
A Novel Role of Connective Tissue Growth Factor in the Regulation of the Epithelial Phenotype
 , , , ,
, , , , 
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
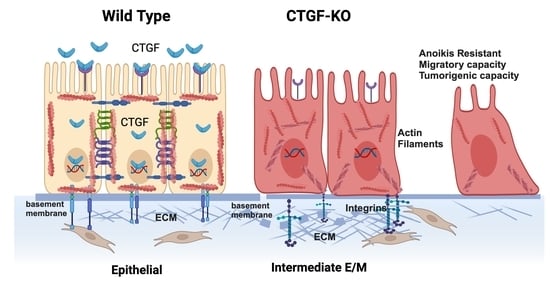
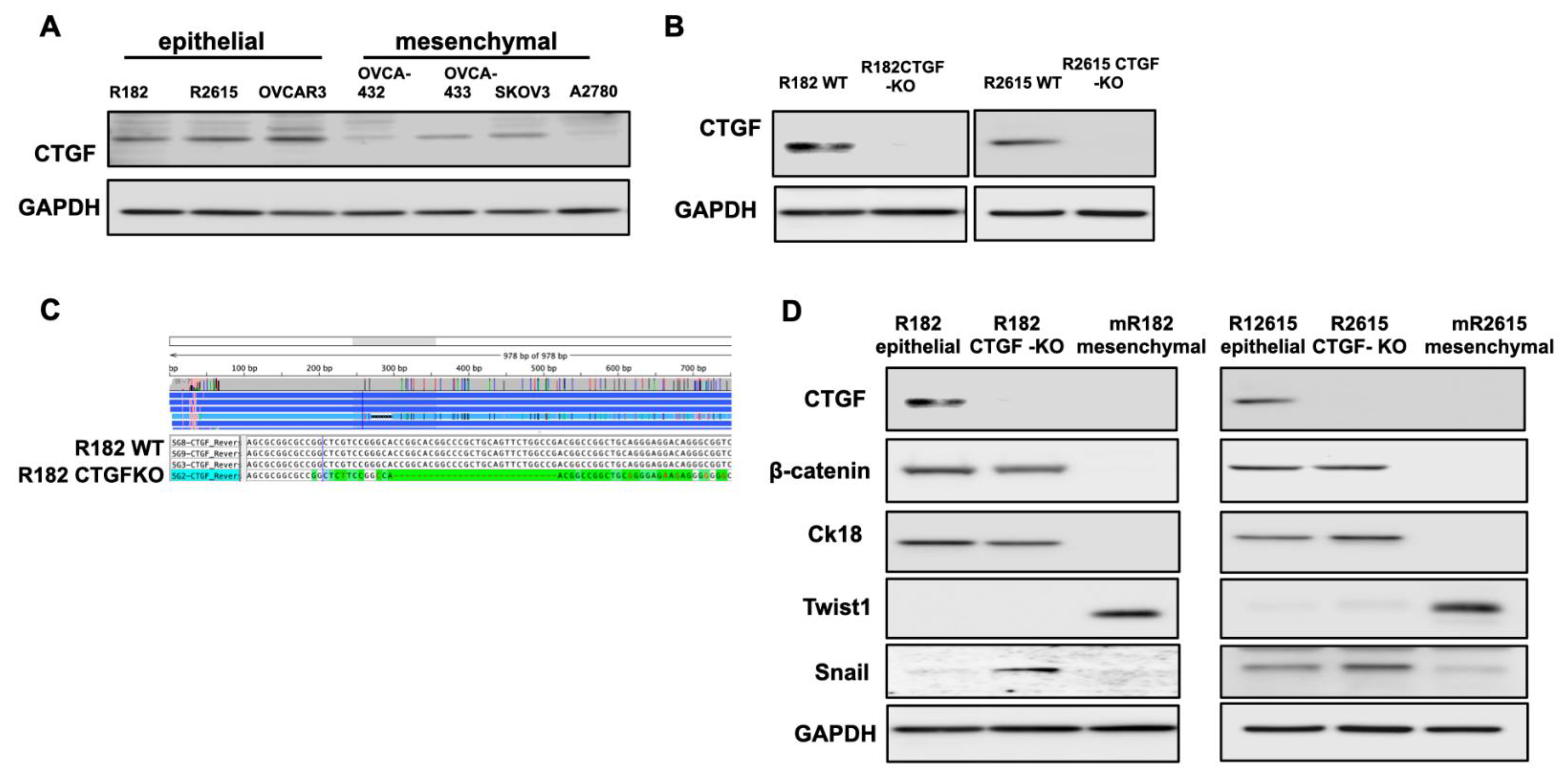
3.1. Differential Expression of CTGF during Early EMT
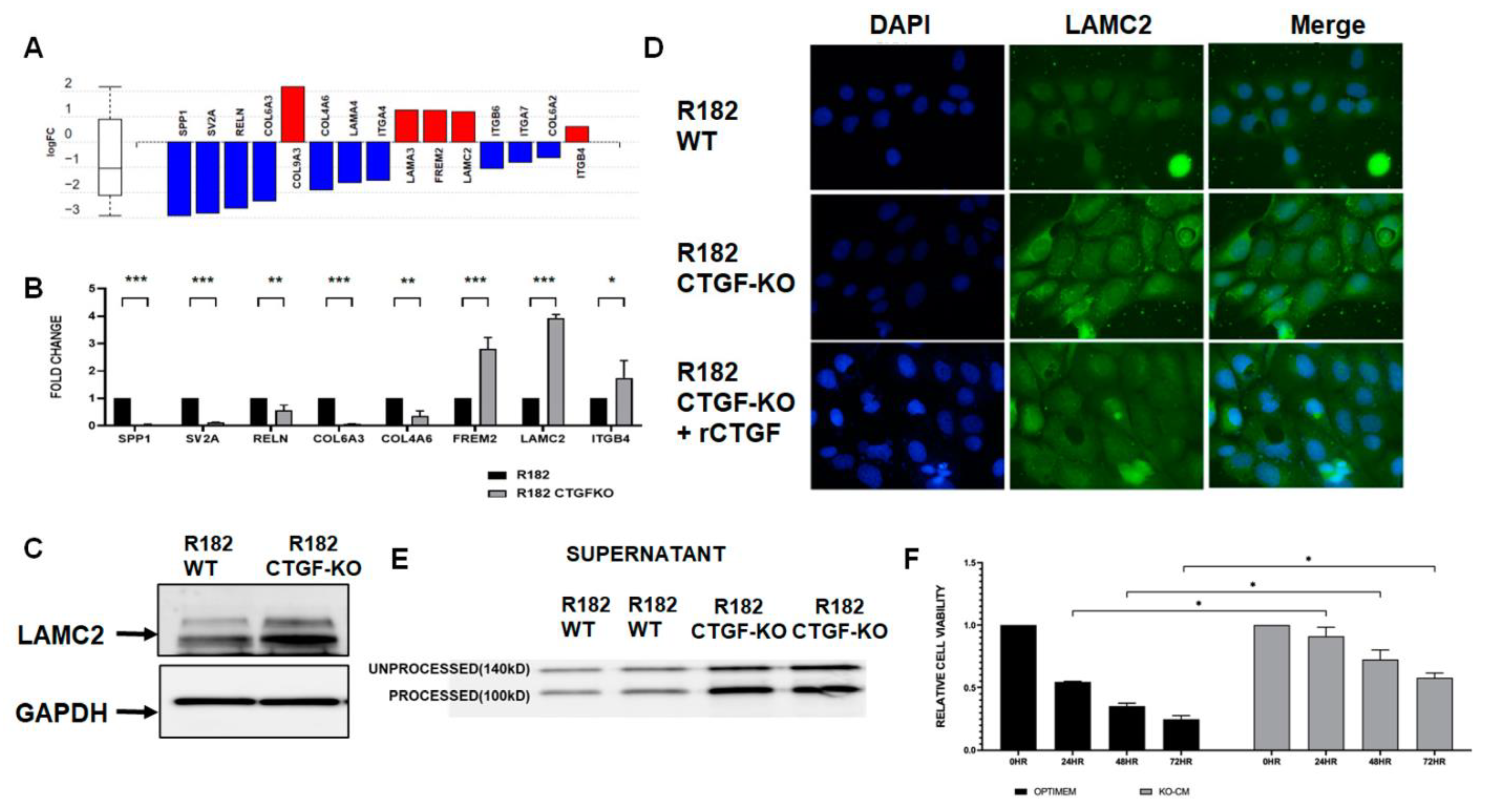
3.2. Loss of CTGF Reprograms ECM in Ovarian Cancer Cells
3.3. Inhibition of CTGF Expression Is Associated with Changes on the Transcriptome of OCCs
3.4. Loss of CTGF Alters Extracellular Matrix
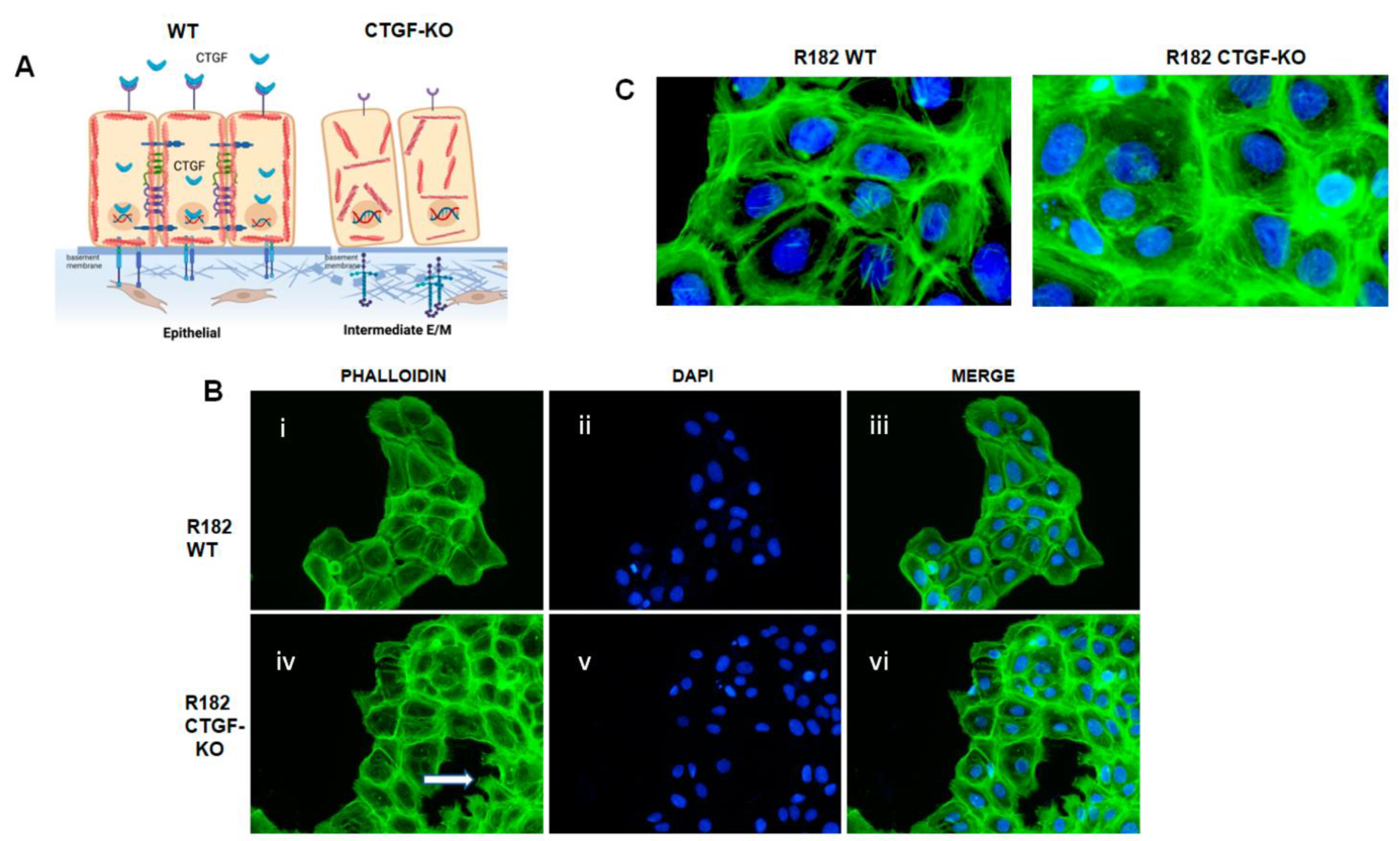
3.5. Loss of CTGF Is Associated with Cytoskeleton Remodeling
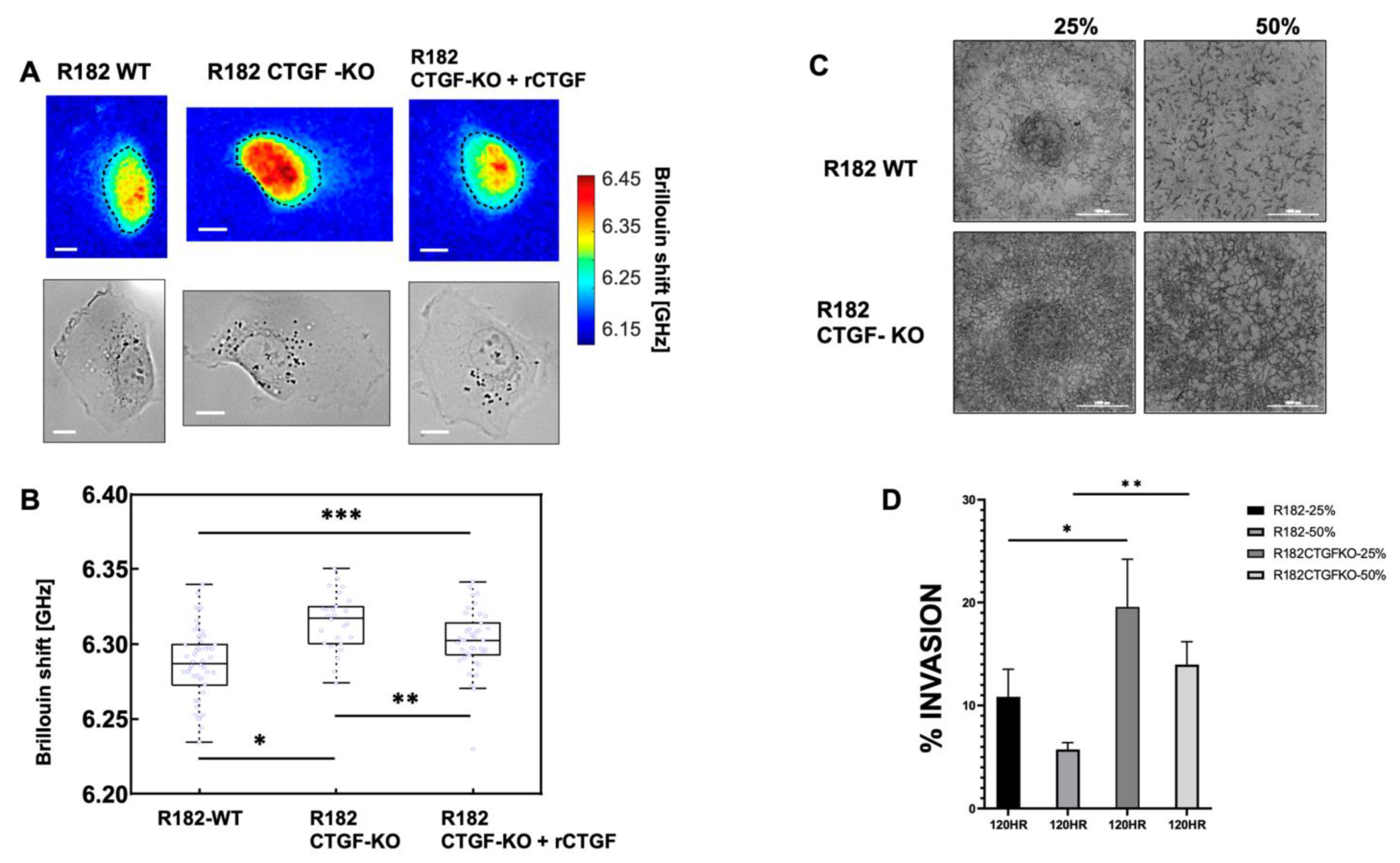
3.6. Loss of CTGF Alters Cell Stiffness
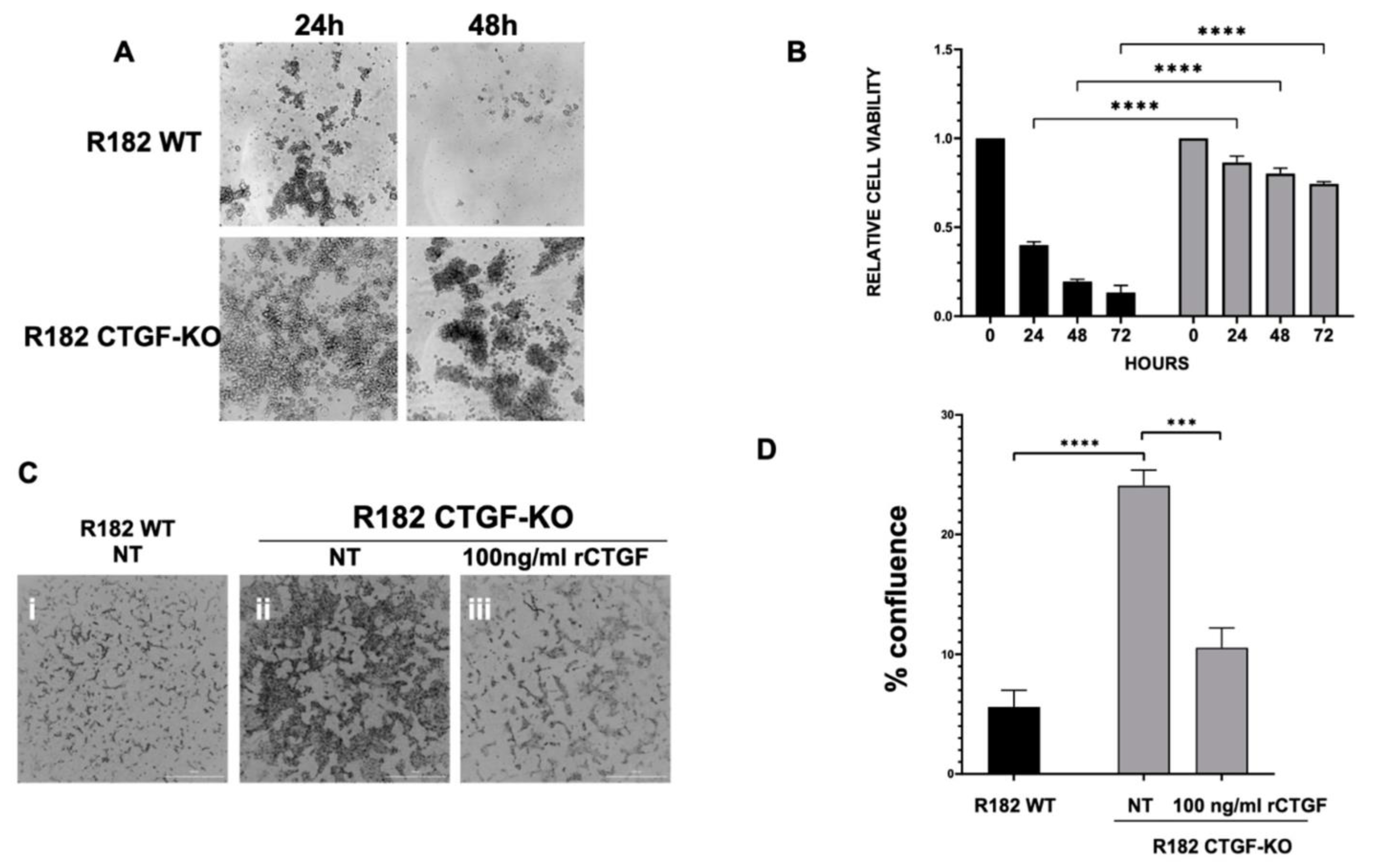
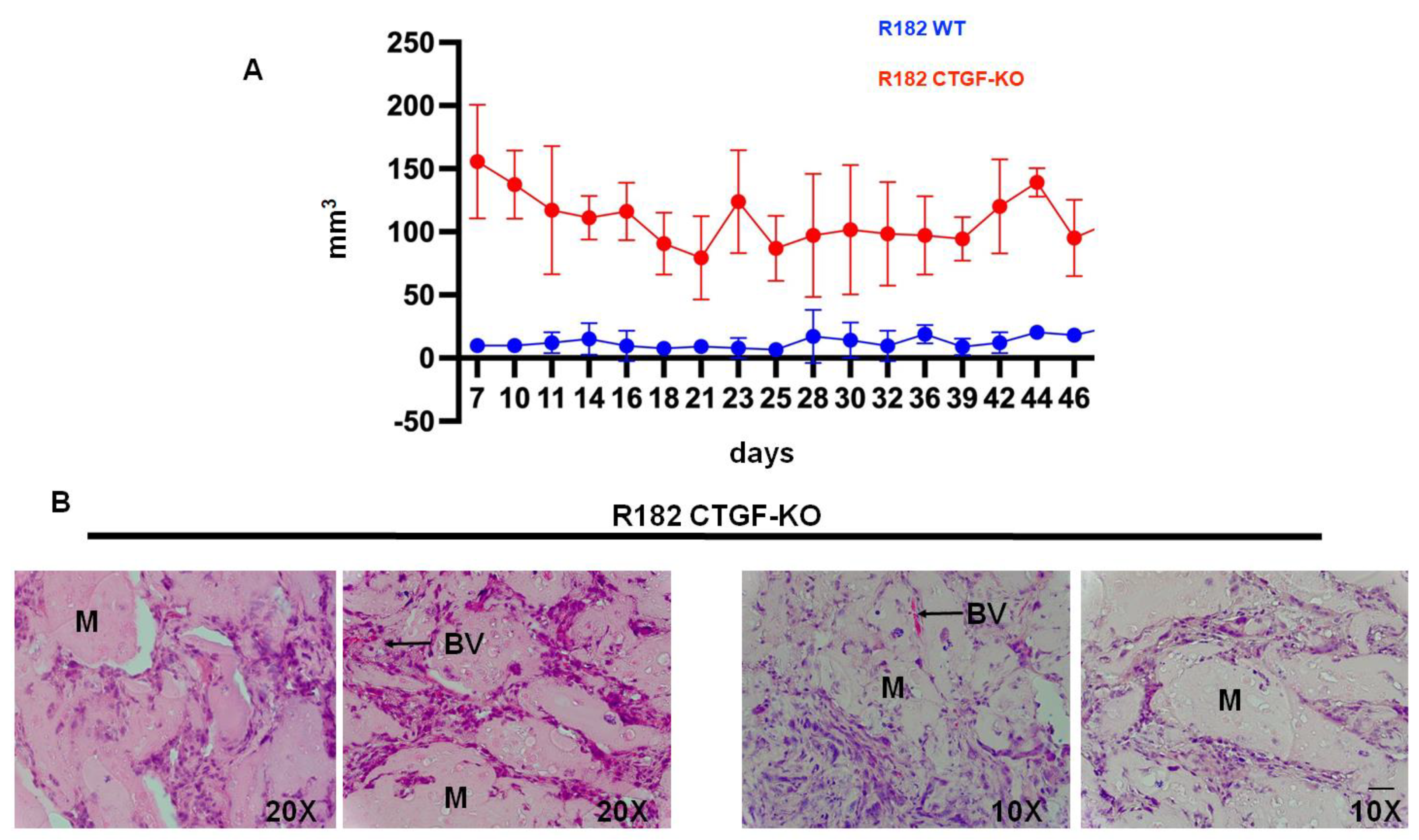
3.7. Lack of CTFG Provides Tumorigenic Capacity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Debnath, P.; Huirem, R.S.; Dutta, P.; Palchaudhuri, S. Epithelial-mesenchymal transition and its transcription factors. Biosci. Rep. 2022, 42, BSR20211754. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Cancer Facts and Figures 2019. Available online: https://www.google.com.hk/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&cad=rja&uact=8&ved=2ahUKEwiho5jStcCBAxUZilYBHWuFBIoQFnoECB4QAQ&url=https%3A%2F%2Fwww.cancer.org%2Fcontent%2Fdam%2Fcancer-org%2Fresearch%2Fcancer-facts-and-statistics%2Fannual-cancer-facts-and-figures%2F2019%2Fcancer-facts-and-figures-2019.pdf&usg=AOvVaw0_gORaOsEKfLUJ4F7PCvT8&opi=89978449 (accessed on 12 November 2020).
- Shih, I.M.; Wang, Y.; Wang, T.L. The Origin of Ovarian Cancer Species and Precancerous Landscape. Am. J. Pathol. 2021, 191, 26–39. [Google Scholar] [CrossRef]
- Soong, T.R.; Kolin, D.L.; Teschan, N.J.; Crum, C.P. Back to the Future? The Fallopian Tube, Precursor Escape and a Dualistic Model of High-Grade Serous Carcinogenesis. Cancers 2018, 10, 468. [Google Scholar] [CrossRef] [PubMed]
- Yang-Hartwich, Y.; Gurrea-Soteras, M.; Sumi, N.; Joo, W.D.; Holmberg, J.C.; Craveiro, V.; Alvero, A.B.; Mor, G. Ovulation and extra-ovarian origin of ovarian cancer. Sci. Rep. 2014, 4, 6116. [Google Scholar] [CrossRef]
- Alvero, A.B.; Kim, D.; Lima, E.; Sumi, N.J.; Lee, J.S.; Cardenas, C.; Pitruzzello, M.; Silasi, D.A.; Buza, N.; Fahmy, T.; et al. Novel approach for the detection of intraperitoneal micrometastasis using an ovarian cancer mouse model. Sci. Rep. 2017, 7, 40989. [Google Scholar] [CrossRef] [PubMed]
- Yeger, H.; Perbal, B. CCN family of proteins: Critical modulators of the tumor cell microenvironment. J. Cell Commun. Signal. 2016, 10, 229–240. [Google Scholar] [CrossRef]
- Yeger, H.; Perbal, B. The CCN family of genes: A perspective on CCN biology and therapeutic potential. J. Cell Commun. Signal. 2007, 1, 159–164. [Google Scholar] [CrossRef]
- Hendesi, H.; Barbe, M.F.; Safadi, F.F.; Monroy, M.A.; Popoff, S.N. Integrin mediated adhesion of osteoblasts to connective tissue growth factor (CTGF/CCN2) induces cytoskeleton reorganization and cell differentiation. PLoS ONE 2015, 10, e0115325. [Google Scholar] [CrossRef]
- Tzanakakis, G.; Kavasi, R.M.; Voudouri, K.; Berdiaki, A.; Spyridaki, I.; Tsatsakis, A.; Nikitovic, D. Role of the extracellular matrix in cancer-associated epithelial to mesenchymal transition phenomenon. Dev. Dyn. 2018, 247, 368–381. [Google Scholar] [CrossRef]
- Scott, L.E.; Weinberg, S.H.; Lemmon, C.A. Mechanochemical Signaling of the Extracellular Matrix in Epithelial-Mesenchymal Transition. Front. Cell Dev. Biol. 2019, 7, 135. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, C.; Montagna, M.K.; Pitruzzello, M.; Lima, E.; Mor, G.; Alvero, A.B. Adipocyte microenvironment promotes Bclxl expression and confers chemoresistance in ovarian cancer cells. Apoptosis 2017, 22, 558–569. [Google Scholar] [CrossRef]
- Alvero, A.B.; Heaton, A.; Lima, E.; Pitruzzello, M.; Sumi, N.; Yang-Hartwich, Y.; Cardenas, C.; Steinmacher, S.; Silasi, D.A.; Brown, D.; et al. TRX-E-002-1 Induces c-Jun-Dependent Apoptosis in Ovarian Cancer Stem Cells and Prevents Recurrence In Vivo. Mol. Cancer Ther. 2016, 15, 1279–1290. [Google Scholar] [CrossRef]
- Tedja, R.; Alvero, A.B.; Fox, A.; Cardenas, C.; Pitruzzello, M.; Chehade, H.; Bawa, T.; Adzibolosu, N.; Gogoi, R.; Mor, G. Generation of Stable Epithelial-Mesenchymal Hybrid Cancer Cells with Tumorigenic Potential. Cancers 2023, 15, 684. [Google Scholar] [CrossRef]
- Li, J.; Alvero, A.B.; Nuti, S.; Tedja, R.; Roberts, C.M.; Pitruzzello, M.; Li, Y.; Xiao, Q.; Zhang, S.; Gan, Y.; et al. CBX7 binds the E-box to inhibit TWIST-1 function and inhibit tumorigenicity and metastatic potential. Oncogene 2020, 39, 3965–3979. [Google Scholar] [CrossRef] [PubMed]
- Artymovich, K.; Appledorn, D.M. A multiplexed method for kinetic measurements of apoptosis and proliferation using live-content imaging. Methods Mol. Biol. 2015, 1219, 35–42. [Google Scholar] [PubMed]
- Chehade, H.; Purandare, N.; Fox, A.; Adzibolosu, N.; Jayee, S.; Singh, A.; Tedja, R.; Gogoi, R.; Aras, S.; Grossman, L.I.; et al. MNRR1 is a driver of ovarian cancer progression. Transl. Oncol. 2023, 29, 101623. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Ahsan, S.; Draghici, S. Identifying Significantly Impacted Pathways and Putative Mechanisms with iPathwayGuide. Curr. Protoc. Bioinform. 2017, 57, 7–15. [Google Scholar] [CrossRef]
- Draghici, S.; Khatri, P.; Tarca, A.L.; Amin, K.; Done, A.; Voichita, C.; Georgescu, C.; Romero, R. A systems biology approach for pathway level analysis. Genome Res. 2007, 17, 1537–1545. [Google Scholar] [CrossRef]
- Tarca, A.L.; Draghici, S.; Khatri, P.; Hassan, S.S.; Mittal, P.; Kim, J.S.; Kim, C.J.; Kusanovic, J.P.; Romero, R. A novel signaling pathway impact analysis. Bioinformatics 2009, 25, 75–82. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Tedja, R.; Roberts, C.M.; Alvero, A.B.; Cardenas, C.; Yang-Hartwich, Y.; Spadinger, S.; Pitruzzello, M.; Yin, G.; Glackin, C.A.; Mor, G. Protein kinase Calpha-mediated phosphorylation of Twist1 at Ser-144 prevents Twist1 ubiquitination and stabilizes it. J. Biol. Chem. 2019, 294, 5082–5093. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Scarcelli, G. Mapping mechanical properties of biological materials via an add-on Brillouin module to confocal microscopes. Nat. Protoc. 2021, 16, 1251–1275. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.H.; Beppu, T.; Kurose, A.; Yamauchi, K.; Sugawara, A.; Suzuki, M.; Ogawa, A.; Sawai, T. Neoplastic cells and proliferating endothelial cells express connective tissue growth factor (CTGF) in glioblastoma. Neurol. Res. 2002, 24, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Ramazani, Y.; Knops, N.; Elmonem, M.A.; Nguyen, T.Q.; Arcolino, F.O.; van den Heuvel, L.; Levtchenko, E.; Kuypers, D.; Goldschmeding, R. Connective tissue growth factor (CTGF) from basics to clinics. Matrix Biol. 2018, 68–69, 44–66. [Google Scholar] [CrossRef] [PubMed]
- Beaufort, C.M.; Helmijr, J.C.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van IJcken, W.F.J.; Heine, A.A.; Smid, M.; et al. Ovarian cancer cell line panel (OCCP): Clinical importance of in vitro morphological subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, L.; Kim, M.K.; Lyle, L.T.; Bunch, K.P.; House, C.D.; Ning, F.; Noonan, A.M.; Annunziata, C.M. Characterization of ovarian cancer cell lines as in vivo models for preclinical studies. Gynecol. Oncol. 2016, 142, 332–340. [Google Scholar] [CrossRef]
- Liu, M.; Mor, G.; Cheng, H.; Xiang, X.; Hui, P.; Rutherford, T.; Yin, G.; Rimm, D.L.; Holmberg, J.; Alvero, A.; et al. High frequency of putative ovarian cancer stem cells with CD44/CK19 coexpression is associated with decreased progression-free intervals in patients with recurrent epithelial ovarian cancer. Reprod. Sci. 2013, 20, 605–615. [Google Scholar] [CrossRef]
- Taki, M.; Abiko, K.; Baba, T.; Hamanishi, J.; Yamaguchi, K.; Murakami, R.; Yamanoi, K.; Horikawa, N.; Hosoe, Y.; Nakamura, E.; et al. Snail promotes ovarian cancer progression by recruiting myeloid-derived suppressor cells via CXCR2 ligand upregulation. Nat. Commun. 2018, 9, 1685. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef]
- Frisch, S.M.; Schaller, M.; Cieply, B. Mechanisms that link the oncogenic epithelial-mesenchymal transition to suppression of anoikis. J. Cell Sci. 2013, 126, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Izdebska, M.; Zielinska, W.; Halas-Wisniewska, M.; Grzanka, A. Involvement of Actin and Actin-Binding Proteins in Carcinogenesis. Cells 2020, 9, 2245. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Kuang, D.; Zhang, B.; Song, G. Cell stiffness determined by atomic force microscopy and its correlation with cell motility. Biochim. et Biophys. Acta 2016, 1860, 1953–1960. [Google Scholar] [CrossRef] [PubMed]
- Scarcelli, G.; Polacheck, W.J.; Nia, H.T.; Patel, K.; Grodzinsky, A.J.; Kamm, R.D.; Yun, S.H. Noncontact three-dimensional mapping of intracellular hydromechanical properties by Brillouin microscopy. Nat. Methods 2015, 12, 1132–1134. [Google Scholar] [CrossRef]
- Zhang, J.; Nou, X.A.; Kim, H.; Scarcelli, G. Brillouin flow cytometry for label-free mechanical phenotyping of the nucleus. Lab Chip 2017, 17, 663–670. [Google Scholar] [CrossRef]
- Prevedel, R.; Diz-Munoz, A.; Ruocco, G.; Antonacci, G. Brillouin microscopy: An emerging tool for mechanobiology. Nat. Methods 2019, 16, 969–977. [Google Scholar] [CrossRef]
- Conrad, C.; Gray, K.M.; Stroka, K.M.; Rizvi, I.; Scarcelli, G. Mechanical Characterization of 3D Ovarian Cancer Nodules Using Brillouin Confocal Microscopy. Cell Mol. Bioeng. 2019, 12, 215–226. [Google Scholar] [CrossRef]
- Jolly, M.K.; Ware, K.E.; Gilja, S.; Somarelli, J.A.; Levine, H. EMT and MET: Necessary or permissive for metastasis? Mol. Oncol. 2017, 11, 755–769. [Google Scholar] [CrossRef]
- Domogatskaya, A.; Rodin, S.; Tryggvason, K. Functional diversity of laminins. Annu. Rev. Cell Dev. Biol. 2012, 28, 523–553. [Google Scholar] [CrossRef]
- Sala, M.; Ros, M.; Saltel, F. A Complex and Evolutive Character: Two Face Aspects of ECM in Tumor Progression. Front. Oncol. 2020, 10, 1620. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Luo, W. Nasopharyngeal carcinoma ecology theory: Cancer as multidimensional spatiotemporal “unity of ecology and evolution” pathological ecosystem. Theranostics 2023, 13, 1607–1631. [Google Scholar] [CrossRef] [PubMed]
- Lipson, K.E.; Wong, C.; Teng, Y.; Spong, S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair 2012, 5, S24. [Google Scholar] [CrossRef]
- Fu, T.; Liu, J.X.; Xie, J.; Gao, Z.; Yang, Z. LAMC2 as a prognostic biomarker in human cancer: A systematic review and meta-analysis. BMJ Open 2022, 12, e063682. [Google Scholar] [CrossRef] [PubMed]
- Garg, M.; Kanojia, D.; Okamoto, R.; Jain, S.; Madan, V.; Chien, W.; Sampath, A.; Ding, L.W.; Xuan, M.; Said, J.W.; et al. Laminin-5gamma-2 (LAMC2) is highly expressed in anaplastic thyroid carcinoma and is associated with tumor progression, migration, and invasion by modulating signaling of EGFR. J. Clin. Endocrinol. Metab. 2014, 99, E62–E72. [Google Scholar] [CrossRef]
- Cave, D.D.; Buonaiuto, S.; Sainz, B., Jr.; Fantuz, M.; Mangini, M.; Carrer, A.; Di Domenico, A.; Iavazzo, T.T.; Andolfi, G.; Cortina, C.; et al. LAMC2 marks a tumor-initiating cell population with an aggressive signature in pancreatic cancer. J. Exp. Clin. Cancer Res. 2022, 41, 315. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Konecny, G.E.; Wang, C.; Hamidi, H.; Winterhoff, B.; Kalli, K.R.; Dering, J.; Ginther, C.; Chen, H.W.; Dowdy, S.; Cliby, W.; et al. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. J. Natl. Cancer Inst. 2014, 106, dju249. [Google Scholar] [CrossRef]
- Wang, C.; Armasu, S.M.; Kalli, K.R.; Maurer, M.J.; Heinzen, E.P.; Keeney, G.L.; Cliby, W.A.; Oberg, A.L.; Kaufmann, S.H.; Goode, E.L. Pooled Clustering of High-Grade Serous Ovarian Cancer Gene Expression Leads to Novel Consensus Subtypes Associated with Survival and Surgical Outcomes. Clin. Cancer Res. 2017, 23, 4077–4085. [Google Scholar] [CrossRef]
- Chen, C.N.; Chang, C.C.; Lai, H.S.; Jeng, Y.M.; Chen, C.I.; Chang, K.J.; Lee, P.H.; Lee, H. Connective tissue growth factor inhibits gastric cancer peritoneal metastasis by blocking integrin alpha3beta1-dependent adhesion. Gastric Cancer 2015, 18, 504–515. [Google Scholar] [CrossRef]
- Tsai, H.C.; Su, H.L.; Huang, C.Y.; Fong, Y.C.; Hsu, C.J.; Tang, C.H. CTGF increases matrix metalloproteinases expression and subsequently promotes tumor metastasis in human osteosarcoma through down-regulating miR-519d. Oncotarget 2014, 5, 3800. [Google Scholar] [CrossRef]
- Jiang, C.G.; Lv, L.; Liu, F.R.; Wang, Z.N.; Na, D.; Li, F.; Li, J.B.; Sun, Z.; Xu, H.M. Connective tissue growth factor is a positive regulator of epithelial-mesenchymal transition and promotes the adhesion with gastric cancer cells in human peritoneal mesothelial cells. Cytokine 2013, 61, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.R.; Chang, C.C.; Chen, R.J.; Jeng, Y.M.; Liang, J.T.; Lee, P.H.; Chang, K.J.; Kuo, M.L. Connective tissue growth factor acts as a therapeutic agent and predictor for peritoneal carcinomatosis of colorectal cancer. Clin. Cancer Res. 2011, 17, 3077–3088. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhong, J.; Zhao, Z.; Sheng, J.; Wang, J.; Liu, J.; Cui, K.; Chang, J.; Zhao, H.; Wong, S. Epithelial derived CTGF promotes breast tumor progression via inducing EMT and collagen I fibers deposition. Oncotarget 2015, 6, 25320–25338. [Google Scholar] [CrossRef] [PubMed]
- Barbolina, M.V.; Adley, B.P.; Kelly, D.L.; Shepard, J.; Fought, A.J.; Scholtens, D.; Penzes, P.; Shea, L.D.; Stack, M.S. Downregulation of connective tissue growth factor by three-dimensional matrix enhances ovarian carcinoma cell invasion. Int. J. Cancer 2009, 125, 816–825. [Google Scholar] [CrossRef]
- Fu, M.; Peng, D.; Lan, T.; Wei, Y.; Wei, X. Multifunctional regulatory protein connective tissue growth factor (CTGF): A potential therapeutic target for diverse diseases. Acta Pharm. Sin. B 2022, 12, 1740–1760. [Google Scholar] [CrossRef]
- Kim, H.; Son, S.; Ko, Y.; Shin, I. CTGF regulates cell proliferation, migration, and glucose metabolism through activation of FAK signaling in triple-negative breast cancer. Oncogene 2021, 40, 2667–2681. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gogoi, R.P.; Galoforo, S.; Fox, A.; Morris, C.; Ramos, H.; Gogoi, V.K.; Chehade, H.; Adzibolosu, N.K.; Shi, C.; Zhang, J.; et al. A Novel Role of Connective Tissue Growth Factor in the Regulation of the Epithelial Phenotype. Cancers 2023, 15, 4834. https://doi.org/10.3390/cancers15194834
Gogoi RP, Galoforo S, Fox A, Morris C, Ramos H, Gogoi VK, Chehade H, Adzibolosu NK, Shi C, Zhang J, et al. A Novel Role of Connective Tissue Growth Factor in the Regulation of the Epithelial Phenotype. Cancers. 2023; 15(19):4834. https://doi.org/10.3390/cancers15194834
Chicago/Turabian StyleGogoi, Radhika P., Sandra Galoforo, Alexandra Fox, Colton Morris, Harry Ramos, Vir K. Gogoi, Hussein Chehade, Nicholas K. Adzibolosu, Chenjun Shi, Jitao Zhang, and et al. 2023. "A Novel Role of Connective Tissue Growth Factor in the Regulation of the Epithelial Phenotype" Cancers 15, no. 19: 4834. https://doi.org/10.3390/cancers15194834
APA StyleGogoi, R. P., Galoforo, S., Fox, A., Morris, C., Ramos, H., Gogoi, V. K., Chehade, H., Adzibolosu, N. K., Shi, C., Zhang, J., Tedja, R., Morris, R., Alvero, A. B., & Mor, G. (2023). A Novel Role of Connective Tissue Growth Factor in the Regulation of the Epithelial Phenotype. Cancers, 15(19), 4834. https://doi.org/10.3390/cancers15194834