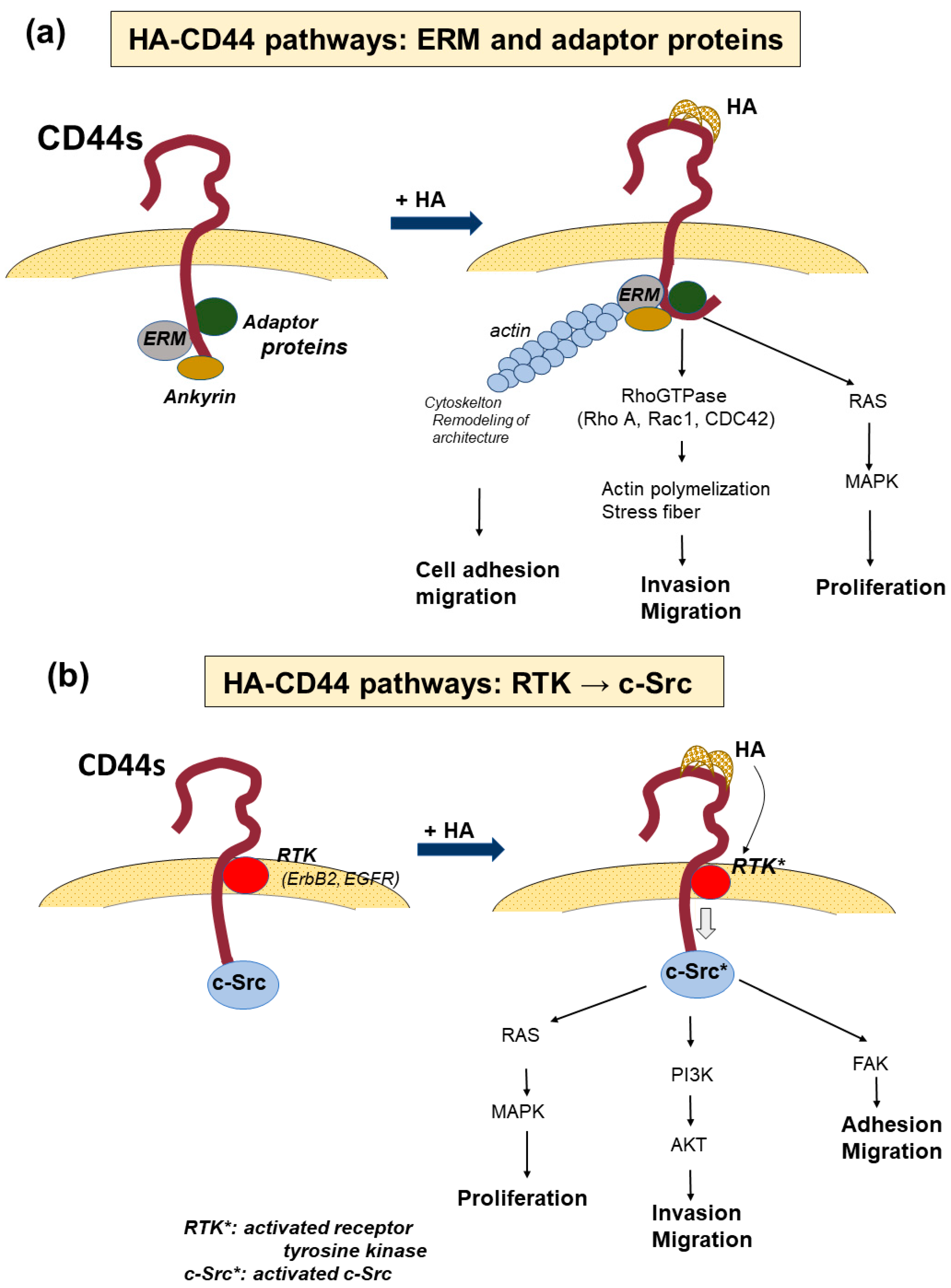
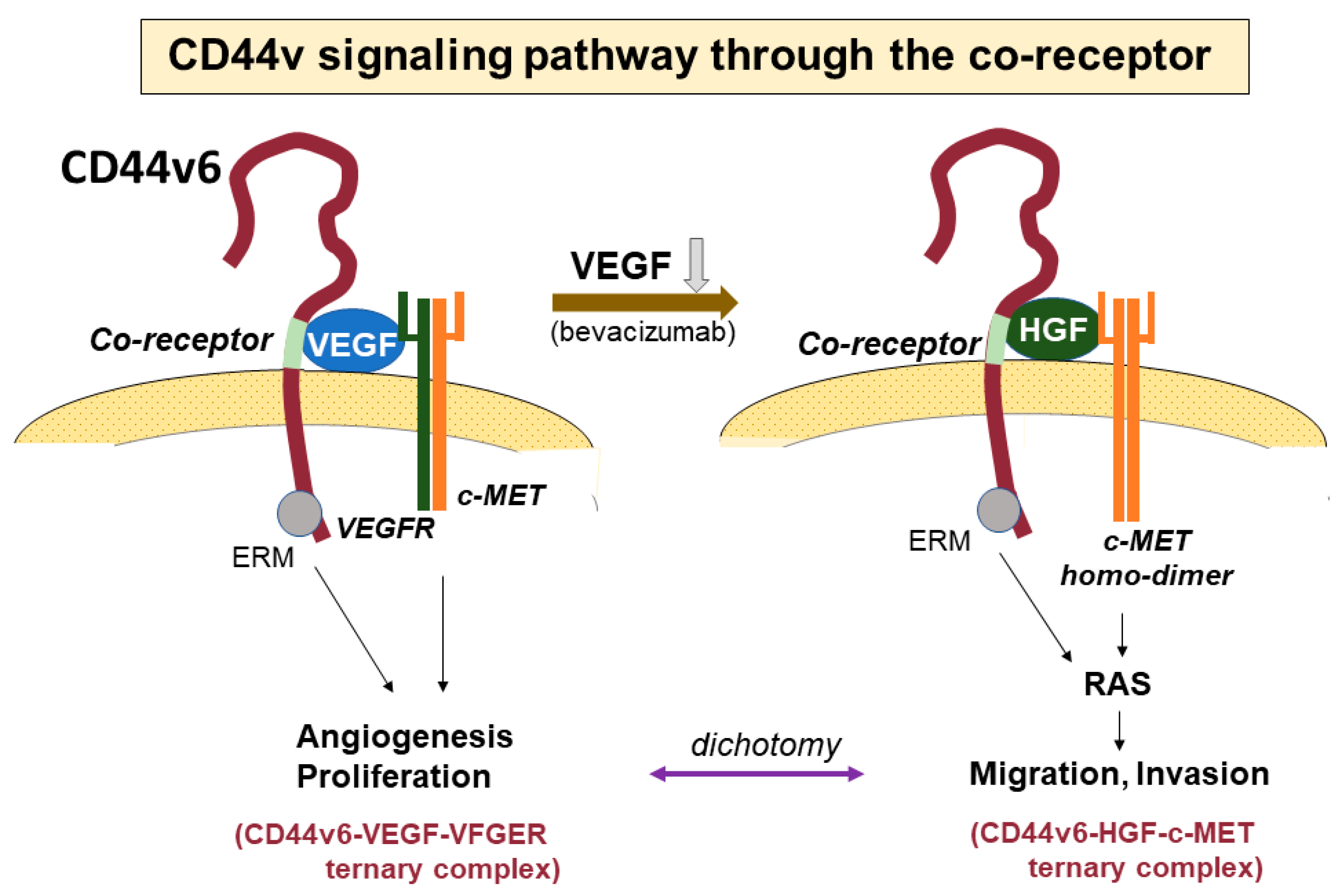
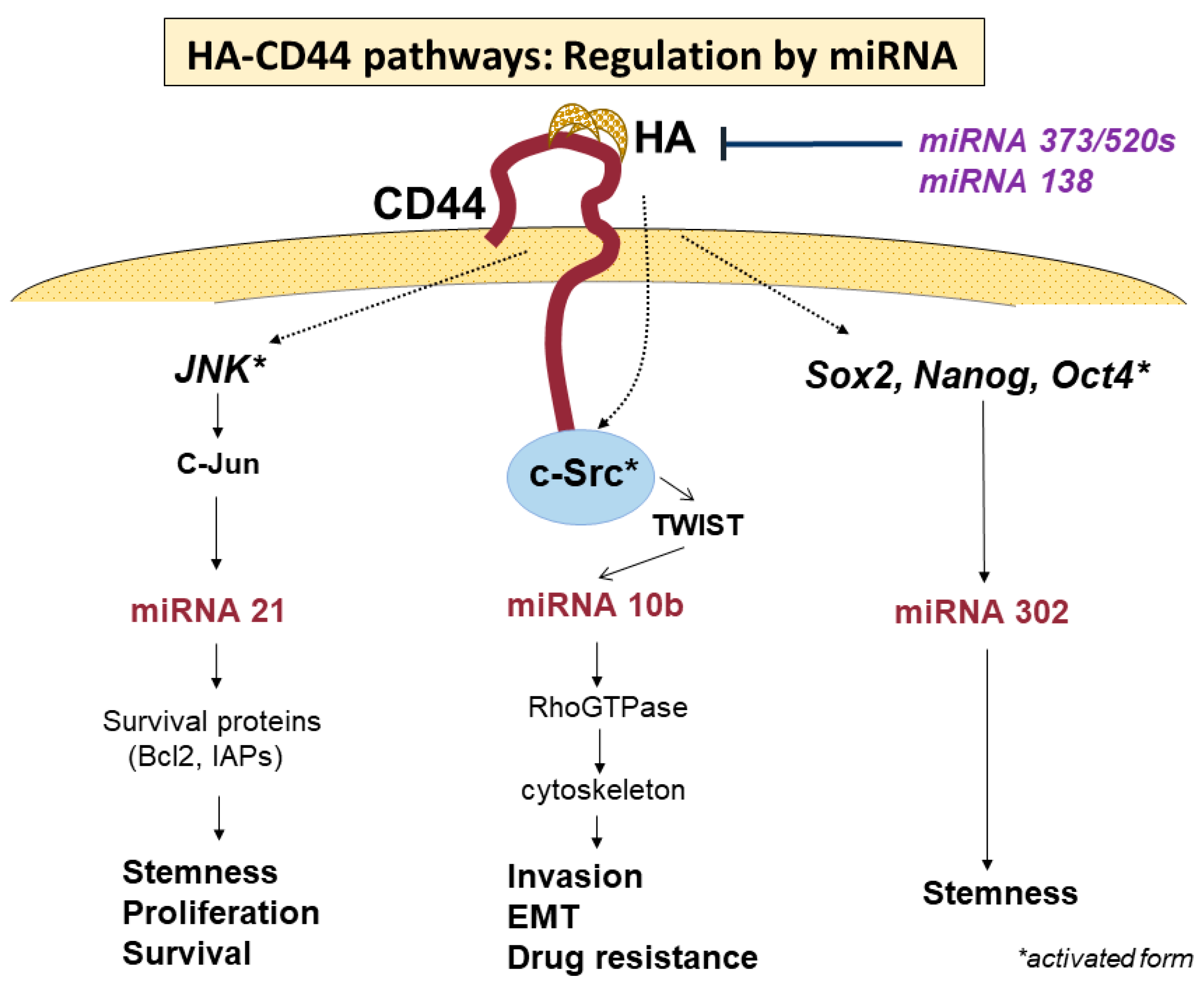
4. Signaling Pathways via CD44-HA Interactions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| microRNA | Regulation | Activated or Inhibitted Cellular Function of CD44 | References |
|---|---|---|---|
| miR-21 | positive | Activation: Proliferation, stemness, invasion, chemotherapy resistant | [76,77,78,80,81,82] |
| Inhibition: senescence, apoptosis | [83] | ||
| miR-10b | positive | Activation: Invasion, migration, EMT, drug resistance | [61,84,85,86,93] |
| miR-302 | positive | Activation: stemness | [87,88,89,90,92] |
| miR-373 | negative | Inhibition: proliferation, invasion | [94] |
| miR-520s | |||
| miR-138 | negative | Inhibition: proliferation, cell cycle, migration, wound-healing ability | [95] |
5. CD44-Promoted Tumor Invasion in GSCs
6. CD44-Promoted Tumor Proliferation in GBM (GSCs)
7. Roles of CD44 in Phenotypic Transition in GBM Progression
8. Molecular Mechanism Underlying GBM Recurrence: From the Perspective of the Dual Activity of CD44: Invasion and Proliferation
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Yu, T.S.; Mckay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Gallagher, J.; Myers, J.T.; Li, M.; Vasanji, A.; McLendon, R.E.; Hjelmeland, A.B.; Huang, A.Y.; Rich, J.N. Direct in vivo evidence for tumor propagation by glioblastoma cancer stem cells. PLoS ONE 2011, 6, e24807. [Google Scholar] [CrossRef] [PubMed]
- Schonberg, D.L.; Lubelski, D.; Miller, T.E.; Rich, J.N. Brain tumor stem cells: Molecular characteristics and their impact on therapy. Mol. Aspects Med. 2014, 39, 82–101. [Google Scholar] [CrossRef] [PubMed]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [PubMed]
- Beier, D.; Scriefer, B.; Brawanski, K.; Hau, P.; Weis, J.; Schulz, J.B.; Beier, C.P. Efficacy of clinically relevant temozolomide dosing schemes in glioblastoma cancer stem cell lines. J. Neuro Oncol. 2012, 109, 45–52. [Google Scholar] [CrossRef]
- Codrici, E.; Enciu, A.M.; Popescu, I.D.; Mihai, S.; Tanase, C. Glioma stem cells and their microenvironments: Providers of challenging therapeutic targets. Stem Cells Int. 2016, 2016, 5728438. [Google Scholar] [CrossRef]
- Schiffer, D.; Annovazzi, L.; Casalone, C.; Corona, C.; Mellai, M. Glioblastoma: Microenvironment and niche concept. Cancers 2019, 11, 5. [Google Scholar] [CrossRef]
- Chaudhry, G.E.; Akim, A.; Naveed Zafar, M.; Safdar, N.; Sung, Y.Y.; Muhammad, T.S.T. Understanding hyaluronan receptor (CD44) interaction, HA-CD44 activated potential targets in cancer therapeutics. Adv. Pharm. Bull. 2021, 11, 426–438. [Google Scholar] [CrossRef]
- Ponta, H.; Sherman, L.; Herrich, P.A. CD44 from adhesion molecules to signaling regulators. Nat. Rev. Mol. Cell Biol. 2003, 4, 33–45. [Google Scholar] [CrossRef]
- Naor, D.; Nedvetzkis, S.; Golan, I.; Melnik, L.; Faitelson, Y. CD44 in cancer. Crit. Rev. Clin. Lab. Sci. 2002, 39, 527–579. [Google Scholar] [CrossRef] [PubMed]
- Hirata, K.; Suzuki, H.; Imaeda, H.; Matsuzaki, J.; Tsugawa, H.; Nagano, O.; Asakura, K.; Saya, H.; Hibi, T. CD44 variant 9 expression in primary early gastric cancer as a predictive marker for recurrence. Br. J. Cancer 2013, 109, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Yae, T.; Tsuchihashi, K.; Ishimoto, T.; Motohara, T.; Yoshikawa, M.; Yoshida, G.J.; Wada, T.; Masuko, T.; Mogushi, K.; Tanaka, H.; et al. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat. Commun. 2012, 3, 883. [Google Scholar] [CrossRef]
- Ozawa, M.; Ichikawa, Y.; Zheng, Y.W.; Oshima, T.; Miyata, H.; Nakazawa, K.; Guan, H.B.; Shiozawa, M.; Akaike, M.; Watanabe, K.; et al. Prognostic significance of CD44 variant 2 upregulation in colorectal cancer. Br. J. Cancer 2014, 111, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Pietras, A.; Katz, A.M.; Ekström, E.J.; Wee, B.; Halliday, J.J.; Pitter, K.L.; Werbeck, J.L.; Amankulor, N.M.; Huse, J.T.; Holland, E.C. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell 2014, 14, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Sahgal, A.; Sanghera, P.; Tsao, M.N.; Davey, P.; Lam, K.; Symons, S.; Aviv, R.; Perry, J.R. Glioblastoma: Patterns of recurrence and efficacy of salvage treatments. Can. J. Neurol. Sci. 2011, 38, 621–625. [Google Scholar] [CrossRef]
- Giese, A.; Kucinski, T.; Knopp, U.; Goldbrunner, R.; Hamel, W.; Mehdorn, H.M.; Tonn, J.C.; Hilt, D.; Westphal, M. Patterns of recurrence following local chemotherapy with biodegradable carmustine (BCNU) implants in patients with glioblastoma. J. Neuro Oncol. 2004, 66, 351–360. [Google Scholar] [CrossRef]
- Hatzikirou, H.; Basanta, D.; Simon, M.; Schaller, K.; Deutsch, A. ‘Go or grow’: The key to the emergence of invasion in tumour progression? Math. Med. Biol. 2012, 29, 49–65. [Google Scholar] [CrossRef]
- Giese, A.; Loo, M.A.; Tran, N.; Haskett, D.; Coons, S.W.; Berens, M.E. Dichotomy of astrocytoma migration and proliferation. Int. J. Cancer 1996, 67, 275–282. [Google Scholar] [CrossRef]
- Herrich, P.; Morrison, H.; Sleeman, J.; Orian-Rousseau, V.; Konig, H.; Weg-Remers, S.; Ponta, H. CD44 acts both as a growth- and invasiveness-promoting molecule and as a tumor-suppressing cofactor. Ann. N. Y. Acad. Sci. 2000, 910, 106–118. [Google Scholar] [CrossRef]
- Lesley, J.; Hyman, R. CD44 structure and function. Front. Biosci. 1998, 3, d616–d630. [Google Scholar] [CrossRef] [PubMed]
- Prochazka, L.; Tesarik, R.; Turanek, J. Regulation of alternative splicing of CD44 in cancer. Cell Signal. 2014, 26, 2234–2239. [Google Scholar] [CrossRef]
- Iczkowski, K.A. Cell adhesion molecule CD44: Its functional roles in prostate cancer. Am. J. Transl. Res. 2010, 3, 1–7. [Google Scholar] [PubMed]
- Bennett, K.L.; Jackson, D.G.; Simon, J.C.; Tanczos, E.; Peach, R.; Modrell, B.; Stamenkovic, I.; Plowman, G.; Aruffo, A. CD44 isoforms containing exon V3 are responsible for the presentation of heparin-binding growth factor. J. Cell Biol. 1995, 128, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Tremmel, M.; Matzke, A.; Albrecht, I.; Laib, A.M.; Olaku, V.; Ballmer-Hofer, K.; Christofori, G.; Héroult, M.; Augustin, H.G.; Ponta, H.; et al. A CD44v6 peptide reveals a role of CD44 in VEGFR-2 signaling and angiogenesis. Blood 2009, 114, 5236–5244. [Google Scholar] [CrossRef]
- Medina, M.; Dotti, C.G. RIPped out by presenilin-dependent γ-secretase. Cell Signal. 2003, 15, 829–841. [Google Scholar] [CrossRef]
- Murakami, D.; Okamoto, I.; Nagano, O.; Kawano, Y.; Tomita, T.; Iwatsubo, T.; De Strooper, B.; Yumoto, E.; Saya, H. Presenilin-dependent gamma-secretase activity mediates the intramembranous cleavage of CD44. Oncogene 2003, 22, 1511–1516. [Google Scholar] [CrossRef]
- Nagano, O.; Saya, H. Mechanism and biological significance of CD44 cleavage. Cancer Sci. 2004, 95, 830–835. [Google Scholar] [CrossRef]
- Nagano, O.; Murakami, D.; Hartmann, D.; De Strooper, B.; Saftig, P.; Iwatsubo, T.; Nakajima, M.; Shinohara, M.; Saya, H. Cell-matrix interaction via CD44 is independently regulated by different metalloproteinases activated in response to extracellular Ca2+ influx and PKC activation. J. Cell Biol. 2004, 165, 893–902. [Google Scholar] [CrossRef]
- Senbanjo, L.T.; Aljohani, H.; Majumdar, S.; Chellaiah, M.A. Characterization of CD44 intracellular domain interaction with RUNX2 in PC3 human prostate cancer cells. Cell. Commun. Signal. 2019, 17, 80. [Google Scholar] [CrossRef]
- Miletti-Gonzaalez, K.E.; Murphy, K.; Kumaran, M.N.; Ravindranath, A.K.; Wernyj, R.P.; Kaur, S.; Miles, G.D.; Lim, E.; Chan, R.; Chekmareva, M.; et al. Identification of function for CD44 intracytoplasmic domain (CD44-ICD): Modulation of matrix metalloproteinase 9 (MMP-9) transcription via novel promoter response element. J. Biol. Chem. 2012, 287, 18995–19007. [Google Scholar] [CrossRef] [PubMed]
- Yohansson, E.; Grassi, E.S.; Pantazopoulou, V.; Tong, B.; Lindgren, D.; Berg, T.J.; Pietras, E.J.; Axelson, H.; Pietras, A. CD44 Interacts with HIF-2α to modulate the hypoxic phenotype of perinecrotic and perivascular glioma cells. Cell Rep. 2017, 20, 1641–1653. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Nishida, N.; Umemoto, R.; Suzuki, M.; Takeda, M.; Terasawa, H.; Kitayama, J.; Matsumoto, M.; Hayasaka, H.; Miyasaka, M.; et al. Two-state conformations in the hyaluronan-binding domain regulate CD44 adhesiveness under flow condition. Structure 2010, 18, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; He, Y.; Zang, H.; Liu, Y.; Wang, W.; Du, Y.; Gao, F. Selective killing of breast cancer cells expressing activated CD44 using CD44 ligand-coated nanoparticles in vitro and in vivo. Oncotarget 2015, 6, 15283–15296. [Google Scholar] [CrossRef]
- Lesley, J.; Hyman, R. CD44 can be activated to function as an hyaluronic acid receptor in normal murine T cells. Eur. J. Immunol. 1992, 22, 2719–2723. [Google Scholar] [CrossRef] [PubMed]
- Moll, J.; Khaldoyanidi, S.; Sleeman, J.P.; Achtnich, M.; Preus, I.; Ponta, H.; Herrlich, P. Two different functions for CD44 proteins in human myelopoiesis. J. Clin. Investig. 1998, 102, 1024–1034. [Google Scholar] [CrossRef]
- Guo, Q.; Yang, C.; Gao, F. The state of CD44 activation in cancer progression and therapeutic targeting. FEBS J. 2022, 289, 7970–7986. [Google Scholar] [CrossRef]
- Spadea, A.; Rios de la Rosa, J.M.; Tirella, A.; Ashford, M.B.; Williams, K.J.; Stratford, I.J.; Tirelli, N.; Mehibel, M. Evaluating the efficiency of hyaluronic acid for tumor targeting via CD44. Mol. Pharm. 2019, 16, 2481–2493. [Google Scholar] [CrossRef]
- Guadagno, E.; Borrelli, G.; Califano, M.; Cali, G.; Solari, D.; Del Basso De Caro, M. Immunohistochemical expression of stem cell markers CD44 and nestin in glioblastoma: Evaluation of their prognostic significance. Pathol. Res. Pract. 2016, 212, 825–832. [Google Scholar] [CrossRef]
- Si, D.; Yin, F.; Peng, J.; Zhang, G. High expression of CD44 predicts a poor prognosis in glioblastomas. Cancer Manag. Res. 2020, 12, 769–775. [Google Scholar] [CrossRef]
- Nishikawa, M.; Inoue, A.; Ohnishi, T.; Kohno, S.; Ohue, S.; Matsumoto, S.; Suehiro, S.; Yamashita, D.; Ozaki, S.; Watanabe, H.; et al. Significance of glioma stem-like cells in the tumor periphery that express high levels of CD44 in tumor invasion, early progression, and poor prognosis in glioblastoma. Stem Cells Int. 2018, 23, 5387041. [Google Scholar] [CrossRef]
- Sawant, S.; Ahire, C.; Dongre, H.; Joshi, S.; Jamghare, S.; Rane, P.; Kane, S.; Chaukar, D. Prognostic significance of elevated serum CD44 levels in patients with oral squamous cell carcinoma. J. Oral Pathol. Med. 2018, 47, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Karan Križanac, D.; Krasić Arapović, A.; Skočibušić, S.; Pintarić, I.; Trgo, G.; Tomić, S. CD44 Immunoexpression is Unfavorable Predictor in Ovarian Serous Cancer. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.H.; Wang, Y.C.; Chou, Y.C.; Yu, M.H.; Chao, T.K. The combination of aldehyde dehydrogenase 1 (ALDH1) and CD44 is associated with poor outcomes in endometrial cancer. PLoS ONE 2018, 13, e0206685. [Google Scholar] [CrossRef]
- Xie, X.; Huang, X.; Tang, H.; Ye, F.; Yang, L.; Guo, X.; Tian, Z.; Xie, X.; Peng, C.; Xie, X. Diallyl Disulfide inhibits breast cancer stem cell progression and glucose metabolism by targeting CD44/PKM2/AMPK signaling. Curr. Cancer Drug Targets 2018, 18, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, T.; Lu, D.; Zhen, J.; Zhang, L. CD44 overexpression related to lymph node metastasis and poor prognosis of pancreatic cancer. Int. J. Biol. Markers 2018, 33, 308–313. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Q.; Wang, Q.; Wang, Y.; Chen, J. Prognostic significance of CD24 and CD44 in breast cancer: A meta-analysis. Int. J. Biol. Markers 2017, 32, e75–e82. [Google Scholar] [CrossRef]
- Tsidulko, A.Y.; Kazanskaya, G.M.; Kostromskaya, D.V.; Aidagulova, S.V.; Kiselev, R.S.; Volkov, A.M.; Kobozev, V.V.; Gaitan, A.S.; Krivoshapkin, A.L.; Grigorieva, E.V. Prognostic relevance of NG2/CSPG4, CD44 and Ki-67 in patients with glioblastoma. Tumour Biol. 2017, 39, 1010428317724282. [Google Scholar] [CrossRef]
- Taniguchi, D.; Saeki, H.; Nakashima, Y.; Kudou, K.; Nakanishi, R.; Kubo, N.; Ando, K.; Oki, E.; Oda, Y.; Maehara, Y. CD44v9 is associated with epithelial-mesenchymal transition and poor outcomes in esophageal squamous cell carcinoma. Cancer Med. 2018, 7, 6258–6268. [Google Scholar] [CrossRef]
- Yamakawa, Y.; Kusuhara, M.; Terashima, M.; Kinugasa, Y.; Sugino, T.; Abe, M.; Mochizuki, T.; Hatakeyama, K.; Kami, K.; Yamaguchi, K. CD44 variant 9 expression as a predictor for gastric cancer recurrence: Immunohistochemical and metabolomic analysis of surgically resected tissues. Biomed Res. 2017, 38, 41–52. [Google Scholar] [CrossRef]
- Todaro, M.; Gaggianesi, M.; Catalano, V.; Benfante, A.; Iovino, F.; Biffoni, M.; Apuzzo, T.; Sperduti, I.; Volpe, S.; Cocorullo, G.; et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell 2014, 14, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Cozzi, P.J.; Hao, J.L.; Beretov, J.; Chang, L.; Duan, W.; Shigdar, S.; Delprado, W.J.; Graham, P.H.; Bucci, J.; et al. CD44 variant 6 associated with prostate cancer metastasis and chemo-/radioresistance. Prostate 2014, 74, 602–617. [Google Scholar] [CrossRef] [PubMed]
- Gotoda, T.; Matsumura, Y.; Kondo, H.; Saitoh, D.; Shimada, Y.; Kosuge, T.; Kanai, Y.; Kakizoe, T. Expression of CD44 variants and its association with survival in pancreatic cancer. Jpn. J. Cancer Res. 1998, 89, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Jijiwa, M.; Demir, H.; Gupta, S.; Leung, C.; Joshi, K.; Orozco, N.; Huang, T.; Yildiz, V.O.; Shibahara, I.; de Jesus, J.A.; et al. CD44v6 regulates growth of brain tumor stem cells partially through the AKT-mediated pathway. PLoS ONE 2011, 6, e24217. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Song, X.; Liu, J.; Li, S.; Gao, W.; Qiu, M.; Yang, C.; Ma, Y.; Chen, Y. Expression of CD44 and the survival in glioma: A meta-analysis. Biosci. Rep. 2020, 40, BSR20200520. [Google Scholar] [CrossRef]
- Inoue, A.; Ohnishi, T.; Nishikawa, M.; Watanabe, H.; Kusakabe, K.; Taniwaki, M.; Yano, H.; Ohtsuka, Y.; Matsumoto, S.; Suehiro, S.; et al. Identification of CD44 as a reliable biomarker for glioblastoma invasion: Based on magnetic resonance imaging and spectroscopic analysis of 5-aminolevulinic acid fluorescence. Biomedicines 2023, 11, 2369. [Google Scholar] [CrossRef]
- Jalkanen, M.; Elenius, K.; Salmivirta, M. Syndecan—A cell surface proteoglycan that selectively binds extracellular effector molecules. Adv. Exp. Med. Biol. 1992, 313, 79–85. [Google Scholar]
- Weber, G.F.; Ashkar, S.; Glimcher, M.J.; Cantor, H. Receptor-ligand interaction between CD44 and osteopontin (Eta-1). Science 1996, 271, 509–512. [Google Scholar] [CrossRef]
- Knutson, J.R.; Iida, J.; Fields, G.B.; McCarthy, J.B. CD44/chondroitin sulfate proteoglycan and alpha 2 beta 1 integrin mediate human melanoma cell migration on type IV collagen and invasion of basement membranes. Mol. Biol. Cell 1996, 7, 383–396. [Google Scholar] [CrossRef]
- Toyama-Sorimachi, N.; Sorimachi, H.; Tobita, Y.; Kitamura, F.; Yagita, H.; Suzuki, K.; Miyasaka, M. A novel ligand for CD44 is serglycin, a hematopoietic cell adherence and activation. J. Biol. Chem. 1995, 270, 7437–7444. [Google Scholar] [CrossRef]
- Toole, B.P.; Slomiany, M.G. Hyaluronan, CD44 and Emmprin: Patners in cancer cell chemoresistance. Drug Resist. Updates 2008, 11, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y. Hyaluronan-mediated CD44 activation of RhoGTPase signaling and cytoskeleton function promotes tumor progression. Semin. Cancer Biol. 2008, 18, 251–259. [Google Scholar] [CrossRef]
- Bourguignon, L.Y. Hyaluronan-mediated CD44 interaction with receptor and non-receptor kinases promotes oncogenic signaling, cytoskeleton activation and tumor progression. In Hyaluronan in Cancer Biology; Stern, R., Ed.; Academic Press: San Diego, CA, USA, 2009; pp. 89–107. [Google Scholar]
- Turley, E.A.; Noble, P.W.; Bourguignon, L.Y. Signaling properties of hyaluronan receptors. J. Biol. Chem. 2002, 277, 4589–4592. [Google Scholar] [CrossRef] [PubMed]
- Slomiany, M.G.; Dai, L.; Bomar, P.A.; Knackstedt, T.J.; Kranc, D.A.; Tolliver, L.; Maria, B.L.; Toole, B.P. Abrogating drug resistance in malignant peripheral nerve sheath tumors by disrupting hyaluronan-CD44 interactions with small hyaluronan oligosaccharides. Cancer Res. 2009, 69, 4992–4998. [Google Scholar] [CrossRef]
- Orian-Rousseau, V.; Morrison, H.; Matzke, A.; Kastilan, T.; Pace, G.; Herrlich, P.; Ponta, H. Hepatocyte growth factor-induced Ras activation requires ERM proteins linked to both CD44v6 and F-actin. Mol. Biol. Cell 2007, 18, 76–83. [Google Scholar] [CrossRef]
- McCarty, J.H. Glioblastoma resistance to anti-VEGF therapy: Has the challenge been MET? Clin. Cancer Res. 2013, 19, 1631–1633. [Google Scholar] [CrossRef]
- Nishikawa, M.; Inoue, A.; Ohnishi, T.; Yano, H.; Kanemura, Y.; Kohno, S.; Ohue, S.; Ozaki, S.; Matsumoto, S.; Suehiro, S.; et al. CD44 expression in the tumor periphery predicts the responsiveness to bevacizumab in the treatment of recurrent glioblastoma. Cancer Med. 2021, 10, 2013–2025. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Lee, H.W.; Kang, H.G.; Kim, H.Y.; Kim, S.J.; Chun, K.H. Cleaved CD44 intracellular domain supports activation of stemness factors and promotes tumorigenesis of breast cancer. Oncotarget 2015, 6, 8709–8721. [Google Scholar] [CrossRef]
- Löfstedt, T.; Fredlund, E.; Holmquist-Mengelbier, L.; Pietras, A.; Ovenberger, M.; Poellinger, L.; Påhlman, S. Hypoxia inducible factor-2alpha in cancer. Cell Cycle 2007, 6, 919–926. [Google Scholar] [CrossRef]
- Okamoto, I.; Kawano, Y.; Murakami, D.; Sasayama, T.; Araki, N.; Miki, T.; Wong, A.J.; Saya, H. Protelytic release of CD44intracellular domain and its role in the CD44 signaling pathway. J. Cell. Biol. 2001, 155, 755–762. [Google Scholar] [CrossRef]
- De Falco, V.; Tamburrino, A.; Ventre, S.; Castellone, M.D.; Malek, M.; Manié, S.N.; Santoro, M. CD44 proteolysis increases CREB phosphorylation and sustains proliferation of thyroid cancer cells. Cancer Res. 2012, 72, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Li, D.; Xun, J.; Zhou, W.; Li, J.; Wang, J.; Liu, C.; Li, X.; Shen, W.; Qiao, H.; et al. CD44ICD promotes breast cancer stemness via PFKFB4-mediated glucose metabolism. Theranostics 2018, 8, 6248–6262. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y. Matrix hyaluronan promotes specific microRNA upregulation leading to drug resistance and tumor progression. Int. J. Mol. Sci. 2016, 17, 517. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Asangani, I.A.; Rasheed, S.A.; Nikolova, D.A.; Leupold, J.H.; Colburn, N.H.; Post, S.; Allgayer, H. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 2008, 27, 2128–2136. [Google Scholar] [CrossRef]
- Xu, L.F.; Wu, Z.P.; Chen, Y.; Zhu, Q.S.; Hamidi, S.; Navab, R. MicroRNA-21 (miR-21) regulates cellular proliferation, invasion, migration, and apoptosis by targeting PTEN, RECK and Bcl-2 in lung squamous carcinoma. PLoS ONE 2014, 9, e103698. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.; Earle, C.; Wong, G.; Spevak, C.C.; Krueger, K. Stem cell marker (Nanog) and Stat-3 signaling promote microRNA-21 expression and chemoresistance in hyaluronan/CD44-activated head and neck squamous cell carcinoma cells. Oncogene 2012, 31, 149–160. [Google Scholar] [CrossRef]
- Fujita, S.; Ito, T.; Mizutani, T.; Minoguchi, S.; Yamamichi, N.; Sakurai, K.; Iba, H. miR-21 Gene expression triggered by AP-1 is sustained through a double-negative feedback mechanism. J. Mol. Biol. 2008, 378, 492–504. [Google Scholar] [CrossRef]
- Chen, L.; Bourguignon, L.Y. Hyaluronan-CD44 interaction promotes c-Jun signaling and miRNA21 expression leading to Bcl-2 expression and chemoresistance in breast cancer cells. Mol. Cancer 2014, 13, 52. [Google Scholar] [CrossRef]
- Gabriely, G.; Wurdinger, T.; Kesarri, S.; Esau, C.C.; Burchard, J.; Linsley, P.S.; Krichevsky, A.M. MicroRNA21 promotes glioma invasion by targeting matrix metalloproteinase regulators. Mol. Cell. Biol. 2008, 28, 5369–5380. [Google Scholar] [CrossRef]
- Luo, G.; Luo, W.; Sun, X.; Lin, J.; Wang, M.; Zhang, Y.; Luo, W.; Zhang, Y. MicroRNA 21 promotes migration and invasion of glioma cells via activation of Sox2 and β catenin signaling. Mol. Med. Rep. 2017, 15, 187–195. [Google Scholar] [CrossRef]
- Shi, L.; Chen, J.; Yang, J.; Pan, T.; Zhang, S.; Wang, Z. MiR-21 protected human glioblastoma U87MG cells from chemotherapeutic drug temozolomide induced apoptosis by decreasing Bax/Bcl-2 ratio and caspase-3 activity. Brain Res. 2010, 1352, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Reinhardt, F.; Pan, E.; Soutschek, J.; Bhat, B.; Marcusson, E.G.; Teruya-Feldstein, J.; Bell, G.W.; Weinberg, R.A. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat. Biotechnol. 2010, 28, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Sasayama, T.; Nishihara, M.; Kondoh, T.; Hosoda, K.; Kohmura, E. MicroRNA-10b is overexpressed in malignant glioma and associated with tumor invasive factors, uPAR and RhoC. Int. J. Cancer 2009, 125, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y.; Wong, G.; Earle, C.; Krueger, K.; Spevak, C.C. Hyaluronan-CD44 interaction promotes c-Src-mediated Twist signaling, microRNA-10b expression, and RhoA/RhoC up-regulation, leading to Rho-kinase-associated cytoskeleton activation and breast tumor cell invasion. J. Biol. Chem. 2010, 285, 36721–36735. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Deng, S.; Zhao, Z.; Zhang, H.; Xiao, J.; Song, W.; Gao, F.; Guan, Y. Oct4 regulates the miR-302 cluster in P19 mouse embryonic carcinoma cells. Mol. Biol. Rep. 2011, 38, 2155–2160. [Google Scholar] [CrossRef]
- Lin, S.L.; Chang, D.C.; Chang-Lin, S.; Lin, C.H.; Wu, D.T.; Chen, D.T.; Ying, S.Y. Mir-302 reprograms human skin cancer cells into a pluripotent ES-cell-like state. RNA 2008, 14, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Clay, M.R.; Tabor, M.; Owen, J.H.; Carey, T.E.; Bradford, C.R.; Wolf, G.T.; Wicha, M.S.; Prince, M.E. Single-marker identification of head and neck squamous cell carcinoma cancer stem cells with aldehyde dehydrogenase. Head Neck 2010, 32, 1195–1201. [Google Scholar] [CrossRef]
- Lin, S.L.; Chang, D.C.; Lin, C.H.; Ying, S.Y.; Leu, D.; Wu, D.T. Regulation of somatic cell reprogramming through inducible mir-302 expression. Nucleic Acids Res. 2011, 39, 1054–1065. [Google Scholar] [CrossRef]
- Chen, Y.C.; Chen, Y.W.; Hsu, H.S.; Tseng, L.M.; Huang, P.I.; Lu, K.H.; Chen, D.T.; Tai, L.K.; Yung, M.C.; Chang, S.C.; et al. Aldehyde dehydrogenase 1 is a putative marker for cancer stem cells in head and neck squamous cancer. Biochem. Biophys. Res. Commun. 2009, 385, 307–313. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.; Wong, G.; Earle, C.; Chen, L. Hyaluronan-CD44v3 interaction with Oct4-Sox2-Nanog promotes miR-302 expression leading to self-renewal, clonal formation, and cisplatin resistance in cancer stem cells from head and neck squamous cell carcinoma. J. Biol. Chem. 2012, 287, 32800–32824. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Teruya-Feldstein, J.; Weinberg, R.A. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 2007, 449, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Wang, K.; Shao, Z.; Lin, Q.; Li, B.; Liu, P. Mir-373/miR-520s-CD44 axis significantly inhibits the growth and invasion of human glioblastoma cells. Arch. Med Res. 2022, 53, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Yeh, M.; Wang, Y.Y.; Yoo, J.; Oh, C.; Otani, Y.; Kang, J.M.; Park, E.S.; Kim, E.; Chung, S.; Jeon, Y.J.; et al. MicroRNA-138 suppresses glioblastoma proliferation through downregulation of CD44. Sci. Rep. 2021, 11, 9219. [Google Scholar] [CrossRef]
- Mooney, K.L.; Choy, W.; Sidhu, S.; Pelargos, P.; Bui, T.T.; Voth, B.; Barnette, N.; Yang, I. The role of CD44 in glioblastoma multiforme. J. Clin. Neurosci. 2016, 34, 1–5. [Google Scholar] [CrossRef]
- Cheng, L.; Wu, Q.; Guryanova, O.A.; Huang, Z.; Huang, Q.; Rich, J.N.; Bao, S. Elevated invasive potential of glioblastoma stem cells. Biochem. Biophys. Res. Commun. 2011, 406, 643–648. [Google Scholar] [CrossRef]
- Chen, J.E.; Lumibao, J.; Blazek, A.; Gaskins, H.R.; Harley, B. Hypoxia activates enhanced invasive potential and endogenous hyaluronic acid production by glioblastoma cells. Biomater. Sci. 2018, 6, 854–862. [Google Scholar] [CrossRef]
- Delpech, B.; Maingonnat, C.; Girard, N.; Chauzy, C.; Maunoury, R.; Olivier, A.; Tayot, J.; Creissard, P. Hyaluronan and hyaluronectin in the extracellular matrix of human brain tumor stroma. Eur. J. Cancer 1993, 29, 1012–1017. [Google Scholar] [CrossRef]
- Nishikawa, M.; Inoue, A.; Ohnishi, T.; Yano, H.; Ozaki, S.; Kanemura, Y.; Suehiro, S.; Ohtsuka, Y.; Kohno, S.; Ohue, S.; et al. Hypoxia-induced phenotypic transition from highly invasive to less invasive tumors in glioma stem-like cells: Significance of CD44 and osteopontin as therapeutic targets in glioblastoma. Transl. Oncol. 2021, 14, 101137. [Google Scholar] [CrossRef]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell migration: Integrating signals from front to back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, P.; Yang, L.; Yu, X.; Ye, X.; Yang, J.; Qian, C.; Zhang, X.; Cui, Y.H.; Bian, X.W. Activation of toll-like receptor 2 promotes invasion by upregulating MMPs in glioma stem cells. Am. J. Transl. Res. 2015, 7, 607–615. [Google Scholar] [PubMed]
- Meldolesi, J. Pharmacology of the cell/matrix form of adhesion. Pharm. Res. 2016, 107, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Ellert-Miklaszewska, A.; Poleszak, K.; Pasierbinska, M.; Kaminska, B. Integrin Signaling in Glioma Pathogenesis: From Biology to Therapy. Int. J. Mol. Sci. 2020, 21, 888. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, V.P.; Moura Neto, V.; Mentlein, R. Glioma infiltration and extracellular matrix: Key players and modulators. Glia 2018, 66, 1542–1565. [Google Scholar] [CrossRef] [PubMed]
- Onishi, M.; Ichikawa, T.; Kurozumi, K.; Date, I. Angiogenesis and invasion in glioma. Brain Tumor Pathol. 2011, 28, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Binder, Z.A.; Kim, S.H.J.; Wu, P.H.; Giri, A.; Gallia, G.L.; Pardo, A.; Wirtz, D. The role of integrin v and CD44 in GBM migration in human brain. bioRxiv, 2019; preprint. [Google Scholar] [CrossRef]
- Wolf, K.J.; Shukla, P.; Springer, K.; Lee, S.; Coombes, J.D.; Choy, C.J.; Kenny, S.J.; Xu, K.; Kumar, S. A mode of cell adhesion and migration facilitated by CD44-dependent microtentacles. Proc. Natl. Acad. Sci. USA 2020, 117, 11432–11443. [Google Scholar] [CrossRef]
- Kim, Y.; Kumar, S. CD44-mediated adhesion to hyaluronic acid contributes to mechanosensing and invasive motility. Mol. Cancer Res. 2014, 12, 1416–1429. [Google Scholar] [CrossRef]
- So, J.S.; Kim, H.; Han, K.S. Mechanisms of invasion in glioblastoma: Extracellular matrix, Ca2+ signaling, and glutamate. Front. Cell. Neurosci. 2021, 15, 663092. [Google Scholar] [CrossRef]
- Giese, A.; Bjerkvig, R.; Berens, M.E.; Westphal, M. Cost of migration: Invasion of malignant gliomas and implications for treatment. J. Clin. Oncol. 2003, 21, 1624–1636. [Google Scholar] [CrossRef]
- Xu, Y.; Stamenkovic, I.; Yu, Q. CD44 attenuates activation of the Hippo signaling pathway and is a prime therapeutic target for glioblastoma. Cancer Res. 2010, 70, 2455–2464. [Google Scholar] [CrossRef]
- Bai, Y.; Liu, Y.J.; Wang, H.; Xu, Y.; Stamenkovic, I.; Yu, Q. Inhibition of the hyaluronan-CD44 interaction by merlin contributes to the tumor suppressor activity of merlin. Oncogene 2007, 26, 836–850. [Google Scholar] [CrossRef]
- Ibrahim, H.M.; AbdElbary, A.M.; Mohamed, S.Y.; Elwan, A.; Abdelhamid, M.I.; Ibrahim, A. Prognostic value of cyclin D1 and CD44 expression in gastric adenocarcinoma. J. Gastrointest. Cancer 2019, 50, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Daniel, P.M.; Filiz, G.; Tymmms, M.J.; Ramsay, R.G.; Kaye, A.H.; Stylli, S.S.; Mantamadiotis, T. Intratumor MAPK and PI3K signaling pathway heterogeneity in glioblastoma tissue correlates with CREB signaling and distinct target gene signatures. Exp. Mol. Pathol. 2018, 105, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Qazi, M.A.; Vora, P.; Venugopal, C.; Sidhu, S.S.; Moffat, J.; Swanton, C.; Singh, S.K. Intratumoral heterogeneity: Pathways to treatment resistance and relapse in human glioblastoma. Ann. Oncol. 2017, 28, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Parker, N.R.; Khong, P.; Parkinson, J.F.; Howell, V.M.; Wheeler, H.R. Molecular heterogeneity in glioblastoma: Potential clinical implications. Front. Oncol. 2015, 5, 55. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Segerman, A.; Niklasson, M.; Haglund, C.; Bergström, T.; Jarvius, M.; Xie, Y.; Westermark, A.; Sönmez, D.; Hermansson, A.; Kastemar, M.; et al. Clonal variation in drug and radiation response among glioma-initiating cells is linked to proneural-mesenchymal transition. Cell Rep. 2016, 11, 2994–3009. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; Decarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef]
- Takashima, Y.; Kawaguchi, A.; Yamanaka, R. Promising prognosis marker candidates on the status of epithelial-mesenchymal transition and glioma stem cells in glioblastoma. Cells 2019, 8, 1312. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Cerchia, L.; Pegoraro, S.; Sgarra, R.; Manfioletti, G. Proneural-Mesenchymal Transition: Phenotypic Plasticity to Acquire Multitherapy Resistance in Glioblastoma. Int. J. Mol. Sci. 2019, 20, 2746. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Slomiany, M.G.; Grass, G.D.; Robertson, A.D.; Yang, X.Y.; Maria, B.L.; Beeson, C.; Toole, B.P. Hyaluronan, CD44, and emmprin regulate lactate efflux and membrane localization of monocarboxylate transporters in human breast carcinoma cells. Cancer Res. 2009, 69, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, B.P. Activation of beta-catenin and Akt pathways by Twist are critical for the maintenance of EMT associated cancer stem cell-like characters. BMC Cancer 2011, 11, 49. [Google Scholar] [CrossRef]
- Jang, B.I.; Li, Y.; Graham, D.Y.; Cen, P. The role of CD44 in the pathogenesis, diagnosis, and therapy of gastric cancer. Gut Liver 2011, 5, 397–405. [Google Scholar] [CrossRef]
- Li, L.; Liu, C.; Amato, R.J.; Chang, J.T.; Du, G.; Li, W. CDKL2 promotes epithelial-mesenchymal transition and breast cancer progression. Oncotarget 2014, 5, 10840–10853. [Google Scholar] [CrossRef]
- Lee, J.K.; Joo, K.M.; Lee, J.; Yoon, Y.; Nam, D.H. Targeting the epithelial to mesenchymal transition in glioblastoma: The emerging role of MET signaling. OncoTargets Ther. 2014, 7, 1933–1944. [Google Scholar]
- Godar, S.; Ince, T.A.; Bell, G.W.; Feldser, D.; Donaher, J.L.; Bergh, J.; Liu, A.; Miu, K.; Watnick, R.S.; Reinhardt, F.; et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell 2008, 134, 62–73. [Google Scholar] [CrossRef]
- Park, J.; Schwarzbauer, J.E. Mammary epithelial cell interactions with fibronectin stimulate epithelial-mesenchymal transition. Oncogene 2014, 33, 1649–1657. [Google Scholar] [CrossRef]
- Way, T.D.; Huang, J.T.; Chou, C.H.; Huang, C.H.; Yang, M.H.; Ho, C.T. Emodin represses TWIST1-induced epithelial-mesenchymal transitions in head and neck squamous cell carcinoma cells by inhibiting the beta-catenin and Akt pathways. Eur. J. Cancer 2014, 50, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Masui, T.; Ota, I.; Yook, J.I.; Mikami, S.; Yane, K.; Yamanaka, T.; Hosoi, H. Snail-induced epithelial-mesenchymal transition promotes cancer stem cell-like phenotype in head and neck cancer cells. Int. J. Oncol. 2014, 44, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Ning, Z.; Zeng, J.; Fan, J.; Zhou, J.; Zhang, T.; Zhang, L.; Chen, Y.; Gao, Y.; Wang, B.; et al. Silibinin inhibits beta-catenin/ZEB1 signaling and suppresses bladder cancer metastasis via dual-blocking epithelial-mesenchymal transition and stemness. Cell Signal. 2013, 25, 2625–2633. [Google Scholar] [CrossRef]
- Bhat-Nakshatri, P.; Appaiah, H.; Ballas, C.; Pick-Franke, P.; Goulet, R., Jr.; Badve, S.; Srour, E.F.; Nakshatri, H. SLUG/SNAI2 and tumor necrosis factor generate breast cells with CD44+/CD24− phenotype. BMC Cancer 2010, 10, 411. [Google Scholar] [CrossRef]
- Ju, S.Y.; Chiou, S.H.; Su, Y. Maintenance of the stemness in CD44(+) HCT-15 and HCT-116 human colon cancer cells requires miR-203 suppression. Stem Cell Res. 2014, 12, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Shang, Z.; Cai, Q.; Zhang, M.; Zhu, S.; Ma, Y.; Sun, L.; Jiang, N.; Tian, J.; Niu, X.; Chen, J.; et al. A switch from CD44(+) cell to EMT cell drives the metastasis of prostate cancer. Oncotarget 2015, 6, 1202–1216. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Park, Y.S.; Kim, H.J.; Kim, C.H.; Lim, S.W.; Huh, J.W.; Lee, J.H.; Kim, H.R. CD44 enhances the epithelial-mesenchymal transition in association with colon cancer invasion. Int. J. Oncol. 2012, 41, 211–218. [Google Scholar]
- Zubeldia, I.G.; Bleau, A.M.; Redrado, M.; Serrano, D.; Agliano, A.; Gil-Puig, C.; Vidal-Vanaclocha, F.; Lecanda, J.; Calvo, A. Epithelial to mesenchymal transition and cancer stem cell phenotypes leading to liver metastasis are abrogated by the novel TGFβ1-targeting peptides P17 and P144. Exp. Cell Res. 2013, 319, 12–22. [Google Scholar] [CrossRef]
- Joseph, J.V.; Conroy, S.; Tomar, T.; Eggens-Meijer, E.; Bhat, K.; Copray, S.; Walenlamp, A.M.; Boddeke, E.; Balasubramamyian, V.; Wagemakers, A.M.; et al. TGF-β is an inducer of ZEB1-dependent mesenchymal transdifferentiation in glioblastoma that is associated with tumor invasion. Cell Death Dis. 2014, 5, e1443. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.; Singleton, P.A.; Zhu, H.; Zhou, B. Hyaluronan promotes signaling interaction between CD44 and the transforming growth factor beta receptor I in metastatic breast tumor cells. J. Biol. Chem. 2002, 277, 39703–39712. [Google Scholar] [CrossRef]
- Luo, D.; Xu, X.; Li, J.; Chen, C.; Wang, F.; Xie, Y.; Li, F. The PDK1/c Jun pathway activated by TGF β induces EMT and promotes proliferation and invasion in human glioblastoma. Int. J. Oncol. 2018, 53, 2067–2080. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Iwata, R.; Lee, J.H.; Hayashi, M.; Dianzani, U.; Ofune, K.; Maruyama, M.; Oe, S.; Ito, T.; Hashiba, T.; Yoshimura, K.; et al. ICOSLG-mediated regulatory T-cell expansion and IL-10 production promote progression of glioblastoma. Neuro Oncol. 2020, 22, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Chen, C.; Chang, K.; Karnad, A.; Jagirdar, J.; Kumar, A.P.; Freeman, J.W. CD44 expression level and isoform contributes to pancreatic cancer cell plasticity, invasiveness and response to therapy. Clin. Cancer Res. 2016, 22, 5592–5604. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.L.; Reinke, L.M.; Damerow, M.S.; Perez, D.; Chodosh, L.A.; Yang, J.; Cheng, C. CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J. Clin. Investig. 2011, 121, 1064–1074. [Google Scholar] [CrossRef]
- Dhruv, H.D.; McDonough Winslow, W.S.; Armstrong, B.; Tuncali, S.; Eschbacher, J.; Kislin, K.; Loftus, J.C.; Tran, N.L.; Berens, M.E. Reciprocal activation of transcription factors underlies the dichotomy between proliferation and invasion of glioma cells. PLoS ONE 2013, 8, e72134. [Google Scholar] [CrossRef]
- Jackson, J.S.; Golding, J.P.; Chapon, C.; Jones, W.A.; Bhakoo, K.K. Homing of stem cells to sites of inflammatory brain injury after intracerebral and intravenous administration: A longitudinal imaging study. Stem Cell Res. Ther. 2010, 1, 17. [Google Scholar] [CrossRef]
- Mehta, S. Editorial: The Role of Microenvironment in the Homing, Maintenance, and Release of Glioma Stem-Like Cells. Front Oncol. 2018, 8, 7. [Google Scholar] [CrossRef]
- Nieto, M.A.; Hung, R.Y.J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Luond, F.; Sugiyama, N.; Bill, R.; Bomes, L.; Hager, C.; Tang, F.; Santacroce, N.; Beisel, C.; Ivanek, R.; Bürglin, T.; et al. Distinct contributions of partial and full EMT to breast cancer malignancy. Dev. Cell 2021, 56, 3203–3221. [Google Scholar] [CrossRef]
- Manfioletti, G.; Fedele, M. Epithelial-mesenchymal transition (EMT) 2021. Int. J. Mol. Sci. 2022, 23, 5848. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hu, Z.; Horta, C.A.; Yang, J. Regulation of epithelial-mesenchymal transition by tumor microenvironmental signals and its implication in cancer therapeutics. Semin. Cancer Biol. 2023, 88, 46–66. [Google Scholar] [CrossRef] [PubMed]
- Mortezaee, K. Hypoxia induces core-to-edge transition of progressive tumoral cells: A critical review on differential yet corroborative roles for HIF-1α and HIF-2α. Life Sci. 2020, 242, 114145. [Google Scholar] [CrossRef]
- Bar, E.E. Glioblastoma, cancer stem cells and hypoxia. Brain Pathol. 2011, 21, 119–129. [Google Scholar] [CrossRef]
- Chakhoyan, A.; Guillamo, J.S.; Collet, S.; Kauffmann, F.; Delcroix, N.; Lechapt-Zalcman, E.; Constans, J.M.; Petit, E.; MacKenzie, E.T.; Barré, L.; et al. FMISO-PET-derived brain oxygen tension maps: Application to glioblastoma and less aggressive gliomas. Sci. Rep. 2017, 7, 10210. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef]
- Boyd, N.H.; Tran, A.N.; Bernstock, J.D.; Etminan, T.; Jones, A.B.; Gillespie, G.Y.; Friedman, G.K.; Hjelmeland, A.B. Glioma stem cells and their roles within the hypoxic tumor microenvironment. Theranostics 2021, 11, 665–683. [Google Scholar] [CrossRef]
- Tam, S.Y.; Wu, V.W.C.; Law, H.K.W. Hypoxia-Induced Epithelial-Mesenchymal Transition in Cancers: HIF-1α and beyond. Front. Oncol. 2020, 10, 486. [Google Scholar] [CrossRef]
- Cruickshank, G.S.; Rampling, R. Peri-tumoral hypoxia in human brain:preoperative of the tissue oxygen tension around malignant brain tumours. Acta Neurochir. Suppl. 1994, 60, 375–377. [Google Scholar]
- Kolliopoulos, C.; Ali, M.M.; Castillejo-Lopez, C.; Heldin, C.H.; Heldin, P. CD44 depletion in glioblastoma cells suppresses growth and stemness and induces senescence. Cancers 2022, 14, 3747. [Google Scholar] [CrossRef] [PubMed]
- Heddleston, J.M.; Li, Z.; MeLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar] [CrossRef] [PubMed]
- Musah-Eroje, A.; Watson, S. Adaptive changes of glioblastoma cells following exposure to hypoxic (1% Oxygen) tumour microenvironment. Int. J. Mol. Sci. 2019, 20, 2091. [Google Scholar] [CrossRef] [PubMed]

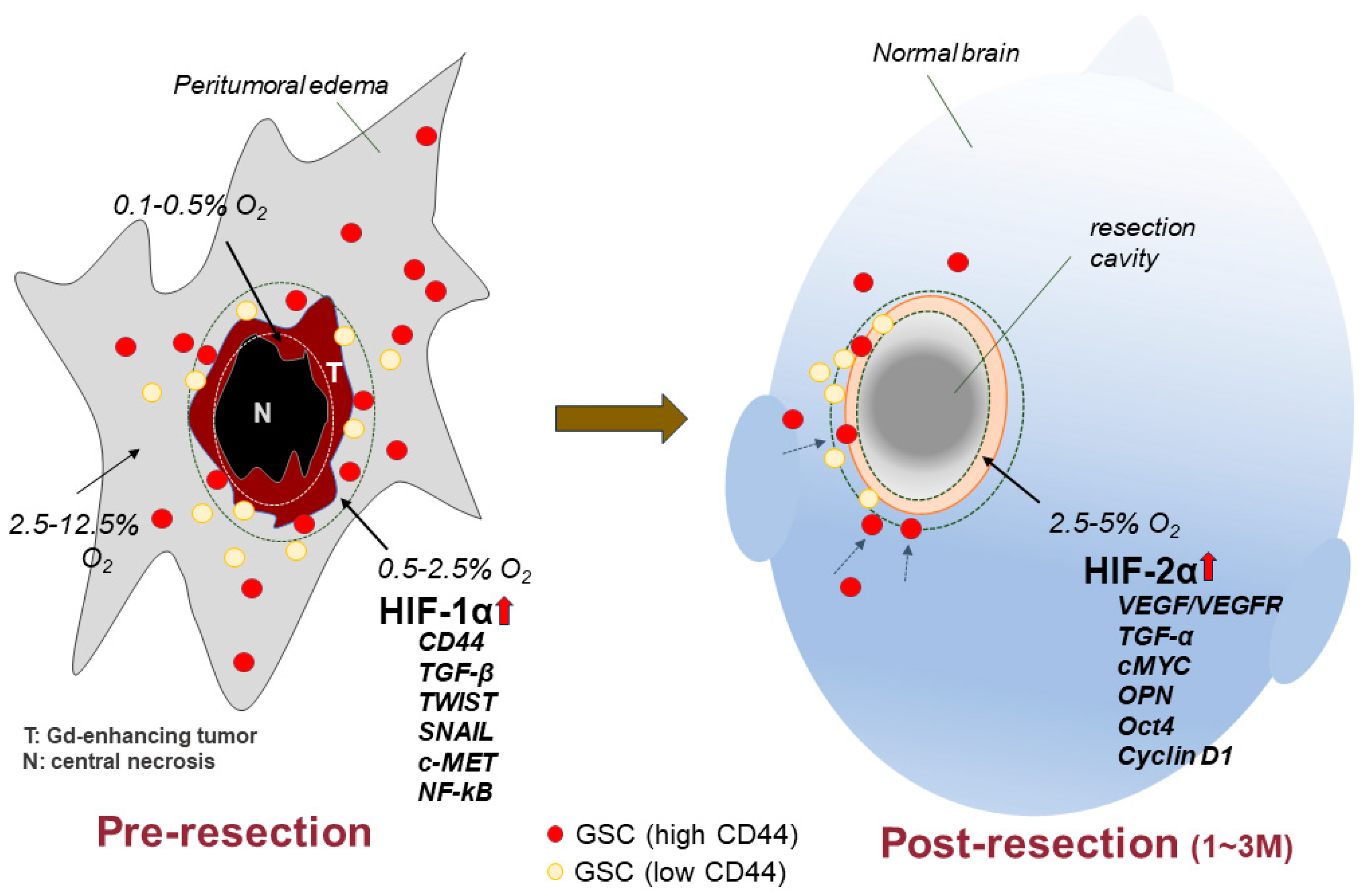
| Gene | Transcription Factor | Cell Function | O2 Condition | References |
|---|---|---|---|---|
| MMP-9 | RUNX2 | Invasion | Normoxia | [30] |
| OPN | (Runt-related transcription factor 2) | Migration | Hypoxia | |
| Metastasis | ||||
| CD44 | CIRE | Invasion | Normoxia | [31] |
| (CD44ICD response element) | Proliferation | |||
| Angiogenesis | ||||
| CyclinD1 | CREB | Proliferation | Normoxia | [72,73] |
| PDK1 | (cAMP response element-binding protein) | Aerobic glycolysis | Hypoxia | |
| PFKFB4 | ||||
| SOX2 | HIF-2α | Stemness | Hypoxia | [32,69] |
| NANOG | (hypoxia-inducible factor-2α) | |||
| OCT4 | ||||
| c-MYC | Proliferation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inoue, A.; Ohnishi, T.; Nishikawa, M.; Ohtsuka, Y.; Kusakabe, K.; Yano, H.; Tanaka, J.; Kunieda, T. A Narrative Review on CD44’s Role in Glioblastoma Invasion, Proliferation, and Tumor Recurrence. Cancers 2023, 15, 4898. https://doi.org/10.3390/cancers15194898
Inoue A, Ohnishi T, Nishikawa M, Ohtsuka Y, Kusakabe K, Yano H, Tanaka J, Kunieda T. A Narrative Review on CD44’s Role in Glioblastoma Invasion, Proliferation, and Tumor Recurrence. Cancers. 2023; 15(19):4898. https://doi.org/10.3390/cancers15194898
Chicago/Turabian StyleInoue, Akihiro, Takanori Ohnishi, Masahiro Nishikawa, Yoshihiro Ohtsuka, Kosuke Kusakabe, Hajime Yano, Junya Tanaka, and Takeharu Kunieda. 2023. "A Narrative Review on CD44’s Role in Glioblastoma Invasion, Proliferation, and Tumor Recurrence" Cancers 15, no. 19: 4898. https://doi.org/10.3390/cancers15194898
APA StyleInoue, A., Ohnishi, T., Nishikawa, M., Ohtsuka, Y., Kusakabe, K., Yano, H., Tanaka, J., & Kunieda, T. (2023). A Narrative Review on CD44’s Role in Glioblastoma Invasion, Proliferation, and Tumor Recurrence. Cancers, 15(19), 4898. https://doi.org/10.3390/cancers15194898