
Advantages of the Combinatorial Molecular Targeted Therapy of Head and Neck Cancer—A Step before Anakoinosis-Based Personalized Treatment


Abstract
Simple Summary
Abstract
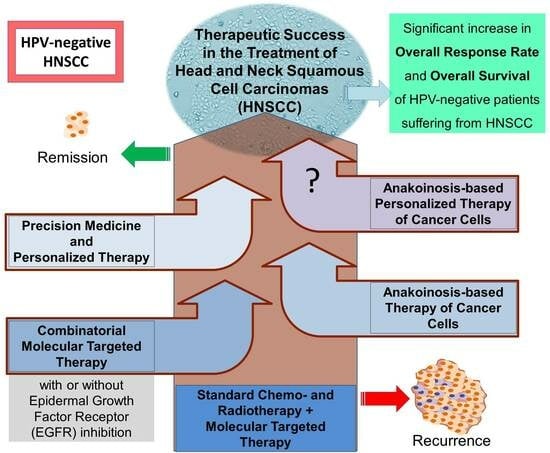
1. Introduction
2. Molecularly Targeted HNSCC Therapy
2.1. Epidermal Growth Factor Receptor (EGFR) Pathway
2.2. Farnesylation of RAS
2.3. RAS/RAF/MAPK Pathway
2.4. PI3K/Akt/mTOR Pathway
2.5. Other Receptor Tyrosine Kinases and Their Downstream Signaling Pathways
2.6. Cancer Stem Cell-Related Signaling Pathways
2.7. Defective Immune Response, Dysregulated Energy Metabolism, and Other Targets for HNSCC Therapy
3. Attempts at Combinatorial Targeted Therapy
3.1. EGRF in the Center of Attention
3.2. Combined Molecular Targeted Therapy Omitting EGFR
4. Limitations of Targeted Therapy
5. Anakoinosis-Based Cancer Therapy
6. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef]
- Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; Dandona, L.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A systematic analysis for the global burden of disease study. JAMA Oncol. 2017, 3, 524–548. [Google Scholar] [CrossRef]
- Mandal, R.; Şenbabaoğlu, Y.; Desrichard, A.; Havel, J.J.; Dalin, M.G.; Riaz, N.; Lee, K.-W.; Ganly, I.; Hakimi, A.A.; Chan, T.A.; et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight 2016, 1, e89829. Available online: https://insight.jci.org/articles/view/89829 (accessed on 15 October 2022). [CrossRef]
- Sun, Y.; Wang, Z.; Qiu, S.; Wang, R. Therapeutic strategies of different HPV status in Head and Neck Squamous Cell Carcinoma. Int. J. Biol. Sci. 2021, 17, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hecht, S.S. Carcinogenic components of tobacco and tobacco smoke: A 2022 update. Food Chem. Toxicol. 2022, 165, 113179. [Google Scholar] [CrossRef] [PubMed]
- Straif, K.; Warnakulasuriya, S. Carcinogenicity of smokeless tobacco: Evidence from studies in humans & experimental animals. Indian J. Med. Res. 2018, 148, 681–686. [Google Scholar] [CrossRef]
- Pai, S.I.; Westra, W.H. Molecular Pathology of Head and Neck Cancer: Implications for Diagnosis, Prognosis, and Treatment. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 49–70. [Google Scholar] [CrossRef]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef]
- Sherr, C.J.; McCormick, F. The RB and p53 pathways in cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar] [CrossRef]
- Stein, A.P.; Saha, S.; Kraninger, J.L.; Swick, A.D.; Yu, M.; Lambert, P.F.; Kimple, R.J. Prevalence of Human Papillomavirus in Oropharyngeal Cancer. Cancer J. 2015, 21, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Michaud, D.S.; Langevin, S.M.; Eliot, M.; Nelson, H.H.; Pawlita, M.; McClean, M.D.; Kelsey, K.T. High-risk HPV types and head and neck cancer. Int. J. Cancer 2014, 135, 1653–1661. [Google Scholar] [CrossRef]
- Szymonowicz, K.A.; Chen, J. Biological and clinical aspects of HPV-related cancers. Cancer Biol. Med. 2020, 17, 864–878. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Stratton, M.R. Mutational signatures: The patterns of somatic mutations hidden in cancer genomes. Curr. Opin. Genet. Dev. 2014, 24, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Mingard, C.; Battey, J.N.D.; Takhaveev, V.; Blatter, K.; Hürlimann, V.; Sierro, N.; Ivanov, N.V.; Sturla, S.J. Dissection of Cancer Mutational Signatures with Individual Components of Cigarette Smoking. Chem. Res. Toxicol. 2023, 36, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef]
- Faden, D.L.; Thomas, S.; Cantalupo, P.G.; Agrawal, N.; Myers, J.; DeRisi, J. Multi-modality analysis supports APOBEC as a major source of mutations in head and neck squamous cell carcinoma. Oral Oncol. 2017, 74, 8–14. [Google Scholar] [CrossRef]
- Faden, D.L.; Kuhs, K.A.L.; Lin, M.; Langenbucher, A.; Pinheiro, M.; Yeager, M.; Cullen, M.; Boland, J.F.; Steinberg, M.; Bass, S.; et al. APOBEC Mutagenesis Is Concordant between Tumor and Viral Genomes in HPV-Positive Head and Neck Squamous Cell Carcinoma. Viruses 2021, 13, 1666. [Google Scholar] [CrossRef]
- Vieira, V.C.; Leonard, B.; White, E.A.; Starrett, G.J.; Temiz, N.A.; Lorenz, L.D.; Lee, D.; Soares, M.A.; Lambert, P.F.; Howley, P.M.; et al. Human Papillomavirus E6 Triggers Upregulation of the Antiviral and Cancer Genomic DNA Deaminase APOBEC3B. mBio 2014, 5, e02234-14. [Google Scholar] [CrossRef] [PubMed]
- Vigneswaran, N.; Williams, M.D. Epidemiologic Trends in Head and Neck Cancer and Aids in Diagnosis. Oral Maxillofac. Surg. Clin. N. Am. 2014, 26, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tân, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human Papillomavirus and Survival of Patients with Oropharyngeal Cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.K.; Engels, E.A.; Pfeiffer, R.M.; Hernandez, B.Y.; Xiao, W.; Kim, E.; Jiang, B.; Goodman, M.T.; Sibug-Saber, M.; Cozen, W.; et al. Human Papillomavirus and Rising Oropharyngeal Cancer Incidence in the United States. J. Clin. Oncol. 2011, 29, 4294–4301. [Google Scholar] [CrossRef] [PubMed]
- Farah, C.S. Molecular landscape of head and neck cancer and implications for therapy. Ann. Transl. Med. 2021, 9, 915. [Google Scholar] [CrossRef]
- Cunningham, D.; Humblet, Y.; Siena, S.; Khayat, D.; Bleiberg, H.; Santoro, A.; Bets, D.; Mueser, M.; Harstrick, A.; Verslype, C.; et al. Cetuximab Monotherapy and Cetuximab plus Irinotecan in Irinotecan-Refractory Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 351, 337–345. [Google Scholar] [CrossRef]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus Cetuximab for Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef]
- Kalyankrishna, S.; Grandis, J.R. Epidermal Growth Factor Receptor Biology in Head and Neck Cancer. J. Clin. Oncol. 2006, 24, 2666–2672. [Google Scholar] [CrossRef]
- Rehmani, H.S.; Issaeva, N. EGFR in head and neck squamous cell carcinoma: Exploring possibilities of novel drug combinations. Ann. Transl. Med. 2020, 8, 813. [Google Scholar] [CrossRef]
- Vermorken, J.; Specenier, P. Cetuximab: Its unique place in head and neck cancer treatment. Biol. Targets Ther. 2013, 2013, 77–90. [Google Scholar] [CrossRef]
- Saltz, L.; Easley, C.; Kirkpatrick, P. Panitumumab. Nat. Rev. Drug Discov. 2006, 5, 987–988. [Google Scholar] [CrossRef] [PubMed]
- William, W.N., Jr.; Tsao, A.S.; Feng, L.; Ginsberg, L.E.; Lee, J.J.; Kies, M.S.; Glisson, B.S.; Kim, E.S. Single Arm, Phase II Study of Cisplatin, Docetaxel, and Erlotinib in Patients with Recurrent and/or Metastatic Head and Neck Squamous Cell Carcinomas. Oncologist 2018, 23, e526–e549. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, T.; Ohba, M.; Ohmori, T. Molecular-Targeted Therapies for Epidermal Growth Factor Receptor and Its Resistance Mechanisms. Int. J. Mol. Sci. 2017, 18, 2420. [Google Scholar] [CrossRef]
- Parker, J.A.; Mattos, C. The K-Ras, N-Ras, and H-Ras Isoforms: Unique Conformational Preferences and Implications for Targeting Oncogenic Mutants. Cold Spring Harb. Perspect. Med. 2017, 8, a031427. [Google Scholar] [CrossRef]
- Ho, A.L.; Brana, I.; Haddad, R.; Bauman, J.; Bible, K.; Oosting, S.; Wong, D.J.; Ahn, M.-J.; Boni, V.; Even, C.; et al. Tipifarnib in Head and Neck Squamous Cell Carcinoma with HRAS Mutations. J. Clin. Oncol. 2021, 39, 1856–1864. [Google Scholar] [CrossRef]
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2016, 109, 314–341. [Google Scholar] [CrossRef] [PubMed]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef]
- Eblen, S.T. Extracellular-Regulated Kinases: Signaling from Ras to ERK Substrates to Control Biological Outcomes. In Advances in Cancer Research; Elsevier: New York, NY, USA, 2018; Volume 138, pp. 99–142. [Google Scholar] [CrossRef]
- Mousa, A. Sorafenib in the treatment of advanced hepatocellular carcinoma. Saudi J. Gastroenterol. 2008, 14, 40–42. [Google Scholar] [CrossRef]
- Elser, C.; Siu, L.L.; Winquist, E.; Agulnik, M.; Pond, G.R.; Chin, S.F.; Francis, P.; Cheiken, R.; Elting, J.; McNabola, A.; et al. Phase II Trial of Sorafenib in Patients with Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck or Nasopharyngeal Carcinoma. J. Clin. Oncol. 2007, 25, 3766–3773. [Google Scholar] [CrossRef]
- Williamson, S.K.; Moon, J.; Huang, C.H.; Guaglianone, P.P.; LeBlanc, M.; Wolf, G.T.; Urba, S.G. Phase II Evaluation of Sorafenib in Advanced and Metastatic Squamous Cell Carcinoma of the Head and Neck: Southwest Oncology Group Study S0420. J. Clin. Oncol. 2010, 28, 3330–3335. [Google Scholar] [CrossRef]
- Dilmaghani, N.A.; Safaroghli-Azar, A.; Pourbagheri-Sigaroodi, A.; Bashash, D. The PI3K/Akt/mTORC signaling axis in head and neck squamous cell carcinoma: Possibilities for therapeutic interventions either as single agents or in combination with conventional therapies. IUBMB Life 2021, 73, 618–642. [Google Scholar] [CrossRef]
- Furet, P.; Guagnano, V.; Fairhurst, R.A.; Imbach-Weese, P.; Bruce, I.; Knapp, M.; Fritsch, C.; Blasco, F.; Blanz, J.; Aichholz, R.; et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg. Med. Chem. Lett. 2013, 23, 3741–3748. [Google Scholar] [CrossRef]
- Narayan, P.; Prowell, T.M.; Gao, J.J.; Fernandes, L.L.; Li, E.; Jiang, X.; Qiu, J.; Fan, J.; Song, P.; Yu, J.; et al. FDA Approval Summary: Alpelisib Plus Fulvestrant for Patients with HR-positive, HER2-negative, PIK3CA-mutated, Advanced or Metastatic Breast Cancer. Clin. Cancer Res. 2020, 27, 1842–1849. [Google Scholar] [CrossRef]
- Dan, H.C.; Baldwin, A.S. Differential Involvement of IκB Kinases α and β in Cytokine- and Insulin-Induced Mammalian Target of Rapamycin Activation Determined by Akt. J. Immunol. 2008, 180, 7582–7589. [Google Scholar] [CrossRef]
- Patel, J.; Nguyen, S.A.; Ogretmen, B.; Gutkind, J.S.; Nathan, C.; Day, T. mTOR inhibitor use in head and neck squamous cell carcinoma: A meta-analysis on survival, tumor response, and toxicity. Laryngoscope Investig. Otolaryngol. 2020, 5, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Freier, K.; Schwaenen, C.; Sticht, C.; Flechtenmacher, C.; Mühling, J.; Hofele, C.; Radlwimmer, B.; Lichter, P.; Joos, S. Recurrent FGFR1 amplification and high FGFR1 protein expression in oral squamous cell carcinoma (OSCC). Oral Oncol. 2007, 43, 60–66. [Google Scholar] [CrossRef]
- Koole, K.; Brunen, D.; van Kempen, P.M.; Noorlag, R.; de Bree, R.; Lieftink, C.; van Es, R.J.; Bernards, R.; Willems, S.M. FGFR1 Is a Potential Prognostic Biomarker and Therapeutic Target in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2016, 22, 3884–3893. [Google Scholar] [CrossRef] [PubMed]
- Brands, R.C.; Knierim, L.M.; De Donno, F.; Steinacker, V.; Hartmann, S.; Seher, A.; Kübler, A.C.; Müller-Richter, U.D. Targeting VEGFR and FGFR in head and neck squamous cell carcinoma in vitro. Oncol. Rep. 2017, 38, 1877–1885. [Google Scholar] [CrossRef]
- Hsu, H.-W.; Wall, N.R.; Hsueh, C.-T.; Kim, S.; Ferris, R.L.; Chen, C.-S.; Mirshahidi, S. Combination antiangiogenic therapy and radiation in head and neck cancers. Oral Oncol. 2014, 50, 19–26. [Google Scholar] [CrossRef]
- Argiris, A.; Kotsakis, A.P.; Hoang, T.; Worden, F.P.; Savvides, P.; Gibson, M.K.; Gyanchandani, R.; Blumenschein, G.R., Jr.; Chen, H.X.; Grandis, J.R.; et al. Cetuximab and bevacizumab: Preclinical data and phase II trial in recurrent or metastatic squamous cell carcinoma of the head and neck. Ann. Oncol. 2013, 24, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.E.; Davis, D.W.; Karrison, T.G.; Seiwert, T.Y.; Wong, S.J.; Nattam, S.; Kozloff, M.F.; Clark, J.I.; Yan, D.-H.; Liu, W.; et al. Erlotinib and bevacizumab in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck: A phase I/II study. Lancet Oncol. 2009, 10, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.-Z.; Chen, Q.; Zhang, W.-Y.; Zhang, H.-H.; Ma, Y.; Zhang, S.-Z.; Fang, J.; Yu, C.-H. PDGF signaling pathway in hepatic fibrosis pathogenesis and therapeutics. Mol. Med. Rep. 2017, 16, 7879–7889. [Google Scholar] [CrossRef]
- Lin, L.-H.; Lin, J.-S.; Yang, C.-C.; Cheng, H.-W.; Chang, K.-W.; Liu, C.-J. Overexpression of Platelet-Derived Growth Factor and Its Receptor Are Correlated with Oral Tumorigenesis and Poor Prognosis in Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2020, 21, 2360. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.D.; Rotunno, S.; Riedel, F.; Anders, C.; Erben, P.; Hofheinz, R.D.; Faber, A.; Thorn, C.; Sommer, J.U.; Hörmann, A.; et al. Synergistic effects of imatinib and carboplatin on VEGF, PDGF and PDGF-Rα/ß expression in squamous cell carcinoma of the head and neck in vitro. Int. J. Oncol. 2011, 38, 1001–1012. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Raj, S.; Kesari, K.K.; Kumar, A.; Rathi, B.; Sharma, A.; Gupta, P.K.; Jha, S.K.; Jha, N.K.; Slama, P.; Roychoudhury, S.; et al. Molecular mechanism(s) of regulation(s) of c-MET/HGF signaling in head and neck cancer. Mol. Cancer 2022, 21, 31. [Google Scholar] [CrossRef]
- Ariyawutyakorn, W.; Saichaemchan, S.; Varella-Garcia, M. Understanding and Targeting MET Signaling in Solid Tumors—Are We There Yet? J. Cancer 2016, 7, 633–649. [Google Scholar] [CrossRef]
- Wang, D.; Lu, Y.; Nannapaneni, S.; Griffith, C.C.; Steuer, C.; Qian, G.; Wang, X.; Chen, Z.; Patel, M.; El-Deiry, M.; et al. Combinatorial approaches targeting the EGFR family and c-Met in SCCHN. Oral Oncol. 2020, 112, 105074. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Macha, M.A.; Matta, A.; Kaur, J.; Chauhan, S.S.; Thakar, A.; Shukla, N.K.; Gupta, S.D.; Ralhan, R. Prognostic significance of nuclear pSTAT3 in oral cancer. Head Neck 2011, 33, 482–489. [Google Scholar] [CrossRef]
- Stegeman, H.; Kaanders, J.H.; Verheijen, M.M.; Peeters, W.J.; Wheeler, D.L.; Iida, M.; Grénman, R.; van der Kogel, A.J.; Span, P.N.; Bussink, J. Combining radiotherapy with MEK1/2, STAT5 or STAT6 inhibition reduces survival of head and neck cancer lines. Mol. Cancer 2013, 12, 133. [Google Scholar] [CrossRef]
- Prince, M.E.; Ailles, L.E. Cancer Stem Cells in Head and Neck Squamous Cell Cancer. J. Clin. Oncol. 2008, 26, 2871–2875. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Liu, L.; Zhang, S.; Yang, X.; Wang, Y. Cancer stem cell biomarkers for head and neck squamous cell carcinoma: A bioinformatic analysis. Oncol. Rep. 2018, 40, 3843–3851. [Google Scholar] [CrossRef]
- Gunduz, M.; Gunduz, E.; Tamagawa, S.; Enomoto, K.; Hotomi, M. Cancer Stem Cells in Oropharyngeal Cancer. Cancers 2021, 13, 3878. [Google Scholar] [CrossRef]
- Leong, K.G.; Karsan, A. Recent insights into the role of Notch signaling in tumorigenesis. Blood 2006, 107, 2223–2233. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.-X.; Zhang, J.; Wang, J.; et al. Exome Sequencing of Head and Neck Squamous Cell Carcinoma Reveals Inactivating Mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef] [PubMed]
- Nyman, P.E.; Buehler, D.; Lambert, P.F. Loss of Function of Canonical Notch Signaling Drives Head and Neck Carcinogenesis. Clin. Cancer Res. 2018, 24, 6308–6318. [Google Scholar] [CrossRef]
- Sun, W.; Gaykalova, D.A.; Ochs, M.F.; Mambo, E.; Arnaoutakis, D.; Liu, Y.; Loyo, M.; Agrawal, N.; Howard, J.; Li, R.; et al. Activation of the NOTCH Pathway in Head and Neck Cancer. Cancer Res. 2014, 74, 1091–1104. [Google Scholar] [CrossRef]
- Lee, S.H.; Do, I.S.; Lee, H.J.; Kang, H.J.; Koo, B.S.; Lim, Y.C. Notch1 signaling contributes to stemness in head and neck squamous cell carcinoma. Lab. Investig. 2016, 96, 508–516. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2016, 36, 1461–1473. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/β-catenin signalling: Function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target. Ther. 2022, 7, 3. [Google Scholar] [CrossRef]
- Paluszczak, J. The Significance of the Dysregulation of Canonical Wnt Signaling in Head and Neck Squamous Cell Carcinomas. Cells 2020, 9, 723. [Google Scholar] [CrossRef] [PubMed]
- Kleszcz, R.; Szymańska, A.; Krajka-Kuźniak, V.; Baer-Dubowska, W.; Paluszczak, J. Inhibition of CBP/β-catenin and porcupine attenuates Wnt signaling and induces apoptosis in head and neck carcinoma cells. Cell. Oncol. 2019, 42, 505–520. [Google Scholar] [CrossRef] [PubMed]
- Bitgood, M.J.; McMahon, A.P. Hedgehog and Bmp Genes Are Coexpressed at Many Diverse Sites of Cell–Cell Interaction in the Mouse Embryo. Dev. Biol. 1995, 172, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.; Litingtung, Y.; Lee, E.; Young, K.E.; Corden, J.L.; Westphal, H.; Beachy, P.A. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 1996, 383, 407–413. [Google Scholar] [CrossRef]
- Rudin, C.M. Vismodegib. Clin. Cancer Res. 2012, 18, 3218–3222. [Google Scholar] [CrossRef]
- Cierpikowski, P.; Lis-Nawara, A.; Bar, J. Sonic Hedgehog is a novel prognostic biomarker in patients with oral squamous cell carcinoma. Neoplasma 2021, 68, 867–874. [Google Scholar] [CrossRef]
- Noman, A.S.M.; Parag, R.R.; Rashid, M.I.; Rahman, M.Z.; Chowdhury, A.A.; Sultana, A.; Jerin, C.; Siddiqua, A.; Rahman, L.; Shirin, A.; et al. Widespread expression of Sonic hedgehog (Shh) and Nrf2 in patients treated with cisplatin predicts outcome in resected tumors and are potential therapeutic targets for HPV-negative head and neck cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920911229. [Google Scholar] [CrossRef]
- Enzenhofer, E.; Parzefall, T.; Haymerle, G.; Schneider, S.; Kadletz, L.; Heiduschka, G.; Pammer, J.; Oberndorfer, F.; Wrba, F.; Loader, B.; et al. Impact of Sonic Hedgehog Pathway Expression on Outcome in HPV Negative Head and Neck Carcinoma Patients after Surgery and Adjuvant Radiotherapy. PLoS ONE 2016, 11, e0167665. [Google Scholar] [CrossRef]
- Lu, X.; Wang, Z.; Huang, H.; Wang, H. Hedgehog signaling promotes multidrug resistance by regulation of ABC transporters in oral squamous cell carcinoma. J. Oral Pathol. Med. 2020, 49, 897–906. [Google Scholar] [CrossRef]
- Cierpikowski, P.; Lis-Nawara, A.; BAR, J. SHH Expression Is Significantly Associated with Cancer Stem Cell Markers in Oral Squamous Cell Carcinoma. Anticancer Res. 2021, 41, 5405–5413. [Google Scholar] [CrossRef]
- Patni, A.P.; Harishankar, M.K.; Joseph, J.P.; Sreeshma, B.; Jayaraj, R.; Devi, A. Comprehending the crosstalk between Notch, Wnt and Hedgehog signaling pathways in oral squamous cell carcinoma—Clinical implications. Cell. Oncol. 2021, 44, 473–494. [Google Scholar] [CrossRef]
- Fu, V.; Plouffe, S.W.; Guan, K.-L. The Hippo pathway in organ development, homeostasis, and regeneration. Curr. Opin. Cell Biol. 2017, 49, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Otsubo, K.; Maehama, T.; Mimori, K.; Suzuki, A. Capturing the mammalian Hippo: Elucidating its role in cancer. Cancer Sci. 2013, 104, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Oshimori, N. Cancer stem cells and their niche in the progression of squamous cell carcinoma. Cancer Sci. 2020, 111, 3985–3992. [Google Scholar] [CrossRef]
- Pang, X.; Tang, Y.L.; Liang, X.H. Transforming growth factor-β signaling in head and neck squamous cell carcinoma: Insights into cellular responses (Review). Oncol. Lett. 2018, 16, 4799–4806. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ding, X.; Bai, X.; Zhou, Z.; Liu, Y.; Zhang, X.; Yu, J.; Hu, M. The current advances and future directions of PD-1/PD-L1 blockade in head and neck squamous cell carcinoma (HNSCC) in the era of immunotherapy. Int. Immunopharmacol. 2023, 120, 110329. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Longoria, T.C.; Tewari, K.S. Evaluation of the pharmacokinetics and metabolism of pembrolizumab in the treatment of melanoma. Expert Opin. Drug Metab. Toxicol. 2016, 12, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Larkins, E.; Blumenthal, G.M.; Yuan, W.; He, K.; Sridhara, R.; Subramaniam, S.; Zhao, H.; Liu, C.; Yu, J.; Goldberg, K.B.; et al. FDA Approval Summary: Pembrolizumab for the Treatment of Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma with Disease Progression on or After Platinum-Containing Chemotherapy. Oncologist 2017, 22, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Pascale, R.M.; Calvisi, D.F.; Simile, M.M.; Feo, C.F.; Feo, F. The Warburg Effect 97 Years after Its Discovery. Cancers 2020, 12, 2819. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Inohara, H.; Nakagawa, T. Targeting metabolic pathways for head and neck cancers therapeutics. Cancer Metastasis Rev. 2017, 36, 503–514. [Google Scholar] [CrossRef]
- Hsieh, Y.-T.; Chen, Y.-F.; Lin, S.-C.; Chang, K.-W.; Li, W.-C. Targeting Cellular Metabolism Modulates Head and Neck Oncogenesis. Int. J. Mol. Sci. 2019, 20, 3960. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2021, 21, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Nikolova, O.; Basom, R.S.; Mitchell, R.M.; Shaw, R.; Moser, R.D.; Park, H.; Gurley, K.E.; Kao, M.C.; Green, C.L.; et al. Functional Precision Medicine Identifies Novel Druggable Targets and Therapeutic Options in Head and Neck Cancer. Clin. Cancer Res. 2018, 24, 2828–2843. [Google Scholar] [CrossRef] [PubMed]
- Cole, D.W.; Svider, P.F.; Shenouda, K.G.; Lee, P.B.; Yoo, N.G.; McLeod, T.M.; Mutchnick, S.A.; Yoo, G.H.; Kaufman, R.J.; Callaghan, M.U.; et al. Targeting the unfolded protein response in head and neck and oral cavity cancers. Exp. Cell Res. 2019, 382, 111386. [Google Scholar] [CrossRef]
- Gaździcka, J.; Gołąbek, K.; Strzelczyk, J.K.; Ostrowska, Z. Epigenetic Modifications in Head and Neck Cancer. Biochem. Genet. 2019, 58, 213–244. [Google Scholar] [CrossRef]
- Kordbacheh, F.; Farah, C.S. Current and Emerging Molecular Therapies for Head and Neck Squamous Cell Carcinoma. Cancers 2021, 13, 5471. [Google Scholar] [CrossRef]
- Van Harten, A.M.; Brakenhoff, R.H. Targeted Treatment of Head and Neck (Pre)Cancer: Preclinical Target Identification and Development of Novel Therapeutic Applications. Cancers 2021, 13, 2774. [Google Scholar] [CrossRef]
- Romanowska, K.; Sobecka, A.; Rawłuszko-Wieczorek, A.A.; Suchorska, W.M.; Golusiński, W. Head and Neck Squamous Cell Carcinoma: Epigenetic Landscape. Diagnostics 2020, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Shah, P.A.; Johnson, F.M. Novel Systemic Treatment Modalities Including Immunotherapy and Molecular Targeted Therapy for Recurrent and Metastatic Head and Neck Squamous Cell Carcinoma. Int. J. Mol. Sci. 2022, 23, 7889. [Google Scholar] [CrossRef] [PubMed]
- Zaryouh, H.; Van Loenhout, J.; Peeters, M.; Vermorken, J.B.; Lardon, F.; Wouters, A. Co-Targeting the EGFR and PI3K/Akt Pathway to Overcome Therapeutic Resistance in Head and Neck Squamous Cell Carcinoma: What about Autophagy? Cancers 2022, 14, 6128. [Google Scholar] [CrossRef] [PubMed]
- Mock, A.; Plath, M.; Moratin, J.; Tapken, M.J.; Jäger, D.; Krauss, J.; Fröhling, S.; Hess, J.; Zaoui, K. EGFR and PI3K Pathway Activities Might Guide Drug Repurposing in HPV-Negative Head and Neck Cancers. Front. Oncol. 2021, 11, 678966. [Google Scholar] [CrossRef] [PubMed]
- Bozec, A.; Ebran, N.; Radosevic-Robin, N.; Chamorey, E.; Ben Yahia, H.; Marcie, S.; Gautier, M.; Penault-Llorca, F.; Milano, G. Combination of phosphotidylinositol-3-kinase targeting with cetuximab and irradiation: A preclinical study on an orthotopic xenograft model of head and neck cancer. Head Neck 2016, 39, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Michmerhuizen, N.L.; Leonard, E.; Matovina, C.; Harris, M.; Herbst, G.; Kulkarni, A.; Zhai, J.; Jiang, H.; Carey, T.E.; Brenner, J.C. Rationale for Using Irreversible Epidermal Growth Factor Receptor Inhibitors in Combination with Phosphatidylinositol 3-Kinase Inhibitors for Advanced Head and Neck Squamous Cell Carcinoma. Mol. Pharmacol. 2019, 95, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Zaryouh, H.; De Pauw, I.; Baysal, H.; Pauwels, P.; Peeters, M.; Vermorken, J.B.; Lardon, F.; Wouters, A. The Role of Akt in Acquired Cetuximab Resistant Head and Neck Squamous Cell Carcinoma: An In Vitro Study on a Novel Combination Strategy. Front. Oncol. 2021, 11, 697967. [Google Scholar] [CrossRef]
- Bozec, A.; Ebran, N.; Radosevic-Robin, N.; Sudaka, A.; Monteverde, M.; Toussan, N.; Etienne-Grimaldi, M.-C.; Nigro, C.L.; Merlano, M.; Penault-Llorca, F.; et al. Combination of mTOR and EGFR targeting in an orthotopic xenograft model of head and neck cancer. Laryngoscope 2015, 126, E156–E163. [Google Scholar] [CrossRef]
- Swick, A.D.; Prabakaran, P.J.; Miller, M.C.; Javaid, A.M.; Fisher, M.M.; Sampene, E.; Ong, I.M.; Hu, R.; Iida, M.; Nickel, K.P.; et al. Cotargeting mTORC and EGFR Signaling as a Therapeutic Strategy in HNSCC. Mol. Cancer Ther. 2017, 16, 1257–1268. [Google Scholar] [CrossRef]
- Lin, X.; Liao, J.; Yang, Z.; Fan, X.; Cullen, K.J.; Chen, L.; Dan, H. Inhibition of cisplatin-resistant head and neck squamous cell carcinoma by combination of Afatinib with PD0325901, a MEK inhibitor. Am. J. Cancer Res. 2019, 9, 1282–1292. [Google Scholar]
- Yamaguchi, K.; Iglesias-Bartolomé, R.; Wang, Z.; Callejas-Valera, J.L.; Amornphimoltham, P.; Molinolo, A.A.; Cohen, E.E.; Califano, J.A.; Lippman, S.M.; Luo, J.; et al. A synthetic-lethality RNAi screen reveals an ERK-mTOR co-targeting pro-apoptotic switch in PIK3CA+ oral cancers. Oncotarget 2016, 7, 10696–10709. [Google Scholar] [CrossRef] [PubMed]
- Michmerhuizen, N.L.; Leonard, E.; Kulkarni, A.; Brenner, J.C. Differential compensation mechanisms define resistance to PI3K inhibitors in PIK3CA amplified HNSCC. Otorhinolaryngol. Neck Surg. 2016, 1, 44–50. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Michmerhuizen, N.L.; Ludwig, M.L.; Birkeland, A.C.; Nimmagadda, S.; Zhai, J.; Wang, J.; Jewell, B.M.; Genouw, D.; Remer, L.; Kim, D.; et al. Small molecule profiling to define synergistic EGFR inhibitor combinations in head and neck squamous cell carcinoma. Head Neck 2022, 44, 1192–1205. [Google Scholar] [CrossRef]
- Bozec, A.; Sudaka, A.; Toussan, N.; Fischel, J.-L.; Etienne-Grimaldi, M.-C.; Milano, G. Combination of sunitinib, cetuximab and irradiation in an orthotopic head and neck cancer model. Ann. Oncol. 2009, 20, 1703–1707. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Stabile, L.P.; Gubish, C.T.; Gooding, W.E.; Grandis, J.R.; Siegfried, J.M. Dual Blockade of EGFR and c-Met Abrogates Redundant Signaling and Proliferation in Head and Neck Carcinoma Cells. Clin. Cancer Res. 2011, 17, 4425–4438. [Google Scholar] [CrossRef]
- Bonner, J.A.; Trummell, H.Q.; Bonner, A.B.; Willey, C.D.; Bredel, M.; Yang, E.S. Enhancement of Cetuximab-Induced Radiosensitization by JAK-1 Inhibition. BMC Cancer 2015, 15, 673. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, Z.; Ding, X.; Dong, Y.; Zhang, W.; Zhong, Y.; Gu, W.; Wu, Y.; Song, X. Combined Erlotinib and PF-03084014 treatment contributes to synthetic lethality in head and neck squamous cell carcinoma. Cell Prolif. 2017, 51, e12424. [Google Scholar] [CrossRef]
- Kleszcz, R.; Frąckowiak, M.; Dorna, D.; Paluszczak, J. Combinations of PRI-724 Wnt/β-Catenin Pathway Inhibitor with Vismodegib, Erlotinib, or HS-173 Synergistically Inhibit Head and Neck Squamous Cancer Cells. Int. J. Mol. Sci. 2023, 24, 10448. [Google Scholar] [CrossRef]
- Liebig, H.; Günther, G.; Kolb, M.; Mozet, C.; Boehm, A.; Dietz, A.; Wichmann, G. Reduced proliferation and colony formation of head and neck squamous cell carcinoma (HNSCC) after dual targeting of EGFR and hedgehog pathways. Cancer Chemother. Pharmacol. 2017, 79, 411–420. [Google Scholar] [CrossRef]
- Bedi, A.; Chang, X.; Noonan, K.; Pham, V.; Bedi, R.; Fertig, E.J.; Considine, M.; Califano, J.A.; Borrello, I.; Chung, C.H.; et al. Inhibition of TGF-β Enhances the In Vivo Antitumor Efficacy of EGF Receptor–Targeted Therapy. Mol. Cancer Ther. 2012, 11, 2429–2439. [Google Scholar] [CrossRef]
- Ando, T.; Arang, N.; Wang, Z.; Costea, D.E.; Feng, X.; Goto, Y.; Izumi, H.; Gilardi, M.; Ando, K.; Gutkind, J.S. EGFR Regulates the Hippo pathway by promoting the tyrosine phosphorylation of MOB1. Commun. Biol. 2021, 4, 1237. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liao, J.; Yang, Z.; Choi, E.Y.; Lapidus, R.G.; Liu, X.; Cullen, K.J.; Dan, H. Co-targeting EGFR and IKKβ/NF-κB signalling pathways in head and neck squamous cell carcinoma: A potential novel therapy for head and neck squamous cell cancer. Br. J. Cancer 2018, 120, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Sinto, M.S.; Thomas, S.; Kannan, S. Combinatorial treatment with Gefitinib and Bay11-7085 sensitizes primary Gefitinib-resistant OSCC cells by influencing the EGFR- NFκB signaling axis. Med. Oncol. 2021, 38, 110. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Le Tourneau, C.; Licitra, L.; Ahn, M.-J.; Soria, A.; Machiels, J.-P.; Mach, N.; Mehra, R.; Zhang, P.; Cheng, J.; et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): A randomised, open-label, phase 3 study. Lancet 2019, 393, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Biau, J.; Sun, X.; Sire, C.; Martin, L.; Alfonsi, M.; Prevost, J.; Modesto, A.; Lafond, C.; Tourani, J.; et al. Pembrolizumab versus cetuximab concurrent with radiotherapy in patients with locally advanced squamous cell carcinoma of head and neck unfit for cisplatin (GORTEC 2015-01 PembroRad): A multicenter, randomized, phase II trial. Ann. Oncol. 2022, 34, 101–110. [Google Scholar] [CrossRef] [PubMed]
- She, L.; Tian, K.; Han, J.; Zuo, W.; Wang, Z.; Zhang, N. Pembrolizumab with or without chemotherapy versus cetuximab plus chemotherapy to treat recurrent or metastatic head and neck squamous cell carcinoma: An updated KEYNOTE-048 based cost-effectiveness analysis. Oral Oncol. 2022, 129, 105871. [Google Scholar] [CrossRef]
- Sacco, A.G.; Chen, R.; Worden, F.P.; Wong, D.J.L.; Adkins, D.; Swiecicki, P.; Chai-Ho, W.; Oppelt, P.; Ghosh, D.; Bykowski, J.; et al. Pembrolizumab plus cetuximab in patients with recurrent or metastatic head and neck squamous cell carcinoma: An open-label, multi-arm, non-randomised, multicentre, phase 2 trial. Lancet Oncol. 2021, 22, 883–892. [Google Scholar] [CrossRef]
- Sobhakumari, A.; Orcutt, K.P.; Love-Homan, L.; Kowalski, C.E.; Parsons, A.D.; Knudson, C.M.; Simons, A.L. 2-Deoxy-d-glucose Suppresses the In Vivo Antitumor Efficacy of Erlotinib in Head and Neck Squamous Cell Carcinoma Cells. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2016, 24, 55–64. [Google Scholar] [CrossRef]
- Schoenwaelder, N.; Salewski, I.; Engel, N.; Krause, M.; Schneider, B.; Müller, M.; Riess, C.; Lemcke, H.; Skorska, A.; Grosse-Thie, C.; et al. The Individual Effects of Cyclin-Dependent Kinase Inhibitors on Head and Neck Cancer Cells—A Systematic Analysis. Cancers 2021, 13, 2396. [Google Scholar] [CrossRef]
- Frederick, B.A.; Gupta, R.; Atilano-Roque, A.; Su, T.T.; Raben, D. Combined EGFR1 and PARP1 Inhibition Enhances the Effect of Radiation in Head and Neck Squamous Cell Carcinoma Models. Radiat. Res. 2020, 194, 519–531. [Google Scholar] [CrossRef]
- Chien, M.-H.; Yang, W.-E.; Yang, Y.-C.; Ku, C.-C.; Lee, W.-J.; Tsai, M.-Y.; Lin, C.-W.; Yang, S.-F. Dual Targeting of the p38 MAPK-HO-1 Axis and cIAP1/XIAP by Demethoxycurcumin Triggers Caspase-Mediated Apoptotic Cell Death in Oral Squamous Cell Carcinoma Cells. Cancers 2020, 12, 703. [Google Scholar] [CrossRef] [PubMed]
- Hoellein, A.; Pickhard, A.; von Keitz, F.; Schoeffmann, S.; Piontek, G.; Rudelius, M.; Baumgart, A.; Wagenpfeil, S.; Peschel, C.; Dechow, T.; et al. Aurora Kinase Inhibition Overcomes Cetuximab Resistance in Squamous Cell Cancer of the Head and Neck. Oncotarget 2011, 2, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Kleszcz, R.; Skalski, M.; Krajka-Kuźniak, V.; Paluszczak, J. The inhibitors of KDM4 and KDM6 histone lysine demethylases enhance the anti-growth effects of erlotinib and HS-173 in head and neck cancer cells. Eur. J. Pharm. Sci. 2021, 166, 105961. [Google Scholar] [CrossRef] [PubMed]
- Jubran, M.R.; Vilenski, D.; Flashner-Abramson, E.; Shnaider, E.; Vasudevan, S.; Rubinstein, A.M.; Meirovitz, A.; Sharon, S.; Polak, D.; Kravchenko-Balasha, N. Overcoming resistance to EGFR monotherapy in HNSCC by identification and inhibition of individualized cancer processes. Theranostics 2022, 12, 1204–1219. [Google Scholar] [CrossRef]
- Javaid, S.; Schaefer, A.; Goodwin, C.M.; Nguyen, V.V.; Massey, F.L.; Pierobon, M.; Gambrell-Sanders, D.; Waters, A.M.; Lambert, K.N.; Diehl, J.N.; et al. Concurrent Inhibition of ERK and Farnesyltransferase Suppresses the Growth of HRAS Mutant Head and Neck Squamous Cell Carcinoma. Mol. Cancer Ther. 2022, 21, 762–774. [Google Scholar] [CrossRef]
- Holzhauser, S.; Wild, N.; Zupancic, M.; Ursu, R.G.; Bersani, C.; Näsman, A.; Kostopoulou, O.N.; Dalianis, T. Targeted Therapy with PI3K and FGFR Inhibitors on Human Papillomavirus Positive and Negative Tonsillar and Base of Tongue Cancer Lines with and Without Corresponding Mutations. Front. Oncol. 2021, 11, 640490. [Google Scholar] [CrossRef]
- Sambandam, V.; Frederick, M.J.; Shen, L.; Tong, P.; Rao, X.; Peng, S.; Singh, R.; Mazumdar, T.; Huang, C.; Li, Q.; et al. PDK1 Mediates NOTCH1-Mutated Head and Neck Squamous Carcinoma Vulnerability to Therapeutic PI3K/mTOR Inhibition. Clin. Cancer Res. 2019, 25, 3329–3340. [Google Scholar] [CrossRef]
- Piha-Paul, S.; Munster, P.; Hollebecque, A.; Argilés, G.; Dajani, O.; Cheng, J.; Wang, R.; Swift, A.; Tosolini, A.; Gupta, S. Results of a phase 1 trial combining ridaforolimus and MK-0752 in patients with advanced solid tumours. Eur. J. Cancer 2015, 51, 1865–1873. [Google Scholar] [CrossRef]
- Kleszcz, R.; Paluszczak, J. The combinatorial inhibition of Wnt signaling and Akt kinase is beneficial for reducing the survival and glycolytic activity of tongue cancer cells. J. Oral Pathol. Med. 2021, 51, 231–239. [Google Scholar] [CrossRef]
- Kleszcz, R.; Paluszczak, J. The Wnt Signaling Pathway Inhibitors Improve the Therapeutic Activity of Glycolysis Modulators against Tongue Cancer Cells. Int. J. Mol. Sci. 2022, 23, 1248. [Google Scholar] [CrossRef]
- Ku, B.M.; Yi, S.Y.; Koh, J.; Bae, Y.-H.; Sun, J.-M.; Lee, S.-H.; Ahn, J.S.; Park, K.; Ahn, M.-J. The CDK4/6 inhibitor LY2835219 has potent activity in combination with mTOR inhibitor in head and neck squamous cell carcinoma. Oncotarget 2016, 7, 14803–14813. [Google Scholar] [CrossRef]
- Kostopoulou, O.N.; Zupancic, M.; Pont, M.; Papin, E.; Lukoseviciute, M.; Mikelarena, B.A.; Holzhauser, S.; Dalianis, T. Targeted Therapy of HPV Positive and Negative Tonsillar Squamous Cell Carcinoma Cell Lines Reveals Synergy between CDK4/6, PI3K and Sometimes FGFR Inhibitors, but Rarely between PARP and WEE1 Inhibitors. Viruses 2022, 14, 1372. [Google Scholar] [CrossRef] [PubMed]
- Byskata, K.; Lukoseviciute, M.; Tuti, F.; Zupancic, M.; Kostopoulou, O.N.; Holzhauser, S.; Dalianis, T. Targeted Therapy with PI3K, PARP, and WEE1 Inhibitors and Radiotherapy in HPV Positive and Negative Tonsillar Squamous Cell Carcinoma Cell Lines Reveals Synergy while Effects with APR-246 Are Limited. Cancers 2022, 15, 93. [Google Scholar] [CrossRef] [PubMed]
- Ruicci, K.M.; Pinto, N.; Khan, M.I.; Yoo, J.; Fung, K.; MacNeil, D.; Mymryk, J.S.; Barrett, J.W.; Nichols, A.C. ERK-TSC2 signalling in constitutively-active HRAS mutant HNSCC cells promotes resistance to PI3K inhibition. Oral Oncol. 2018, 84, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Massarelli, E.; Lin, H.; Ginsberg, L.E.; Tran, H.T.; Lee, J.J.; Canales, J.R.; Williams, M.D.; Blumenschein, G.R., Jr.; Lu, C.; Heymach, J.V.; et al. Phase II trial of everolimus and erlotinib in patients with platinum-resistant recurrent and/or metastatic head and neck squamous cell carcinoma. Ann. Oncol. 2015, 26, 1476–1480. [Google Scholar] [CrossRef]
- Ferrarotto, R.; William, W.N.; Tseng, J.E.; Marur, S.; Shin, D.M.; Murphy, B.; Cohen, E.E.; Thomas, C.Y.; Willey, R.; Cosaert, J.; et al. Randomized phase II trial of cixutumumab alone or with cetuximab for refractory recurrent/metastatic head and neck squamous cell carcinoma. Oral Oncol. 2018, 82, 83–90. [Google Scholar] [CrossRef]
- Jin, H.; Wang, L.; Bernards, R. Rational combinations of targeted cancer therapies: Background, advances and challenges. Nat. Rev. Drug Discov. 2022, 22, 213–234. [Google Scholar] [CrossRef]
- Tímár, J.; Uhlyarik, A. On-Target Side Effects of Targeted Therapeutics of Cancer. Pathol. Oncol. Res. 2022, 28, 1610694. [Google Scholar] [CrossRef]
- Rudmann, D.G. On-target and Off-target-based Toxicologic Effects. Toxicol. Pathol. 2012, 41, 310–314. [Google Scholar] [CrossRef]
- Hampton, T. New Insight on Preventing EGFR Inhibitor–Induced Adverse Effects. JAMA 2020, 323, 814. [Google Scholar] [CrossRef]
- Galimont-Collen, A.; Vos, L.; Lavrijsen, A.; Ouwerkerk, J.; Gelderblom, H. Classification and management of skin, hair, nail and mucosal side-effects of epidermal growth factor receptor (EGFR) inhibitors. Eur. J. Cancer 2007, 43, 845–851. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, H.; Xu, Z.; Yang, B.; Luo, P.; He, Q. Molecular basis for class side effects associated with PI3K/AKT/mTOR pathway inhibitors. Expert Opin. Drug Metab. Toxicol. 2019, 15, 767–774. [Google Scholar] [CrossRef]
- Pagès, A.; Foulon, S.; Zou, Z.; Lacroix, L.; Lemare, F.; de Baère, T.; Massard, C.; Soria, J.-C.; Bonastre, J. The cost of molecular-guided therapy in oncology: A prospective cost study alongside the MOSCATO trial. Anesthesia Analg. 2017, 19, 683–690. [Google Scholar] [CrossRef]
- Wilson, L.E.; Greiner, M.A.; Altomare, I.; Rotter, J.; Dinan, M.A. Rapid rise in the cost of targeted cancer therapies for Medicare patients with solid tumors from 2006 to 2015. J. Geriatr. Oncol. 2020, 12, 375–380. [Google Scholar] [CrossRef]
- Seo, M.K.; Cairns, J. Do cancer biomarkers make targeted therapies cost-effective? A systematic review in metastatic colorectal cancer. PLoS ONE 2018, 13, e0204496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhang, J.; Gao, Y.; Li, Y.; Li, Y. Strategies for targeting undruggable targets. Expert Opin. Drug Discov. 2021, 17, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Chaudhary, P.; Quinn, A.; Su, D. Barriers for cancer clinical trial enrollment: A qualitative study of the perspectives of healthcare providers. Contemp. Clin. Trials Commun. 2022, 28, 100939. [Google Scholar] [CrossRef] [PubMed]
- Polverini, P.; D’silva, N.; Lei, Y. Precision Therapy of Head and Neck Squamous Cell Carcinoma. J. Dent. Res. 2018, 97, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Kaidar-Person, O.; Gil, Z.; Billan, S. Precision medicine in head and neck cancer. Drug Resist. Updat. 2018, 40, 13–16. [Google Scholar] [CrossRef]
- Cao, M.; Shi, E.; Wang, H.; Mao, L.; Wu, Q.; Li, X.; Liang, Y.; Yang, X.; Wang, Y.; Li, C. Personalized Targeted Therapeutic Strategies against Oral Squamous Cell Carcinoma. An Evidence-Based Review of Literature. Int. J. Nanomed. 2022, 2022, 4293–4306. [Google Scholar] [CrossRef]
- Koll, L.; Gül, D.; Elnouaem, M.I.; Raslan, H.; Ramadan, O.R.; Knauer, S.K.; Strieth, S.; Hagemann, J.; Stauber, R.H.; Khamis, A. Exploiting Vitamin D Receptor and Its Ligands to Target Squamous Cell Carcinomas of the Head and Neck. Int. J. Mol. Sci. 2023, 24, 4675. [Google Scholar] [CrossRef] [PubMed]
- Heudobler, D.; Lüke, F.; Vogelhuber, M.; Klobuch, S.; Pukrop, T.; Herr, W.; Gerner, C.; Pantziarka, P.; Ghibelli, L.; Reichle, A. Anakoinosis: Correcting Aberrant Homeostasis of Cancer Tissue—Going Beyond Apoptosis Induction. Front. Oncol. 2019, 9, 1408. [Google Scholar] [CrossRef] [PubMed]
- Heudobler, D.; Rechenmacher, M.; Lüke, F.; Vogelhuber, M.; Pukrop, T.; Herr, W.; Ghibelli, L.; Gerner, C.; Reichle, A. Peroxisome Proliferator-Activated Receptors (PPAR)γ Agonists as Master Modulators of Tumor Tissue. Int. J. Mol. Sci. 2018, 19, 3540. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, A.; Carré, M.; Pasquier, E. Metronomics: Intrinsic Anakoinosis Modulator? Front. Pharmacol. 2018, 9, 689. [Google Scholar] [CrossRef] [PubMed]
- Lüke, F.; Harrer, D.C.; Pantziarka, P.; Pukrop, T.; Ghibelli, L.; Gerner, C.; Reichle, A.; Heudobler, D. Drug Repurposing by Tumor Tissue Editing. Front. Oncol. 2022, 12, 900985. [Google Scholar] [CrossRef]
- Bruni, E.; Reichle, A.; Scimeca, M.; Bonanno, E.; Ghibelli, L. Lowering Etoposide Doses Shifts Cell Demise from Caspase-Dependent to Differentiation and Caspase-3-Independent Apoptosis via DNA Damage Response, Inducing AML Culture Extinction. Front. Pharmacol. 2018, 9, 1307. [Google Scholar] [CrossRef]
- Gou, Q.; Gong, X.; Jin, J.; Shi, J.; Hou, Y. Peroxisome proliferator-activated receptors (PPARs) are potential drug targets for cancer therapy. Oncotarget 2017, 8, 60704–60709. [Google Scholar] [CrossRef]
- Walter, I.; Schulz, U.; Vogelhuber, M.; Wiedmann, K.; Endlicher, E.; Klebl, F.; Andreesen, R.; Herr, W.; Ghibelli, L.; Hackl, C.; et al. Communicative reprogramming non-curative hepatocellular carcinoma with low-dose metronomic chemotherapy, COX-2 inhibitor and PPAR-gamma agonist: A phase II trial. Med. Oncol. 2017, 34, 192. [Google Scholar] [CrossRef]
- Heudobler, D.; Schulz, C.; Fischer, J.R.; Staib, P.; Wehler, T.; Südhoff, T.; Schichtl, T.; Wilke, J.; Hahn, J.; Lüke, F.; et al. A Randomized Phase II Trial Comparing the Efficacy and Safety of Pioglitazone, Clarithromycin and Metronomic Low-Dose Chemotherapy with Single-Agent Nivolumab Therapy in Patients with Advanced Non-small Cell Lung Cancer Treated in Second or Further Line (ModuLung). Front. Pharmacol. 2021, 12, 599598. [Google Scholar] [CrossRef]
- Majchrzak-Celińska, A.; Misiorek, J.O.; Kruhlenia, N.; Przybyl, L.; Kleszcz, R.; Rolle, K.; Krajka-Kuźniak, V. COXIBs and 2,5-dimethylcelecoxib counteract the hyperactivated Wnt/β-catenin pathway and COX-2/PGE2/EP4 signaling in glioblastoma cells. BMC Cancer 2021, 21, 493. [Google Scholar] [CrossRef]
- Papierska, K.; Krajka-Kuźniak, V.; Paluszczak, J.; Kleszcz, R.; Skalski, M.; Studzińska-Sroka, E.; Baer-Dubowska, W. Lichen-Derived Depsides and Depsidones Modulate the Nrf2, NF-κB and STAT3 Signaling Pathways in Colorectal Cancer Cells. Molecules 2021, 26, 4787. [Google Scholar] [CrossRef]
- Narożna, M.; Krajka-Kuźniak, V.; Kleszcz, R.; Baer-Dubowska, W. Indomethacin and Diclofenac Hybrids with Oleanolic Acid Oximes Modulate Key Signaling Pathways in Pancreatic Cancer Cells. Int. J. Mol. Sci. 2022, 23, 1230. [Google Scholar] [CrossRef]
- Krajka-Kuźniak, V.; Bednarczyk-Cwynar, B.; Paluszczak, J.; Szaefer, H.; Narożna, M.; Zaprutko, L.; Baer-Dubowska, W. Oleanolic acid oxime derivatives and their conjugates with aspirin modulate the NF-κB-mediated transcription in HepG2 hepatoma cells. Bioorganic Chem. 2019, 93, 103326. [Google Scholar] [CrossRef]
- Narożna, M.; Krajka-Kuźniak, V.; Bednarczyk-Cwynar, B.; Kleszcz, R.; Kujawski, J.; Baer-Dubowska, W. The Effect of Novel Oleanolic Acid Oximes Conjugated with Indomethacin on the Nrf2-ARE And NF-κB Signaling Pathways in Normal Hepatocytes and Human Hepatocellular Cancer Cells. Pharmaceuticals 2020, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Krajka-Kuźniak, V.; Bednarczyk-Cwynar, B.; Narożna, M.; Szaefer, H.; Baer-Dubowska, W. Morpholide derivative of the novel oleanolic oxime and succinic acid conjugate diminish the expression and activity of NF-κB and STATs in human hepatocellular carcinoma cells. Chem. Interactions 2019, 311, 108786. [Google Scholar] [CrossRef] [PubMed]
- Narożna, M.; Krajka-Kuźniak, V.; Bednarczyk-Cwynar, B.; Kucińska, M.; Kleszcz, R.; Kujawski, J.; Piotrowska-Kempisty, H.; Plewiński, A.; Murias, M.; Baer-Dubowska, W. Conjugation of Diclofenac with Novel Oleanolic Acid Derivatives Modulate Nrf2 and NF-κB Activity in Hepatic Cancer Cells and Normal Hepatocytes Leading to Enhancement of Its Therapeutic and Chemopreventive Potential. Pharmaceuticals 2021, 14, 688. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Platonova, N.; Giannandrea, D.; Palano, M.T.; Basile, A.; Chiaramonte, R. Re-establishing Apoptosis Competence in Bone Associated Cancers via Communicative Reprogramming Induced Through Notch Signaling Inhibition. Front. Pharmacol. 2019, 10, 145. [Google Scholar] [CrossRef] [PubMed]
- Kattner, A.-S.; Holler, E.; Herr, W.; Reichle, A.; Wolff, D.; Heudobler, D. Successful Treatment of Early Relapsed High-Risk AML After Allogeneic Hematopoietic Stem Cell Transplantation with Biomodulatory Therapy. Front. Oncol. 2020, 10, 443. [Google Scholar] [CrossRef] [PubMed]
- Heudobler, D.; Klobuch, S.; Thomas, S.; Hahn, J.; Herr, W.; Reichle, A. Cutaneous Leukemic Infiltrates Successfully Treated with Biomodulatory Therapy in a Rare Case of Therapy-Related High Risk MDS/AML. Front. Pharmacol. 2018, 9, 1279. [Google Scholar] [CrossRef]
- Lüke, F.; Harrer, D.C.; Hahn, J.; Grube, M.; Pukrop, T.; Herr, W.; Reichle, A.; Heudobler, D. Continuous Complete Remission in Two Patients with Acute Lymphoblastic Leukemia and Severe Fungal Infection Following Short-Term, Dose-Reduced Chemotherapy. Front. Pharmacol. 2021, 12, 599552. [Google Scholar] [CrossRef] [PubMed]
- Lüke, F.; Harrer, D.C.; Menhart, K.; Wolff, D.; Holler, E.; Hellwig, D.; Herr, W.; Grube, M.; Vogelhuber, M.; Reichle, A.; et al. Biomodulatory Treatment Regimen, MEPED, Rescues Relapsed and Refractory Classic Hodgkin’s Disease. Front. Pharmacol. 2021, 12, 599561. [Google Scholar] [CrossRef] [PubMed]
- Harrer, D.C.; Jakob, M.; Vogelhuber, M.; Lüke, F.; Utpatel, K.; Corbacioglu, S.; Herr, W.; Reichle, A.; Heudobler, D. Biomodulatory therapy induces durable remissions in multi-system Langerhans cell histiocytosis. Leuk. Lymphoma 2022, 63, 2858–2868. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Ghibelli, L.; Reichle, A. A Computational Model of Tumor Growth and Anakoinosis. Front. Pharmacol. 2019, 10, 287. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Second Target in Combination | Combination EGFR inh. + Second inh. | General Effect and Experimental Model * | Reference |
|---|---|---|---|
| PI3K (phosphoinositide 3-kinase) | cetuximab + buparlisib (+ radiotherapy) | improved tumor inhibition via double/triple combination (in vivo) | [106] |
| afatinib + HS-173 | synergistic anticancer activity (ex vivo) | [107] | |
| Akt kinase | cetuximab + MK2206 | partly synergistic anticancer activity (in vitro) | [108] |
| mTOR (mammalian target of rapamycin) | cetuximab + temsirolimus | synergistic tumor growth inhibition (in vivo) | [109] |
| cetuximab + AZD8055 | improved reduction of tumor growth (in vivo) | [110] | |
| MAPK (mitogen-activated protein kinases) | afatinib + PD0325901 | prevention of single-compound-related resistance and synergistic reduction of cancer cells survival (in vitro) | [111] |
| FGFR (fibroblast growth factor receptor) | gefitinib + AZD4547 | synergistic reduction of cell proliferation (in vitro) | [48] |
| gefitinib + BGJ398 | synergistic tumor growth inhibition (in vivo) | [114] | |
| VEGFR (vascular endothelial growth factor receptor) | cetuximab + bevacizumab | improved anticancer activity (in vitro) and reduction of tumor growth (in vivo) | [51] |
| erlotinib + bevacizumab | better outcomes in patients with R/M HNSCC ** (clinical trial) | [52] | |
| VEGFR and PDGFR (platelet-derived growth factor receptor) | cetuximab + sunitinib + radiotherapy | improved reduction of tumor growth (in vivo) | [115] |
| HGF/c-MET (hepatocyte growth factor/mesenchymal-epithelial- transition factor) | gefitinib + crizotinib or gefitinib + SU11274 | reduced cell proliferation, migration and invasion (in vitro), and reduced tumor growth (in vivo) | [116] |
| JAK/STAT (Janus kinase/ signal transducer and activator of transcription) | cetuximab + JAK1i | improved anticancer activity (in vitro) | [117] |
| NOTCH | erlotinib + PF-03084014 | reduced proliferation and invasion, synergy in inhibition of PI3K pathway (in vitro), and improved reduction of tumor growth (in vivo) | [118] |
| Wnt (canonical) | erlotinib + PRI-724 | synergy in reduction of cell proliferation and migration, accelerated apoptosis (in vitro) | [119] |
| Hedgehog | cetuximab + vismodegib | improved anticancer activity (in vitro and ex vivo) | [120] |
| TGF-β (transforming growth factor-β) | cetuximab + antibody against TGF-β | improved reduction of tumor growth (in vivo) | [121] |
| NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) | gefitinib + CmpdA | improved anticancer activity (in vitro) | [123] |
| gefitinib + Bay117085 | improved anticancer activity (in vitro) and reduced tumor growth (in vivo) | [124] | |
| PD-1/PD-L1 (programmed cell death 1/programmed cell death ligand 1) | cetuximab + pembrolizumab | better outcomes in patients with R/M HNSCC ** (clinical trial) | [128] |
| Glycolysis | erlotinib + 2-deoxyglucose | reduced cell viability (in vitro) and loss of effects due to tumor-rescue autophagy induction (in vivo) | [129] |
| CDK (cyclin-dependent kinase) | cetuximab + palbociclib | synergistic viability reduction (in vitro) | [130] |
| PARP1 (poly (adenosine diphosphate-ribose) polymerase-1)) | cetuximab + olaparib + radiotherapy | improved anticancer activity (in vitro) and reduced tumor growth (in vivo) | [131] |
| XIAP (X-linked inhibitor of apoptosis protein) | gefitinib + demethoxycurcumin | activation of cell cycle arrest and induction of apoptosis (in vitro) | [132] |
| Aurora kinase | cetuximab + R763 | accelerated induction of apoptosis and cell cycle checkpoint activation (in vitro) | [133] |
| KDM (histone lysine demethylase) | erlotinib + ML324 (inhibitor of KDM4) or erlotinib + GSK-J4 (inhibitor of KDM6) | synergistic inhibition of cell viability and activation of apoptosis (in vitro) | [134] |
| First Molecular Target /Drug Name/ | Second Molecular Target /Drug Name/ | General Effect and Experimental Model * | Reference |
|---|---|---|---|
| RAS farnesylation /tipifarnib/ | PI3K pathway (phosphoinositide 3-kinase) /alpelisib/ | increased sensitivity of cancer cells to tipifarnib (in vitro) | [136] |
| MAPK pathway (mitogen-activated protein kinase) /SCH772984/ | |||
| PI3K /alpelisib/ | FGFR (fibroblast growth factor receptor) /erdafitinib/ | synergistic reduction of cell viability (in vitro) | [137] |
| PI3K /HS-173/ | KDM4 (histone lysine demethylase 4) /ML324/ | synergistic inhibition of cell viability and induction of apoptosis (in vitro) | [134] |
| KDM6 (histone lysine demethylase 6) /GSK-J4/ | |||
| PI3K /GSK2126458/ | NOTCH /NOTCH1 mutant cells/ | improved and predicable response to PI3K pathway inhibitors (in vitro) | [138] |
| mTOR (mammalian target of rapamycin) /ridaforolimus/ | NOTCH (MK-0752) | partial response to treatment, problems with side effects in maximum tolerated dose (clinical trial) | [139] |
| mTOR /everolimus/ | CDK 4/6 (cyclin-dependent kinase 4/6) /LY2835219/ | improved anticancer activity (in vitro) and synergistic tumor growth inhibition (in vivo) | [142] |
| CDK 4/6 /PD-0332991/ | PI3K /BYL719/ | synergistic reduction of cell viability (in vitro) | [143] |
| FGFR /NJ-42756493/ | |||
| PARP (poly (adenosine diphosphate-ribose) polymerase)) /BMN-673/ (+ radiotherapy) | PI3K /BYL719/ (+ radiotherapy) | synergistic reduction of cell viability by drugs combination, lack of additional effect of irradiation (in vitro) | [144] |
| Wnt (canonical) /PRI-724/ | PI3K /HS-173/ | synergy in reduction of cell viability and migration rate (in vitro) | [119] |
| Hedgehog /vismodegib/ | |||
| Akt kinase /Akt kinase inhibitor X/ | Wnt (canonical) /PRI-724/ | reduced viability of cells growing in 2D and 3D culture, decreased glycolytic activity-glucose intake and lactate release (in vitro) | [140] |
| Wnt (canonical and non-canonical) /IWP-O1/ | |||
| Glycolysis /2-deoksyglucose/ and /lonidamine/ | Wnt (canonical) /PRI-724/ | reduced cell viability, decreased glycolytic activity—glucose intake and lactate release (in vitro) | [141] |
| Wnt (canonical and non-canonical) /IWP-O1/ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kleszcz, R. Advantages of the Combinatorial Molecular Targeted Therapy of Head and Neck Cancer—A Step before Anakoinosis-Based Personalized Treatment. Cancers 2023, 15, 4247. https://doi.org/10.3390/cancers15174247
Kleszcz R. Advantages of the Combinatorial Molecular Targeted Therapy of Head and Neck Cancer—A Step before Anakoinosis-Based Personalized Treatment. Cancers. 2023; 15(17):4247. https://doi.org/10.3390/cancers15174247
Chicago/Turabian StyleKleszcz, Robert. 2023. "Advantages of the Combinatorial Molecular Targeted Therapy of Head and Neck Cancer—A Step before Anakoinosis-Based Personalized Treatment" Cancers 15, no. 17: 4247. https://doi.org/10.3390/cancers15174247
APA StyleKleszcz, R. (2023). Advantages of the Combinatorial Molecular Targeted Therapy of Head and Neck Cancer—A Step before Anakoinosis-Based Personalized Treatment. Cancers, 15(17), 4247. https://doi.org/10.3390/cancers15174247