

Unraveling the Anticancer Potential of Statins: Mechanisms and Clinical Significance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Anticancer Mechanisms of Statins
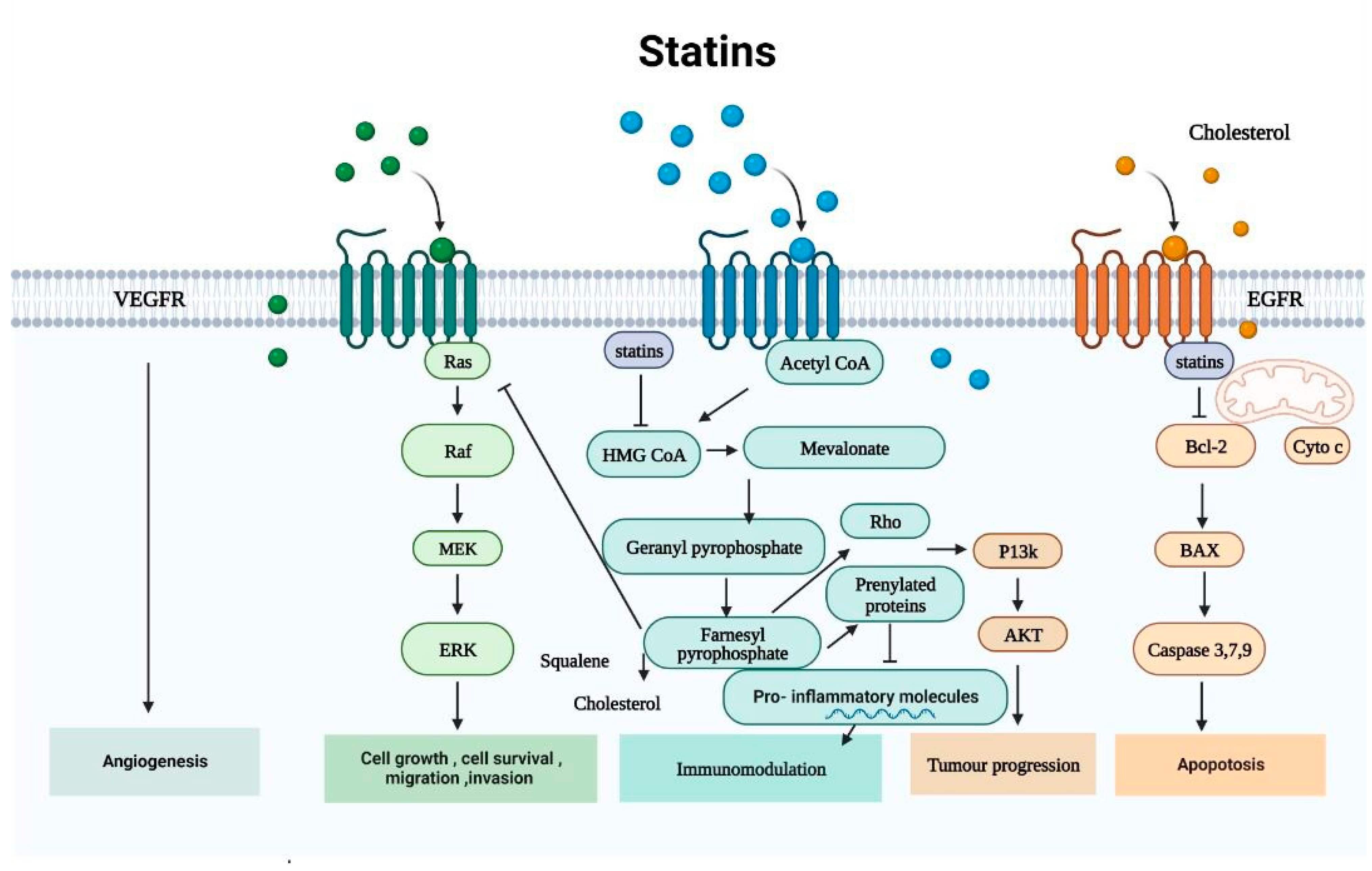
2.1. The Mevalonate Pathway
2.1.1. Non-Cholesterol-Mediated Pathways
2.1.2. Cholesterol-Mediated Pathways
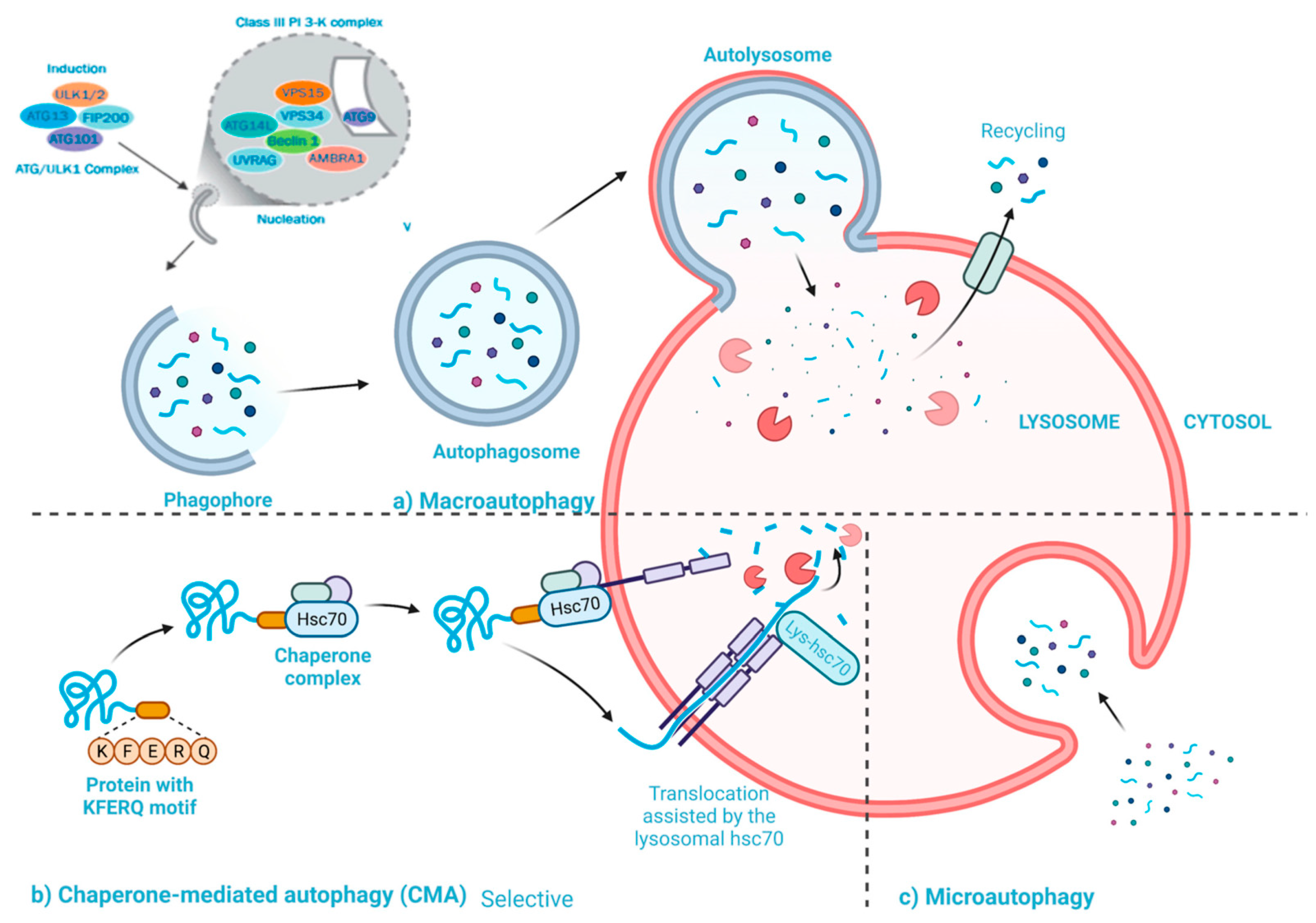
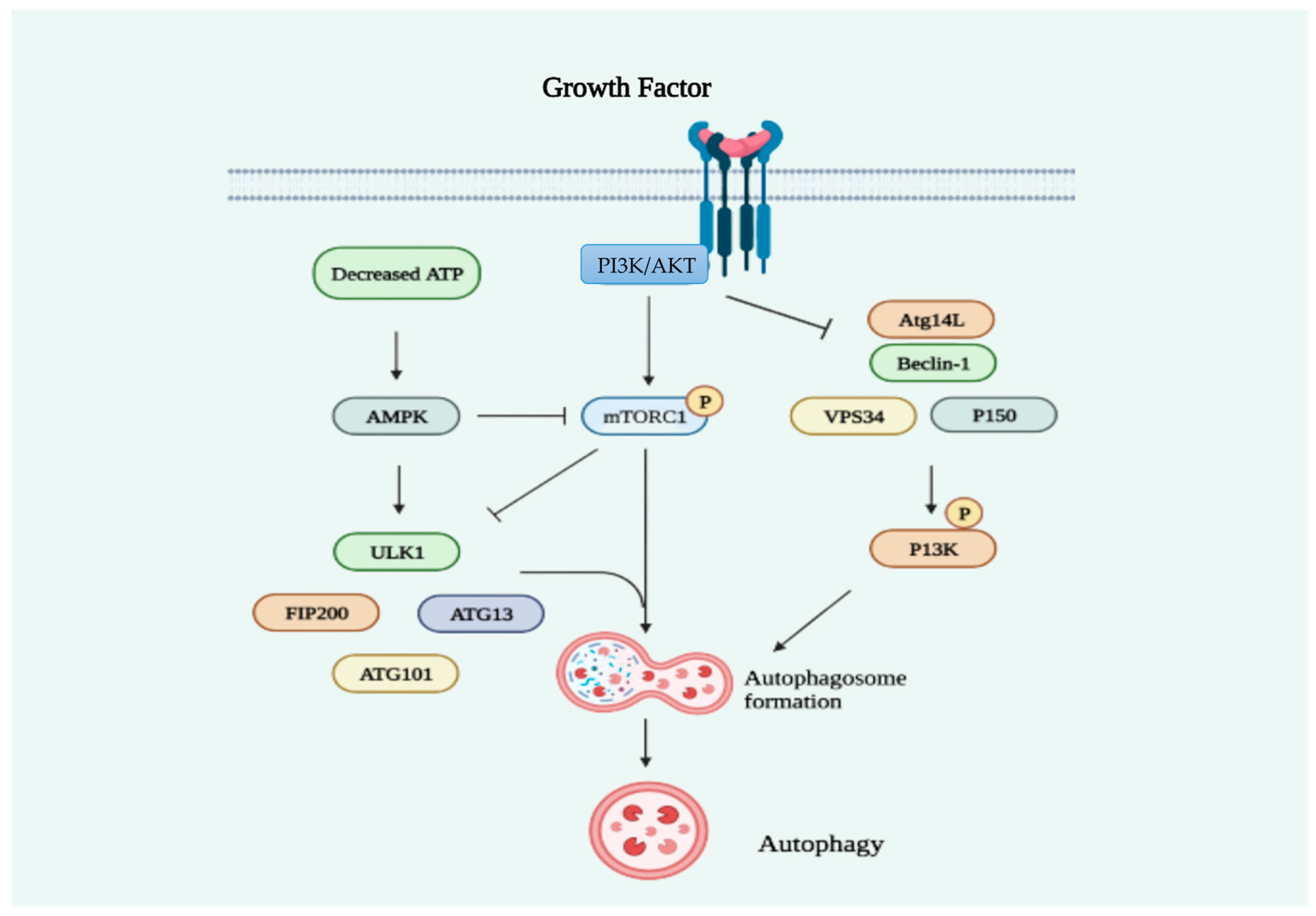
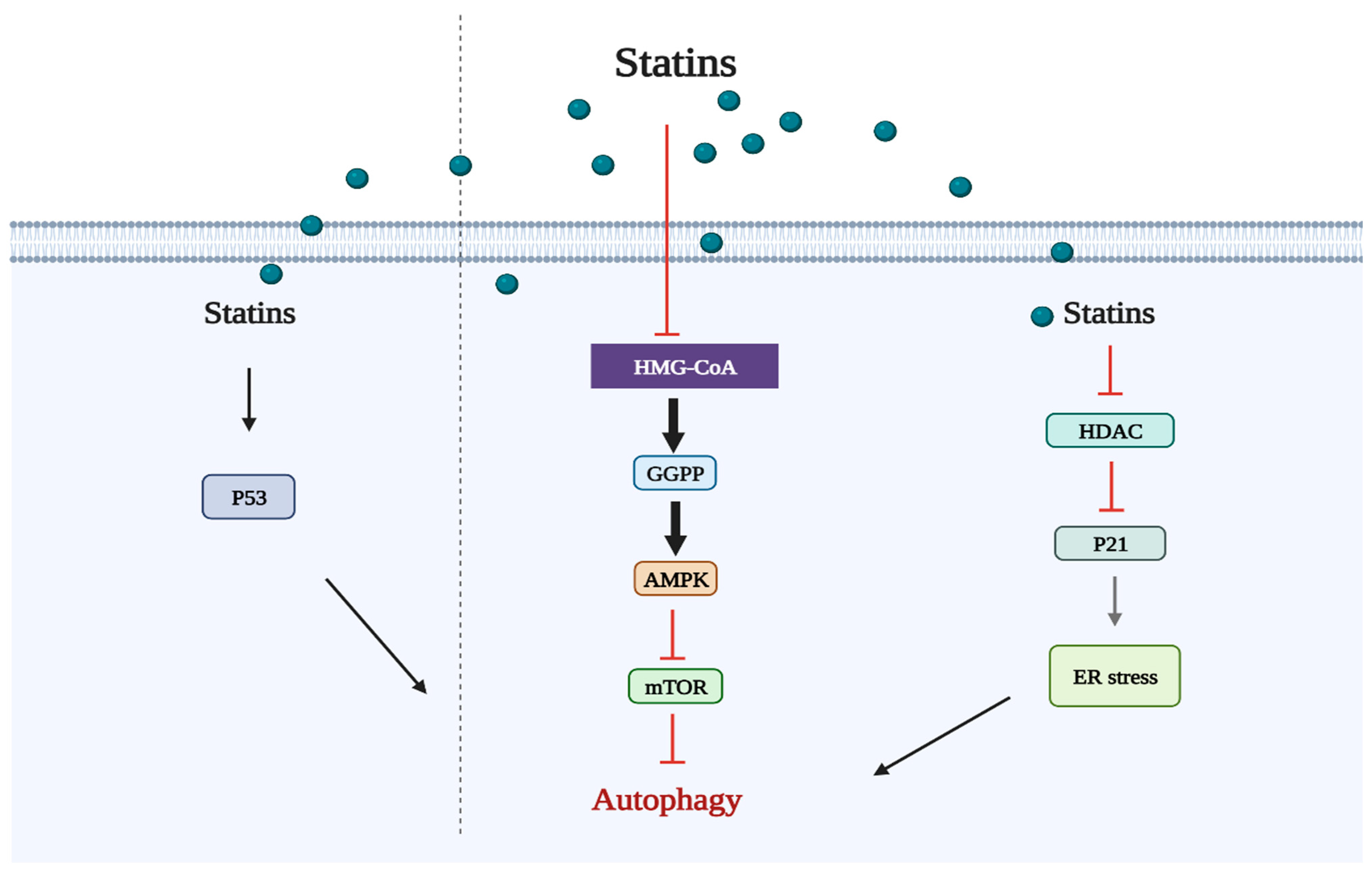
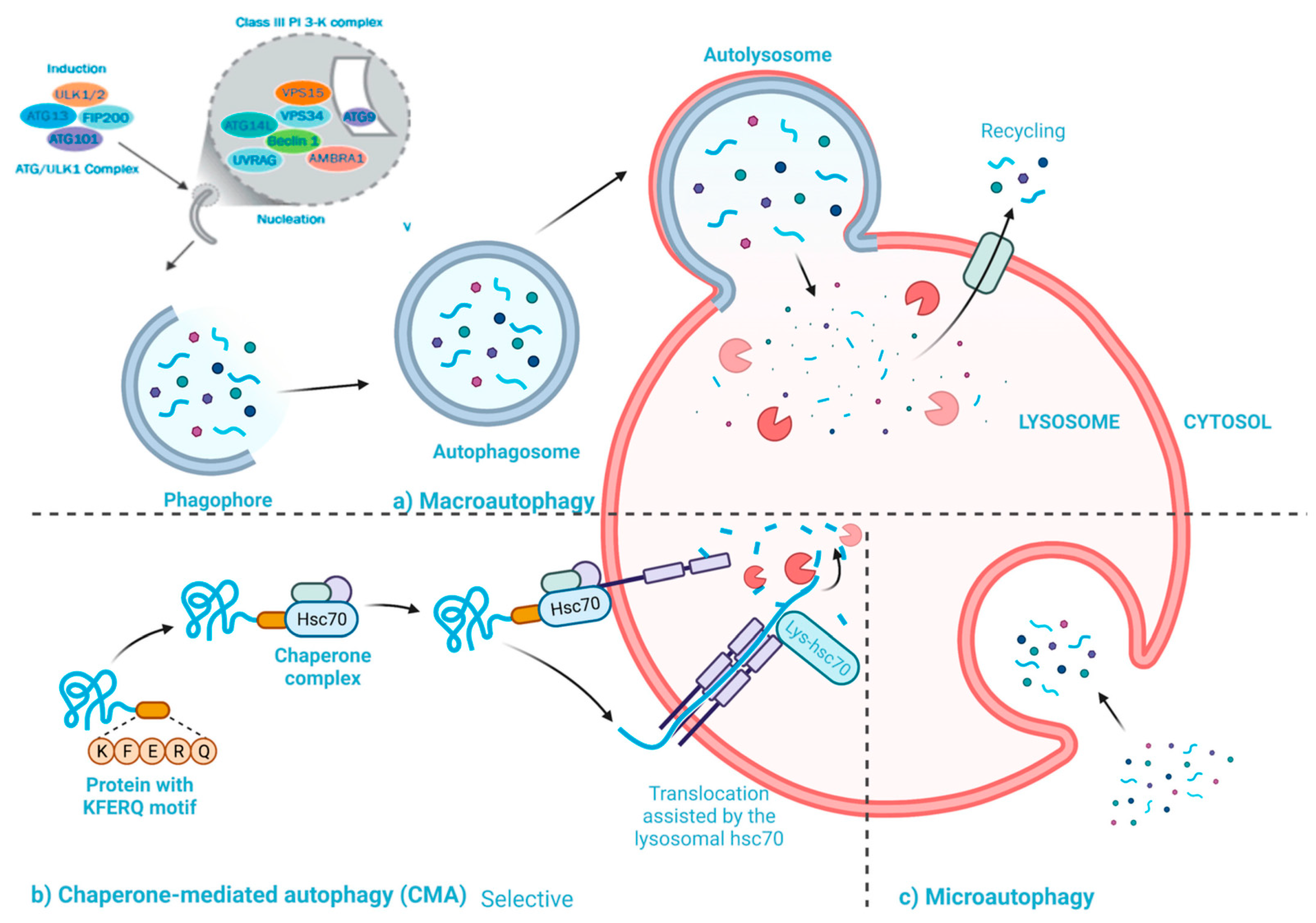
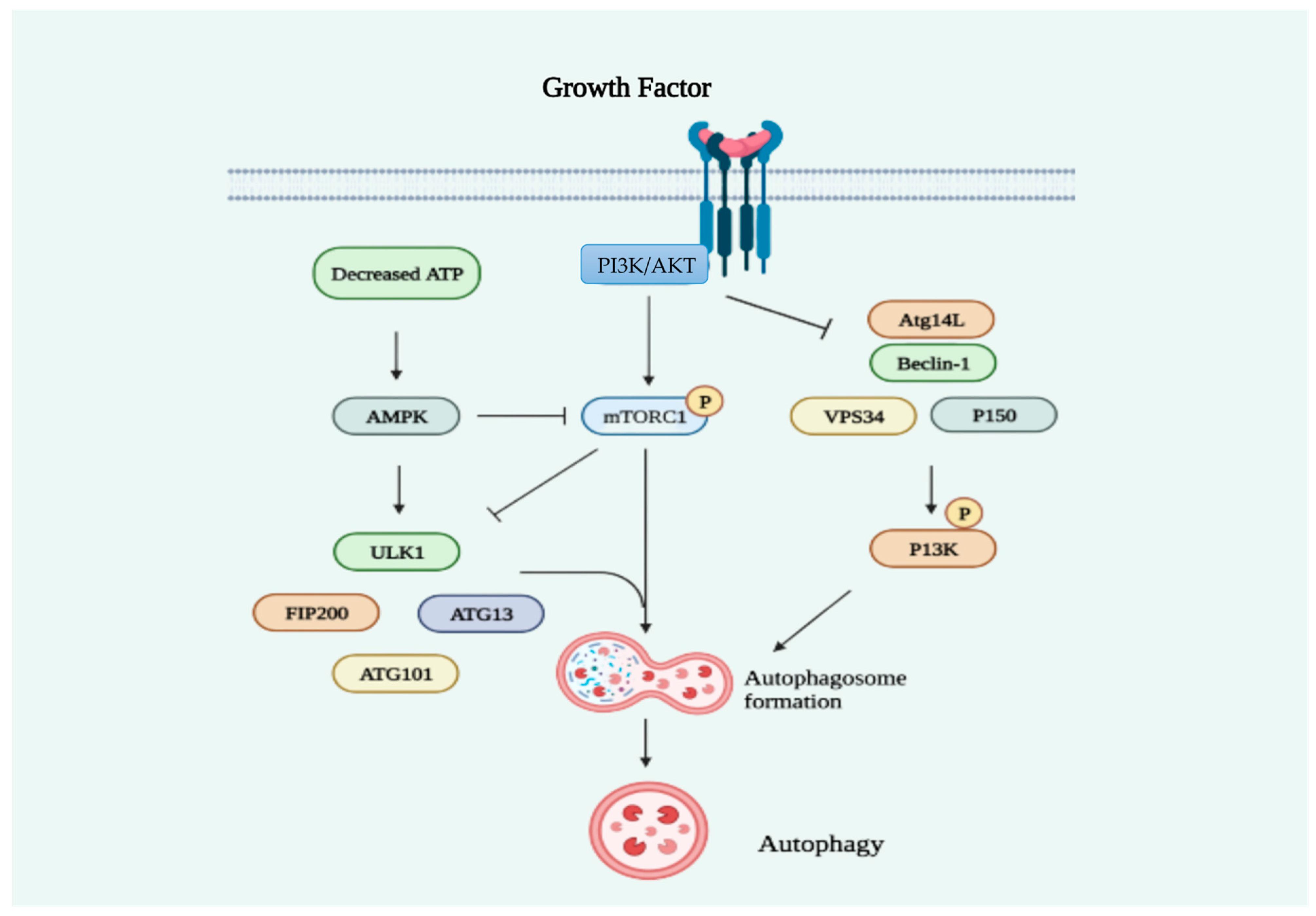
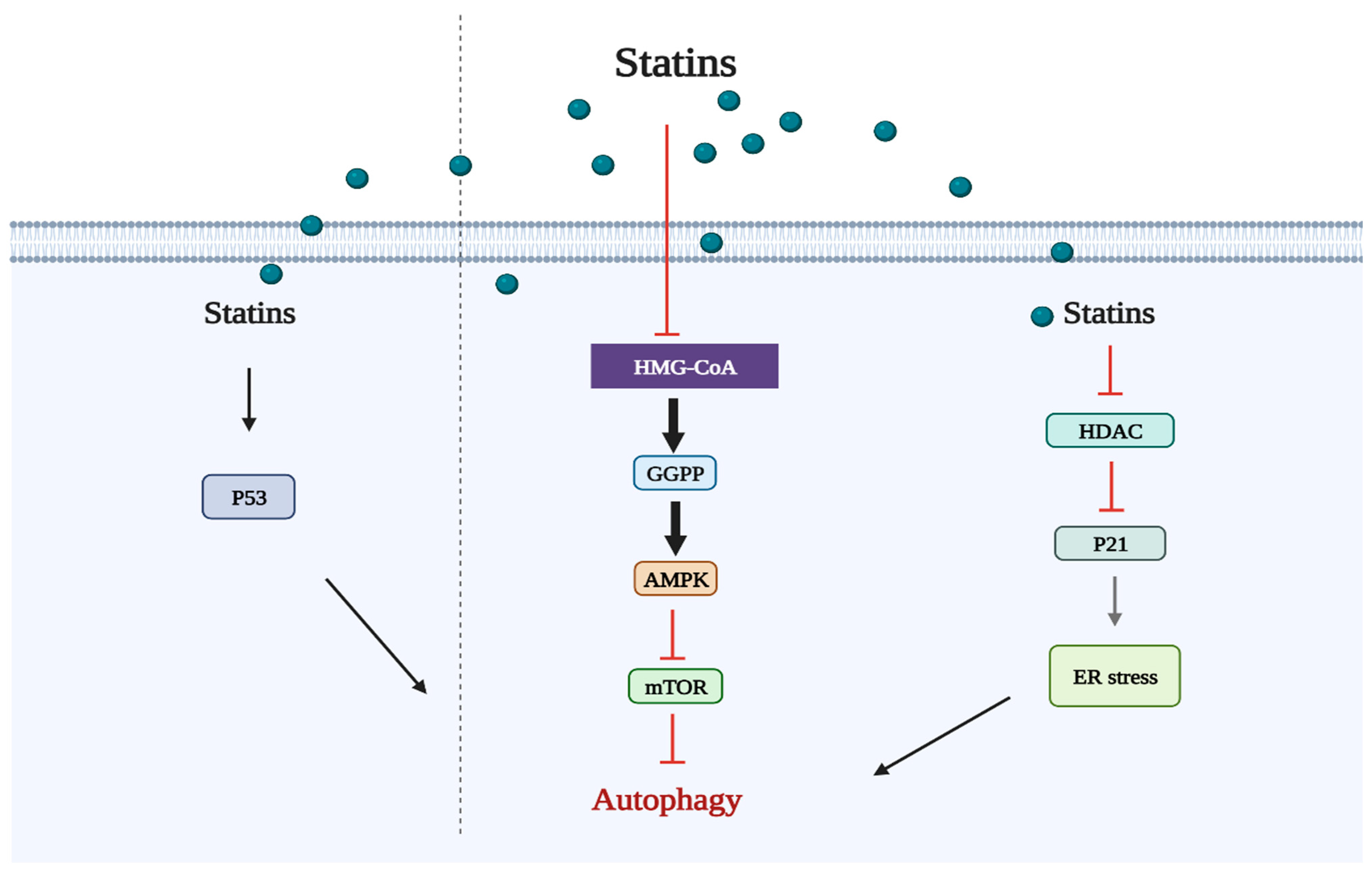
2.2. Statins Regulate Autophagy
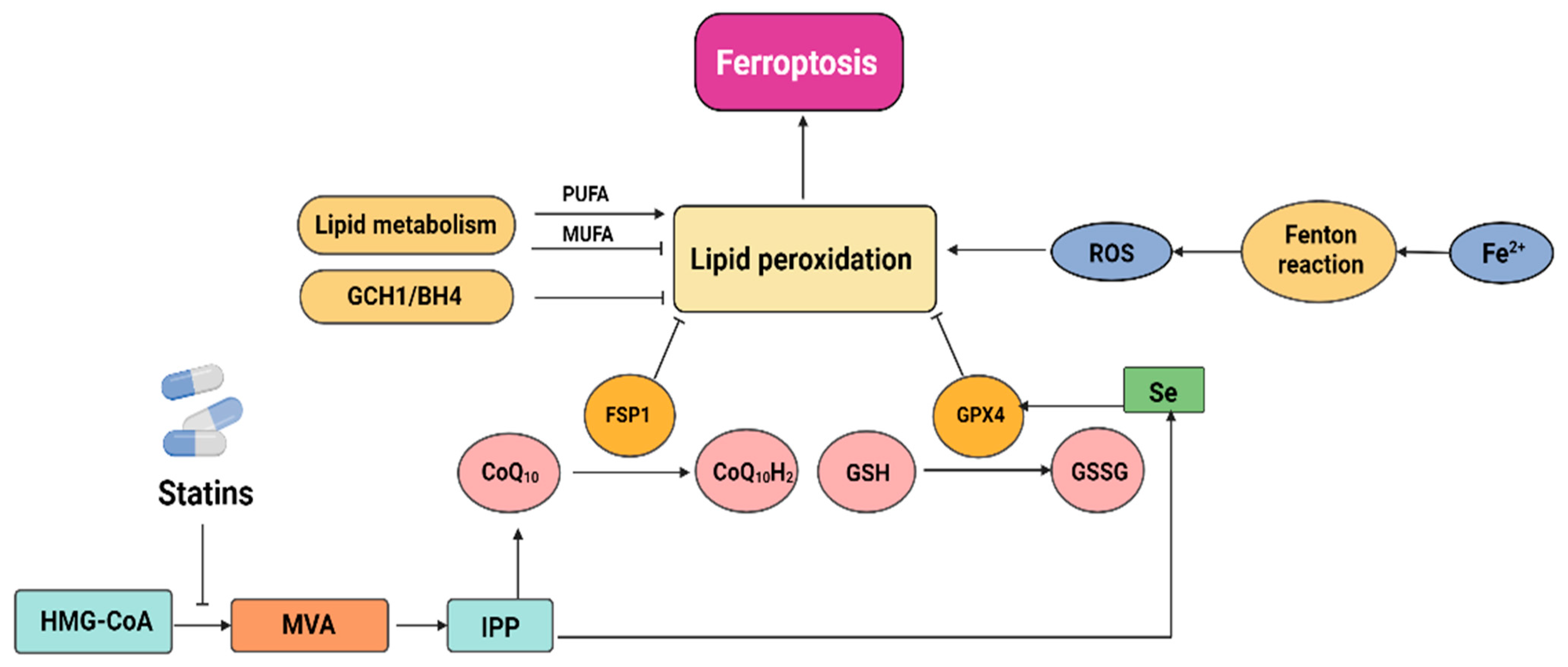
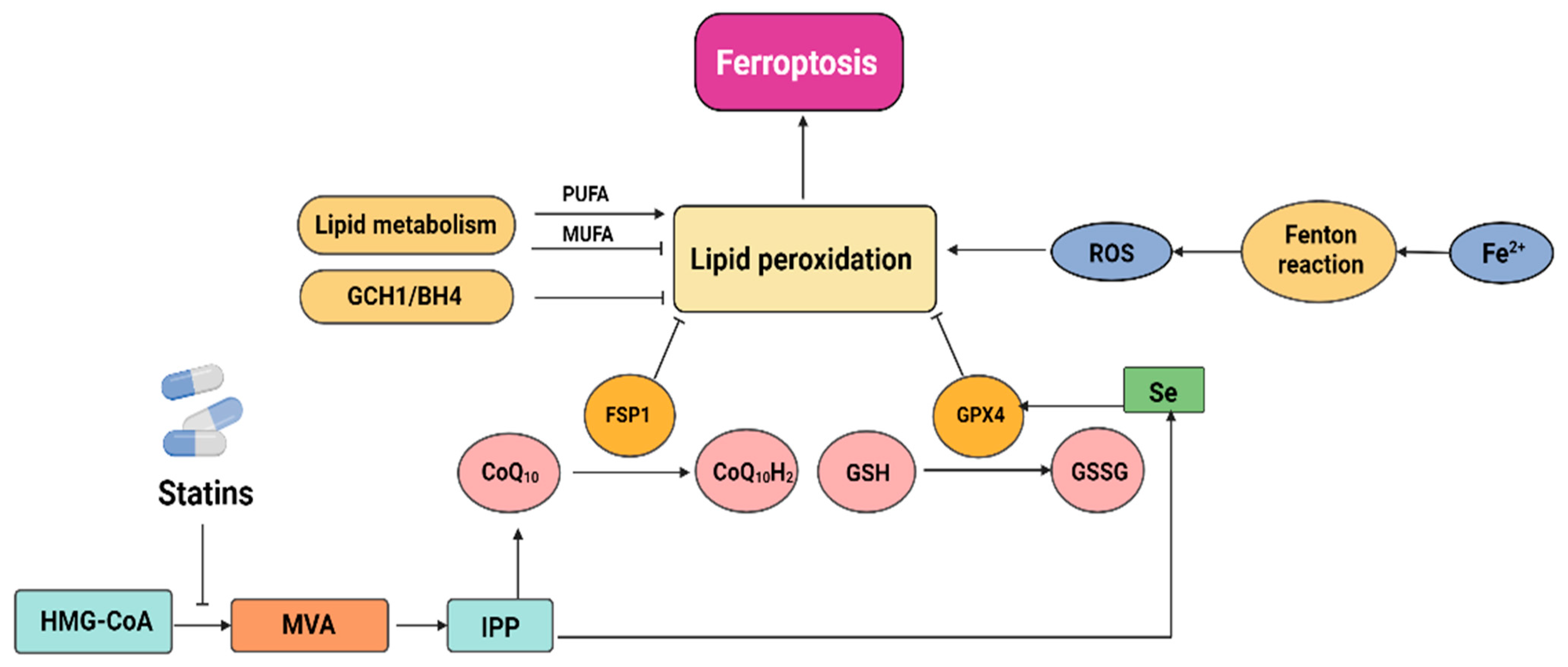
2.3. Statins Induce Ferroptosis
2.4. Statins Induce Pyroptosis
2.5. Statins Target the Tumor Microenvironment
3. Statin Anticancer In Vitro Studies
4. Statin Anticancer In Vivo Studies
5. Clinical Application of Statins
5.1. Epidemiology
5.2. Meta-Analysis
5.3. Observational Studies
5.4. Randomized Controlled Trials (RCTs)
5.5. Pharmacoepidemiological Studies
5.6. Mendelian Randomization
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Endo, A.; Tsujita, Y.; Kuroda, M.; Tanzawa, K. Inhibition of Cholesterol Synthesis in vitro and in vivo by ML-236A and ML-236B, Competitive Inhibitors of 3-Hydroxy-3-methylglutaryl-Coenzyme A Reductase. Eur. J. Biochem. 1977, 77, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Carrer, A.; Trefely, S.; Zhao, S.; Campbell, S.L.; Norgard, R.J.; Schultz, K.C.; Sidoli, S.; Parris, J.L.D.; Affronti, H.C.; Sivanand, S. Acetyl-CoA Metabolism Supports Multistep Pancreatic TumorigenesisRoles of Acetyl-CoA Metabolism in Pancreatic Tumorigenesis. Cancer Discov. 2019, 9, 416–435. [Google Scholar] [CrossRef]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 2012, 367, 1792–1802. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, M.I.; de Wolf, E.; Jawad, M.J.; Richardson, A. The poor design of clinical trials of statins in oncology may explain their failure–lessons for drug repurposing. Cancer Treat. Rev. 2018, 69, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Hu, J.-W.; He, X.-R.; Jin, W.-L.; He, X.-Y. Statins: A repurposed drug to fight cancer. J. Exp. Clin. Cancer Res. 2021, 40, 241. [Google Scholar] [CrossRef]
- Göbel, A.; Rauner, M.; Hofbauer, L.C.; Rachner, T.D. Cholesterol and beyond-The role of the mevalonate pathway in cancer biology. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2020, 1873, 188351. [Google Scholar] [CrossRef]
- Juarez, D.; Fruman, D.A. Targeting the Mevalonate Pathway in Cancer. Trends Cancer 2021, 7, 525–540. [Google Scholar] [CrossRef]
- Jeong, A.; Suazo, K.F.; Wood, W.G.; Distefano, M.D.; Li, L. Isoprenoids and protein prenylation: Implications in the pathogenesis and therapeutic intervention of Alzheimer’s disease. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 279–310. [Google Scholar] [CrossRef]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef]
- Brown, D.N.; Caffa, I.; Cirmena, G.; Piras, D.; Garuti, A.; Gallo, M.; Alberti, S.; Nencioni, A.; Ballestrero, A.; Zoppoli, G. Squalene epoxidase is a bona fide oncogene by amplification with clinical relevance in breast cancer. Sci. Rep. 2016, 6, 19435. [Google Scholar] [CrossRef] [PubMed]
- Narwal, V.; Deswal, R.; Batra, B.; Kalra, V.; Hooda, R.; Sharma, M.; Rana, J.S. Cholesterol biosensors: A review. Steroids 2019, 143, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Min, W.; Wang, J.; Wang, L.; Li, Y.; Zhou, N.; Yang, Z.; Qian, Q. Effects of mevalonate kinase interference on cell differentiation, apoptosis, prenylation and geranylgeranylation of human keratinocytes are attenuated by farnesyl pyrophosphate or geranylgeranyl pyrophosphate. Exp. Ther. Med. 2020, 19, 2861–2870. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Shen, H.; Huang, H.; Yang, Z.; Zhou, Y.; Zhao, G. Cholesterol-lowering drug pitavastatin targets lung cancer and angiogenesis via suppressing prenylation-dependent Ras/Raf/MEK and PI3K/Akt/mTOR signaling. Anticancer Drugs 2020, 31, 377–384. [Google Scholar] [CrossRef] [PubMed]
- White, C.P. On the occurrence of crystals in tumors. J. Pathol. Bacteriol. 1909, 13, 3. [Google Scholar] [CrossRef]
- Dessì, S.; Batetta, B.; Pulisci, D.; Spano, O.; Anchisi, C.; Tessitore, L.; Costelli, P.; Baccino, F.M.; Aroasio, E.; Pani, P. Cholesterol content in tumor tissues is inversely associated with high-density lipoprotein cholesterol in serum in patients with gastrointestinal cancer. Cancer 1994, 73, 253–258. [Google Scholar] [CrossRef]
- Kolanjiappan, K.; Ramachandran, C.R.; Manoharan, S. Biochemical changes in tumor tissues of oral cancer patients. Clin. Biochem. 2003, 36, 61–65. [Google Scholar] [CrossRef]
- Gu, J.; Zhu, N.; Li, H.-F.; Zhao, T.-J.; Zhang, C.-J.; Liao, D.-F.; Qin, L. Cholesterol homeostasis and cancer: A new perspective on the low-density lipoprotein receptor. Cell. Oncol. 2022, 45, 709–728. [Google Scholar] [CrossRef]
- Likus, W.; Siemianowicz, K.; Bieńk, K.; Pakuła, M.; Pathak, H.; Dutta, C.; Wang, Q.; Shojaei, S.; Assaraf, Y.G.; Ghavami, S. Could drugs inhibiting the mevalonate pathway also target cancer stem cells? Drug Resist. Updates 2016, 25, 13–25. [Google Scholar] [CrossRef]
- Cruz, P.M.R.; Mo, H.; McConathy, W.J.; Sabnis, N.; Lacko, A.G. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: A review of scientific findings, relevant to future cancer therapeutics. Front. Pharmacol. 2013, 4, 119. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef]
- Moossavi, M.; Parsamanesh, N.; Bahrami, A.; Atkin, S.L.; Sahebkar, A. Role of the NLRP3 inflammasome in cancer. Mol. Cancer 2018, 17, 158. [Google Scholar] [CrossRef]
- Bae, J.Y.; Lee, S.-W.; Shin, Y.-H.; Lee, J.-H.; Jahng, J.W.; Park, K. P2X7 receptor and NLRP3 inflammasome activation in head and neck cancer. Oncotarget 2017, 8, 48972. [Google Scholar] [CrossRef]
- Du, Q.; Wang, Q.; Fan, H.; Wang, J.; Liu, X.; Wang, H.; Wang, Y.; Hu, R. Dietary cholesterol promotes AOM-induced colorectal cancer through activating the NLRP3 inflammasome. Biochem. Pharmacol. 2016, 105, 42–54. [Google Scholar] [CrossRef]
- Weichand, B.; Popp, R.; Dziumbla, S.; Mora, J.; Strack, E.; Elwakeel, E.; Frank, A.-C.; Scholich, K.; Pierre, S.; Syed, S.N. S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via NLRP3/IL-1β. J. Exp. Med. 2017, 214, 2695–2713. [Google Scholar] [CrossRef]
- Mok, E.H.K.; Lee, T.K.W. The pivotal role of the dysregulation of cholesterol homeostasis in cancer: Implications for therapeutic targets. Cancers 2020, 12, 1410. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.; Cheng, X.; Wu, X.; Yoo, J.Y.; Cheng, C.; Guo, J.Y.; Mo, X.; Ru, P.; Hurwitz, B.; Kim, S.-H. Inhibition of SOAT1 Suppresses Glioblastoma Growth via Blocking SREBP-1–Mediated LipogenesisTargeting SOAT1 to Treat Glioblastoma. Clin. Cancer Res. 2016, 22, 5337–5348. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Sun, A.; Zhao, Y.; Ying, W.; Sun, H.; Yang, X.; Xing, B.; Sun, W.; Ren, L.; Hu, B. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature 2019, 567, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Saraon, P.; Trudel, D.; Kron, K.; Dmitromanolakis, A.; Trachtenberg, J.; Bapat, B.; van der Kwast, T.; Jarvi, K.A.; Diamandis, E.P. Evaluation and prognostic significance of ACAT1 as a marker of prostate cancer progression. Prostate 2014, 74, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Tveten, K.; Berge, K.E.; Leren, T.P. PCSK9-mediated degradation of the LDL receptor generates a 17 kDa C-terminal LDL receptor fragment. J. Lipid Res. 2013, 54, 1560–1566. [Google Scholar] [CrossRef]
- Athavale, D.; Chouhan, S.; Pandey, V.; Mayengbam, S.S.; Singh, S.; Bhat, M.K. Hepatocellular carcinoma-associated hypercholesterolemia: Involvement of proprotein-convertase-subtilisin-kexin type-9 (PCSK9). Cancer Metab. 2018, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Ouvrier, A.; Cadet, R.; Vernet, P.; Laillet, B.; Chardigny, J.-M.; Lobaccaro, J.-M.A.; Drevet, J.R.; Saez, F. LXR and ABCA1 control cholesterol homeostasis in the proximal mouse epididymis in a cell-specific manner. J. Lipid Res. 2009, 50, 1766–1775. [Google Scholar] [CrossRef] [PubMed]
- Bi, D.-P.; Yin, C.-H.; Zhang, X.-Y.; Yang, N.-N.; Xu, J.-Y. MiR-183 functions as an oncogene by targeting ABCA1 in colon cancer. Oncol. Rep. 2016, 35, 2873–2879. [Google Scholar] [CrossRef]
- Su, C.; Huang, D.P.; Liu, J.W.; Liu, W.Y.; Cao, Y.O. miR-27a-3p regulates proliferation and apoptosis of colon cancer cells by potentially targeting BTG1. Oncol. Lett. 2019, 18, 2825–2834. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Feng, F.; Wang, J.; Ren, M.; Shi, Z.; Mao, X.; Zhang, H.; Ju, X. Liver X receptor activation reduces gastric cancer cell proliferation by suppressing Wnt signalling via LXR β relocalization. J. Cell. Mol. Med. 2019, 23, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Nedelcu, D.; Watanabe, M.; Jao, C.; Kim, Y.; Liu, J.; Salic, A. Cellular cholesterol directly activates smoothened in hedgehog signaling. Cell 2016, 166, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.L.; Lo, H.-W. Hedgehog pathway and GLI1 isoforms in human cancer. Discov. Med. 2012, 13, 105. [Google Scholar]
- Gordon, R.E.; Zhang, L.; Peri, S.; Kuo, Y.-M.; Du, F.; Egleston, B.L.; Ng, J.M.Y.; Andrews, A.J.; Astsaturov, I.; Curran, T. Statins Synergize with Hedgehog Pathway Inhibitors for Treatment of MedulloblastomaStatins Effectively Inhibit Medulloblastoma Growth. Clin. Cancer Res. 2018, 24, 1375–1388. [Google Scholar] [CrossRef]
- Fan, Q.; Gong, T.; Zheng, C.; Ng, J.M.Y.; Chen, J.; Myers, C.; Hensley, H.; Curran, T.; Yang, Z. Statins repress hedgehog signaling in medulloblastoma with no bone toxicities. Oncogene 2021, 40, 2258–2272. [Google Scholar] [CrossRef]
- Ghaderi, A.; Vahdati-Mashhadian, N.; Oghabian, Z.; Moradi, V.; Afshari, R.; Mehrpour, O. Thallium exists in opioid poisoned patients. DARU J. Pharm. Sci. 2015, 23, 39. [Google Scholar] [CrossRef]
- Kerr, S.-L.; Mathew, C.; Ghildyal, R. Rhinovirus and Cell Death. Viruses 2021, 13, 629. [Google Scholar] [CrossRef]
- Galluzzi, L.; Green, D.R. Autophagy-independent functions of the autophagy machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef]
- Narita, M.; Young, A.R.J.; Narita, M. Autophagy facilitates oncogene-induced senescence. Autophagy 2009, 5, 1046–1047. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Autophagy and cellular immune responses. Immunity 2013, 39, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Kenific, C.M.; Debnath, J. Cellular and metabolic functions for autophagy in cancer cells. Trends Cell Biol. 2015, 25, 37–45. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- Araki, M.; Motojima, K. Hydrophobic statins induce autophagy in cultured human rhabdomyosarcoma cells. Biochem. Biophys. Res. Commun. 2008, 367, 462–467. [Google Scholar] [CrossRef]
- Yang, Z.; Su, Z.; DeWitt, J.P.; Xie, L.; Chen, Y.; Li, X.; Han, L.; Li, D.; Xia, J.; Zhang, Y. Fluvastatin prevents lung adenocarcinoma bone metastasis by triggering autophagy. EBioMedicine 2017, 19, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Toepfer, N.; Childress, C.; Parikh, A.; Rukstalis, D.; Yang, W. Atorvastatin induces autophagy in prostate cancer PC3 cells through activation of LC3 transcription. Cancer Biol. Ther. 2011, 12, 691–699. [Google Scholar] [CrossRef]
- Misirkic, M.; Janjetovic, K.; Vucicevic, L.; Tovilovic, G.; Ristic, B.; Vilimanovich, U.; Harhaji-Trajkovic, L.; Sumarac-Dumanovic, M.; Micic, D.; Bumbasirevic, V. Inhibition of AMPK-dependent autophagy enhances in vitro antiglioma effect of simvastatin. Pharmacol. Res. 2012, 65, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Vilimanovich, U.; Bosnjak, M.; Bogdanovic, A.; Markovic, I.; Isakovic, A.; Kravic-Stevovic, T.; Mircic, A.; Trajkovic, V.; Bumbasirevic, V. Statin-mediated inhibition of cholesterol synthesis induces cytoprotective autophagy in human leukemic cells. Eur. J. Pharmacol. 2015, 765, 415–428. [Google Scholar] [CrossRef]
- Yang, P.-M.; Liu, Y.-L.; Lin, Y.-C.; Shun, C.-T.; Wu, M.-S.; Chen, C.-C. Inhibition of autophagy enhances anticancer effects of atorvastatin in digestive malignancies. Cancer Res. 2010, 70, 7699–7709. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Esparza, Y.C.; Wu, S.; Huang, L.; Buquet, C.; Shen, R.; Sanchez-Gonzalez, B.; García Latorre, E.A.; Boyer, O.; Varin, R.; Jimenez-Zamudio, L.A. Synergistic promoting effects of pentoxifylline and simvastatin on the apoptosis of triple-negative MDA-MB-231 breast cancer cells. Int. J. Oncol. 2018, 52, 1246–1254. [Google Scholar] [CrossRef] [PubMed]
- Asakura, K.; Izumi, Y.; Yamamoto, M.; Yamauchi, Y.; Kawai, K.; Serizawa, A.; Mizushima, T.; Ohmura, M.; Kawamura, M.; Wakui, M. The cytostatic effects of lovastatin on ACC-MESO-1 cells. J. Surg. Res. 2011, 170, e197–e209. [Google Scholar] [CrossRef] [PubMed]
- Wojtkowiak, J.W.; Sane, K.M.; Kleinman, M.; Sloane, B.F.; Reiners, J.J.; Mattingly, R.R. Aborted autophagy and nonapoptotic death induced by farnesyl transferase inhibitor and lovastatin. J. Pharmacol. Exp. Ther. 2011, 337, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.-B.; Zhang, J.-W.; Gao, J.-B.; Qi, Y.-W.; Gao, Y.; Xu, L.; Ma, Y.; Wei, Z.-Z. Atorvastatin induces autophagy in MDA-MB-231 breast cancer cells. Ultrastruct. Pathol. 2018, 42, 409–415. [Google Scholar] [CrossRef]
- Shi, Y.; Felley-Bosco, E.; Marti, T.M.; Stahel, R.A. Differential effects of lovastatin on cisplatin responses in normal human mesothelial cells versus cancer cells: Implication for therapy. PLoS ONE 2012, 7, e45354. [Google Scholar] [CrossRef]
- Hombach-Klonisch, S.; Mehrpour, M.; Shojaei, S.; Harlos, C.; Pitz, M.; Hamai, A.; Siemianowicz, K.; Likus, W.; Wiechec, E.; Toyota, B.D. Glioblastoma and chemoresistance to alkylating agents: Involvement of apoptosis, autophagy, and unfolded protein response. Pharmacol. Ther. 2018, 184, 13–41. [Google Scholar] [CrossRef]
- Shojaei, S.; Koleini, N.; Samiei, E.; Aghaei, M.; Cole, L.K.; Alizadeh, J.; Islam, M.I.; Vosoughi, A.; Albokashy, M.; Butterfield, Y. Simvastatin increases temozolomide-induced cell death by targeting the fusion of autophagosomes and lysosomes. FEBS J. 2020, 287, 1005–1034. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Cao, Y.; Cao, W.; Jia, Y.; Lu, N. The Application of Ferroptosis in Diseases. Pharmacol. Res. 2020, 159, 104919. [Google Scholar] [CrossRef] [PubMed]
- Basuli, D.; Tesfay, L.; Deng, Z.; Paul, B.; Yamamoto, Y.; Ning, G.; Xian, W.; McKeon, F.; Lynch, M.; Crum, C.P. Iron addiction: A novel therapeutic target in ovarian cancer. Oncogene 2017, 36, 4089–4099. [Google Scholar] [CrossRef] [PubMed]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Müller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kössl, J. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent. Sci. 2019, 6, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Angeli, J.P.F.; Conrad, M. Selenium and GPX4, a vital symbiosis. Free Radic. Biol. Med. 2018, 127, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Wallach, D.; Kang, T.-B.; Dillon, C.P.; Green, D.R. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science 2016, 352, aaf2154. [Google Scholar] [CrossRef]
- Wallach, D.; Kang, T.-B.; Kovalenko, A. Concepts of tissue injury and cell death in inflammation: A historical perspective. Nat. Rev. Immunol. 2014, 14, 51–59. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.-C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Aachoui, Y.; Sagulenko, V.; Miao, E.A.; Stacey, K.J. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr. Opin. Microbiol. 2013, 16, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Zhou, B.; Zhang, J.; Liu, X.; Chen, H.; Ai, Y.; Cheng, K.; Sun, R.; Zhou, D.; Han, J.; Wu, Q. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018, 28, 1171–1185. [Google Scholar] [CrossRef]
- Wu, L.-M.; Wu, S.-G.; Chen, F.; Wu, Q.; Wu, C.-M.; Kang, C.-M.; He, X.; Zhang, R.-Y.; Lu, Z.-F.; Li, X.-H. Atorvastatin inhibits pyroptosis through the lncRNA NEXN-AS1/NEXN pathway in human vascular endothelial cells. Atherosclerosis 2020, 293, 26–34. [Google Scholar] [CrossRef]
- Chen, A.O.; Chen, Z.; Zhou, Y.; Wu, Y.; Xia, Y.; Lu, D.; Fan, M.; Li, S.; Chen, J.; Sun, A. Rosuvastatin protects against coronary microembolization-induced cardiac injury via inhibiting NLRP3 inflammasome activation. Cell Death Dis. 2021, 12, 78. [Google Scholar] [CrossRef]
- Merlo, L.M.F.; Pepper, J.W.; Reid, B.J.; Maley, C.C. Cancer as an evolutionary and ecological process. Nat. Rev. Cancer 2006, 6, 924–935. [Google Scholar] [CrossRef]
- Mehibel, M.; Ortiz-Martinez, F.; Voelxen, N.; Boyers, A.; Chadwick, A.; Telfer, B.A.; Mueller-Klieser, W.; West, C.M.; Critchlow, S.E.; Williams, K.J. Statin-induced metabolic reprogramming in head and neck cancer: A biomarker for targeting monocarboxylate transporters. Sci. Rep. 2018, 8, 16804. [Google Scholar] [CrossRef]
- Jin, H.; He, Y.; Zhao, P.; Hu, Y.; Tao, J.; Chen, J.; Huang, Y. Targeting lipid metabolism to overcome EMT-associated drug resistance via integrin β3/FAK pathway and tumor-associated macrophage repolarization using legumain-activatable delivery. Theranostics 2019, 9, 265. [Google Scholar] [CrossRef]
- Galland, S.; Martin, P.; Fregni, G.; Letovanec, I.; Stamenkovic, I. Attenuation of the pro-inflammatory signature of lung cancer-derived mesenchymal stromal cells by statins. Cancer Lett. 2020, 484, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, A.R.; Trenton, N.J.; Debeb, B.G.; Larson, R.; Ruffell, B.; Chu, K.; Hittelman, W.; Diehl, M.; Reuben, J.M.; Ueno, N.T. Mesenchymal stem cells and macrophages interact through IL-6 to promote inflammatory breast cancer in pre-clinical models. Oncotarget 2016, 7, 82482. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Bi, E.; Lu, Y.; Su, P.; Huang, C.; Liu, L.; Wang, Q.; Yang, M.; Kalady, M.F.; Qian, J. Cholesterol induces CD8+ T cell exhaustion in the tumor microenvironment. Cell Metab. 2019, 30, 143–156. [Google Scholar] [CrossRef]
- McGregor, G.H.; Campbell, A.D.; Fey, S.K.; Tumanov, S.; Sumpton, D.; Blanco, G.R.; Mackay, G.; Nixon, C.; Vazquez, A.; Sansom, O.J. Targeting the Metabolic Response to Statin-Mediated Oxidative Stress Produces a Synergistic Antitumor ResponseMEK Inhibition Synergizes with Statins for Cancer Treatment. Cancer Res. 2020, 80, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Taccioli, C.; Sorrentino, G.; Zannini, A.; Caroli, J.; Beneventano, D.; Anderlucci, L.; Lolli, M.; Bicciato, S.; Del Sal, G. MDP, a database linking drug response data to genomic information, identifies dasatinib and statins as a combinatorial strategy to inhibit YAP/TAZ in cancer cells. Oncotarget 2015, 6, 38854. [Google Scholar] [CrossRef]
- Iannelli, F.; Roca, M.S.; Lombardi, R.; Ciardiello, C.; Grumetti, L.; De Rienzo, S.; Moccia, T.; Vitagliano, C.; Sorice, A.; Costantini, S. Synergistic antitumor interaction of valproic acid and simvastatin sensitizes prostate cancer to docetaxel by targeting CSCs compartment via YAP inhibition. J. Exp. Clin. Cancer Res. 2020, 39, 213. [Google Scholar] [CrossRef]
- Di Bello, E.; Zwergel, C.; Mai, A.; Valente, S. The innovative potential of statins in cancer: New targets for new therapies. Front. Chem. 2020, 8, 516. [Google Scholar] [CrossRef]
- Matusewicz, L.; Meissner, J.; Toporkiewicz, M.; Sikorski, A.F. The effect of statins on cancer cells. Tumor Biol. 2015, 36, 4889–4904. [Google Scholar] [CrossRef]
- Menter, D.G.; Ramsauer, V.P.; Harirforoosh, S.; Chakraborty, K.; Yang, P.; Hsi, L.; Newman, R.A.; Krishnan, K. Differential effects of pravastatin and simvastatin on the growth of tumor cells from different organ sites. PLoS ONE 2011, 6, e28813. [Google Scholar] [CrossRef]
- Bai, F.; Yu, Z.; Gao, X.; Gong, J.; Fan, L.; Liu, F. Simvastatin induces breast cancer cell death through oxidative stress up-regulating miR-140-5p. Aging 2019, 11, 3198. [Google Scholar] [CrossRef]
- Graaf, M.R.; Richel, D.J.; van Noorden, C.J.F.; Guchelaar, H.-J. Effects of statins and farnesyltransferase inhibitors on the development and progression of cancer. Cancer Treat. Rev. 2004, 30, 609–641. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, S.; Northrop, W.; Ellison, G.; Sabapathy, T.; Melton, P.E.; Newsholme, P.; Mamotte, C.D.S. Statins do not directly inhibit the activity of major epigenetic modifying enzymes. Cancers 2019, 11, 516. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, S.; Hayashi, H.; Kinoshita, K.; Abe, M.; Kuroki, H.; Tokunaga, R.; Tomiyasu, S.; Tanaka, H.; Sugita, H.; Arita, T. Statins inhibit tumor progression via an enhancer of zeste homolog 2-mediated epigenetic alteration in colorectal cancer. Int. J. Cancer 2014, 135, 2528–2536. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Gan, Y.-H. Inhibiting HDAC1 enhances the anti-cancer effects of statins through downregulation of GGTase-Iβ expression. Int. J. Mol. Sci. 2017, 18, 1010. [Google Scholar] [CrossRef]
- Lin, Z.; Zhang, Z.; Jiang, X.; Kou, X.; Bao, Y.; Liu, H.; Sun, F.; Ling, S.; Qin, N.; Jiang, L. Mevastatin blockade of autolysosome maturation stimulates LBH589-induced cell death in triple-negative breast cancer cells. Oncotarget 2017, 8, 17833. [Google Scholar] [CrossRef]
- Nagayama, D.; Saiki, A.; Shirai, K. The Anti-Cancer Effect of Pitavastatin May Be a Drug-Specific Effect: Subgroup Analysis of the TOHO-LIP Study. Vasc. Health Risk Manag. 2021, 17, 169–173. [Google Scholar] [CrossRef]
- Sheikholeslami, K.; Ali Sher, A.; Lockman, S.; Kroft, D.; Ganjibakhsh, M.; Nejati-Koshki, K.; Shojaei, S.; Ghavami, S.; Rastegar, M. Simvastatin induces apoptosis in medulloblastoma brain tumor cells via mevalonate cascade prenylation substrates. Cancers 2019, 11, 994. [Google Scholar] [CrossRef]
- Longo, J.; van Leeuwen, J.E.; Elbaz, M.; Branchard, E.; Penn, L.Z. Statins as Anticancer Agents in the Era of Precision MedicineStatins as Precision Anticancer Therapeutics. Clin. Cancer Res. 2020, 26, 5791–5800. [Google Scholar] [CrossRef]
- Beckwitt, C.H.; Brufsky, A.; Oltvai, Z.N.; Wells, A. Statin drugs to reduce breast cancer recurrence and mortality. Breast Cancer Res. 2018, 20, 144. [Google Scholar] [CrossRef]
- Grabarek, B.O.; Boroń, D.; Morawiec, E.; Michalski, P.; Palazzo-Michalska, V.; Pach, Ł.; Dziuk, B.; Świder, M.; Zmarzły, N. Crosstalk between statins and cancer prevention and therapy: An update. Pharmaceuticals 2021, 14, 1220. [Google Scholar] [CrossRef]
- Lau, C.P.Y.; Fung, C.S.H.; Wong, K.C.; Wang, Y.; Huang, L.; Tsui, S.K.W.; Lee, O.K.; Kumta, S.M. Simvastatin possesses antitumor and differentiation-promoting properties that affect stromal cells in giant cell tumor of bone. J. Orthop. Res. 2020, 38, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Borrego, M.C.; Fuentes-Fayos, A.C.; Herrera-Martínez, A.D.; Venegas-Moreno, E.; Fernando, L.; Fanciulli, A.; Moreno-Moreno, P.; Alhambra-Expósito, M.R.; Barrera-Martín, A.; Dios, E. Statins directly regulate pituitary cell function and exert antitumor effects in pituitary tumors. Neuroendocrinology 2020, 110, 1028–1041. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, W.; Ning, J.; Wang, J.; Lang, Y.; Jin, X.; Zhu, K.; Wang, X.; Li, X.; Yang, F. Simvastatin suppresses proliferation and migration in non-small cell lung cancer via pyroptosis. Int. J. Biol. Sci. 2018, 14, 406. [Google Scholar] [CrossRef]
- Duarte, J.A.; de Barros, A.L.B.; Leite, E.A. The potential use of simvastatin for cancer treatment: A review. Biomed. Pharmacother. 2021, 141, 111858. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, Y.; Sekine, Y.; Kato, H.; Furuya, Y.; Koike, H.; Suzuki, K. Simvastatin up-regulates annexin A10 that can inhibit the proliferation, migration, and invasion in androgen-independent human prostate cancer cells. Prostate 2017, 77, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Kamel, W.A.; Sugihara, E.; Nobusue, H.; Yamaguchi-Iwai, S.; Onishi, N.; Maki, K.; Fukuchi, Y.; Matsuo, K.; Muto, A.; Saya, H. Simvastatin-induced apoptosis in osteosarcoma cells: A key role of RhoA-AMPK/p38 MAPK signaling in antitumor activity. Mol. Cancer Ther. 2017, 16, 182–192. [Google Scholar] [CrossRef]
- Rennó, A.L.; Alves-Júnior, M.J.; Rocha, R.M.; De Souza, P.C.; de Souza, V.B.; Jampietro, J.; Vassallo, J.; Hyslop, S.; Anhê, G.F.; de Moraes Schenka, N.G. Decreased Expression of Stem Cell Markers by Simvastatin in 7, 12-dimethylbenz (a) anthracene (DMBA)–induced Breast Cancer. Toxicol. Pathol. 2015, 43, 400–410. [Google Scholar] [CrossRef]
- Karimi, B.; Ashrafi, M.; Shomali, T.; Yektaseresht, A. Therapeutic effect of simvastatin on DMBA-induced breast cancer in mice. Fundam. Clin. Pharmacol. 2019, 33, 84–93. [Google Scholar] [CrossRef]
- Jang, H.J.; Hong, E.M.; Park, S.W.; Byun, H.W.; Koh, D.H.; Choi, M.H.; Kae, S.H.; Lee, J. Statin induces apoptosis of human colon cancer cells and downregulation of insulin-like growth factor 1 receptor via proapoptotic ERK activation. Oncol. Lett. 2016, 12, 250–256. [Google Scholar] [CrossRef]
- Zhao, J.; Xu, C.; Yao, J.; Yu, C.; Liao, L.; Dong, J. Statins and thyroid carcinoma: A meta-analysis. Cell. Physiol. Biochem. 2018, 47, 1422–1431. [Google Scholar] [CrossRef]
- Liu, H.; Wang, Z.; Li, Y.; Li, W.; Chen, Y. Simvastatin prevents proliferation and bone metastases of lung adenocarcinoma in vitro and in vivo. Neoplasma 2013, 60, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, M.; Amiri, S.; Pecic, S.; Machaj, F.; Rosik, J.; Łos, M.J.; Alizadeh, J.; Mahdian, R.; da Silva Rosa, S.C.; Schaafsma, D. Pleiotropic effects of statins: A focus on cancer. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165968. [Google Scholar] [CrossRef] [PubMed]
- Barbalata, C.I.; Tefas, L.R.; Achim, M.; Tomuta, I.; Porfire, A.S. Statins in risk-reduction and treatment of cancer. World J. Clin. Oncol. 2020, 11, 573. [Google Scholar] [CrossRef] [PubMed]
- Poynter, J.N.; Gruber, S.B.; Higgins, P.D.R.; Almog, R.; Bonner, J.D.; Rennert, H.S.; Low, M.; Greenson, J.K.; Rennert, G. Statins and the risk of colorectal cancer. N. Engl. J. Med. 2005, 352, 2184–2192. [Google Scholar] [CrossRef] [PubMed]
- Farwell, W.R.; Scranton, R.E.; Lawler, E.V.; Lew, R.A.; Brophy, M.T.; Fiore, L.D.; Gaziano, J.M. The association between statins and cancer incidence in a veterans population. J. Natl. Cancer Inst. 2008, 100, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Mei, Z.; Liang, M.; Li, L.; Zhang, Y.; Wang, Q.; Yang, W. Effects of statins on cancer mortality and progression: A systematic review and meta-analysis of 95 cohorts including 1,111,407 individuals. Int. J. Cancer 2017, 140, 1068–1081. [Google Scholar] [CrossRef]
- Farooqi, M.A.M.; Malhotra, N.; Mukherjee, S.D.; Sanger, S.; Dhesy-Thind, S.K.; Ellis, P.; Leong, D.P. Statin therapy in the treatment of active cancer: A systematic review and meta-analysis of randomized controlled trials. PLoS ONE 2018, 13, e0209486. [Google Scholar] [CrossRef]
- Ahmadi, Y.; Karimian, R.; Panahi, Y. Effects of statins on the chemoresistance—The antagonistic drug-drug interactions versus the anti-cancer effects. Biomed. Pharmacother. 2018, 108, 1856–1865. [Google Scholar] [CrossRef]
- Sławińska, A.; Kandefer-Szerszeń, M. The anticancer properties of statins. Adv. Hyg. Exp. Med. 2008, 62, 393–404. [Google Scholar]
- Alizadeh, J.; Zeki, A.A.; Mirzaei, N.; Tewary, S.; Rezaei Moghadam, A.; Glogowska, A.; Nagakannan, P.; Eftekharpour, E.; Wiechec, E.; Gordon, J.W. Mevalonate cascade inhibition by simvastatin induces the intrinsic apoptosis pathway via depletion of isoprenoids in tumor cells. Sci. Rep. 2017, 7, 44841. [Google Scholar] [CrossRef]
- Zhou, C.; Zhong, X.; Gao, P.; Wu, Z.; Shi, J.; Guo, Z.; Wang, Z.; Song, Y. Statin use and its potential therapeutic role in esophageal cancer: A systematic review and meta-analysis. Cancer Manag. Res. 2019, 11, 5655. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.U.; Chang, J.; Jeon, C.Y.; Pandol, S.J.; Huang, B.; Ngor, E.W.; Difronzo, A.L.; Cooper, R.M. Impact of statin use on survival in patients undergoing resection for early-stage pancreatic cancer. Am. J. Gastroenterol. 2015, 110, 1233. [Google Scholar] [CrossRef] [PubMed]
- Friis, S.; Poulsen, A.H.; Johnsen, S.P.; McLaughlin, J.K.; Fryzek, J.P.; Dalton, S.O.; Sørensen, H.T.; Olsen, J.H. Cancer risk among statin users: A population-based cohort study. Int. J. Cancer 2005, 114, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Kuoppala, J.; Lamminpää, A.; Pukkala, E. Statins and cancer: A systematic review and meta-analysis. Eur. J. Cancer 2008, 44, 2122–2132. [Google Scholar] [CrossRef]
- Taylor, M.L.; Wells, B.J.; Smolak, M.J. Statins and cancer: A meta-analysis of case-control studies. Eur. J. Cancer Prev. 2008, 17, 259–268. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Fowkes, F.G.R.; Belch, J.F.F.; Ogawa, H.; Warlow, C.P.; Meade, T.W. Effect of daily aspirin on long-term risk of death due to cancer: Analysis of individual patient data from randomised trials. Lancet 2011, 377, 31–41. [Google Scholar] [CrossRef]
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20 536 high-risk individuals: A randomised placebocontrolled trial. Lancet 2002, 360, 7–22. [Google Scholar] [CrossRef]
- Gorin, A.; Gabitova, L.; Astsaturov, I. Regulation of cholesterol biosynthesis and cancer signaling. Curr. Opin. Pharmacol. 2012, 12, 710–716. [Google Scholar] [CrossRef]
- Heart Protection Study Collaborative Group. Effects on 11-year mortality and morbidity of lowering LDL cholesterol with simvastatin for about 5 years in 20 536 high-risk individuals: A randomised controlled trial. Lancet 2011, 378, 2013–2020. [Google Scholar] [CrossRef]
- Kim, M.K.; Myung, S.K.; Tran, B.T.; Park, B. Statins and risk of cancer: A meta-analysis of randomized, double-blind, placebo-controlled trials. Indian J. Cancer 2017, 54, 470–477. [Google Scholar]
- Joharatnam-Hogan, N.; Alexandre, L.; Yarmolinsky, J.; Lake, B.; Capps, N.; Martin, R.M.; Ring, A.; Cafferty, F.; Langley, R.E. Statins as potential chemoprevention or therapeutic agents in cancer: A model for evaluating repurposed drugs. Curr. Oncol. Rep. 2021, 23, 29. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Denmeade, S.R. Fatty acid synthesis in prostate cancer: Vulnerability or epiphenomenon? Cancer Res. 2021, 81, 4385. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, S.; Menju, T.; Takahashi, K.; Miyata, R.; Chen-Yoshikawa, T.F.; Sonobe, M.; Yoshizawa, A.; Sabe, H.; Sato, T.; Date, H. Statins may have double-edged effects in patients with lung adenocarcinoma after lung resection. Cancer Manag. Res. 2019, 11, 3419–3432. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P. A review on pharmacodynamics of Ashtamangal ghrita and its uses in mental and physical growth in children. J. Pharmacogn. Phytochem. 2019, 8, 3809–3812. [Google Scholar]
- Langan, S.M.; Schmidt, S.A.J.; Wing, K.; Ehrenstein, V.; Nicholls, S.G.; Filion, K.B.; Klungel, O.; Petersen, I.; Sorensen, H.T.; Dixon, W.G. The reporting of studies conducted using observational routinely collected health data statement for pharmacoepidemiology (RECORD-PE). BMJ 2018, 363, k3532. [Google Scholar] [CrossRef]
- Coyle, C.; Cafferty, F.H.; Rowley, S.; MacKenzie, M.; Berkman, L.; Gupta, S.; Pramesh, C.S.; Gilbert, D.; Kynaston, H.; Cameron, D. ADD-ASPIRIN: A phase III, double-blind, placebo controlled, randomised trial assessing the effects of aspirin on disease recurrence and survival after primary therapy in common non-metastatic solid tumours. Contemp. Clin. Trials 2016, 51, 56–64. [Google Scholar] [CrossRef]
- Jankowski, J.A.Z.; De Caestecker, J.; Love, S.B.; Reilly, G.; Watson, P.; Sanders, S.; Ang, Y.; Morris, D.; Bhandari, P.; Brooks, C. Esomeprazole and aspirin in Barrett’s oesophagus (AspECT): A randomised factorial trial. Lancet 2018, 392, 400–408. [Google Scholar] [CrossRef]
- Soni, P.D.; Hartman, H.E.; Dess, R.T.; Abugharib, A.; Allen, S.G.; Feng, F.Y.; Zietman, A.L.; Jagsi, R.; Schipper, M.J.; Spratt, D.E. Comparison of population-based observational studies with randomized trials in oncology. J. Clin. Oncol. 2019, 37, 1209. [Google Scholar] [CrossRef]
- Dickerman, B.A.; García-Albéniz, X.; Logan, R.W.; Denaxas, S.; Hernán, M.A. Avoidable flaws in observational analyses: An application to statins and cancer. Nat. Med. 2019, 25, 1601–1606. [Google Scholar] [CrossRef]
- Orho-Melander, M.; Hindy, G.; Borgquist, S.; Schulz, C.-A.; Manjer, J.; Melander, O.; Stocks, T. Blood lipid genetic scores, the HMGCR gene and cancer risk: A Mendelian randomization study. Int. J. Epidemiol. 2018, 47, 495–505. [Google Scholar] [CrossRef]
- Singh, R.S.; Chaudhary, D.K.; Mohan, A.; Kumar, P.; Chaturvedi, C.P.; Ecelbarger, C.M.; Godbole, M.M.; Tiwari, S. Greater efficacy of atorvastatin versus a non-statin lipid-lowering agent against renal injury: Potential role as a histone deacetylase inhibitor. Sci. Rep. 2016, 6, 38034. [Google Scholar] [CrossRef] [PubMed]
- Herrett, E.; Gallagher, A.M.; Bhaskaran, K.; Forbes, H.; Mathur, R.; Van Staa, T.; Smeeth, L. Data resource profile: Clinical practice research datalink (CPRD). Int. J. Epidemiol. 2015, 44, 827–836. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaky, M.Y.; Fan, C.; Zhang, H.; Sun, X.-F. Unraveling the Anticancer Potential of Statins: Mechanisms and Clinical Significance. Cancers 2023, 15, 4787. https://doi.org/10.3390/cancers15194787
Zaky MY, Fan C, Zhang H, Sun X-F. Unraveling the Anticancer Potential of Statins: Mechanisms and Clinical Significance. Cancers. 2023; 15(19):4787. https://doi.org/10.3390/cancers15194787
Chicago/Turabian StyleZaky, Mohamed Y., Chuanwen Fan, Huan Zhang, and Xiao-Feng Sun. 2023. "Unraveling the Anticancer Potential of Statins: Mechanisms and Clinical Significance" Cancers 15, no. 19: 4787. https://doi.org/10.3390/cancers15194787
APA StyleZaky, M. Y., Fan, C., Zhang, H., & Sun, X.-F. (2023). Unraveling the Anticancer Potential of Statins: Mechanisms and Clinical Significance. Cancers, 15(19), 4787. https://doi.org/10.3390/cancers15194787