
A Novel Allogeneic Rituximab-Conjugated Gamma Delta T Cell Therapy for the Treatment of Relapsed/Refractory B-Cell Lymphoma

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Antibodies, Cell Lines, and Mice
2.2. ACE1831 Generation
2.3. Flow Cytometry Analysis
2.4. In Vitro Cytotoxicity
2.5. Animal Studies
2.6. Computational Interpretation of the Cell Surface Proteome
2.7. Jurkat-NFAT-Luc Reporter Assay
2.8. Statistical Analysis
3. Results
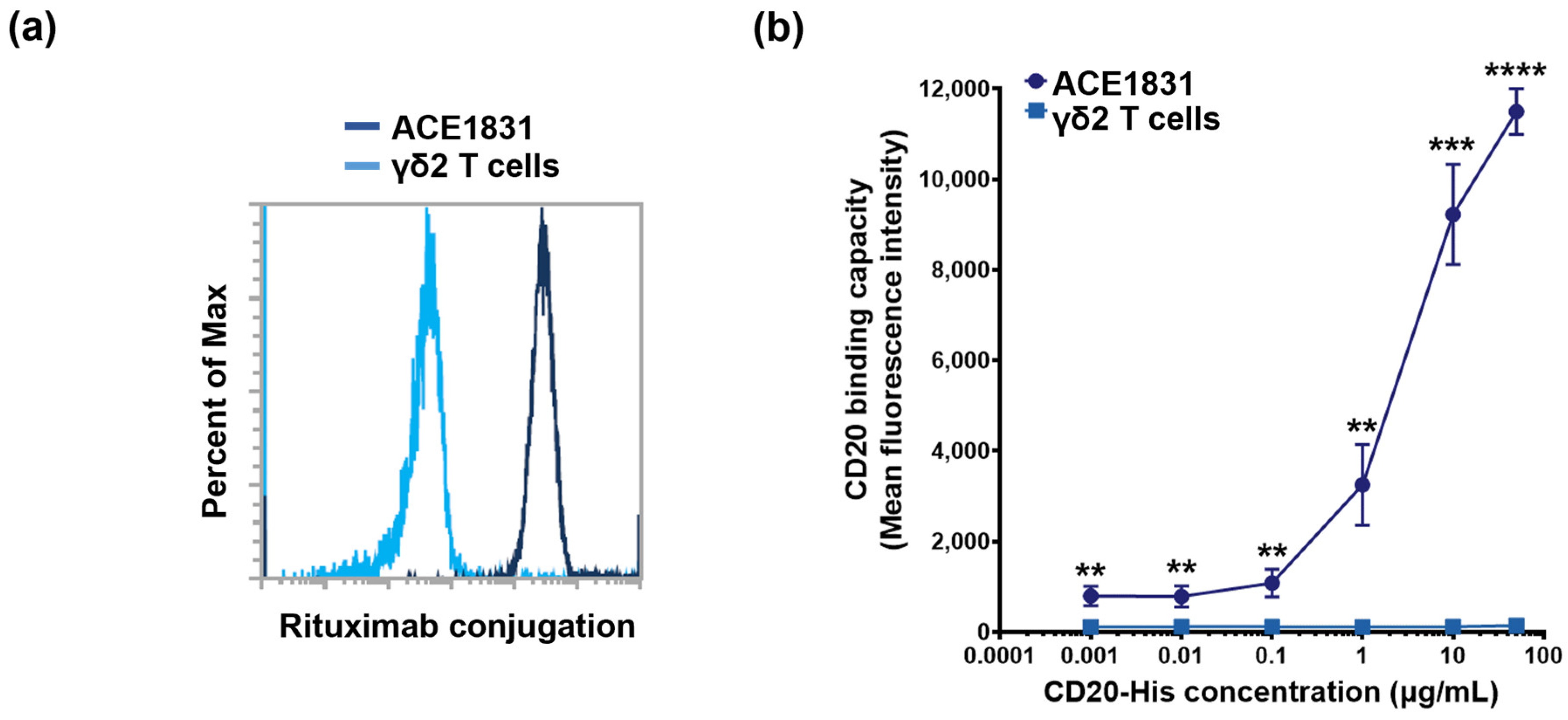
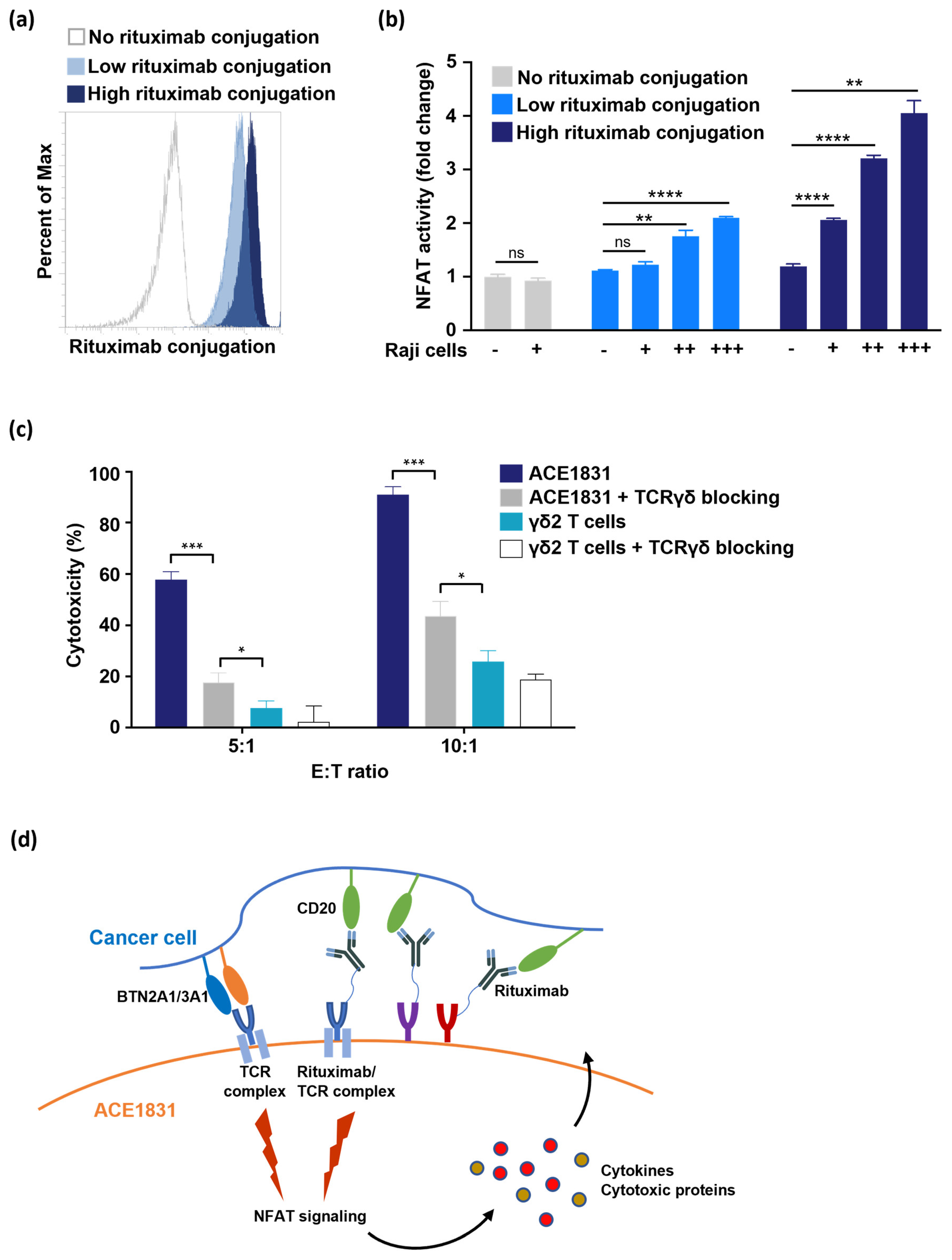
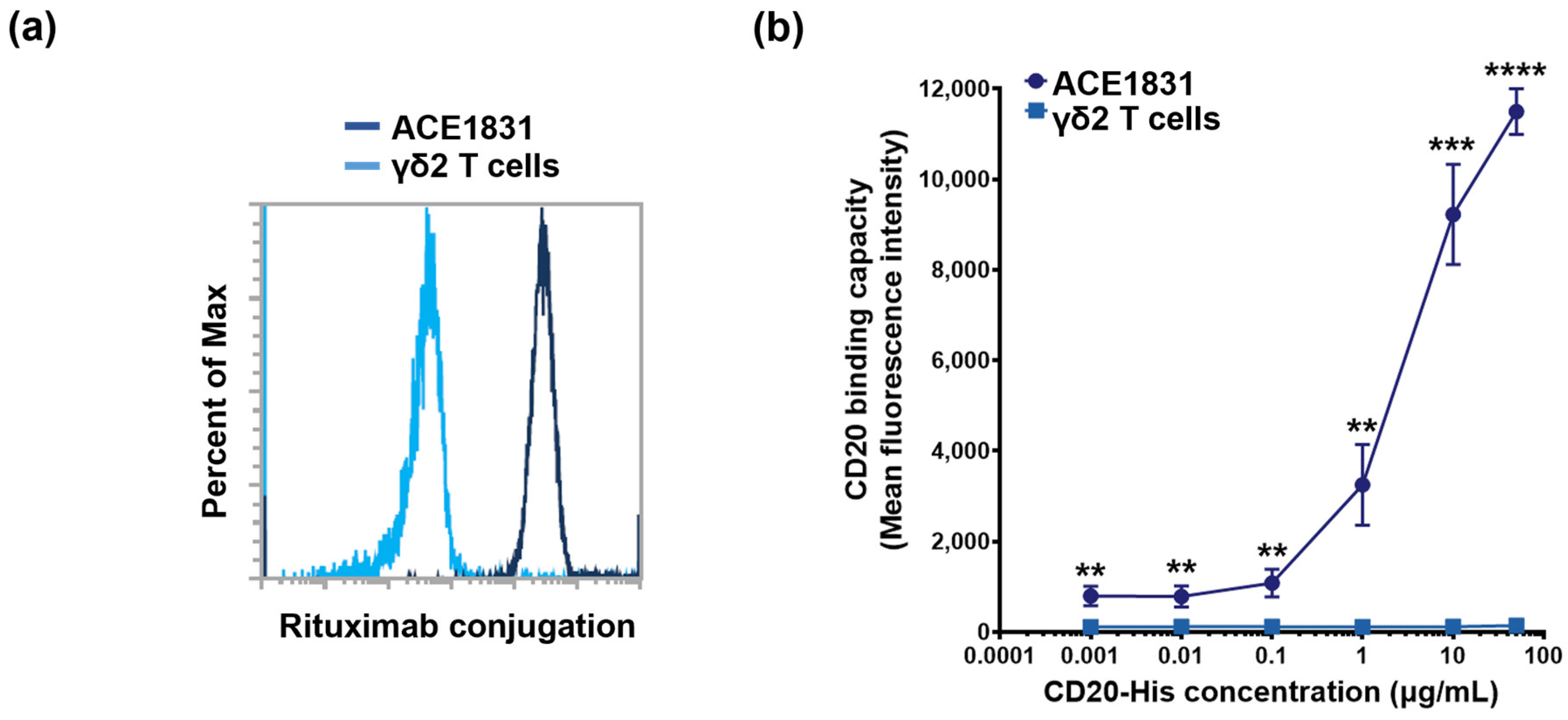
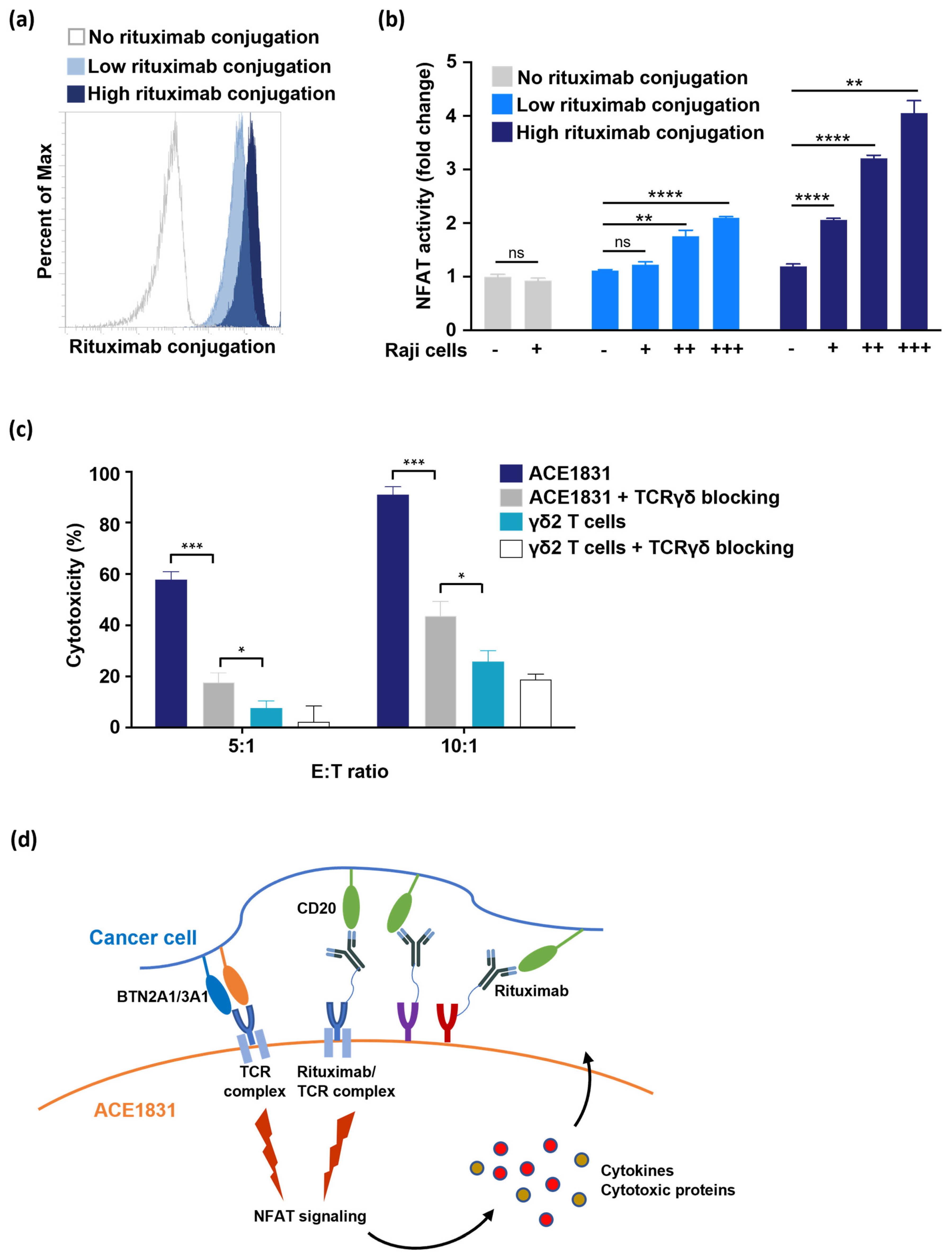
3.1. Rituximab Conjugation on γδ2 T Cells Using ACC Technology
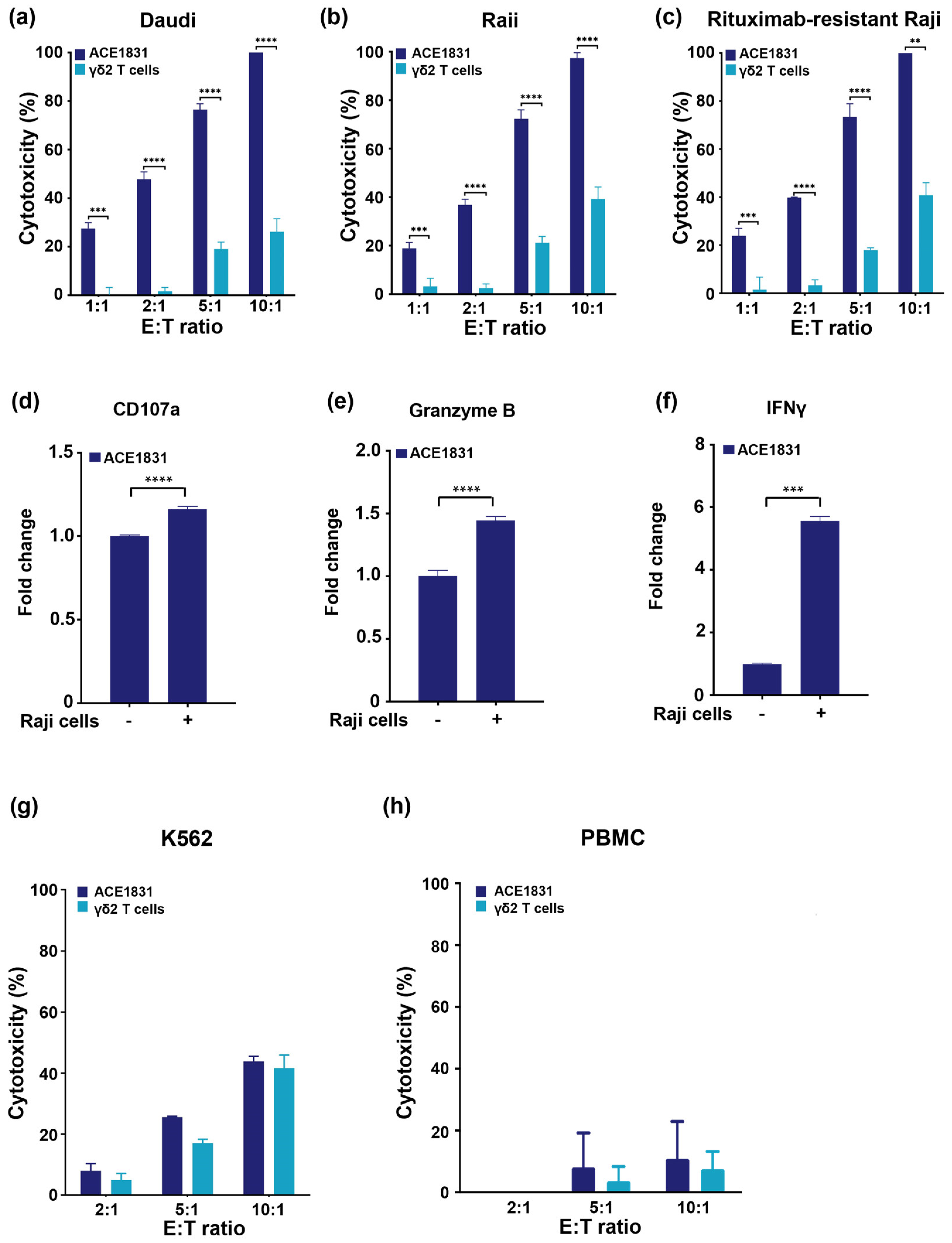
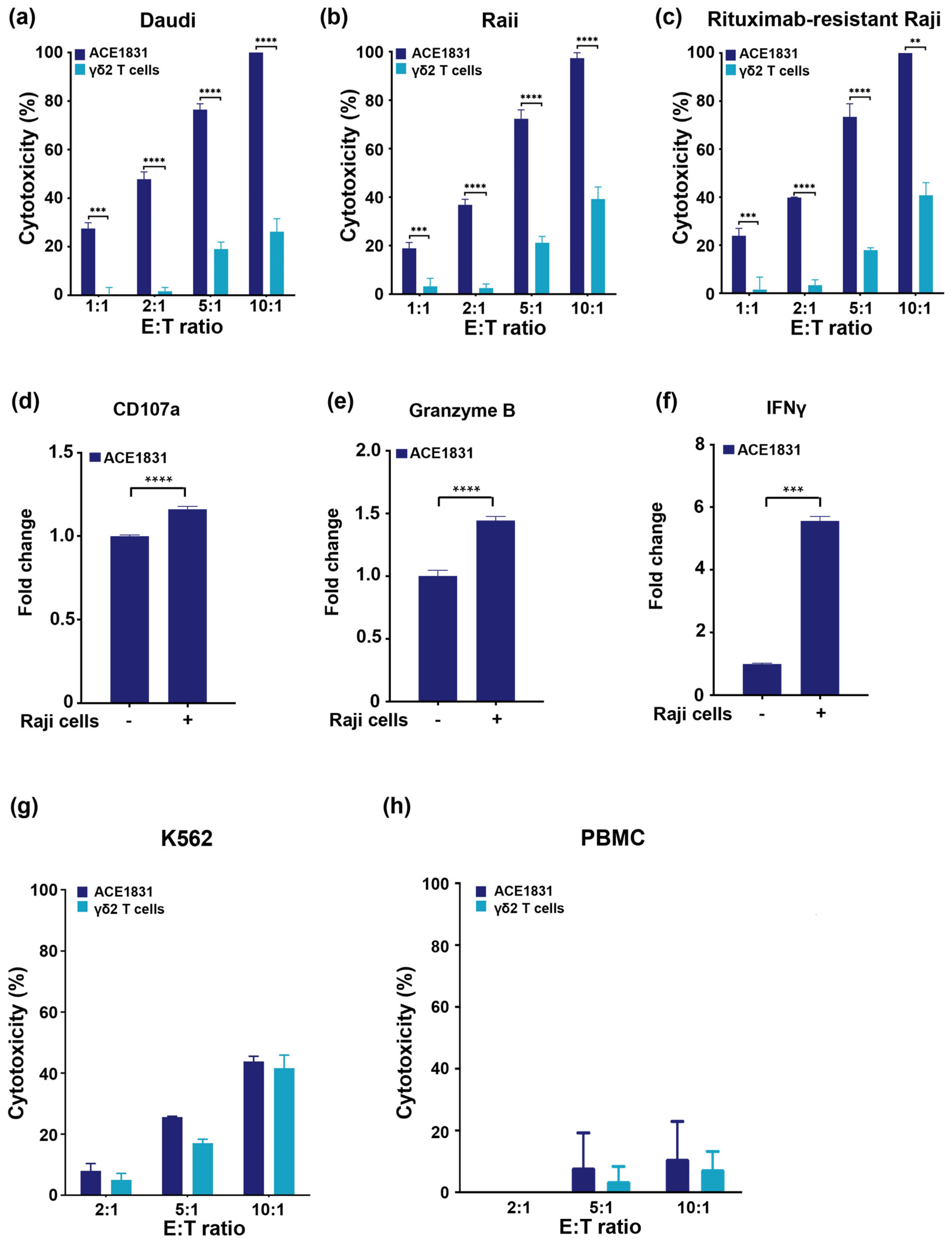
3.2. Enhanced Cytotoxicity of ACE1831 against CD20-Expressing Cancer Cells
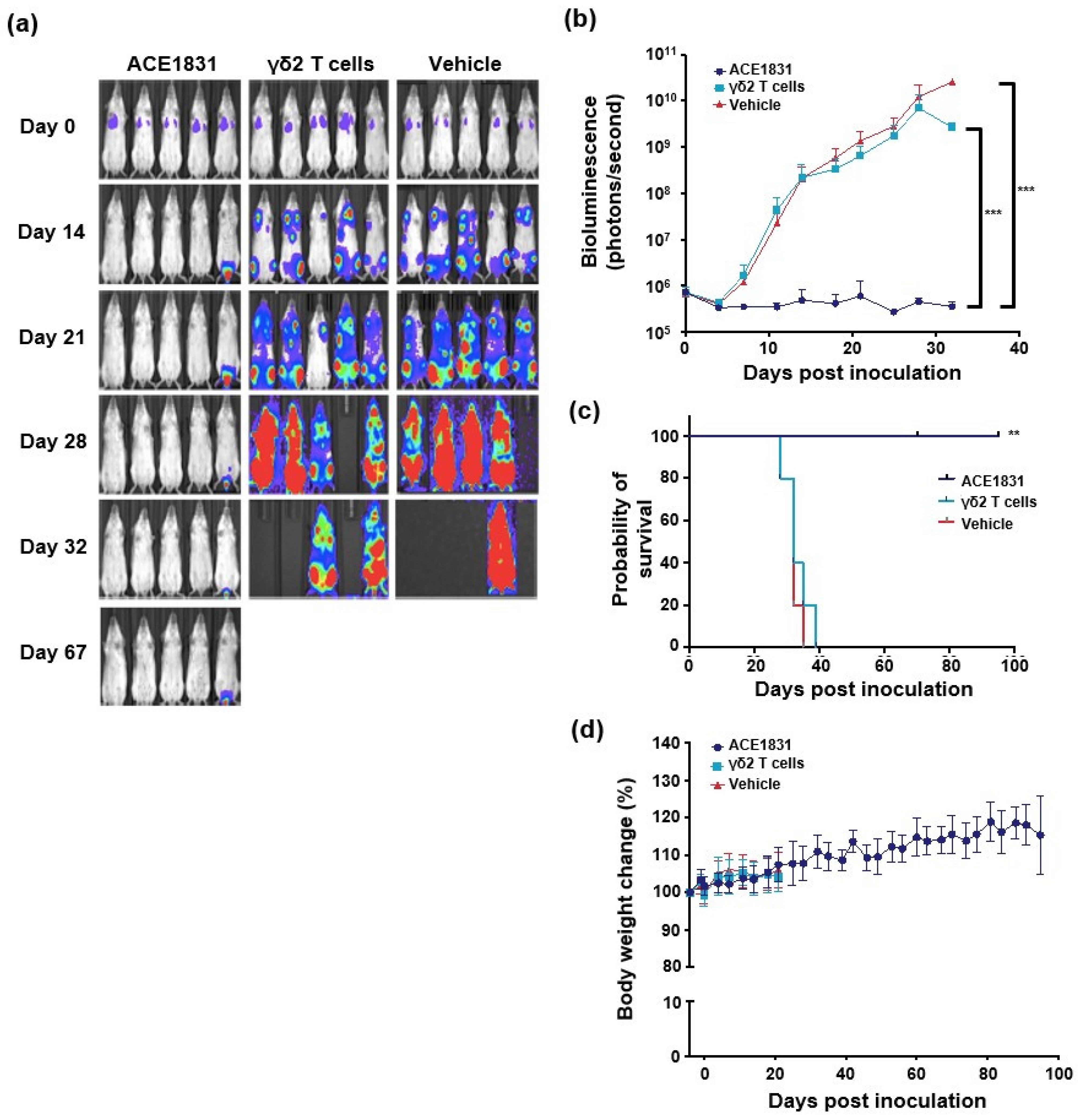
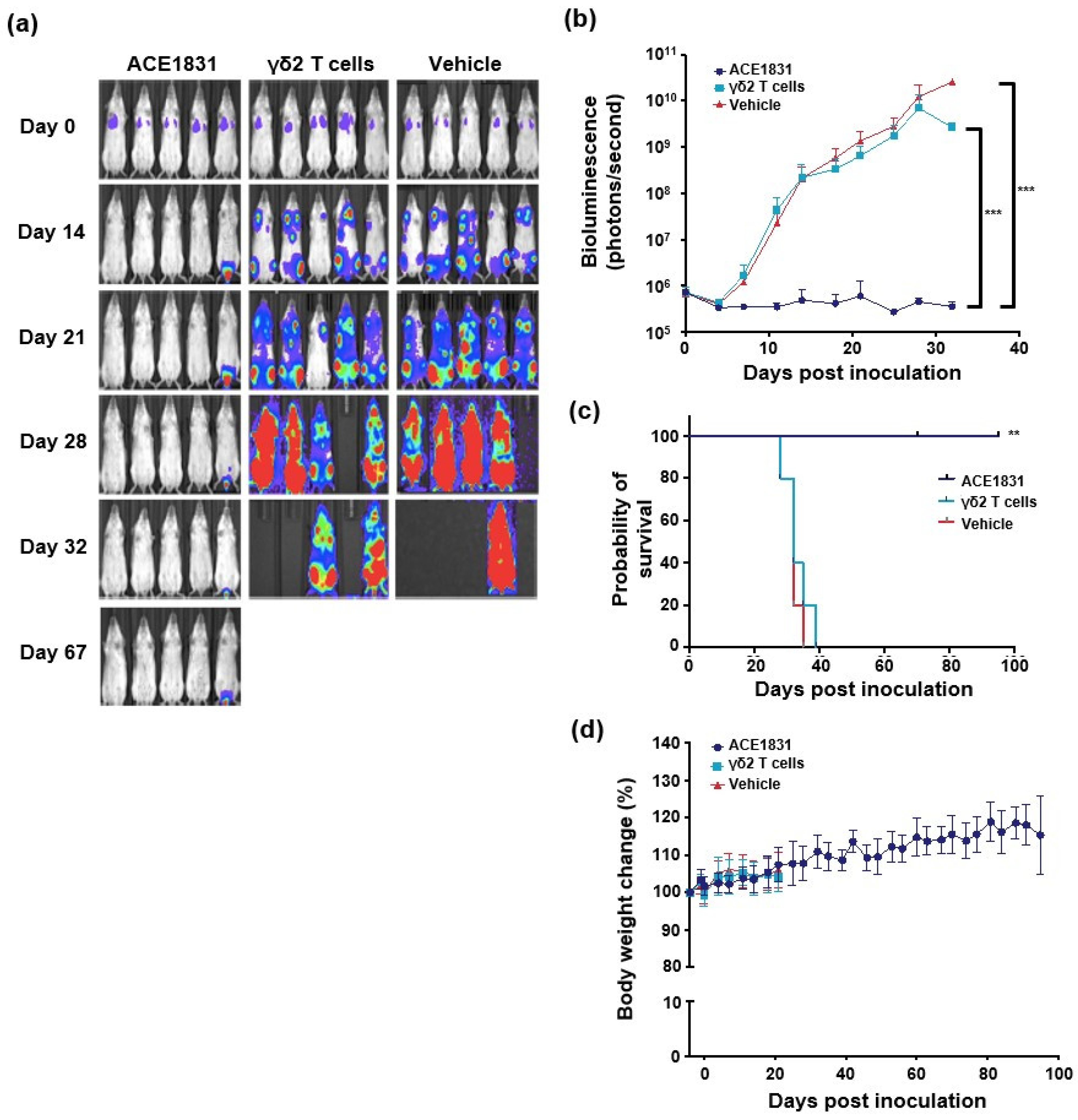
3.3. In Vivo Potency of ACE1831 against CD20-Expressing Cancer Cells
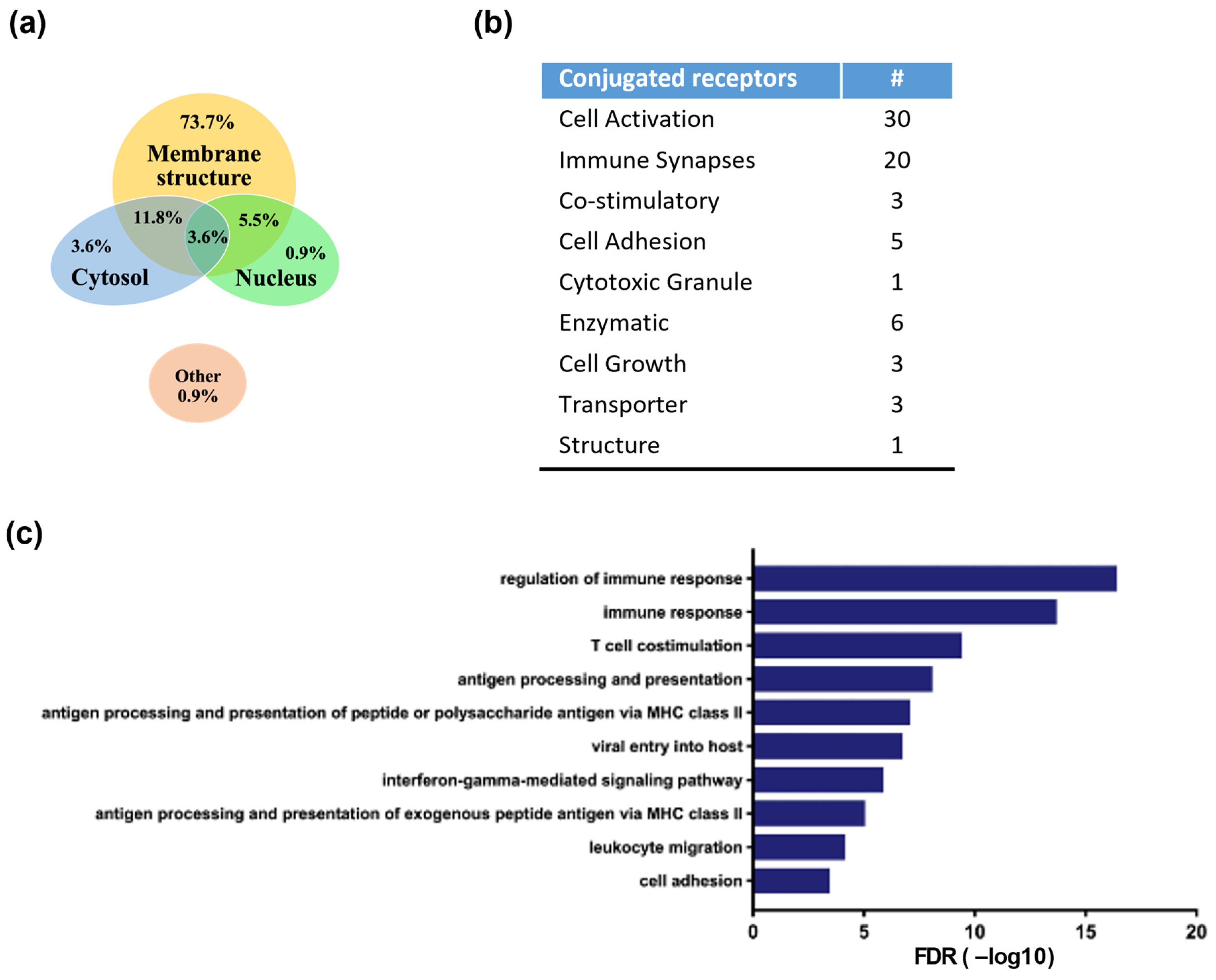
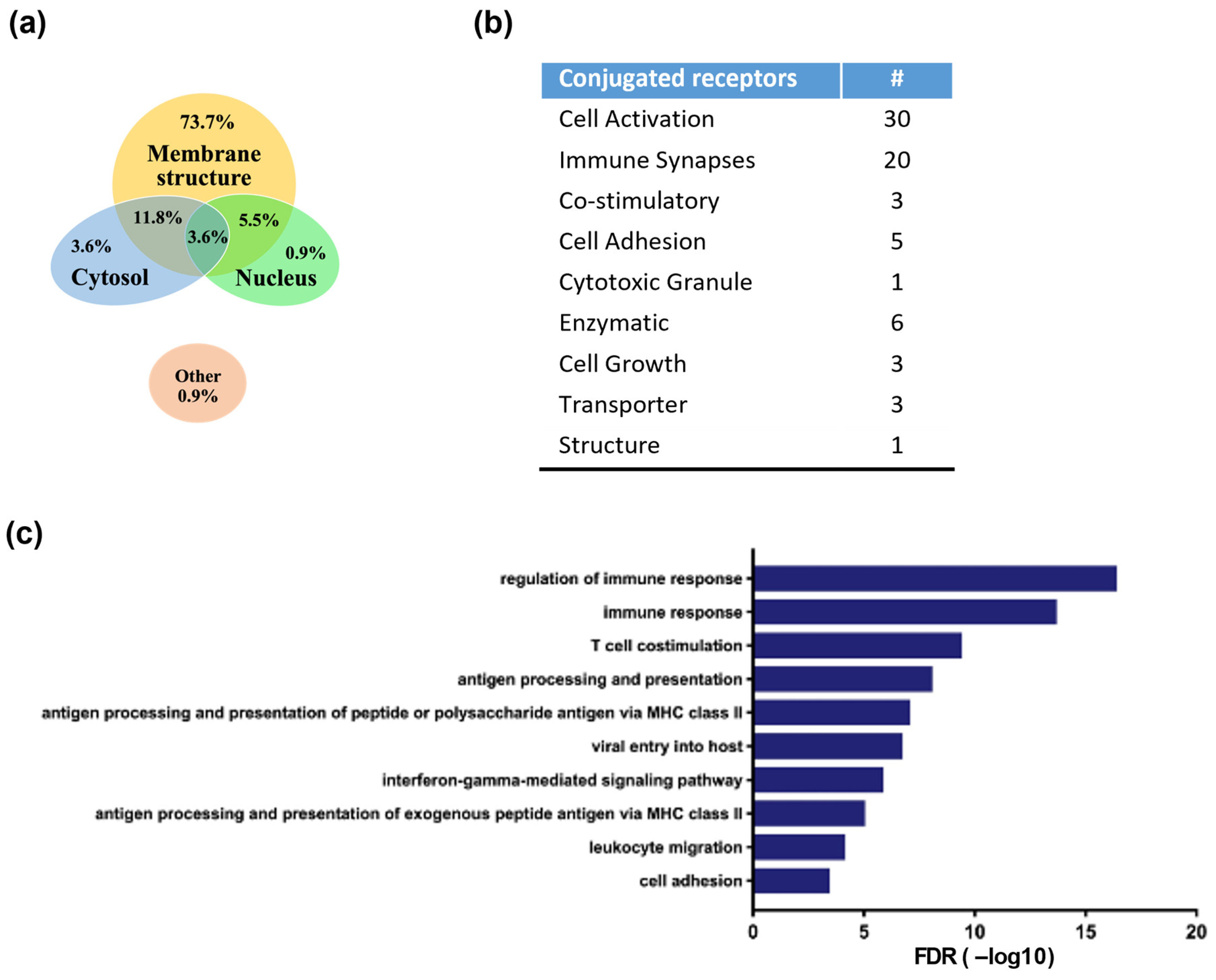
3.4. Identification of Antibody-Linked Surface Proteins of ACE1831
3.5. T Cell Activation and Cytotoxicity Mediated by Antigen Recognition of ACC-Linked Antibody
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Radestad, E.; Wikell, H.; Engstrom, M.; Watz, E.; Sundberg, B.; Thunberg, S.; Uzunel, M.; Mattsson, J.; Uhlin, M. Alpha/beta T-cell depleted grafts as an immunological booster to treat graft failure after hematopoietic stem cell transplantation with HLA-matched related and unrelated donors. J. Immunol. Res. 2014, 2014, 578741. [Google Scholar] [CrossRef] [PubMed]
- Graham, C.; Jozwik, A.; Pepper, A.; Benjamin, R. Allogeneic CAR-T Cells: More than Ease of Access? Cells 2018, 7, 155. [Google Scholar] [CrossRef] [PubMed]
- Ribot, J.C.; Lopes, N.; Silva-Santos, B. γδ T cells in tissue physiology and surveillance. Nat. Rev. Immunol. 2021, 21, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Meyer, C.; Bonneville, M. γδ T cells: First line of defense and beyond. Annu. Rev. Immunol. 2014, 32, 121–155. [Google Scholar] [CrossRef]
- Yazdanifar, M.; Barbarito, G.; Bertaina, A.; Airoldi, I. γδ T Cells: The Ideal Tool for Cancer Immunotherapy. Cells 2020, 9, 1305. [Google Scholar] [CrossRef]
- Zhao, Y.; Niu, C.; Cui, J. γδ T cells: Friend or foe in cancer development? J. Transl. Med. 2018, 16, 3. [Google Scholar] [CrossRef]
- Lamb, L.S.J.; Lopez, R.D. γδ T cells: A new frontier for immunotherapy? Biol. Blood Marrow. Transplant. 2005, 11, 161–168. [Google Scholar] [CrossRef]
- Handgretinger, R.; Schilbach, K. The potential role of gammadelta T cells after allogeneic HCT for leukemia. Blood 2018, 131, 1063–1072. [Google Scholar] [CrossRef]
- Minculescu, L.; Sengelov, H. The role of gamma delta T cells in haematopoietic stem cell transplantation. Scand J. Immunol. 2015, 81, 459–468. [Google Scholar] [CrossRef]
- Hoeres, T.; Smetak, M.; Pretscher, D.; Wilhelm, M. Improving the Efficiency of Vgamma9Vdelta2 T-Cell Immunotherapy in Cancer. Front. Immunol. 2018, 9, 800. [Google Scholar] [CrossRef]
- Xu, Y.; Xiang, Z.; Alnaggar, M.; Kouakanou, L.; Li, J.; He, J.; Yang, J.; Hu, Y.; Chen, Y.; Lin, L.; et al. Allogeneic Vγ9Vδ2 T-cell immunotherapy exhibits promising clinical safety and prolongs the survival of patients with late-stage lung or liver cancer. Cell Mol. Immunol. 2021, 18, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Rischer, M.; Pscherer, S.; Duwe, S.; Vormoor, J.; Jurgens, H.; Rossig, C. Human gammadelta T cells as mediators of chimaeric-receptor redirected anti-tumour immunity. Br. J. Haematol. 2004, 126, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Capsomidis, A.; Benthall, G.; Van Acker, H.H.; Fisher, J.; Kramer, A.M.; Abeln, Z.; Majani, Y.; Gileadi, T.; Wallace, R.; Gustafsson, K.; et al. Chimeric Antigen Receptor-Engineered Human Gamma Delta T Cells: Enhanced Cytotoxicity with Retention of Cross Presentation. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 354–365. [Google Scholar] [CrossRef]
- Deniger, D.C.; Switzer, K.; Mi, T.; Maiti, S.; Hurton, L.; Singh, H.; Huls, H.; Olivares, S.; Lee, D.A.; Champlin, R.E.; et al. Bispecific T-cells expressing polyclonal repertoire of endogenous gammadelta T-cell receptors and introduced CD19-specific chimeric antigen receptor. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 638–647. [Google Scholar] [CrossRef]
- Sletten, E.M.; Bertozzi, C.R. Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. Engl. 2009, 48, 6974–6998. [Google Scholar] [CrossRef]
- Saxon, E.; Bertozzi, C.R. Cell surface engineering by a modified Staudinger reaction. Science 2000, 287, 2007–2010. [Google Scholar] [CrossRef]
- Chandra, R.A.; Douglas, E.S.; Mathies, R.A.; Bertozzi, C.R.; Francis, M.B. Programmable cell adhesion encoded by DNA hybridization. Angew. Chem. Int. Ed. Engl. 2006, 45, 896–901. [Google Scholar] [CrossRef]
- Hsiao, S.C.; Shum, B.J.; Onoe, H.; Douglas, E.S.; Gartner, Z.J.; Mathies, R.A.; Bertozzi, C.R.; Francis, M.B. Direct cell surface modification with DNA for the capture of primary cells and the investigation of myotube formation on defined patterns. Langmuir ACS J. Surf. Colloids 2009, 25, 6985–6991. [Google Scholar] [CrossRef]
- Frank, M.J.; Olsson, N.; Huang, A.; Tang, S.W.; Negrin, R.S.; Elias, J.E.; Meyer, E.H. A novel antibody-cell conjugation method to enhance and characterize cytokine-induced killer cells. Cytotherapy 2020, 22, 135–143. [Google Scholar] [CrossRef]
- Li, H.K.; Hsiao, C.W.; Yang, S.H.; Yang, H.P.; Wu, T.S.; Lee, C.Y.; Lin, Y.L.; Pan, J.; Cheng, Z.F.; Lai, Y.D.; et al. A Novel off-the-Shelf Trastuzumab-Armed NK Cell Therapy (ACE1702) Using Antibody-Cell-Conjugation Technology. Cancers 2021, 13, 2724. [Google Scholar] [CrossRef]
- Olejniczak, S.H.; Hernandez-Ilizaliturri, F.J.; Clements, J.L.; Czuczman, M.S. Acquired resistance to rituximab is associated with chemotherapy resistance resulting from decreased Bax and Bak expression. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Alter, G.; Malenfant, J.M.; Altfeld, M. CD107a as a functional marker for the identification of natural killer cell activity. J. Immunol. Methods 2004, 294, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Betts, M.R.; Brenchley, J.M.; Price, D.A.; De Rosa, S.C.; Douek, D.C.; Roederer, M.; Koup, R.A. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J. Immunol. Methods 2003, 281, 65–78. [Google Scholar] [CrossRef]
- Seidel, U.J.; Vogt, F.; Grosse-Hovest, L.; Jung, G.; Handgretinger, R.; Lang, P. γδ T Cell-Mediated Antibody-Dependent Cellular Cytotoxicity with CD19 Antibodies Assessed by an Impedance-Based Label-Free Real-Time Cytotoxicity Assay. Front. Immunol. 2014, 5, 618. [Google Scholar] [CrossRef]
- De Bruin, R.C.G.; Lougheed, S.M.; van der Kruk, L.; Stam, A.G.; Hooijberg, E.; Roovers, R.C.; van Bergen En Henegouwen, P.M.P.; Verheul, H.M.W.; de Gruijl, T.D.; van der Vliet, H.J. Highly specific and potently activating Vgamma9Vdelta2-T cell specific nanobodies for diagnostic and therapeutic applications. Clin. Immunol. 2016, 169, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Hogan, P.G. Calcium-NFAT transcriptional signalling in T cell activation and T cell exhaustion. Cell Calcium 2017, 63, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Cano, C.E.; Pasero, C.; De Gassart, A.; Kerneur, C.; Gabriac, M.; Fullana, M.; Granarolo, E.; Hoet, R.; Scotet, E.; Rafia, C.; et al. BTN2A1, an immune checkpoint targeting Vγ9Vδ2 T cell cytotoxicity against malignant cells. Cell Rep. 2021, 36, 109359. [Google Scholar] [CrossRef]
- Dutta, I.; Postovit, L.M.; Siegers, G.M. Apoptosis Induced via γδ T Cell Antigen Receptor “Blocking” Antibodies: A Cautionary Tale. Front. Immunol. 2017, 8, 776. [Google Scholar] [CrossRef]
- Takayama, Y.; Kusamori, K.; Nishikawa, M. Click Chemistry as a Tool for Cell Engineering and Drug Delivery. Molecules 2019, 24, 172. [Google Scholar] [CrossRef]
- Kim, E.; Koo, H. Biomedical applications of copper-free click chemistry: In vitro, in vivo, and ex vivo. Chem. Sci. 2019, 10, 7835–7851. [Google Scholar] [CrossRef]
- Hill, J.A.; Seo, S.K. How I prevent infections in patients receiving CD19-targeted chimeric antigen receptor T cells for B-cell malignancies. Blood 2020, 136, 925–935. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, H.K. Function of gammadelta T cells in tumor immunology and their application to cancer therapy. Exp. Mol. Med. 2021, 53, 318–327. [Google Scholar] [CrossRef]
- Rozenbaum, M.; Meir, A.; Aharony, Y.; Itzhaki, O.; Schachter, J.; Bank, I.; Jacoby, E.; Besser, M.J. Gamma-Delta CAR-T Cells Show CAR-Directed and Independent Activity Against Leukemia. Front. Immunol. 2020, 11, 1347. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Chen, C.; Li, Z.; Zhu, S.; Tay, J.C.; Zhang, X.; Zha, S.; Zeng, J.; Tan, W.K.; Liu, X.; et al. Large-scale expansion of Vgamma9Vdelta2 T cells with engineered K562 feeder cells in G-Rex vessels and their use as chimeric antigen receptor-modified effector cells. Cytotherapy 2018, 20, 420–435. [Google Scholar] [CrossRef] [PubMed]
- Bonneville, M.; O’Brien, R.L.; Born, W.K. γδ T cell effector functions: A blend of innate programming and acquired plasticity. Nat. Rev. Immunol. 2010, 10, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Deniger, D.C.; Moyes, J.S.; Cooper, L.J. Clinical applications of γδ T cells with multivalent immunity. Front. Immunol. 2014, 5, 636. [Google Scholar] [CrossRef]
- Legut, M.; Cole, D.K.; Sewell, A.K. The promise of γδ T cells and the γδ T cell receptor for cancer immunotherapy. Cell. Mol. Immunol. 2015, 12, 656–668. [Google Scholar] [CrossRef]
- Kabelitz, D.; Serrano, R.; Kouakanou, L.; Peters, C.; Kalyan, S. Cancer immunotherapy with γδ T cells: Many paths ahead of us. Cell. Mol. Immunol. 2020, 17, 925–939. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Piha-Paul, S.A.; Mahalingam, D.; Mulcahy, M.F.; Kalyan, A.; Li, H.-K.; Wu, E.; Kurman, M.; Lee, S.; Lin, Y.-L.; Tang, S.-W.; et al. ACE1702, a first-in-class, off-the-shelf, selected natural killer cell [oNK] product using antibody cell conjugation technology [ACC], with pre-clinical and early clinical activity in HER2 < 3+ tumors. Ann. Oncol. 2021, 32, S851. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.-K.; Wu, T.-S.; Kuo, Y.-C.; Hsiao, C.-W.; Yang, H.-P.; Lee, C.-Y.; Leng, P.-J.; Chiang, Y.-J.; Cheng, Z.-F.; Yang, S.-H.; et al. A Novel Allogeneic Rituximab-Conjugated Gamma Delta T Cell Therapy for the Treatment of Relapsed/Refractory B-Cell Lymphoma. Cancers 2023, 15, 4844. https://doi.org/10.3390/cancers15194844
Li H-K, Wu T-S, Kuo Y-C, Hsiao C-W, Yang H-P, Lee C-Y, Leng P-J, Chiang Y-J, Cheng Z-F, Yang S-H, et al. A Novel Allogeneic Rituximab-Conjugated Gamma Delta T Cell Therapy for the Treatment of Relapsed/Refractory B-Cell Lymphoma. Cancers. 2023; 15(19):4844. https://doi.org/10.3390/cancers15194844
Chicago/Turabian StyleLi, Hao-Kang, Tai-Sheng Wu, Yi-Chiu Kuo, Ching-Wen Hsiao, Hsiu-Ping Yang, Chia-Yun Lee, Pei-Ju Leng, Yun-Jung Chiang, Zih-Fei Cheng, Sen-Han Yang, and et al. 2023. "A Novel Allogeneic Rituximab-Conjugated Gamma Delta T Cell Therapy for the Treatment of Relapsed/Refractory B-Cell Lymphoma" Cancers 15, no. 19: 4844. https://doi.org/10.3390/cancers15194844
APA StyleLi, H.-K., Wu, T.-S., Kuo, Y.-C., Hsiao, C.-W., Yang, H.-P., Lee, C.-Y., Leng, P.-J., Chiang, Y.-J., Cheng, Z.-F., Yang, S.-H., Lin, Y.-L., Chen, L.-Y., Chen, C.-S., Chen, Y.-J., Hsiao, S.-C., & Tang, S.-W. (2023). A Novel Allogeneic Rituximab-Conjugated Gamma Delta T Cell Therapy for the Treatment of Relapsed/Refractory B-Cell Lymphoma. Cancers, 15(19), 4844. https://doi.org/10.3390/cancers15194844