
The Evolving Pathways of the Efficacy of and Resistance to CDK4/6 Inhibitors in Breast Cancer


Simple Summary
Abstract
1. Introduction
2. Selective CDK4/6i
2.1. Selective CDK4/6i Approved to Treat Breast Cancer
2.1.1. Palbociclib
2.1.2. Ribociclib
2.1.3. Abemaciclib
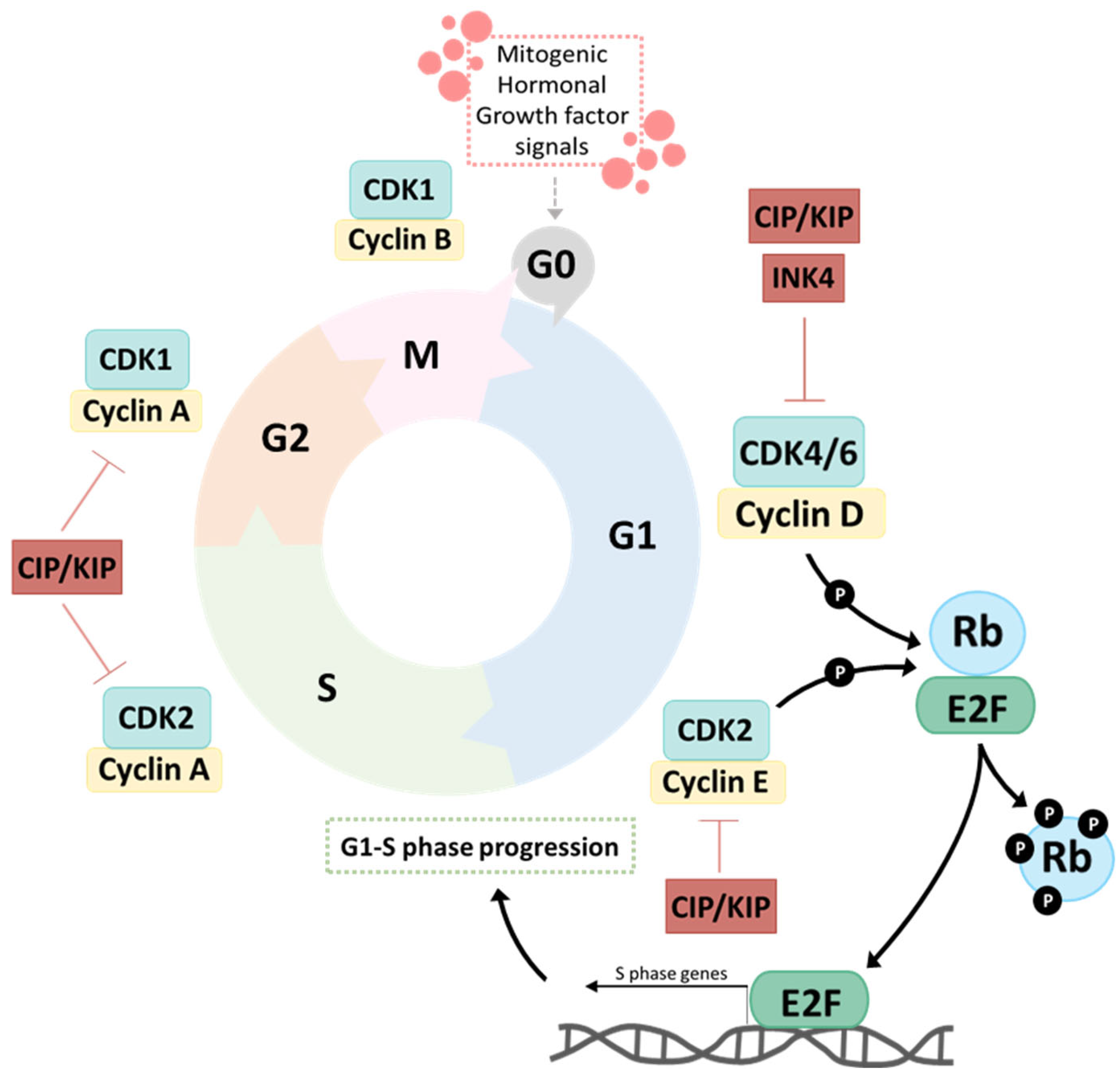
2.2. Mechanism of Action of CDK4/6i
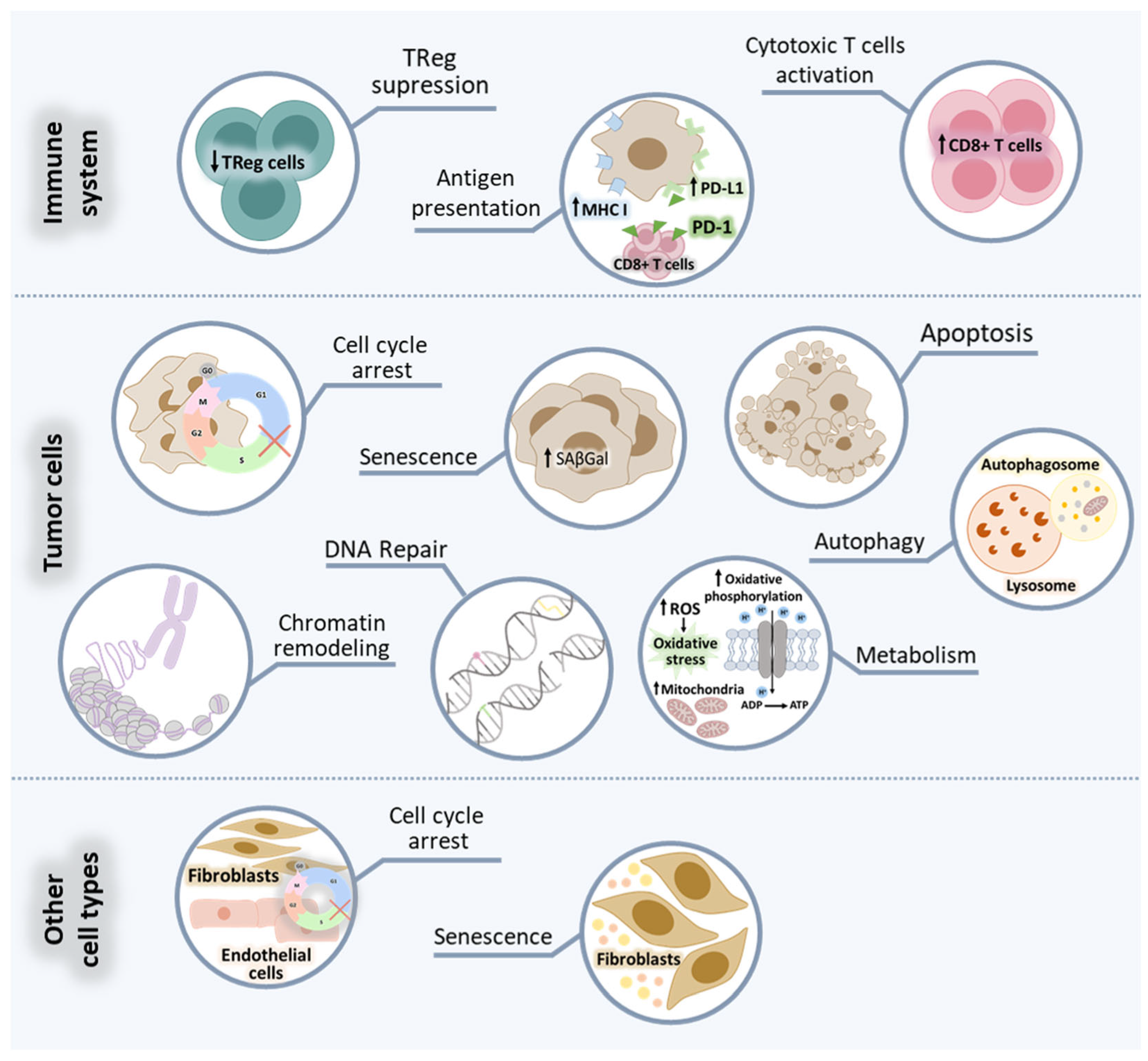
2.2.1. Effect on Tumor Cells
2.2.2. Effect on Immune Cells
2.2.3. Effect on Other Cell Types
3. Mechanisms of Resistance to CDK4/6i and Possible Strategies to Overcome Them
3.1. Mechanisms of Resistance to CDK4/6i
3.1.1. Retinoblastoma Protein (Rb)
3.1.2. Cyclin D–CDK4–CDK6 Axis
3.1.3. Cyclin E–CDK2 Axis
3.1.4. CDK7
3.1.5. INK4 and CIP/KIP Families of CDK Suppressors
3.1.6. Other Cell Cycle Regulators
WEE1
MDM2-TP53
APC/C-FZR1
AP-1
TK1
Aurora Kinase A
c-Myc
c-MET/FAK
3.1.7. Hormone Receptors and HER2
3.1.8. PI3K-AKT-mTOR Pathway
3.1.9. FGFR Pathway
3.1.10. MAPK-ERK Pathway
3.1.11. RANK-RANKL Pathway
3.1.12. Autophagy
3.1.13. TGF-β and EMT
3.1.14. ABC Transporters
3.2. Emerging Strategies to Overcome Resistance to CDK4/6i
3.2.1. Endocrine Therapy
3.2.2. PI3K-AKT-mTOR Inhibitors
3.2.3. FGFR Inhibitors
3.2.4. Immunotherapy
3.2.5. Chemotherapy
3.2.6. CDK7 Inhibitors
3.2.7. BCL2 Inhibitors
3.2.8. Aurora Kinase A Inhibitors
3.2.9. Histone Deacetylase (HDAC) Inhibitors

{kind=link}
{kind=link}
| Therapy | Clinical Setting | Phase | Intervention | Trial Identifier | Status * | Results |
|---|---|---|---|---|---|---|
| Endocrine therapy | Disease progression under ET+ CDK4/6i. | II | Fulvestrant or exemestane vs. fulvestrant or exemestane + ribociclib | MAINTAIN (NCT02632045) | Completed | Fulvestrant or exemestane + ribociclib increased PFS compared to ET alone [223]. |
| Disease progression under ET. | Ib/II | Bazedoxifene + palbociclib | NCT02448771 | Completed | Clinical benefit, complete/partial response or stable disease were observed [227]. | |
| Disease progression under ET. | I | LSZ102 vs. LSZ102 + ribociclib vs. LSZ102 + alpelisib | NCT02734615 | Completed | LSZ102 well tolerated alone or in combination. Preliminary clinical activity observed in combination groups [224]. | |
| Disease progression under ET + CDK4/6i with ESR1 mutations. | III | Elacestrant vs. fulvestrant or AI | EMERALD (NCT03778931) | Active, not recruiting | PFS improvement in elacestrant arm [225]. | |
| Disease progression under ET. | I | Rintodestrant vs. rintodestrant + palbociclib | NCT03455270 | Active, not recruiting | Rintodestrant demonstrated a safety/tolerability profile as monotherapy or combined with palbociclib [251]. | |
| Disease progression under ET + CDK4/6i. | III | ET vs. imlunestrant vs. imlunestrant + abemaciclib | EMBER-3 (NCT04975308) | Recruiting | N/A | |
| Disease progression under AI + Palbociclib or Abemaciclib with ESR1 Mutations. | III | Camizestrant + palbociclib or abemaciclib vs. AI + palbociclib or abemaciclib | SERENA-6 (NCT04964934) | Recruiting | N/A | |
| Disease progression under ET + CDK4/6i. | Ib/II | Giredestrant vs. giredestrant + abemaciclib or ipatasertib or inavolisib or ribociclib or everolimus or samuraciclib or atezolizumab or abemaciclib + atezolizumab | MORPHEUS (NCT04802759) | Recruiting | N/A | |
| Disease progression under ET + CDK4/6i. | III | Giredestrant + everolimus vs. everolimus + exemestane | NCT05306340 | Recruiting | NA | |
| Disease progression under AI + palbociclib or ribociclib with ESR1 mutations. | III | Lasofoxifene + abemaciclib vs. fulvestrant + abemacicclib | ELAINE III (NCT05696626) | Not yet recruiting | N/A | |
| PI3K-AKT-mTOR inhibitors | Disease progression under ET or ET + CDK4/6i. | I/II | Exemestane + ribociclib + everolimus | TRINITI-1 (NCT02732119) | Completed | Triple combination with clinical benefit at 24 weeks [233]. |
| Disease progression under AI. | III | Fulvestrant vs. fulvestrant + capivasertib | CAPItello-291 (NCT04305496) | Active, not recruiting | Capivasertib + fulvestrant increased PFS compared with fulvestrant alone [231]. | |
| Disease progression under AI + CDK4/6i with PIK3CA mutations. | II | Alpelisib + fulvestrant or letrozole | BYLieve (NCT03056755) | Active, not recruiting | Alpelisib demonstrated clinical activity in combination with fulvestrant or letrozole [252]. | |
| Disease progression under ET or ET+ CDK4/6i. | Ib/II | Ipatasertib + fulvestrant or letrozole vs. ipatasertib + fulvestrant or letrozole + palbociclib | TAKTIC (NCT03959891) | Active, not recruiting | Triple combination well tolerated. Partial response or stable disease observed [230]. | |
| Disease progression under AI + CDK4/6i with PIK3CA Mutations. | III | Fulvestrant vs. alpelisib + fulvestrant | EPIK-B5 (NCT05038735) | Recruiting | N/A | |
| Disease progression under ET + CDK4/6i. | I | Inavolisib + letrozole/fulvestrant | NCT03006172 | Recruiting | NA | |
| Disease progression under AI + CDK4/6i. | III | Fulvestrant vs. ipatasertib + fulvestrant | NCT04650581 | Recruiting | NA | |
| FGFR inhibitors | Disease progression under ET + CDK4/6i with FGFR amplification. | Ib | Erdafitinib + fulvestrant + palbociclib | NCT03238196 | Active, not recruiting | Increased PFS in patients with high levels of FGFR1 amplification [234]. |
| Immunotherapy | Disease progression under AI + CDK4/6i. | II | Fulvestrant vs. fulvestrant + palbociclib vs. fulvestrant + palbociclib + avelumab | PACE (NCT03147287) | Active, not recruiting | Increased PFS in the triple-combination group [240]. |
| Disease progression under ET + CDK4/6i with PD-L1 Expression. | III | CT vs. CT + pembrolizumab | KEYNOTE-B49 (NCT04895358) | Recruiting | N/A | |
| Chemotherapy | Disease progression under ET + CDK4/6i. | II | Pembrolizumab + paclitaxel | TATEN (NCT04251169) | Active, not recruiting | N/A |
| Disease progression under ET + CDK4/6i. | I | ASTX727 + talazoparib | NCT04134884 | Recruiting | N/A | |
| Disease progression under ET + CDK4/6i. | I/II | CT7001 + fulvestrant | NCT03363893 | Completed | Tolerable safety [253]. | |
| CDK7 inhibitors | Disease progression under ET + CDK4/6i. | I | SY-5609 + fulvestrant | NCT04247126 | Active, not recruiting | N/A |
| Disease progression under CDK4/6i. | II | Fulvestrant vs. fulventrant + venetoclax | VERONICA (NCT03584009) | Completed | No significant difference in PFS [246]. | |
| BCL2 inhibitors | Disease progression under ET with BCL2 expression. | Ib | Letrozole + palbociclib + venetoclax | PALVEN (NCT03900884) | Recruiting | N/A |
| Disease progression under ET + CDK4/6i. | Ib | Erbumine vs. erbumine + ET | NCT03955939 | Completed | N/A | |
| Aurora kinase A inhibitors | Patients pretreated with CDK4/6i. | I | Xentuzumab + abemaciclib | NCT03099174 | Active, not recruiting | NA |
| IGF inhibitors | Disease progression under ET + CDK4/6i. | II | PF-06873600 vs. PF-06873600 + ET | NCT03519178 | PF-06873600 vs. PF-06873600 + ET | N/A |
| CDK4/6 inhibitors | Disease progression under AI or tamoxifen ± LHRHa + CDK4/6i. | II | Palbociclib + fulvestrant | NCT04318223 | Palbociclib + fulvestrant | N/A |
| Clinical benefit of 1st line palbociclib + ET. | II | Palbociclib rechallenge + ET | PALMIRA (NCT03809988) | Clinical benefit of 1st line Palbociclib + ET | N/A | |
| Disease progression under ET + CDK4/6i. | III | Abemaciclib + fulvestrant | Post-MONARCH (NCT05169567) | Recruiting | NA | |
| ADC | Disease progression under ET + CDK4/6i and CT. | III | Sacituzumab govitecan vs. CT | TROPiCS-02 (NCT03901339) | Active, not recruiting | Increased OS in the sacituzumab govitecan group [254]. |
| Chemotherapy naïve disease progression under ET with HR+/Her-2 low or ultralow. | III | T-DXD vs. investigator choice chemotherapy | DB-06 (NCT04494425) | Recruiting | NA | |
| Endocrine resistant disease. | III | Dato-Dxd | TROPION Breast01 (NCT04494425) | Recruiting | NA |
4. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Nurse, P.; Thuriaux, P.; Nasmyth, K. Genetic control of the cell division cycle in the fission yeast Schizosaccharomyces pombe. Mol. Gen. Genet. 1976, 146, 167–178. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Mortimer, R.K.; Culotti, J.; Culotti, M. Genetic Control of the Cell Division Cycle in Yeast: V. Genetic Analysis of cdc Mutants. Genetics 1973, 74, 267–286. [Google Scholar] [CrossRef]
- Evans, T.; Rosenthal, E.T.; Youngblom, J.; Distel, D.; Hunt, T. Cyclin: A protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell 1983, 33, 389–396. [Google Scholar] [CrossRef]
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2022, 23, 74–88. [Google Scholar] [CrossRef]
- Bury, M.; Le Calve, B.; Ferbeyre, G.; Blank, V.; Lessard, F. New Insights into CDK Regulators: Novel Opportunities for Cancer Therapy. Trends Cell Biol. 2021, 31, 331–344. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Sicinski, P.; Donaher, J.L.; Parker, S.B.; Li, T.; Fazeli, A.; Gardner, H.; Haslam, S.Z.; Bronson, R.T.; Elledge, S.J.; Weinberg, R.A. Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell 1995, 82, 621–630. [Google Scholar] [CrossRef]
- Fantl, V.; Stamp, G.; Andrews, A.; Rosewell, I.; Dickson, C. Mice lacking cyclin D1 are small and show defects in eye and mammary gland development. Genes. Dev. 1995, 9, 2364–2372. [Google Scholar] [CrossRef]
- Yu, Q.; Sicinska, E.; Geng, Y.; Ahnstrom, M.; Zagozdzon, A.; Kong, Y.; Gardner, H.; Kiyokawa, H.; Harris, L.N.; Stal, O.; et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell 2006, 9, 23–32. [Google Scholar] [CrossRef]
- Yu, Q.; Geng, Y.; Sicinski, P. Specific protection against breast cancers by cyclin D1 ablation. Nature 2001, 411, 1017–1021. [Google Scholar] [CrossRef]
- Choi, Y.J.; Li, X.; Hydbring, P.; Sanda, T.; Stefano, J.; Christie, A.L.; Signoretti, S.; Look, A.T.; Kung, A.L.; von Boehmer, H.; et al. The requirement for cyclin D function in tumor maintenance. Cancer Cell 2012, 22, 438–451. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Buckley, M.F.; Sweeney, K.J.; Hamilton, J.A.; Sini, R.L.; Manning, D.L.; Nicholson, R.I.; deFazio, A.; Watts, C.K.; Musgrove, E.A.; Sutherland, R.L. Expression and amplification of cyclin genes in human breast cancer. Oncogene 1993, 8, 2127–2133. [Google Scholar] [PubMed]
- Spring, L.M.; Wander, S.A.; Andre, F.; Moy, B.; Turner, N.C.; Bardia, A. Cyclin-dependent kinase 4 and 6 inhibitors for hormone receptor-positive breast cancer: Past, present, and future. Lancet 2020, 395, 817–827. [Google Scholar] [CrossRef]
- Gennari, A.; Andre, F.; Barrios, C.H.; Cortes, J.; de Azambuja, E.; DeMichele, A.; Dent, R.; Fenlon, D.; Gligorov, J.; Hurvitz, S.A.; et al. ESMO Clinical Practice Guideline for the diagnosis, staging and treatment of patients with metastatic breast cancer. Ann. Oncol. 2021, 32, 1475–1495. [Google Scholar] [CrossRef]
- Lu, Y.S.; Im, S.A.; Colleoni, M.; Franke, F.; Bardia, A.; Cardoso, F.; Harbeck, N.; Hurvitz, S.; Chow, L.; Sohn, J.; et al. Updated Overall Survival of Ribociclib plus Endocrine Therapy versus Endocrine Therapy Alone in Pre- and Perimenopausal Patients with HR+/HER2(−) Advanced Breast Cancer in MONALEESA-7: A Phase III Randomized Clinical Trial. Clin. Cancer Res. 2022, 28, 851–859. [Google Scholar] [CrossRef]
- Johnston, S.; Martin, M.; Di Leo, A.; Im, S.A.; Awada, A.; Forrester, T.; Frenzel, M.; Hardebeck, M.C.; Cox, J.; Barriga, S.; et al. MONARCH 3 final PFS: A randomized study of abemaciclib as initial therapy for advanced breast cancer. NPJ Breast Cancer 2019, 5, 5. [Google Scholar] [CrossRef]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Ammazzalorso, A.; Agamennone, M.; De Filippis, B.; Fantacuzzi, M. Development of CDK4/6 Inhibitors: A Five Years Update. Molecules 2021, 26, 1488. [Google Scholar] [CrossRef]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef]
- Goel, S.; Bergholz, J.S.; Zhao, J.J. Targeting CDK4 and CDK6 in cancer. Nat. Rev. Cancer 2022, 22, 356–372. [Google Scholar] [CrossRef]
- Jhaveri, K.; Burris Rd, H.A.; Yap, T.A.; Hamilton, E.; Rugo, H.S.; Goldman, J.W.; Dann, S.; Liu, F.; Wong, G.Y.; Krupka, H.; et al. The evolution of cyclin dependent kinase inhibitors in the treatment of cancer. Expert. Rev. Anticancer Ther. 2021, 21, 1105–1124. [Google Scholar] [CrossRef]
- Shah, A.; Bloomquist, E.; Tang, S.; Fu, W.; Bi, Y.; Liu, Q.; Yu, J.; Zhao, P.; Palmby, T.R.; Goldberg, K.B.; et al. FDA Approval: Ribociclib for the Treatment of Postmenopausal Women with Hormone Receptor-Positive, HER2-Negative Advanced or Metastatic Breast Cancer. Clin. Cancer Res. 2018, 24, 2999–3004. [Google Scholar] [CrossRef]
- Beaver, J.A.; Amiri-Kordestani, L.; Charlab, R.; Chen, W.; Palmby, T.; Tilley, A.; Zirkelbach, J.F.; Yu, J.; Liu, Q.; Zhao, L.; et al. FDA Approval: Palbociclib for the Treatment of Postmenopausal Patients with Estrogen Receptor-Positive, HER2-Negative Metastatic Breast Cancer. Clin. Cancer Res. 2015, 21, 4760–4766. [Google Scholar] [CrossRef]
- Kim, E.S. Abemaciclib: First Global Approval. Drugs 2017, 77, 2063–2070. [Google Scholar] [CrossRef]
- Mills, J.N.; Rutkovsky, A.C.; Giordano, A. Mechanisms of resistance in estrogen receptor positive breast cancer: Overcoming resistance to tamoxifen/aromatase inhibitors. Curr. Opin. Pharmacol. 2018, 41, 59–65. [Google Scholar] [CrossRef]
- Hanker, A.B.; Sudhan, D.R.; Arteaga, C.L. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 2020, 37, 496–513. [Google Scholar] [CrossRef]
- Belachew, E.B.; Sewasew, D.T. Molecular Mechanisms of Endocrine Resistance in Estrogen-Positive Breast Cancer. Front Endocrinol 2021, 12, 599586. [Google Scholar] [CrossRef]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009, 11, R77. [Google Scholar] [CrossRef]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; Andre, F.; Winer, E.P.; et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1738–1748. [Google Scholar] [CrossRef]
- Torres-Guzman, R.; Calsina, B.; Hermoso, A.; Baquero, C.; Alvarez, B.; Amat, J.; McNulty, A.M.; Gong, X.; Boehnke, K.; Du, J.; et al. Preclinical characterization of abemaciclib in hormone receptor positive breast cancer. Oncotarget 2017, 8, 69493–69507. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar] [CrossRef]
- Finn, R.S.; Crown, J.P.; Ettl, J.; Schmidt, M.; Bondarenko, I.M.; Lang, I.; Pinter, T.; Boer, K.; Patel, R.; Randolph, S.; et al. Efficacy and safety of palbociclib in combination with letrozole as first-line treatment of ER-positive, HER2-negative, advanced breast cancer: Expanded analyses of subgroups from the randomized pivotal trial PALOMA-1/TRIO-18. Breast Cancer Res. 2016, 18, 67. [Google Scholar] [CrossRef]
- Finn, R.S.; Rugo, H.S.; Dieras, V.C.; Harbeck, N.; Im, S.A.; Gelmon, K.A.; Walshe, J.M.; Martin, M.; Mac Gregor, M.C.; Bananis, E.; et al. Overall survival (OS) with first-line palbociclib plus letrozole (PAL plus LET) versus placebo plus letrozole (PBO plus LET) in women with estrogen receptor positive/human epidermal growth factor receptor 2-negative advanced breast cancer (ER+/HER2-ABC): Analyses from PALOMA-2. J. Clin. Oncol. 2022, 40, LBA1003. [Google Scholar] [CrossRef]
- Turner, N.C.; Slamon, D.J.; Ro, J.; Bondarenko, I.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 2018, 379, 1926–1936. [Google Scholar] [CrossRef]
- Turner, N.C.; Huang Bartlett, C.; Cristofanilli, M. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2015, 373, 1672–1673. [Google Scholar] [CrossRef]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.A.; Petrakova, K.; Bianchi, G.V.; Esteva, F.J.; Martin, M.; et al. Phase III Randomized Study of Ribociclib and Fulvestrant in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: MONALEESA-3. J. Clin. Oncol. 2018, 36, 2465–2472. [Google Scholar] [CrossRef]
- Tripathy, D.; Im, S.A.; Colleoni, M.; Franke, F.; Bardia, A.; Harbeck, N.; Hurvitz, S.A.; Chow, L.; Sohn, J.; Lee, K.S.; et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): A randomised phase 3 trial. Lancet Oncol. 2018, 19, 904–915. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Hart, L.; Campone, M.; Petrakova, K.; Winer, E.P.; Janni, W.; et al. Overall Survival with Ribociclib plus Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2022, 386, 942–950. [Google Scholar] [CrossRef]
- Im, S.A.; Lu, Y.S.; Bardia, A.; Harbeck, N.; Colleoni, M.; Franke, F.; Chow, L.; Sohn, J.; Lee, K.S.; Campos-Gomez, S.; et al. Overall Survival with Ribociclib plus Endocrine Therapy in Breast Cancer. N. Engl. J. Med. 2019, 381, 307–316. [Google Scholar] [CrossRef]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.A.; Petrakova, K.; Bianchi, G.V.; Esteva, F.J.; Martin, M.; et al. Overall Survival with Ribociclib plus Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 2020, 382, 514–524. [Google Scholar] [CrossRef]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Tredan, O.; Chen, S.C.; Manso, L.; et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef]
- Johnston, S.; Puhalla, S.; Wheatley, D.; Ring, A.; Barry, P.; Holcombe, C.; Boileau, J.F.; Provencher, L.; Robidoux, A.; Rimawi, M.; et al. Randomized Phase II Study Evaluating Palbociclib in Addition to Letrozole as Neoadjuvant Therapy in Estrogen Receptor-Positive Early Breast Cancer: PALLET Trial. J. Clin. Oncol. 2019, 37, 178–189. [Google Scholar] [CrossRef]
- Johnston, S.R.D.; Toi, M.; O’Shaughnessy, J.; Rastogi, P.; Campone, M.; Neven, P.; Huang, C.S.; Huober, J.; Jaliffe, G.G.; Cicin, I.; et al. Abemaciclib plus endocrine therapy for hormone receptor-positive, HER2-negative, node-positive, high-risk early breast cancer (monarchE): Results from a preplanned interim analysis of a randomised, open-label, phase 3 trial. Lancet Oncol. 2023, 24, 77–90. [Google Scholar] [CrossRef]
- Sledge, G.W.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Overall survival of abemaciclib plus fulvestrant in patients with HR+, HER2-advanced breast cancer. Ann. Oncol. 2019, 30, 856. [Google Scholar] [CrossRef]
- Sledge, G.W., Jr.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in Combination With Fulvestrant in Women With HR+/HER2- Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef]
- George, M.A.; Qureshi, S.; Omene, C.; Toppmeyer, D.L.; Ganesan, S. Clinical and Pharmacologic Differences of CDK4/6 Inhibitors in Breast Cancer. Front. Oncol. 2021, 11, 693104. [Google Scholar] [CrossRef]
- Panagiotou, E.; Gomatou, G.; Trontzas, I.P.; Syrigos, N.; Kotteas, E. Cyclin-dependent kinase (CDK) inhibitors in solid tumors: A review of clinical trials. Clin. Transl. Oncol. 2022, 24, 161–192. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Petrakova, K.; Blackwell, K.L.; Winer, E.P.; et al. Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann. Oncol. 2018, 29, 1541–1547. [Google Scholar] [CrossRef]
- Royce, M.; Osgood, C.; Mulkey, F.; Bloomquist, E.; Pierce, W.F.; Roy, A.; Kalavar, S.; Ghosh, S.; Philip, R.; Rizvi, F.; et al. FDA Approval Summary: Abemaciclib With Endocrine Therapy for High-Risk Early Breast Cancer. J. Clin. Oncol. 2022, 40, 1155–1162. [Google Scholar] [CrossRef]
- Wander, S.A.; O’Brien, N.; Litchfield, L.M.; O’Dea, D.; Morato Guimaraes, C.; Slamon, D.J.; Goel, S. Targeting CDK4 and 6 in Cancer Therapy: Emerging Preclinical Insights Related to Abemaciclib. Oncologist 2022, 27, 811–821. [Google Scholar] [CrossRef]
- Dickler, M.N.; Tolaney, S.M.; Rugo, H.S.; Cortes, J.; Dieras, V.; Patt, D.; Wildiers, H.; Hudis, C.A.; O’Shaughnessy, J.; Zamora, E.; et al. MONARCH 1, A Phase II Study of Abemaciclib, a CDK4 and CDK6 Inhibitor, as a Single Agent, in Patients with Refractory HR+/HER2- Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 5218–5224. [Google Scholar] [CrossRef]
- Goetz, M.P.; Toi, M.; Huober, J.; Sohn, J.; Tredan, O.; Park, I.H.; Campone, M.; Chen, S.C.; Sanchez, L.M.M.; Paluch-Shimon, S.; et al. MONARCH 3: Interim overall survival (OS) results of abemaciclib plus a nonsteroidal aromatase inhibitor (NSAI) in patients (pts) with HR+, HER2-advanced breast cancer (ABC). Ann. Oncol. 2022, 33, S1384. [Google Scholar] [CrossRef]
- Al-Saleh, K.A.; Zawahry, H.M.E.; Bounedjar, A.; Oukkal, M.; Saadeddin, A.; Mahfouf, H.; Kamel, B.; Bensalem, A.; Abdel-Razeq, H.; Kandil, A.; et al. Final result for SAFIA trial for neoadjuvant palbociclib in patients with operable luminal breast cancer responding to fulvestrant. J. Clin. Oncol. 2022, 40, 596. [Google Scholar] [CrossRef]
- Prat, A.; Saura, C.; Pascual, T.; Hernando, C.; Munoz, M.; Pare, L.; Gonzalez Farre, B.; Fernandez, P.L.; Galvan, P.; Chic, N.; et al. Ribociclib plus letrozole versus chemotherapy for postmenopausal women with hormone receptor-positive, HER2-negative, luminal B breast cancer (CORALLEEN): An open-label, multicentre, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 33–43. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Martin, M.; Press, M.F.; Chan, D.; Fernandez-Abad, M.; Petru, E.; Rostorfer, R.; Guarneri, V.; Huang, C.S.; Barriga, S.; et al. Potent Cell-Cycle Inhibition and Upregulation of Immune Response with Abemaciclib and Anastrozole in neoMONARCH, Phase II Neoadjuvant Study in HR(+)/HER2(−) Breast Cancer. Clin. Cancer Res. 2020, 26, 566–580. [Google Scholar] [CrossRef]
- Khan, Q.J.; O’Dea, A.; Bardia, A.; Kalinsky, K.; Wisinski, K.B.; O’Regan, R.; Yuan, Y.; Ma, C.X.; Jahanzeb, M.; Trivedi, M.S.; et al. Letrozole + ribociclib versus letrozole + placebo as neoadjuvant therapy for ER+ breast cancer (FELINE trial). J. Clin. Oncol. 2020, 38, 505. [Google Scholar] [CrossRef]
- Ma, C.X.; Gao, F.; Luo, J.; Northfelt, D.W.; Goetz, M.; Forero, A.; Hoog, J.; Naughton, M.; Ademuyiwa, F.; Suresh, R.; et al. NeoPalAna: Neoadjuvant Palbociclib, a Cyclin-Dependent Kinase 4/6 Inhibitor, and Anastrozole for Clinical Stage 2 or 3 Estrogen Receptor-Positive Breast Cancer. Clin. Cancer Res. 2017, 23, 4055–4065. [Google Scholar] [CrossRef]
- Loibl, S.; Marme, F.; Martin, M.; Untch, M.; Bonnefoi, H.; Kim, S.B.; Bear, H.; McCarthy, N.; Mele Olive, M.; Gelmon, K.; et al. Palbociclib for Residual High-Risk Invasive HR-Positive and HER2-Negative Early Breast Cancer-The Penelope-B Trial. J. Clin. Oncol. 2021, 39, 1518–1530. [Google Scholar] [CrossRef]
- Gnant, M.; Dueck, A.C.; Frantal, S.; Martin, M.; Burstein, H.J.; Greil, R.; Fox, P.; Wolff, A.C.; Chan, A.; Winer, E.P.; et al. Adjuvant Palbociclib for Early Breast Cancer: The PALLAS Trial Results (ABCSG-42/AFT-05/BIG-14-03). J. Clin. Oncol. 2022, 40, 282–293. [Google Scholar] [CrossRef]
- Stroyakovskiy, D.; Yardley, D.A.; Huang, C.-S.; Fasching, P.A.; Crown, J.; Bardia, A.; Chia, S.; Im, S.-A.; Martin, M.; Loi, S.; et al. Ribociclib and endocrine therapy as adjuvant treatment in patients with HR+/HER2- early breast cancer: Primary results from the phase III NATALEE trial. J. Clin. Oncol. 2023, 41, LBA500. [Google Scholar] [CrossRef]
- Fassl, A.; Geng, Y.; Sicinski, P. CDK4 and CDK6 kinases: From basic science to cancer therapy. Science 2022, 375, eabc1495. [Google Scholar] [CrossRef]
- Weiss, J.; Goldschmidt, J.; Andric, Z.; Dragnev, K.H.; Gwaltney, C.; Skaltsa, K.; Pritchett, Y.; Antal, J.M.; Morris, S.R.; Daniel, D. Effects of Trilaciclib on Chemotherapy-Induced Myelosuppression and Patient-Reported Outcomes in Patients with Extensive-Stage Small Cell Lung Cancer: Pooled Results from Three Phase II Randomized, Double-Blind, Placebo-Controlled Studies. Clin. Lung Cancer 2021, 22, 449–460. [Google Scholar] [CrossRef]
- Goel, S.; Tan, A.R.; Rugo, H.S.; Aftimos, P.; Andric, Z.; Beelen, A.; Zhang, J.; Yi, J.S.; Malik, R.; O’Shaughnessy, J. Trilaciclib prior to gemcitabine plus carboplatin for metastatic triple-negative breast cancer: Phase III PRESERVE 2. Future Oncol. 2022, 18, 3701–3711. [Google Scholar] [CrossRef]
- Xu, B.; Zhang, Q.; Zhang, P.; Hu, X.; Li, W.; Tong, Z.; Sun, T.; Teng, Y.; Wu, X.; Ouyang, Q.; et al. Dalpiciclib or placebo plus fulvestrant in hormone receptor-positive and HER2-negative advanced breast cancer: A randomized, phase 3 trial. Nat. Med. 2021, 27, 1904–1909. [Google Scholar] [CrossRef]
- Niu, N.; Qiu, F.; Xu, Q.; He, G.; Gu, X.; Guo, W.; Zhang, D.; Li, Z.; Zhao, Y.; Li, Y.; et al. A multicentre single arm phase 2 trial of neoadjuvant pyrotinib and letrozole plus dalpiciclib for triple-positive breast cancer. Nat. Commun. 2022, 13, 7043. [Google Scholar] [CrossRef]
- Klein, M.E.; Kovatcheva, M.; Davis, L.E.; Tap, W.D.; Koff, A. CDK4/6 Inhibitors: The Mechanism of Action May Not Be as Simple as Once Thought. Cancer Cell 2018, 34, 9–20. [Google Scholar] [CrossRef]
- Toogood, P.L.; Harvey, P.J.; Repine, J.T.; Sheehan, D.J.; VanderWel, S.N.; Zhou, H.; Keller, P.R.; McNamara, D.J.; Sherry, D.; Zhu, T.; et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J. Med. Chem. 2005, 48, 2388–2406. [Google Scholar] [CrossRef]
- Fry, D.W.; Harvey, P.J.; Keller, P.R.; Elliott, W.L.; Meade, M.; Trachet, E.; Albassam, M.; Zheng, X.; Leopold, W.R.; Pryer, N.K.; et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther. 2004, 3, 1427–1438. [Google Scholar] [CrossRef]
- DeMichele, A.; Clark, A.S.; Tan, K.S.; Heitjan, D.F.; Gramlich, K.; Gallagher, M.; Lal, P.; Feldman, M.; Zhang, P.; Colameco, C.; et al. CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: Phase II activity, safety, and predictive biomarker assessment. Clin. Cancer Res. 2015, 21, 995–1001. [Google Scholar] [CrossRef]
- Ren, B.; Cam, H.; Takahashi, Y.; Volkert, T.; Terragni, J.; Young, R.A.; Dynlacht, B.D. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes. Dev. 2002, 16, 245–256. [Google Scholar] [CrossRef]
- Robertson, K.D.; Ait-Si-Ali, S.; Yokochi, T.; Wade, P.A.; Jones, P.L.; Wolffe, A.P. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat. Genet. 2000, 25, 338–342. [Google Scholar] [CrossRef]
- Watt, A.C.; Cejas, P.; DeCristo, M.J.; Metzger-Filho, O.; Lam, E.Y.N.; Qiu, X.; BrinJones, H.; Kesten, N.; Coulson, R.; Font-Tello, A.; et al. CDK4/6 inhibition reprograms the breast cancer enhancer landscape by stimulating AP-1 transcriptional activity. Nat. Cancer 2021, 2, 34–48. [Google Scholar] [CrossRef]
- Blanchet, E.; Annicotte, J.S.; Lagarrigue, S.; Aguilar, V.; Clape, C.; Chavey, C.; Fritz, V.; Casas, F.; Apparailly, F.; Auwerx, J.; et al. E2F transcription factor-1 regulates oxidative metabolism. Nat. Cell Biol. 2011, 13, 1146–1152. [Google Scholar] [CrossRef]
- Ziebold, U.; Reza, T.; Caron, A.; Lees, J.A. E2F3 contributes both to the inappropriate proliferation and to the apoptosis arising in Rb mutant embryos. Genes Dev. 2001, 15, 386–391. [Google Scholar] [CrossRef]
- Muller, H.; Bracken, A.P.; Vernell, R.; Moroni, M.C.; Christians, F.; Grassilli, E.; Prosperini, E.; Vigo, E.; Oliner, J.D.; Helin, K. E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes. Dev. 2001, 15, 267–285. [Google Scholar] [CrossRef]
- Crozier, L.; Foy, R.; Mouery, B.L.; Whitaker, R.H.; Corno, A.; Spanos, C.; Ly, T.; Gowen Cook, J.; Saurin, A.T. CDK4/6 inhibitors induce replication stress to cause long-term cell cycle withdrawal. EMBO J. 2022, 41, e108599. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Thangavel, C.; Boopathi, E.; Liu, Y.; McNair, C.; Haber, A.; Perepelyuk, M.; Bhardwaj, A.; Addya, S.; Ertel, A.; Shoyele, S.; et al. Therapeutic Challenge with a CDK 4/6 Inhibitor Induces an RB-Dependent SMAC-Mediated Apoptotic Response in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 1402–1414. [Google Scholar] [CrossRef]
- Salvador-Barbero, B.; Alvarez-Fernandez, M.; Zapatero-Solana, E.; El Bakkali, A.; Menendez, M.D.C.; Lopez-Casas, P.P.; Di Domenico, T.; Xie, T.; VanArsdale, T.; Shields, D.J.; et al. CDK4/6 Inhibitors Impair Recovery from Cytotoxic Chemotherapy in Pancreatic Adenocarcinoma. Cancer Cell 2020, 38, 584. [Google Scholar] [CrossRef]
- Dean, J.L.; McClendon, A.K.; Knudsen, E.S. Modification of the DNA damage response by therapeutic CDK4/6 inhibition. J. Biol. Chem. 2012, 287, 29075–29087. [Google Scholar] [CrossRef]
- Wang, B.; Varela-Eirin, M.; Brandenburg, S.M.; Hernandez-Segura, A.; van Vliet, T.; Jongbloed, E.M.; Wilting, S.M.; Ohtani, N.; Jager, A.; Demaria, M. Pharmacological CDK4/6 inhibition reveals a p53-dependent senescent state with restricted toxicity. EMBO J. 2022, 41, e108946. [Google Scholar] [CrossRef]
- Terenziani, R.; Galetti, M.; La Monica, S.; Fumarola, C.; Zoppi, S.; Alfieri, R.; Digiacomo, G.; Cavazzoni, A.; Cavallo, D.; Corradi, M.; et al. CDK4/6 Inhibition Enhances the Efficacy of Standard Chemotherapy Treatment in Malignant Pleural Mesothelioma Cells. Cancers 2022, 14, 5925. [Google Scholar] [CrossRef]
- Kartika, I.D.; Kotani, H.; Iida, Y.; Koyanagi, A.; Tanino, R.; Harada, M. Protective role of cytoplasmic p21Cip1/Waf1 in apoptosis of CDK4/6 inhibitor-induced senescence in breast cancer cells. Cancer Med. 2021, 10, 8988–8999. [Google Scholar] [CrossRef]
- Gallanis, G.T.; Sharif, G.M.; Schmidt, M.O.; Friedland, B.N.; Battina, R.; Rahhal, R.; Davis, J.E., Jr.; Khan, I.S.; Wellstein, A.; Riegel, A.T. Stromal Senescence following Treatment with the CDK4/6 Inhibitor Palbociclib Alters the Lung Metastatic Niche and Increases Metastasis of Drug-Resistant Mammary Cancer Cells. Cancers 2023, 15, 1908. [Google Scholar] [CrossRef]
- Anders, L.; Ke, N.; Hydbring, P.; Choi, Y.J.; Widlund, H.R.; Chick, J.M.; Zhai, H.; Vidal, M.; Gygi, S.P.; Braun, P.; et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell 2011, 20, 620–634. [Google Scholar] [CrossRef]
- Beykou, M.; Arias-Garcia, M.; Roumeliotis, T.I.; Choudhary, J.S.; Moser, N.; Georgiou, P.; Bakal, C. Proteomic characterisation of triple negative breast cancer cells following CDK4/6 inhibition. Sci. Data 2022, 9, 395. [Google Scholar] [CrossRef]
- Uzhachenko, R.V.; Bharti, V.; Ouyang, Z.; Blevins, A.; Mont, S.; Saleh, N.; Lawrence, H.A.; Shen, C.; Chen, S.C.; Ayers, G.D.; et al. Metabolic modulation by CDK4/6 inhibitor promotes chemokine-mediated recruitment of T cells into mammary tumors. Cell Rep. 2021, 35, 109271. [Google Scholar] [CrossRef]
- Yoshida, A.; Lee, E.K.; Diehl, J.A. Induction of Therapeutic Senescence in Vemurafenib-Resistant Melanoma by Extended Inhibition of CDK4/6. Cancer Res. 2016, 76, 2990–3002. [Google Scholar] [CrossRef]
- Michaud, K.; Solomon, D.A.; Oermann, E.; Kim, J.S.; Zhong, W.Z.; Prados, M.D.; Ozawa, T.; James, C.D.; Waldman, T. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010, 70, 3228–3238. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- Llanos, S.; Megias, D.; Blanco-Aparicio, C.; Hernandez-Encinas, E.; Rovira, M.; Pietrocola, F.; Serrano, M. Lysosomal trapping of palbociclib and its functional implications. Oncogene 2019, 38, 3886–3902. [Google Scholar] [CrossRef]
- Acevedo, M.; Vernier, M.; Mignacca, L.; Lessard, F.; Huot, G.; Moiseeva, O.; Bourdeau, V.; Ferbeyre, G. A CDK4/6-Dependent Epigenetic Mechanism Protects Cancer Cells from PML-induced Senescence. Cancer Res. 2016, 76, 3252–3264. [Google Scholar] [CrossRef]
- Kovatcheva, M.; Liu, D.D.; Dickson, M.A.; Klein, M.E.; O’Connor, R.; Wilder, F.O.; Socci, N.D.; Tap, W.D.; Schwartz, G.K.; Singer, S.; et al. MDM2 turnover and expression of ATRX determine the choice between quiescence and senescence in response to CDK4 inhibition. Oncotarget 2015, 6, 8226–8243. [Google Scholar] [CrossRef]
- Martinez-Zamudio, R.I.; Roux, P.F.; de Freitas, J.; Robinson, L.; Dore, G.; Sun, B.; Belenki, D.; Milanovic, M.; Herbig, U.; Schmitt, C.A.; et al. AP-1 imprints a reversible transcriptional programme of senescent cells. Nat. Cell Biol. 2020, 22, 842–855. [Google Scholar] [CrossRef]
- Han, R.; Li, L.; Ugalde, A.P.; Tal, A.; Manber, Z.; Barbera, E.P.; Chiara, V.D.; Elkon, R.; Agami, R. Functional CRISPR screen identifies AP1-associated enhancer regulating FOXF1 to modulate oncogene-induced senescence. Genome Biol. 2018, 19, 118. [Google Scholar] [CrossRef]
- Vijayaraghavan, S.; Karakas, C.; Doostan, I.; Chen, X.; Bui, T.; Yi, M.; Raghavendra, A.S.; Zhao, Y.; Bashour, S.I.; Ibrahim, N.K.; et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat. Commun. 2017, 8, 15916. [Google Scholar] [CrossRef]
- Santiappillai, N.T.; Abuhammad, S.; Slater, A.; Kirby, L.; McArthur, G.A.; Sheppard, K.E.; Smith, L.K. CDK4/6 Inhibition Reprograms Mitochondrial Metabolism in BRAF(V600) Melanoma via a p53 Dependent Pathway. Cancers 2021, 13, 524. [Google Scholar] [CrossRef]
- Fassl, A.; Brain, C.; Abu-Remaileh, M.; Stukan, I.; Butter, D.; Stepien, P.; Feit, A.S.; Bergholz, J.; Michowski, W.; Otto, T.; et al. Increased lysosomal biomass is responsible for the resistance of triple-negative breast cancers to CDK4/6 inhibition. Sci. Adv. 2020, 6, eabb2210. [Google Scholar] [CrossRef]
- Yin, Q.; Jian, Y.; Xu, M.; Huang, X.; Wang, N.; Liu, Z.; Li, Q.; Li, J.; Zhou, H.; Xu, L.; et al. CDK4/6 regulate lysosome biogenesis through TFEB/TFE3. J. Cell Biol. 2020, 219, e201911036. [Google Scholar] [CrossRef]
- Franco, J.; Balaji, U.; Freinkman, E.; Witkiewicz, A.K.; Knudsen, E.S. Metabolic Reprogramming of Pancreatic Cancer Mediated by CDK4/6 Inhibition Elicits Unique Vulnerabilities. Cell Rep. 2016, 14, 979–990. [Google Scholar] [CrossRef]
- Lorito, N.; Bacci, M.; Smiriglia, A.; Mannelli, M.; Parri, M.; Comito, G.; Ippolito, L.; Giannoni, E.; Bonechi, M.; Benelli, M.; et al. Glucose Metabolic Reprogramming of ER Breast Cancer in Acquired Resistance to the CDK4/6 Inhibitor Palbociclib. Cells 2020, 9, 668. [Google Scholar] [CrossRef]
- Schaer, D.A.; Beckmann, R.P.; Dempsey, J.A.; Huber, L.; Forest, A.; Amaladas, N.; Li, Y.; Wang, Y.C.; Rasmussen, E.R.; Chin, D.; et al. The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep. 2018, 22, 2978–2994. [Google Scholar] [CrossRef]
- Jin, X.; Ding, D.; Yan, Y.; Li, H.; Wang, B.; Ma, L.; Ye, Z.; Ma, T.; Wu, Q.; Rodrigues, D.N.; et al. Phosphorylated RB Promotes Cancer Immunity by Inhibiting NF-kappaB Activation and PD-L1 Expression. Mol. Cell 2019, 73, 22–35.e26. [Google Scholar] [CrossRef]
- Scirocchi, F.; Scagnoli, S.; Botticelli, A.; Di Filippo, A.; Napoletano, C.; Zizzari, I.G.; Strigari, L.; Tomao, S.; Cortesi, E.; Rughetti, A.; et al. Immune effects of CDK4/6 inhibitors in patients with HR(+)/HER2(−) metastatic breast cancer: Relief from immunosuppression is associated with clinical response. eBioMedicine 2022, 79, 104010. [Google Scholar] [CrossRef]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef]
- Peuker, C.A.; Yaghobramzi, S.; Grunert, C.; Keilholz, L.; Gjerga, E.; Hennig, S.; Schaper, S.; Na, I.K.; Keller, U.; Brucker, S.; et al. Treatment with ribociclib shows favourable immunomodulatory effects in patients with hormone receptor-positive breast cancer-findings from the RIBECCA trial. Eur. J. Cancer 2022, 162, 45–55. [Google Scholar] [CrossRef]
- Heckler, M.; Ali, L.R.; Clancy-Thompson, E.; Qiang, L.; Ventre, K.S.; Lenehan, P.; Roehle, K.; Luoma, A.; Boelaars, K.; Peters, V.; et al. Inhibition of CDK4/6 Promotes CD8 T-cell Memory Formation. Cancer Discov. 2021, 11, 2564–2581. [Google Scholar] [CrossRef]
- Lelliott, E.J.; Kong, I.Y.; Zethoven, M.; Ramsbottom, K.M.; Martelotto, L.G.; Meyran, D.; Zhu, J.J.; Costacurta, M.; Kirby, L.; Sandow, J.J.; et al. CDK4/6 Inhibition Promotes Antitumor Immunity through the Induction of T-cell Memory. Cancer Discov. 2021, 11, 2582–2601. [Google Scholar] [CrossRef]
- Zhang, Q.F.; Li, J.; Jiang, K.; Wang, R.; Ge, J.L.; Yang, H.; Liu, S.J.; Jia, L.T.; Wang, L.; Chen, B.L. CDK4/6 inhibition promotes immune infiltration in ovarian cancer and synergizes with PD-1 blockade in a B cell-dependent manner. Theranostics 2020, 10, 10619–10633. [Google Scholar] [CrossRef]
- Cheng, M.; Olivier, P.; Diehl, J.A.; Fero, M.; Roussel, M.F.; Roberts, J.M.; Sherr, C.J. The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999, 18, 1571–1583. [Google Scholar] [CrossRef]
- Ruas, M.; Gregory, F.; Jones, R.; Poolman, R.; Starborg, M.; Rowe, J.; Brookes, S.; Peters, G. CDK4 and CDK6 delay senescence by kinase-dependent and p16INK4a-independent mechanisms. Mol. Cell Biol. 2007, 27, 4273–4282. [Google Scholar] [CrossRef]
- Guan, X.; LaPak, K.M.; Hennessey, R.C.; Yu, C.Y.; Shakya, R.; Zhang, J.; Burd, C.E. Stromal Senescence By Prolonged CDK4/6 Inhibition Potentiates Tumor Growth. Mol. Cancer Res. 2017, 15, 237–249. [Google Scholar] [CrossRef]
- Alhaja, E.; Adan, J.; Pagan, R.; Mitjans, F.; Cascallo, M.; Rodriguez, M.; Noe, V.; Ciudad, C.J.; Mazo, A.; Vilaro, S.; et al. Anti-migratory and anti-angiogenic effect of p16: A novel localization at membrane ruffles and lamellipodia in endothelial cells. Angiogenesis 2004, 7, 323–333. [Google Scholar] [CrossRef]
- Kollmann, K.; Briand, C.; Bellutti, F.; Schicher, N.; Blunder, S.; Zojer, M.; Hoeller, C. The interplay of CDK4 and CDK6 in melanoma. Oncotarget 2019, 10, 1346–1359. [Google Scholar] [CrossRef]
- Chavkin, N.W.; Genet, G.; Poulet, M.; Jeffery, E.D.; Marziano, C.; Genet, N.; Vasavada, H.; Nelson, E.A.; Acharya, B.R.; Kour, A.; et al. Endothelial cell cycle state determines propensity for arterial-venous fate. Nat. Commun. 2022, 13, 5891. [Google Scholar] [CrossRef]
- Herrera-Abreu, M.T.; Palafox, M.; Asghar, U.; Rivas, M.A.; Cutts, R.J.; Garcia-Murillas, I.; Pearson, A.; Guzman, M.; Rodriguez, O.; Grueso, J.; et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res. 2016, 76, 2301–2313. [Google Scholar] [CrossRef]
- Dean, J.L.; Thangavel, C.; McClendon, A.K.; Reed, C.A.; Knudsen, E.S. Therapeutic CDK4/6 inhibition in breast cancer: Key mechanisms of response and failure. Oncogene 2010, 29, 4018–4032. [Google Scholar] [CrossRef]
- Guarducci, C.; Bonechi, M.; Benelli, M.; Biagioni, C.; Boccalini, G.; Romagnoli, D.; Verardo, R.; Schiff, R.; Osborne, C.K.; De Angelis, C.; et al. Cyclin E1 and Rb modulation as common events at time of resistance to palbociclib in hormone receptor-positive breast cancer. NPJ Breast Cancer 2018, 4, 38. [Google Scholar] [CrossRef]
- Malorni, L.; Piazza, S.; Ciani, Y.; Guarducci, C.; Bonechi, M.; Biagioni, C.; Hart, C.D.; Verardo, R.; Di Leo, A.; Migliaccio, I. A gene expression signature of retinoblastoma loss-of-function is a predictive biomarker of resistance to palbociclib in breast cancer cell lines and is prognostic in patients with ER positive early breast cancer. Oncotarget 2016, 7, 68012–68022. [Google Scholar] [CrossRef]
- Roberts, P.J.; Bisi, J.E.; Strum, J.C.; Combest, A.J.; Darr, D.B.; Usary, J.E.; Zamboni, W.C.; Wong, K.K.; Perou, C.M.; Sharpless, N.E. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J. Natl. Cancer Inst. 2012, 104, 476–487. [Google Scholar] [CrossRef]
- Dean, J.L.; McClendon, A.K.; Hickey, T.E.; Butler, L.M.; Tilley, W.D.; Witkiewicz, A.K.; Knudsen, E.S. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle 2012, 11, 2756–2761. [Google Scholar] [CrossRef]
- Turner, N.C.; Liu, Y.; Zhu, Z.; Loi, S.; Colleoni, M.; Loibl, S.; DeMichele, A.; Harbeck, N.; Andre, F.; Bayar, M.A.; et al. Cyclin E1 Expression and Palbociclib Efficacy in Previously Treated Hormone Receptor-Positive Metastatic Breast Cancer. J. Clin. Oncol. 2019, 37, 1169–1178. [Google Scholar] [CrossRef]
- Finn, R.S.; Liu, Y.; Zhu, Z.; Martin, M.; Rugo, H.S.; Dieras, V.; Im, S.A.; Gelmon, K.A.; Harbeck, N.; Lu, D.R.; et al. Biomarker Analyses of Response to Cyclin-Dependent Kinase 4/6 Inhibition and Endocrine Therapy in Women with Treatment-Naive Metastatic Breast Cancer. Clin. Cancer Res. 2020, 26, 110–121. [Google Scholar] [CrossRef]
- Jansen, V.M.; Bhola, N.E.; Bauer, J.A.; Formisano, L.; Lee, K.M.; Hutchinson, K.E.; Witkiewicz, A.K.; Moore, P.D.; Estrada, M.V.; Sanchez, V.; et al. Kinome-Wide RNA Interference Screen Reveals a Role for PDK1 in Acquired Resistance to CDK4/6 Inhibition in ER-Positive Breast Cancer. Cancer Res. 2017, 77, 2488–2499. [Google Scholar] [CrossRef]
- Cai, Z.; Wang, J.; Li, Y.; Shi, Q.; Jin, L.; Li, S.; Zhu, M.; Wang, Q.; Wong, L.L.; Yang, W.; et al. Overexpressed Cyclin D1 and CDK4 proteins are responsible for the resistance to CDK4/6 inhibitor in breast cancer that can be reversed by PI3K/mTOR inhibitors. Sci. China Life Sci. 2023, 66, 94–109. [Google Scholar] [CrossRef]
- Pancholi, S.; Ribas, R.; Simigdala, N.; Schuster, E.; Nikitorowicz-Buniak, J.; Ressa, A.; Gao, Q.; Leal, M.F.; Bhamra, A.; Thornhill, A.; et al. Tumour kinome re-wiring governs resistance to palbociclib in oestrogen receptor positive breast cancers, highlighting new therapeutic modalities. Oncogene 2020, 39, 4781–4797. [Google Scholar] [CrossRef]
- Yang, C.; Li, Z.; Bhatt, T.; Dickler, M.; Giri, D.; Scaltriti, M.; Baselga, J.; Rosen, N.; Chandarlapaty, S. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 2017, 36, 2255–2264. [Google Scholar] [CrossRef]
- Li, Z.; Razavi, P.; Li, Q.; Toy, W.; Liu, B.; Ping, C.; Hsieh, W.; Sanchez-Vega, F.; Brown, D.N.; Da Cruz Paula, A.F.; et al. Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway. Cancer Cell 2018, 34, 893–905.e898. [Google Scholar] [CrossRef]
- Ji, W.; Zhang, W.; Wang, X.; Shi, Y.; Yang, F.; Xie, H.; Zhou, W.; Wang, S.; Guan, X. c-myc regulates the sensitivity of breast cancer cells to palbociclib via c-myc/miR-29b-3p/CDK6 axis. Cell Death Dis. 2020, 11, 760. [Google Scholar] [CrossRef]
- Cornell, L.; Wander, S.A.; Visal, T.; Wagle, N.; Shapiro, G.I. MicroRNA-Mediated Suppression of the TGF-beta Pathway Confers Transmissible and Reversible CDK4/6 Inhibitor Resistance. Cell Rep. 2019, 26, 2667–2680.e2667. [Google Scholar] [CrossRef]
- Iida, M.; Nakamura, M.; Tokuda, E.; Toyosawa, D.; Niwa, T.; Ohuchi, N.; Ishida, T.; Hayashi, S.I. The p21 levels have the potential to be a monitoring marker for ribociclib in breast cancer. Oncotarget 2019, 10, 4907–4918. [Google Scholar] [CrossRef]
- Wander, S.A.; Cohen, O.; Gong, X.; Johnson, G.N.; Buendia-Buendia, J.E.; Lloyd, M.R.; Kim, D.; Luo, F.; Mao, P.; Helvie, K.; et al. The Genomic Landscape of Intrinsic and Acquired Resistance to Cyclin-Dependent Kinase 4/6 Inhibitors in Patients with Hormone Receptor-Positive Metastatic Breast Cancer. Cancer Discov. 2020, 10, 1174–1193. [Google Scholar] [CrossRef]
- Gong, X.; Litchfield, L.M.; Webster, Y.; Chio, L.C.; Wong, S.S.; Stewart, T.R.; Dowless, M.; Dempsey, J.; Zeng, Y.; Torres, R.; et al. Genomic Aberrations that Activate D-type Cyclins Are Associated with Enhanced Sensitivity to the CDK4 and CDK6 Inhibitor Abemaciclib. Cancer Cell 2017, 32, 761–776.e766. [Google Scholar] [CrossRef]
- Gomes, I.; Gallego-Paez, L.M.; Jimenez, M.; Santamaria, P.G.; Mansinho, A.; Sousa, R.; Abreu, C.; Suarez, E.G.; Costa, L.; Casimiro, S. Co-targeting RANK pathway treats and prevents acquired resistance to CDK4/6 inhibitors in luminal breast cancer. Cell Rep. Med. 2023, 4, 101120. [Google Scholar] [CrossRef]
- Palafox, M.; Monserrat, L.; Bellet, M.; Villacampa, G.; Gonzalez-Perez, A.; Oliveira, M.; Braso-Maristany, F.; Ibrahimi, N.; Kannan, S.; Mina, L.; et al. High p16 expression and heterozygous RB1 loss are biomarkers for CDK4/6 inhibitor resistance in ER(+) breast cancer. Nat. Commun. 2022, 13, 5258. [Google Scholar] [CrossRef]
- Patel, P.; Asbach, B.; Shteyn, E.; Gomez, C.; Coltoff, A.; Bhuyan, S.; Tyner, A.L.; Wagner, R.; Blain, S.W. Brk/Protein tyrosine kinase 6 phosphorylates p27KIP1, regulating the activity of cyclin D-cyclin-dependent kinase 4. Mol. Cell Biol. 2015, 35, 1506–1522. [Google Scholar] [CrossRef]
- Grolmusz, V.K.; Chen, J.; Emond, R.; Cosgrove, P.A.; Pflieger, L.; Nath, A.; Moos, P.J.; Bild, A.H. Exploiting collateral sensitivity controls growth of mixed culture of sensitive and resistant cells and decreases selection for resistant cells in a cell line model. Cancer Cell Int. 2020, 20, 253. [Google Scholar] [CrossRef]
- Fallah, Y.; Demas, D.M.; Jin, L.; He, W.; Shajahan-Haq, A.N. Targeting WEE1 Inhibits Growth of Breast Cancer Cells That Are Resistant to Endocrine Therapy and CDK4/6 Inhibitors. Front. Oncol. 2021, 11, 681530. [Google Scholar] [CrossRef]
- Portman, N.; Milioli, H.H.; Alexandrou, S.; Coulson, R.; Yong, A.; Fernandez, K.J.; Chia, K.M.; Halilovic, E.; Segara, D.; Parker, A.; et al. MDM2 inhibition in combination with endocrine therapy and CDK4/6 inhibition for the treatment of ER-positive breast cancer. Breast Cancer Res. 2020, 22, 87. [Google Scholar] [CrossRef]
- The, I.; Ruijtenberg, S.; Bouchet, B.P.; Cristobal, A.; Prinsen, M.B.; van Mourik, T.; Koreth, J.; Xu, H.; Heck, A.J.; Akhmanova, A.; et al. Rb and FZR1/Cdh1 determine CDK4/6-cyclin D requirement in C. elegans and human cancer cells. Nat. Commun. 2015, 6, 5906. [Google Scholar] [CrossRef]
- De Angelis, C.; Nardone, A.; Cataldo, M.; Veeraraghavan, J.; Fu, X.; Giuliano, M.; Malorni, L.; Jeselsohn, R.; Osborne, K.; Schiff, R. Abstract P4-03-05: AP-1 as a potential mediator of resistance to the cyclin-dependent kinase (CDK) 4/6-inhibitor palbociclib in ER-positive endocrine-resistant breast cancer. Cancer Res. 2018, 78, P4-03-05. [Google Scholar] [CrossRef]
- Del Re, M.; Bertolini, I.; Crucitta, S.; Fontanelli, L.; Rofi, E.; De Angelis, C.; Diodati, L.; Cavallero, D.; Gianfilippo, G.; Salvadori, B.; et al. Overexpression of TK1 and CDK9 in plasma-derived exosomes is associated with clinical resistance to CDK4/6 inhibitors in metastatic breast cancer patients. Breast Cancer Res. Tr. 2019, 178, 57–62. [Google Scholar] [CrossRef]
- McCartney, A.; Bonechi, M.; De Luca, F.; Biagioni, C.; Curigliano, G.; Moretti, E.; Minisini, A.M.; Bergqvist, M.; Benelli, M.; Migliaccio, I.; et al. Plasma Thymidine Kinase Activity as a Biomarker in Patients with Luminal Metastatic Breast Cancer Treated with Palbociclib within the TREnd Trial. Clin. Cancer Res. 2020, 26, 2131–2139. [Google Scholar] [CrossRef]
- Goetz, M.P.; Hamilton, E.P.; Campone, M.; Hurvitz, S.A.; Cortes, J.; Johnston, S.R.D.; Jerusalem, G.H.M.; Graham, H.; Wang, H.; Litchfield, L.; et al. Acquired genomic alterations in circulating tumor DNA from patients receiving abemaciclib alone or in combination with endocrine therapy. J. Clin. Oncol. 2020, 38, 3519. [Google Scholar] [CrossRef]
- Freeman-Cook, K.; Hoffman, R.L.; Miller, N.; Almaden, J.; Chionis, J.; Zhang, Q.; Eisele, K.; Liu, C.; Zhang, C.; Huser, N.; et al. Expanding control of the tumor cell cycle with a CDK2/4/6 inhibitor. Cancer Cell 2021, 39, 1404–1421.e11. [Google Scholar] [CrossRef]
- Ji, W.; Shi, Y.; Wang, X.; He, W.; Tang, L.; Tian, S.; Jiang, H.; Shu, Y.; Guan, X. Combined Androgen receptor blockade overcomes the resistance of breast cancer cells to palbociclib. Int. J. Biol. Sci. 2019, 15, 522–532. [Google Scholar] [CrossRef]
- Nayar, U.; Cohen, O.; Kapstad, C.; Cuoco, M.S.; Waks, A.G.; Wander, S.A.; Painter, C.; Freeman, S.; Persky, N.S.; Marini, L.; et al. Acquired HER2 mutations in ER+ metastatic breast cancer confer resistance to estrogen receptor–directed therapies. Nat. Genet. 2019, 51, 207–216. [Google Scholar] [CrossRef]
- Zanudo, J.G.T.; Barroso-Sousa, R.; Jain, E.; Buendia-Buendia, J.; Li, T.; Tayob, N.; Rees, R.; Pereslete, A.; Ferreira, A.R.; Abravanel, D.L.; et al. Genomic and transcriptomic analysis reveals known and novel resistance mechanisms to CDK4/6 inhibitors and sensitivity factors for the response to triplet therapy (palbociclib plus everolimus plus exemestane) in a phase I/IIb study in hormone-receptor positive (HR+)/HER2-metastatic breast cancer (MBC) after progression on a CDK4/6 inhibitor (CDK4/6i). Cancer Res. 2022, 82, P4-01-06. [Google Scholar] [CrossRef]
- O’Leary, B.; Hrebien, S.; Morden, J.P.; Beaney, M.; Fribbens, C.; Huang, X.; Liu, Y.; Bartlett, C.H.; Koehler, M.; Cristofanilli, M.; et al. Early circulating tumor DNA dynamics and clonal selection with palbociclib and fulvestrant for breast cancer. Nat. Commun. 2018, 9, 896. [Google Scholar] [CrossRef]
- O’Leary, B.; Cutts, R.J.; Liu, Y.; Hrebien, S.; Huang, X.; Fenwick, K.; Andre, F.; Loibl, S.; Loi, S.; Garcia-Murillas, I.; et al. The Genetic Landscape and Clonal Evolution of Breast Cancer Resistance to Palbociclib plus Fulvestrant in the PALOMA-3 Trial. Cancer Discov. 2018, 8, 1390–1403. [Google Scholar] [CrossRef]
- Miller, T.W.; Rexer, B.N.; Garrett, J.T.; Arteaga, C.L. Mutations in the phosphatidylinositol 3-kinase pathway: Role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011, 13, 224. [Google Scholar] [CrossRef]
- Alves, C.L.; Ehmsen, S.; Terp, M.G.; Portman, N.; Tuttolomondo, M.; Gammelgaard, O.L.; Hundebol, M.F.; Kaminska, K.; Johansen, L.E.; Bak, M.; et al. Co-targeting CDK4/6 and AKT with endocrine therapy prevents progression in CDK4/6 inhibitor and endocrine therapy-resistant breast cancer. Nat. Commun. 2021, 12, 5112. [Google Scholar] [CrossRef]
- Costa, C.; Wang, Y.; Ly, A.; Hosono, Y.; Murchie, E.; Walmsley, C.S.; Huynh, T.; Healy, C.; Peterson, R.; Yanase, S.; et al. PTEN Loss Mediates Clinical Cross-Resistance to CDK4/6 and PI3Kalpha Inhibitors in Breast Cancer. Cancer Discov. 2020, 10, 72–85. [Google Scholar] [CrossRef]
- Vora, S.R.; Juric, D.; Kim, N.; Mino-Kenudson, M.; Huynh, T.; Costa, C.; Lockerman, E.L.; Pollack, S.F.; Liu, M.; Li, X.; et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell 2014, 26, 136–149. [Google Scholar] [CrossRef]
- Michaloglou, C.; Crafter, C.; Siersbaek, R.; Delpuech, O.; Curwen, J.O.; Carnevalli, L.S.; Staniszewska, A.D.; Polanska, U.M.; Cheraghchi-Bashi, A.; Lawson, M.; et al. Combined Inhibition of mTOR and CDK4/6 Is Required for Optimal Blockade of E2F Function and Long-term Growth Inhibition in Estrogen Receptor-positive Breast Cancer. Mol. Cancer Ther. 2018, 17, 908–920. [Google Scholar] [CrossRef]
- Litchfield, L.M.; Boehnke, K.; Brahmachary, M.; Mur, C.; Bi, C.; Stephens, J.R.; Sauder, J.M.; Gutierrez, S.M.; McNulty, A.M.; Ye, X.S.; et al. Combined inhibition of PIM and CDK4/6 suppresses both mTOR signaling and Rb phosphorylation and potentiates PI3K inhibition in cancer cells. Oncotarget 2020, 11, 1478–1492. [Google Scholar] [CrossRef]
- Occhipinti, G.; Romagnoli, E.; Santoni, M.; Cimadamore, A.; Sorgentoni, G.; Cecati, M.; Giulietti, M.; Battelli, N.; Maccioni, A.; Storti, N.; et al. Sequential or Concomitant Inhibition of Cyclin-Dependent Kinase 4/6 Before mTOR Pathway in Hormone-Positive HER2 Negative Breast Cancer: Biological Insights and Clinical Implications. Front. Genet. 2020, 11, 349. [Google Scholar] [CrossRef]
- Andre, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Darrigues, L.; Pierga, J.Y.; Bernard-Tessier, A.; Bieche, I.; Silveira, A.B.; Michel, M.; Loirat, D.; Cottu, P.; Cabel, L.; Dubot, C.; et al. Circulating tumor DNA as a dynamic biomarker of response to palbociclib and fulvestrant in metastatic breast cancer patients. Breast Cancer Res. 2021, 23, 31. [Google Scholar] [CrossRef]
- Formisano, L.; Lu, Y.; Servetto, A.; Hanker, A.B.; Jansen, V.M.; Bauer, J.A.; Sudhan, D.R.; Guerrero-Zotano, A.L.; Croessmann, S.; Guo, Y.; et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat. Commun. 2019, 10, 1373. [Google Scholar] [CrossRef]
- De Leeuw, R.; McNair, C.; Schiewer, M.J.; Neupane, N.P.; Brand, L.J.; Augello, M.A.; Li, Z.; Cheng, L.C.; Yoshida, A.; Courtney, S.M.; et al. MAPK Reliance via Acquired CDK4/6 Inhibitor Resistance in Cancer. Clin. Cancer Res. 2018, 24, 4201–4214. [Google Scholar] [CrossRef]
- Lanceta, L.; O’Neill, C.; Lypova, N.; Li, X.; Rouchka, E.; Waigel, S.; Gomez-Gutierrez, J.G.; Chesney, J.; Imbert-Fernandez, Y. Transcriptomic Profiling Identifies Differentially Expressed Genes in Palbociclib-Resistant ER+ MCF7 Breast Cancer Cells. Genes 2020, 11, 467. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.H. Non-Smad TGF-beta signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Tobin, N.P.; Sims, A.H.; Lundgren, K.L.; Lehn, S.; Landberg, G. Cyclin D1, Id1 and EMT in breast cancer. BMC Cancer 2011, 11, 417. [Google Scholar] [CrossRef]
- De Gooijer, M.C.; Zhang, P.; Thota, N.; Mayayo-Peralta, I.; Buil, L.C.M.; Beijnen, J.H.; van Tellingen, O. P-glycoprotein and breast cancer resistance protein restrict the brain penetration of the CDK4/6 inhibitor palbociclib. Investig. New Drugs 2015, 33, 1012–1019. [Google Scholar] [CrossRef]
- Fu, H.; Wu, Z.X.; Lei, Z.N.; Teng, Q.X.; Yang, Y.; Ashby, C.R.; Lei, Y.; Lian, Y.; Chen, Z.S. The Resistance of Cancer Cells to Palbociclib, a Cyclin-Dependent Kinase 4/6 Inhibitor, is Mediated by the ABCB1 Transporter. Front. Pharmacol. 2022, 13, 861642. [Google Scholar] [CrossRef]
- Condorelli, R.; Spring, L.; O’Shaughnessy, J.; Lacroix, L.; Bailleux, C.; Scott, V.; Dubois, J.; Nagy, R.J.; Lanman, R.B.; Iafrate, A.J.; et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann. Oncol. 2018, 29, 640–645. [Google Scholar] [CrossRef]
- Xu, B.; Krie, A.; De, P.; Williams, C.; Elsey, R.; Klein, J.; Leyland-Jones, B. Utilizing Tumor and Plasma Liquid Biopsy in Treatment Decision Making for an Estrogen Receptor-Positive Advanced Breast Cancer Patient. Cureus 2017, 9, e1408. [Google Scholar] [CrossRef]
- An, H.X.; Beckmann, M.W.; Reifenberger, G.; Bender, H.G.; Niederacher, D. Gene amplification and overexpression of CDK4 in sporadic breast carcinomas is associated with high tumor cell proliferation. Am. J. Pathol. 1999, 154, 113–118. [Google Scholar] [CrossRef]
- Guo, P.; Pu, T.J.; Chen, S.N.; Qiu, Y.; Zhong, X.R.; Zheng, H.; Chen, L.N.; Ye, F.; Bu, H. CDK4 amplification is associated with distant metastasis and poor clinical outcome in breast cancer. Int. J. Clin. Exp. Pathol. 2016, 9, 6476–6482. [Google Scholar]
- Lundberg, A.; Lindstrom, L.S.; Li, J.; Harrell, J.C.; Darai-Ramqvist, E.; Sifakis, E.G.; Foukakis, T.; Perou, C.M.; Czene, K.; Bergh, J.; et al. The long-term prognostic and predictive capacity of cyclin D1 gene amplification in 2305 breast tumours. Breast Cancer Res 2019, 21, 34. [Google Scholar] [CrossRef]
- Schachter, M.M.; Merrick, K.A.; Larochelle, S.; Hirschi, A.; Zhang, C.; Shokat, K.M.; Rubin, S.M.; Fisher, R.P. A Cdk7-Cdk4 T-loop phosphorylation cascade promotes G1 progression. Mol. Cell 2013, 50, 250–260. [Google Scholar] [CrossRef]
- Fisher, R.P. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J. Cell Sci. 2005, 118, 5171–5180. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, T.; Kwiatkowski, N.; Abraham, B.J.; Lee, T.I.; Xie, S.; Yuzugullu, H.; Von, T.; Li, H.; Lin, Z.; et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 2015, 163, 174–186. [Google Scholar] [CrossRef]
- Matheson, C.J.; Backos, D.S.; Reigan, P. Targeting WEE1 Kinase in Cancer. Trends Pharmacol. Sci. 2016, 37, 872–881. [Google Scholar] [CrossRef]
- Kundu, N.; Brekman, A.; Kim, J.Y.; Xiao, G.; Gao, C.; Bargonetti, J. Estrogen-activated MDM2 disrupts mammary tissue architecture through a p53-independent pathway. Oncotarget 2017, 8, 47916–47930. [Google Scholar] [CrossRef]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Peters, J.M. The anaphase promoting complex/cyclosome: A machine designed to destroy. Nat. Rev. Mol. Cell Biol. 2006, 7, 644–656. [Google Scholar] [CrossRef]
- Ramanujan, A.; Tiwari, S. APC/C and retinoblastoma interaction: Cross-talk of retinoblastoma protein with the ubiquitin proteasome pathway. Biosci. Rep. 2016, 36, e00377. [Google Scholar] [CrossRef]
- Guo, Z.Y.; Hao, X.H.; Tan, F.F.; Pei, X.; Shang, L.M.; Jiang, X.L.; Yang, F. The elements of human cyclin D1 promoter and regulation involved. Clin. Epigenet. 2011, 2, 63–76. [Google Scholar] [CrossRef]
- Bejjani, F.; Evanno, E.; Zibara, K.; Piechaczyk, M.; Jariel-Encontre, I. The AP-1 transcriptional complex: Local switch or remote command? Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 11–23. [Google Scholar] [CrossRef]
- Jagarlamudi, K.K.; Shaw, M. Thymidine kinase 1 as a tumor biomarker: Technical advances offer new potential to an old biomarker. Biomark. Med. 2018, 12, 1035–1048. [Google Scholar] [CrossRef]
- Willems, E.; Dedobbeleer, M.; Digregorio, M.; Lombard, A.; Lumapat, P.N.; Rogister, B. The functional diversity of Aurora kinases: A comprehensive review. Cell Div. 2018, 13, 7. [Google Scholar] [CrossRef]
- Soria-Bretones, I.; Thu, K.L.; Silvester, J.; Cruickshank, J.; El Ghamrasni, S.; Ba-Alawi, W.; Fletcher, G.C.; Kiarash, R.; Elliott, M.J.; Chalmers, J.J.; et al. The spindle assembly checkpoint is a therapeutic vulnerability of CDK4/6 inhibitor-resistant ER(+) breast cancer with mitotic aberrations. Sci. Adv. 2022, 8, eabq4293. [Google Scholar] [CrossRef]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and cancer metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef]
- Pandey, K.; Park, N.; Park, K.-S.; Hur, J.; Cho, Y.B.; Kang, M.; An, H.-J.; Kim, S.; Hwang, S.; Moon, Y.W. Combined CDK2 and CDK4/6 Inhibition Overcomes Palbociclib Resistance in Breast Cancer by Enhancing Senescence. Cancers 2020, 12, 3566. [Google Scholar] [CrossRef]
- Zhang, C.; Stockwell, S.R.; Elbanna, M.; Ketteler, R.; Freeman, J.; Al-Lazikani, B.; Eccles, S.; De Haven Brandon, A.; Raynaud, F.; Hayes, A.; et al. Signalling involving MET and FAK supports cell division independent of the activity of the cell cycle-regulating CDK4/6 kinases. Oncogene 2019, 38, 5905–5920. [Google Scholar] [CrossRef]
- Olmez, I.; Zhang, Y.; Manigat, L.; Benamar, M.; Brenneman, B.; Nakano, I.; Godlewski, J.; Bronisz, A.; Lee, J.; Abbas, T.; et al. Combined c-Met/Trk Inhibition Overcomes Resistance to CDK4/6 Inhibitors in Glioblastoma. Cancer Res. 2018, 78, 4360–4369. [Google Scholar] [CrossRef]
- Finn, R.S.; Aleshin, A.; Slamon, D.J. Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers. Breast Cancer Res. 2016, 18, 17. [Google Scholar] [CrossRef]
- Schiavon, G.; Hrebien, S.; Garcia-Murillas, I.; Cutts, R.J.; Pearson, A.; Tarazona, N.; Fenwick, K.; Kozarewa, I.; Lopez-Knowles, E.; Ribas, R.; et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci. Transl. Med. 2015, 7, 313ra182. [Google Scholar] [CrossRef]
- Gyanchandani, R.; Kota, K.J.; Jonnalagadda, A.R.; Minteer, T.; Knapick, B.A.; Oesterreich, S.; Brufsky, A.M.; Lee, A.V.; Puhalla, S.L. Detection of ESR1 mutations in circulating cell-free DNA from patients with metastatic breast cancer treated with palbociclib and letrozole. Oncotarget 2017, 8, 66901–66911. [Google Scholar] [CrossRef]
- Astvatsaturyan, K.; Yue, Y.; Walts, A.E.; Bose, S. Androgen receptor positive triple negative breast cancer: Clinicopathologic, prognostic, and predictive features. PLoS ONE 2018, 13, e0197827. [Google Scholar] [CrossRef]
- Di Donato, M.; Giovannelli, P.; Cernera, G.; Di Santi, A.; Marino, I.; Bilancio, A.; Galasso, G.; Auricchio, F.; Migliaccio, A.; Castoria, G. Non-genomic androgen action regulates proliferative/migratory signaling in stromal cells. Front. Endocrinol. 2014, 5, 225. [Google Scholar] [CrossRef]
- Balk, S.P.; Knudsen, K.E. AR, the cell cycle, and prostate cancer. Nucl. Recept. Signal 2008, 6, e001. [Google Scholar] [CrossRef]
- Takeshita, T.; Yamamoto, Y.; Yamamoto-Ibusuki, M.; Tomiguchi, M.; Sueta, A.; Murakami, K.; Iwase, H. Clinical significance of plasma cell-free DNA mutations in PIK3CA, AKT1, and ESR1 gene according to treatment lines in ER-positive breast cancer. Mol. Cancer 2018, 17, 67. [Google Scholar] [CrossRef]
- Peng, Y.; Wang, Y.; Zhou, C.; Mei, W.; Zeng, C. PI3K/Akt/mTOR Pathway and Its Role in Cancer Therapeutics: Are We Making Headway? Front. Oncol. 2022, 12, 819128. [Google Scholar] [CrossRef]
- Ding, Z.; Liang, J.; Li, J.; Lu, Y.; Ariyaratna, V.; Lu, Z.; Davies, M.A.; Westwick, J.K.; Mills, G.B. Physical association of PDK1 with AKT1 is sufficient for pathway activation independent of membrane localization and phosphatidylinositol 3 kinase. PLoS ONE 2010, 5, e9910. [Google Scholar] [CrossRef]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Perez-Garcia, J.; Munoz-Couselo, E.; Soberino, J.; Racca, F.; Cortes, J. Targeting FGFR pathway in breast cancer. Breast 2018, 37, 126–133. [Google Scholar] [CrossRef]
- Rochlitz, C.F.; Scott, G.K.; Dodson, J.M.; Liu, E.; Dollbaum, C.; Smith, H.S.; Benz, C.C. Incidence of activating ras oncogene mutations associated with primary and metastatic human breast cancer. Cancer Res. 1989, 49, 357–360. [Google Scholar]
- Raimondi, L.; Raimondi, F.M.; Pietranera, M.; Di Rocco, A.; Di Benedetto, L.; Miele, E.; Lazzeroni, R.; Cimino, G.; Spinelli, G.P. Assessment of Resistance Mechanisms and Clinical Implications in Patients with KRAS Mutated-Metastatic Breast Cancer and Resistance to CDK4/6 Inhibitors. Cancers 2021, 13, 1928. [Google Scholar] [CrossRef]
- Casimiro, S.; Vilhais, G.; Gomes, I.; Costa, L. The Roadmap of RANKL/RANK Pathway in Cancer. Cells 2021, 10, 1978. [Google Scholar] [CrossRef]
- Gomes, I.; de Almeida, B.P.; Damaso, S.; Mansinho, A.; Correia, I.; Henriques, S.; Cruz-Duarte, R.; Vilhais, G.; Felix, P.; Alves, P.; et al. Expression of receptor activator of NFkB (RANK) drives stemness and resistance to therapy in ER+HER2- breast cancer. Oncotarget 2020, 11, 1714–1728. [Google Scholar] [CrossRef]
- Benitez, S.; Cordero, A.; Santamaria, P.G.; Redondo-Pedraza, J.; Rocha, A.S.; Collado-Sole, A.; Jimenez, M.; Sanz-Moreno, A.; Yoldi, G.; Santos, J.C.; et al. RANK links senescence to stemness in the mammary epithelia, delaying tumor onset but increasing tumor aggressiveness. Dev. Cell 2021, 56, 1727–1741.e7. [Google Scholar] [CrossRef]
- De Angelis, C.; Fu, X.; Cataldo, M.L.; Nardone, A.; Pereira, R.; Veeraraghavan, J.; Nanda, S.; Qin, L.; Sethunath, V.; Wang, T.; et al. Activation of the IFN Signaling Pathway is Associated with Resistance to CDK4/6 Inhibitors and Immune Checkpoint Activation in ER-Positive Breast Cancer. Clin. Cancer Res. 2021, 27, 4870–4882. [Google Scholar] [CrossRef]
- Sisinni, L.; Pietrafesa, M.; Lepore, S.; Maddalena, F.; Condelli, V.; Esposito, F.; Landriscina, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Breast Cancer: The Balance between Apoptosis and Autophagy and Its Role in Drug Resistance. Int. J. Mol. Sci. 2019, 20, 857. [Google Scholar] [CrossRef]
- Lefort, S.; Joffre, C.; Kieffer, Y.; Givel, A.M.; Bourachot, B.; Zago, G.; Bieche, I.; Dubois, T.; Meseure, D.; Vincent-Salomon, A.; et al. Inhibition of autophagy as a new means of improving chemotherapy efficiency in high-LC3B triple-negative breast cancers. Autophagy 2014, 10, 2122–2142. [Google Scholar] [CrossRef]
- Chen, S.; Jiang, Y.Z.; Huang, L.; Zhou, R.J.; Yu, K.D.; Liu, Y.; Shao, Z.M. The residual tumor autophagy marker LC3B serves as a prognostic marker in local advanced breast cancer after neoadjuvant chemotherapy. Clin. Cancer Res. 2013, 19, 6853–6862. [Google Scholar] [CrossRef]
- Zelivianski, S.; Cooley, A.; Kall, R.; Jeruss, J.S. Cyclin-dependent kinase 4-mediated phosphorylation inhibits Smad3 activity in cyclin D-overexpressing breast cancer cells. Mol. Cancer Res. 2010, 8, 1375–1387. [Google Scholar] [CrossRef]
- Du, B.; Shim, J.S. Targeting Epithelial-Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef]
- Pradella, D.; Naro, C.; Sette, C.; Ghigna, C. EMT and stemness: Flexible processes tuned by alternative splicing in development and cancer progression. Mol. Cancer 2017, 16, 8. [Google Scholar] [CrossRef]
- Stefan, S.M. Multi-target ABC transporter modulators: What next and where to go? Future Med. Chem. 2019, 11, 2353–2358. [Google Scholar] [CrossRef]
- Parrish, K.E.; Pokorny, J.; Mittapalli, R.K.; Bakken, K.; Sarkaria, J.N.; Elmquist, W.F. Efflux Transporters at the Blood-Brain Barrier Limit Delivery and Efficacy of Cyclin-Dependent Kinase 4/6 Inhibitor Palbociclib (PD-0332991) in an Orthotopic Brain Tumor Model. J. Pharmacol. Exp. Ther. 2015, 355, 264–271. [Google Scholar] [CrossRef]
- Sava, G.P.; Fan, H.; Fisher, R.A.; Lusvarghi, S.; Pancholi, S.; Ambudkar, S.V.; Martin, L.-A.; Charles Coombes, R.; Buluwela, L.; Ali, S. ABC-transporter upregulation mediates resistance to the CDK7 inhibitors THZ1 and ICEC0942. Oncogene 2020, 39, 651–663. [Google Scholar] [CrossRef]
- Gao, Y.; Shen, J.; Choy, E.; Mankin, H.; Hornicek, F.; Duan, Z. Inhibition of CDK4 sensitizes multidrug resistant ovarian cancer cells to paclitaxel by increasing apoptosiss. Cell. Oncol. 2017, 40, 209–218. [Google Scholar] [CrossRef]
- Sorf, A.; Sucha, S.; Morell, A.; Novotna, E.; Staud, F.; Zavrelova, A.; Visek, B.; Wsol, V.; Ceckova, M. Targeting Pharmacokinetic Drug Resistance in Acute Myeloid Leukemia Cells with CDK4/6 Inhibitors. Cancers 2020, 12, 1596. [Google Scholar] [CrossRef]
- Wu, T.; Chen, Z.; To, K.K.W.; Fang, X.; Wang, F.; Cheng, B.; Fu, L. Effect of abemaciclib (LY2835219) on enhancement of chemotherapeutic agents in ABCB1 and ABCG2 overexpressing cells in vitro and in vivo. Biochem. Pharmacol. 2017, 124, 29–42. [Google Scholar] [CrossRef]
- Gupta, P.; Zhang, Y.-K.; Zhang, X.-Y.; Wang, Y.-J.; Lu, K.W.; Hall, T.; Peng, R.; Yang, D.-H.; Xie, N.; Chen, Z.-S. Voruciclib, a Potent CDK4/6 Inhibitor, Antagonizes ABCB1 and ABCG2-Mediated Multi-Drug Resistance in Cancer Cells. Cell. Physiol. Biochem. 2018, 45, 1515–1528. [Google Scholar] [CrossRef]
- Kalinsky, K.; Accordino, M.K.; Chiuzan, C.; Mundi, P.S.; Trivedi, M.S.; Novik, Y.; Tiersten, A.; Raptis, G.; Baer, L.N.; Oh, S.Y.; et al. A randomized, phase II trial of fulvestrant or exemestane with or without ribociclib after progression on anti-estrogen therapy plus cyclin-dependent kinase 4/6 inhibition (CDK 4/6i) in patients (pts) with unresectable or hormone receptor positive (HR+), HER2-negative metastatic breast cancer (MBC): MAINTAIN trial. J. Clin. Oncol. 2022, 40, LBA1004. [Google Scholar] [CrossRef]
- Lloyd, M.R.; Wander, S.A.; Hamilton, E.; Razavi, P.; Bardia, A. Next-generation selective estrogen receptor degraders and other novel endocrine therapies for management of metastatic hormone receptor-positive breast cancer: Current and emerging role. Ther. Adv. Med. Oncol. 2022, 14, 17588359221113694. [Google Scholar] [CrossRef]
- Jhaveri, K.; Juric, D.; Yap, Y.S.; Cresta, S.; Layman, R.M.; Duhoux, F.P.; Terret, C.; Takahashi, S.; Huober, J.; Kundamal, N.; et al. A Phase I Study of LSZ102, an Oral Selective Estrogen Receptor Degrader, with or without Ribociclib or Alpelisib, in Patients with Estrogen Receptor-Positive Breast Cancer. Clin. Cancer Res. 2021, 27, 5760–5770. [Google Scholar] [CrossRef]
- Bidard, F.C.; Kaklamani, V.G.; Neven, P.; Streich, G.; Montero, A.J.; Forget, F.; Mouret-Reynier, M.A.; Sohn, J.H.; Taylor, D.; Harnden, K.K.; et al. Elacestrant (oral selective estrogen receptor degrader) Versus Standard Endocrine Therapy for Estrogen Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: Results From the Randomized Phase III EMERALD Trial. J. Clin. Oncol. 2022, 40, 3246. [Google Scholar] [CrossRef]
- Damodaran, S.; Plourde, P.V.; Moore, H.C.F.; Anderson, I.C.; Portman, D.J. Open-label, phase 2, multicenter study of lasofoxifene (LAS) combined with abemaciclib (Abema) for treating pre- and postmenopausal women with locally advanced or metastatic ER+/HER2− breast cancer and an ESR1 mutation after progression on prior therapies. J. Clin. Oncol. 2022, 40, 1022. [Google Scholar] [CrossRef]
- Tsuji, J.; Li, T.; Grinshpun, A.; Coorens, T.; Russo, D.; Anderson, L.; Rees, R.; Nardone, A.; Patterson, C.; Lennon, N.J.; et al. Clinical Efficacy and Whole-Exome Sequencing of Liquid Biopsies in a Phase IB/II Study of Bazedoxifene and Palbociclib in Advanced Hormone Receptor-Positive Breast Cancer. Clin. Cancer Res. 2022, 28, 5066–5078. [Google Scholar] [CrossRef]
- Rugo, H.S.; Lerebours, F.; Ciruelos, E.; Drullinsky, P.; Ruiz-Borrego, M.; Neven, P.; Park, Y.H.; Prat, A.; Bachelot, T.; Juric, D.; et al. Alpelisib plus fulvestrant in PIK3CA-mutated, hormone receptor-positive advanced breast cancer after a CDK4/6 inhibitor (BYLieve): One cohort of a phase 2, multicentre, open-label, non-comparative study. Lancet Oncol. 2021, 22, 489–498. [Google Scholar] [CrossRef]
- Rugo, H.S.; Lerebours, F.; Juric, D.; Turner, N.; Chia, S.; Drullinsky, P.; Prat, A.; Vazquez, R.V.; Akdere, M.; Arce, C.; et al. Alpelisib plus letrozole in patients with PIK3CA-mutated, hormone-receptor positive (HR plus), human epidermal growth factor receptor-2-negative (HER2-) advanced breast cancer (ABC) previously treated with a cyclin-dependent kinase 4/6 inhibitor (CDK4/6i) + fulvestrant: BYLieve study results. Cancer Res. 2021, 81, PD2-07. [Google Scholar] [CrossRef]
- Wander, S.A.; Juric, D.; Supko, J.G.; Micalizzi, D.S.; Spring, L.; Vidula, N.; Beeler, M.; Habin, K.R.; Viscosi, E.; Fitzgerald, D.M.; et al. Phase Ib trial to evaluate safety and anti-tumor activity of the AKT inhibitor, ipatasertib, in combination with endocrine therapy and a CDK4/6 inhibitor for patients with hormone receptor positive (HR+)/HER2 negative metastatic breast cancer (MBC) (TAKTIC). J. Clin. Oncol. 2020, 38, 1066. [Google Scholar] [CrossRef]
- Turner, N.C.; Oliveira, M.; Howell, S.J.; Dalenc, F.; Cortes, J.; Gomez Moreno, H.L.; Hu, X.; Jhaveri, K.; Krivorotko, P.; Loibl, S.; et al. Capivasertib in Hormone Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2023, 388, 2058–2070. [Google Scholar] [CrossRef]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A., 3rd; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef]
- Bardia, A.; Hurvitz, S.A.; DeMichele, A.; Clark, A.S.; Zelnak, A.; Yardley, D.A.; Karuturi, M.; Sanft, T.; Blau, S.; Hart, L.; et al. Phase I/II Trial of Exemestane, Ribociclib, and Everolimus in Women with HR(+)/HER2(−) Advanced Breast Cancer after Progression on CDK4/6 Inhibitors (TRINITI-1). Clin. Cancer Res. 2021, 27, 4177–4185. [Google Scholar] [CrossRef]
- Mayer, I.A.; Haley, B.B.; Abramson, V.G.; Brufsky, A.; Rexer, B.; Stringer-Reasor, E.; Jhaveri, K.L.; Sanders, M.; Ericsson-Gonzalez, P.I.; Ye, F.; et al. A phase Ib trial of fulvestrant+CDK4/6 inhibitor (CDK4/6i) palbociclib plus pan-FGFR tyrosine kinase inhibitor (TKI) erdafitinib in FGFR-amplified/ER+/HER2-negative metastatic breast cancer (MBC). Cancer Res. 2021, 81, PD1-03. [Google Scholar] [CrossRef]
- Rugo, H.S.; Kabos, P.; Beck, J.T.; Jerusalem, G.; Wildiers, H.; Sevillano, E.; Paz-Ares, L.; Chisamore, M.J.; Chapman, S.C.; Hossain, A.M.; et al. Abemaciclib in combination with pembrolizumab for HR+, HER2- metastatic breast cancer: Phase 1b study. NPJ Breast Cancer 2022, 8, 118. [Google Scholar] [CrossRef]
- Yuan, Y.; Lee, J.S.; Yost, S.E.; Frankel, P.H.; Ruel, C.; Egelston, C.A.; Guo, W.; Padam, S.; Tang, A.; Martinez, N.; et al. Phase I/II trial of palbociclib, pembrolizumab and letrozole in patients with hormone receptor-positive metastatic breast cancer. Eur. J. Cancer 2021, 154, 11–20. [Google Scholar] [CrossRef]
- Santa-Maria, C.A.; Wang, C.; Cimino-Mathews, A.; Roussos-Torres, E.; Connolly, R.M.; Wolff, A.C.; Jaffee, E.M.; Stearns, V. IMMUNe mOdulation in early stage estrogen receptor positive breast cancer treated with neoADjuvant Avelumab, Palbociclib, and Tamoxifen: The ImmunoADAPT study (NCT03573648). Cancer Res. 2019, 79, OT3-02-03. [Google Scholar] [CrossRef]
- Mayer, E.L.; Wander, S.A.; Regan, M.M.; DeMichele, A.; Forero-Torres, A.; Rimawi, M.F.; Ma, C.X.; Cristofanilli, M.; Anders, C.K.; Bartlett, C.H.; et al. Palbociclib after CDK and endocrine therapy (PACE): A randomized phase II study of fulvestrant, palbociclib, and avelumab for endocrine pre-treated ER+/HER2-metastatic breast cancer. J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Mayer, E.L.; Wander, S.A.; Regan, M.M.; DeMichele, A.M.; Forero, A.; Rimawi, M.F.; Ma, C.X.; Cristofanilli, M.; Anders, C.K.; Bartlett, C.H.; et al. Palbociclib after CDK inhibitor and endocrine therapy (PACE): A randomized phase II study of fulvestrant versus palbociclib plus fulvestrant, with and without avelumab, for CDK inhibitor pre-treated HR+/HER2-metastatic breast cancer. Cancer Res. 2018, 78, OT3-05-11. [Google Scholar] [CrossRef]
- Mayer, E.L.; Ren, Y.; Wagle, N.; Mahtani, R.; Ma, C.; DeMichele, A.; Cristofanilli, M.; Meisel, J.; Miller, K.D.; Jolly, T.; et al. Abstract GS3-06: GS3-06 Palbociclib After CDK4/6i and Endocrine Therapy (PACE): A Randomized Phase II Study of Fulvestrant, Palbociclib, and Avelumab for Endocrine Pre-treated ER+/HER2- Metastatic Breast Cancer. Cancer Res. 2023, 83, GS3-06. [Google Scholar] [CrossRef]
- Clark, A.S.; McAndrew, N.P.; Troxel, A.; Feldman, M.; Lal, P.; Rosen, M.; Burrell, J.; Redlinger, C.; Gallagher, M.; Bradbury, A.R.; et al. Combination Paclitaxel and Palbociclib: Results of a Phase I Trial in Advanced Breast Cancer. Clin. Cancer Res. 2019, 25, 2072–2079. [Google Scholar] [CrossRef]
- Ogata, R.; Kishino, E.; Saitoh, W.; Koike, Y.; Kurebayashi, J. Resistance to cyclin-dependent kinase (CDK) 4/6 inhibitors confers cross-resistance to other CDK inhibitors but not to chemotherapeutic agents in breast cancer cells. Breast Cancer 2021, 28, 206–215. [Google Scholar] [CrossRef]
- Sava, G.P.; Fan, H.; Coombes, R.C.; Buluwela, L.; Ali, S. CDK7 inhibitors as anticancer drugs. Cancer Metastasis Rev. 2020, 39, 805–823. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Lok, S.W.; Whittle, J.R.; Vaillant, F.; Teh, C.E.; Lo, L.L.; Policheni, A.N.; Bergin, A.R.T.; Desai, J.; Ftouni, S.; Gandolfo, L.C.; et al. A Phase Ib Dose-Escalation and Expansion Study of the BCL2 Inhibitor Venetoclax Combined with Tamoxifen in ER and BCL2-Positive Metastatic Breast Cancer. Cancer Discov. 2019, 9, 354–369. [Google Scholar] [CrossRef]
- Lindeman, G.J.; Fernando, T.M.; Bowen, R.; Jerzak, K.J.; Song, X.; Decker, T.; Boyle, F.; McCune, S.; Armstrong, A.; Shannon, C.; et al. VERONICA: Randomized Phase II Study of Fulvestrant and Venetoclax in ER-Positive Metastatic Breast Cancer Post-CDK4/6 Inhibitors—Efficacy, Safety, and Biomarker Results. Clin. Cancer Res. 2022, 28, 3256–3267. [Google Scholar] [CrossRef]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, T.; Geng, C.; Zhang, Y.; Zhang, J.; Ning, Z.; Jiang, Z. Exploratory clinical study of chidamide, an oral subtype-selective histone deacetylase inhibitor, in combination with exemestane in hormone receptor-positive advanced breast cancer. Chin. J. Cancer Res. 2018, 30, 605–612. [Google Scholar] [CrossRef]
- Jiang, Z.; Li, W.; Hu, X.; Zhang, Q.; Sun, T.; Cui, S.; Wang, S.; Ouyang, Q.; Yin, Y.; Geng, C.; et al. Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer (ACE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 806–815. [Google Scholar] [CrossRef]
- Zhou, J.; Wu, X.; Zhang, H.; Wang, X.; Yuan, Y.; Zhang, S.; Jiang, Z.; Wang, T. Clinical outcomes of tucidinostat-based therapy after prior CDK4/6 inhibitor progression in hormone receptor-positive heavily pretreated metastatic breast cancer. Breast 2022, 66, 255–261. [Google Scholar] [CrossRef]
- Maglakelidze, M.; Bulat, I.; Ryspayeva, D.; Krastev, B.M.; Gogiladze, M.; Crijanovschi, A.; Aftimos, P.G.; Neven, P.; Pegram, M.D.; Menke, C.W.; et al. Rintodestrant (G1T48), an oral selective estrogen receptor degrader, in combination with palbociclib for ER+/HER2– advanced breast cancer: Phase 1 results. J. Clin. Oncol. 2021, 39, 1063. [Google Scholar] [CrossRef]
- Chia, S.; Neven, P.; Ciruelos, E.M.; Lerebours, F.; Ruiz-Borrego, M.; Drullinsky, P.; Prat, A.; Park, Y.H.; Juric, D.; Turner, N.C.; et al. Alpelisib + endocrine therapy in patients with PIK3CA-mutated, hormone receptor–positive, human epidermal growth factor receptor 2–negative, advanced breast cancer: Analysis of all 3 cohorts of the BYLieve study. J. Clin. Oncol. 2023, 41, 1078. [Google Scholar] [CrossRef]
- Coombes, R.C.; Howell, S.; Lord, S.R.; Kenny, L.; Mansi, J.; Mitri, Z.; Palmieri, C.; Chap, L.I.; Richards, P.; Gradishar, W.; et al. Dose escalation and expansion cohorts in patients with advanced breast cancer in a Phase I study of the CDK7-inhibitor samuraciclib. Nat. Commun. 2023, 14, 4444. [Google Scholar] [CrossRef]
- Rugo, H.S.; Bardia, A.; Marme, F.; Cortes, J.; Schmid, P.; Loirat, D.; Tredan, O.; Ciruelos, E.; Dalenc, F.; Gomez Pardo, P.; et al. Overall survival with sacituzumab govitecan in hormone receptor-positive and human epidermal growth factor receptor 2-negative metastatic breast cancer (TROPiCS-02): A randomised, open-label, multicentre, phase 3 trial. Lancet 2023. [Google Scholar] [CrossRef]


| Compound | Company | Clinical Status | Selectivity (IC50 or Ki) |
|---|---|---|---|
| Palbociclib (PD0332991) | Pfizer | Approved. HR+/HER2− advanced or metastatic BC in combination with ET. | CDK4: 11 nM (IC50) CDK6: 16 nM (IC50) |
| Ribociclib (LEE011) | Novartis | Approved. HR+/HER2− advanced or metastatic BC. | CDK4: 10 nM (IC50) CDK6: 39 nM (IC50) |
| Abemaciclib (LY2835219) | Eli Lilly | Approved. HR+/HER2− advanced or metastatic BC in combination with ET. HR+/HER2− advanced or metastatic BC as monotherapy. Adjuvant therapy for high-risk, early-stage HR+/HER2− BC in combination with ET. | CDK4: 2 nM (IC50) CDK6: 10 nM (IC50) CDK9: 57 nM (IC50) |
| Dalpiciclib (SHR6390) | Jiangsu Hengrui Medicine | In clinical trials. Phase III for HR+/HER2− BC in combination with ET. Phase I/II for multiple tumor types in combination with ET or immunotherapy. | CDK4: 12 nM (IC50) CDK6: 10 nM (IC50) |
| PF-06873600 | Pfizer | In clinical trials. Phase II for metastatic HR+/HER2− and TNBC. | CDK2: 0.09 nM (Ki) CDK4: 0.13 nM (Ki) CDK6: 0.16 nM (Ki) |
| Trilaciclib (G1T28) | G1 Therapeutics | In clinical trials. Phase III for early-stage and metastatic TNBC in combination with chemotherapy. | CDK4: 1 nM (IC50) CDK6: 4 nM (IC50) CDK9: 50 nM (IC50) |
| Lerociclib (G1T38) | G1 Therapeutics | In clinical trials. Phase I/II for HR+/HER2− metastatic BC in combination with ET. | CDK4: 1 nM (IC50) CDK6: 2 nM (IC50) CDK9: 28 nM (IC50) |
| R547 | Hoffmann-La Roche | In clinical trials. Phase I for advanced BC and other solid cancers. | CDK1: 2 nM (Ki) CDK2: 3 nM (Ki) CDK4: 1 nM (Ki) |
| Neoadjuvant Studies | |||||
|---|---|---|---|---|---|
| Phase | BC Stage | Intervention | Trial Identifier | Status * | Results |
| III | Stage II–III | Palbociclib + ET vs. ET | SAFIA (NCT03447132) | Completed | No statistically significant differences in pCR rates [55]. |
| II | Stage I–III | AI vs. AI then AI + palbociclib vs. palbociclib then AI + palbociclib vs. AI + palbociclib | PALLET (NCT02296801) | Completed | Palbociclib + AI (all arms) significantly decreased Ki67 compared to AI alone but did not increase the pCR rate [44]. |
| II | Stage I–III | Ribociclib + AI vs. chemotherapy | CORALLEEN (NCT03248427) | Completed | No significant differences in ROR. Ribociclib + AI was associated with better HRQoL outcomes [56]. |
| II | Stage I–III | AI vs. AI + abemaciclib vs. abemaciclib then AI + abemaciclib | neoMONARCH (NCT02441946) | Completed | AI + abemaciclib or abemaciclib alone significantly decreases Ki67 compared to AI alone [57]. |
| II | Stage II–III | AI vs. ribociclib + AI vs. ET | FELINE (NCT02712723) | Active, not recruiting | PEPI score was equal in AI and ribociclib + AI groups [58]. |
| II | Stage II–III | AI vs. AI then palbociclib + AI | NeoPalAna (NCT01723774) | Active, not recruiting | The CCCA rate was significantly higher after adding palbociclib to AI [59]. |
| II | Stage II–III | AI vs. AI then chemotherapy vs. AI + ribociclib | NEOBLC (NCT03283384) | Active, not recruiting | N/A |
| II | Stage II | Ribociclib + AI | RIBOLARIS (NCT05296746) | Recruiting | N/A |
| Adjuvant Studies | |||||
| Phase | BC Stage | Intervention | Trial Identifier | Status * | Results |
| III | High risk of recurrence | Palbociclib + ET vs. ET | PENELOPE-B (NCT01864746) | Active, not recruiting | Palbociclib in addition to ET did not improve iDFS [60]. |
| III | Stage II–III | Palbociclib + ET vs. ET | PALLAS (NCT02513394) | Active, not recruiting | Palbociclib in addition to ET did not improve iDFS [61]. |
| III | High risk of recurrence | Abemaciclib + ET vs. ET | MonarchE (NCT03155997) | Active, not recruiting | Abemaciclib + ET reduces significantly ROR compared to ET alone [45]. |
| III | Stage II–III | Ribociclib + ET vs. ET | NATALEE (NCT03701334) | Active, not recruiting | Ribociclib + ET improves iDFS compared to ET alone [62]. |
| III | High risk of recurrence | Abemaciclib + ET vs. ET | ADAPTlate (NCT04565054) | Recruiting | N/A |
| III | Intermediate risk of recurrence | Ribociclib + ET vs. chemotherapy | ADAPTcycle (NCT04055493) | Recruiting | N/A |
| II | High risk of recurrence | Palbociclib + ET vs. ET | HIPEx (NCT04247633) | Recruiting | N/A |
| II | stage II–III | Ribociclib + ET vs. ET | LEADER (NCT03285412) | Recruiting | N/A |
| Mechanism/Player | Impact on Response to CDK4/6i | Clinical Potential |
|---|---|---|
| Protein Rb | Impaired Rb function abrogates response [29,118,119,120,121,122,123]. | Prognostic/predictive role not validated [124,125] |
| Cyclin D–CDK4–CDK6 | Upregulation of cyclin D [118,126,127], CDK4 [127,128,129] and/or CDK6 [118,129,130,131,132,133] associated with resistance. | Prognostic/predictive role not validated [18,124]. |
| Cyclin E–CDK2 | Upregulation ofcCyclin E1, cyclin E2 and CDK2 associated with resistance [59,118,120,124,129,134,135]. | Prognostic/predictive role not validated [50,125]. |
| CDK7 | Upregulation of CDK7 associated with resistance [128,136]. | Not assessed. |
| INK4 and CIP/KIP members | p16 overexpression in Rb-proficient models [123,137] and increased phosphorylation of p27 [120,138] associated with decreased sensitivity. | Prognostic/predictive role not validated [124,125]. |
| Other cell cycle regulators | Overexpression of WEE1 or MDM2 associated with intrinsic resistance [139,140]. | Combination with MDM2 inhibitor could abrogate resistance to CDK4/6i and ET in preclinical models [141]. |
| FZR1 KD associated with intrinsic resistance [142]. | Not assessed. | |
| AP-1 and c-Fos increased upon acquired resistance [143]. | AP-1 blockade combined with palbociclib could effectively inhibit cell proliferation [143]. | |
| TK1 overexpression associated with intrinsic resistance [144]. | Potential prognostic value of plasma TKa reported (TREnd, NCT02549430) [145] and under investigation (BioItaLEE, NCT03439046; PYTHIA, NCT02536742). | |
| Amplification of AURKA (Aurora kinase A) found in tumor biopsies from resistant patients [134]. | Not assessed. | |
| Mutated MYC was found in patients treated with abemaciclib plus AI [146] c-MYC induction and cyclin E/CDK2 activity followed CDK4/6i therapy [147]. | Not assessed. | |
| Activated c-MET found in patients treated with abemaciclib plus AI [146]. | Not assessed. | |
| HRs and HER2 | Loss of ER/PR expression [128,129] and activation of AR signaling [148] associated with intrinsic and acquired resistance. HER2 mutations conferred estrogen independence as well as resistance to ET and CDK4/6i [149]. ERBB2 amplification in patients treated with CDK4/6i [134,150]. | Prognostic/predictive role of ESR1 mutations not validated [151,152]. Combination of palbociclib with enzalutamide, a selective AR inhibitor, abrogated resistance in BC cell lines [148]. Anti-HER2 neratinib abrogated resistance to ET+CDK4/6i [149]. |
| PI3K-AKT-mTOR pathway | Activation of PI3K-AKT-mTOR signaling associated with intrinsic and acquired resistance [118,126,134,153,154,155,156,157,158,159]. | Combination of PI3K-AKT-mTORi and CDK4/6i has been proven to overcome/prevent/delay intrinsic and acquired resistance [118,126,155,156,159,160]. PIK3CA mutations in plasma ctDNA [151,161] or increased levels of activated AKT [154] may have predictive potential. |
| FGFR pathway | FGFR1 upregulation associated with acquired resistance [50,128,152,162]. | Combination of CDK4/6i with FGFR inhibitor could abrogate acquired resistance [162]. FGFR1 amplification (MONALEESA-2 and PALOMA-3) [50,152,162] or FGFR2 mutations [134] may have prognostic potential. |
| MAPK-ERK pathway | Activation of MAPK-ERK signaling associated with intrinsic and acquired resistance [128,163]. | Combination of MEK inhibitors with CDK4/6i plus ET was shown to be effective in blocking cells proliferation [162,163]. |
| RANK-RANKL pathway | RANK OE associated with intrinsic resistance; RANK upregulation observed upon acquired resistance [136]. | Combination of CDK4/6i with RANKL inhibitors could abrogate/delay intrinsic and acquired resistance [136]. |
| Autophagy | Upregulation of genes involved in autophagy and an increase in autophagy observed upon treatment [98,164]. Increased lysosomal activity associated with resistance in TNBC [100]. | Combination of autophagy inhibitors with palbociclib-induced proliferation arrest and senescence in preclinical models [98]. Combination of CDK4/6i with lysosomotropic or lysosome destabilizers resulted in increased sensitivity of TNBC cells to CDK4/6i [100]. |
| TGF-β and EMT | CDK4/6i could induce EMT by TGF-β signaling pathway activation [165,166,167]. | Not assessed. |
| ABC transporters | Overexpression of ABCB1 and/or ABCG2 transporters may decrease anticancer efficacy of palbociclib [168,169]. | Not assessed. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, I.; Abreu, C.; Costa, L.; Casimiro, S. The Evolving Pathways of the Efficacy of and Resistance to CDK4/6 Inhibitors in Breast Cancer. Cancers 2023, 15, 4835. https://doi.org/10.3390/cancers15194835
Gomes I, Abreu C, Costa L, Casimiro S. The Evolving Pathways of the Efficacy of and Resistance to CDK4/6 Inhibitors in Breast Cancer. Cancers. 2023; 15(19):4835. https://doi.org/10.3390/cancers15194835
Chicago/Turabian StyleGomes, Inês, Catarina Abreu, Luis Costa, and Sandra Casimiro. 2023. "The Evolving Pathways of the Efficacy of and Resistance to CDK4/6 Inhibitors in Breast Cancer" Cancers 15, no. 19: 4835. https://doi.org/10.3390/cancers15194835
APA StyleGomes, I., Abreu, C., Costa, L., & Casimiro, S. (2023). The Evolving Pathways of the Efficacy of and Resistance to CDK4/6 Inhibitors in Breast Cancer. Cancers, 15(19), 4835. https://doi.org/10.3390/cancers15194835