

The Impact of ETV6-NTRK3 Oncogenic Gene Fusions on Molecular and Signaling Pathway Alterations

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Line Generation
2.3. Biotin Proximity Assay
2.4. Mass Spectrometry Analysis
2.5. Heatmap
2.6. Dotplot and Correlation Plot
2.7. GO Term Analysis
2.8. Luciferase Assays
2.9. Barcoded Mass cytometry
2.10. Blots
2.11. Morphology Microscopy
2.12. Fluorescence Microscopy
3. Results
3.1. EN Variants Are Present in Different Human Cancers and Vary in Their Ability to Transform Cells
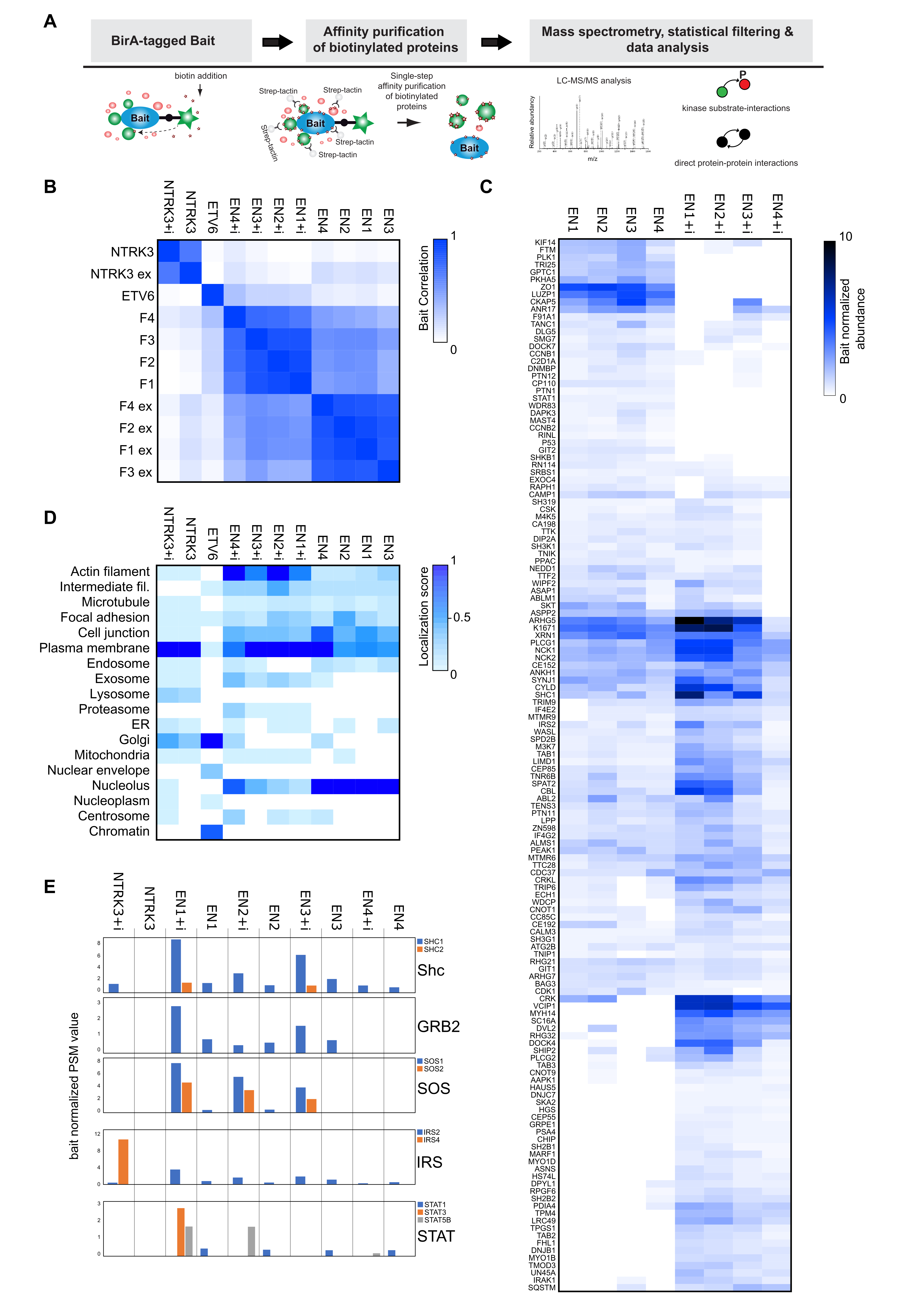
3.2. Interactome Analysis Reveals Newly Acquired Molecular Interactions of EN Oncofusions and Identifies Signaling Pathway Targeted by EN Variants
3.3. Analysis of the EN Interactome Map Reveals Connections to Multiple Signaling Pathways
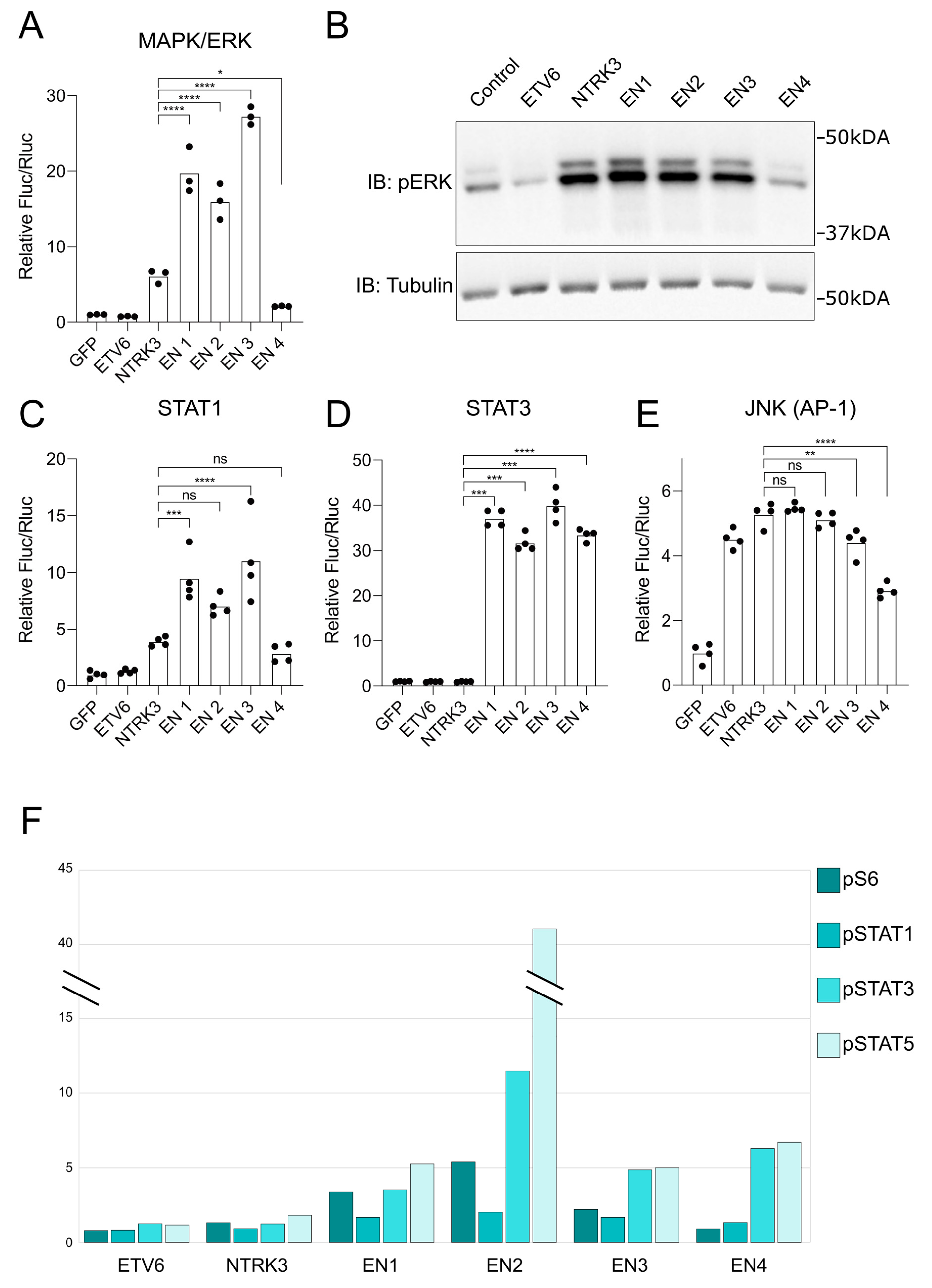
3.4. EN Variants Aberrantly Activate Several Key Signaling Pathways
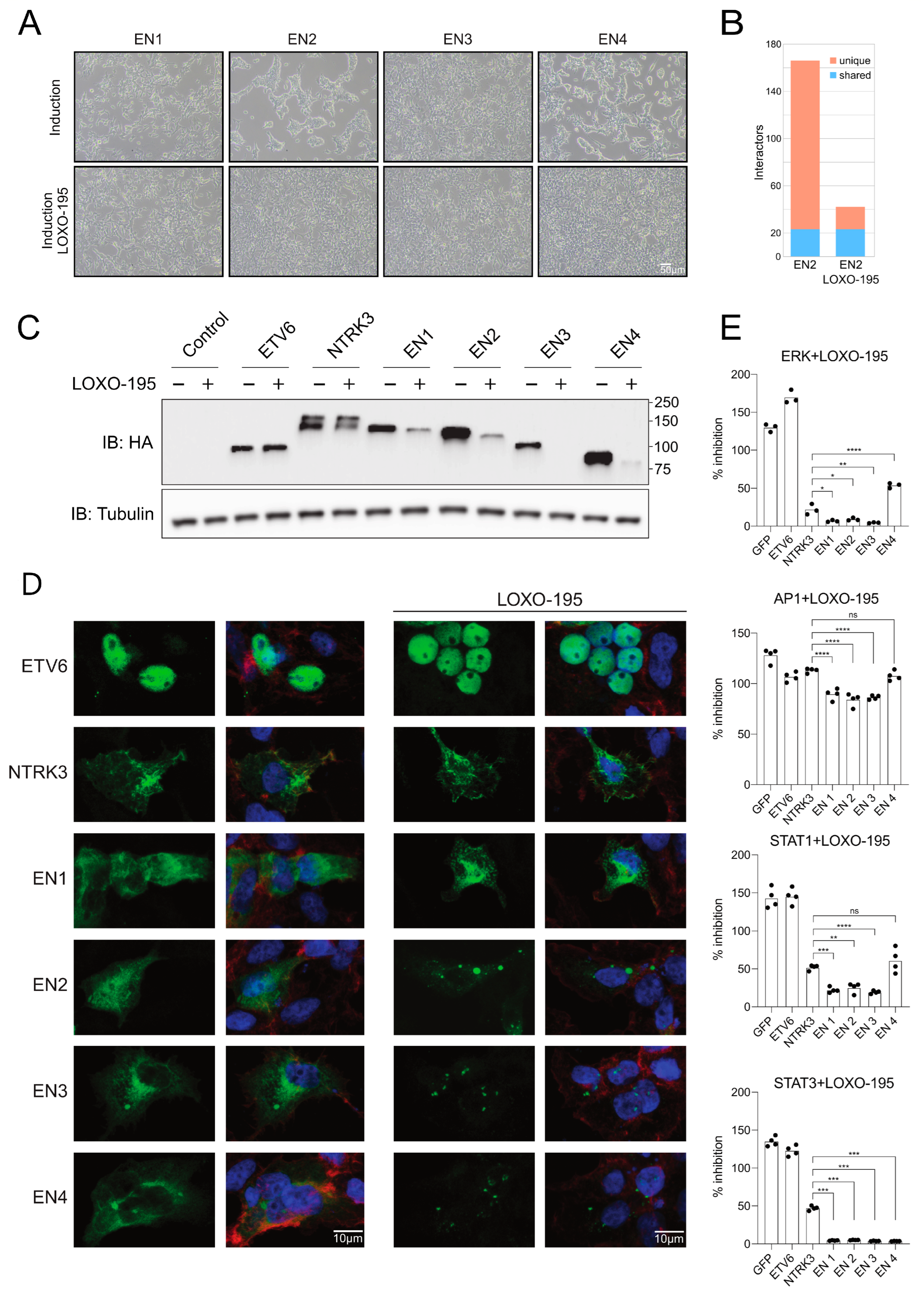
3.5. Selitrectinib Is a Potent Inhibitor of EN-Induced Transformation and Aberrant Pathway Activation
3.6. A Framework for EN2 Activation of Key Signaling Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A Census of Human Cancer Genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Mitelman, F.; Johansson, B.; Mertens, F. The Impact of Translocations and Gene Fusions on Cancer Causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Salokas, K.; Weldatsadik, R.G.; Varjosalo, M. Human Transcription Factor and Protein Kinase Gene Fusions in Human Cancer. Sci. Rep. 2020, 10, 14169. [Google Scholar] [CrossRef]
- Berger, M.F.; Lawrence, M.S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A.Y.; Sboner, A.; Esgueva, R.; Pflueger, D.; Sougnez, C.; et al. The Genomic Complexity of Primary Human Prostate Cancer. Nature 2011, 470, 214–220. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive Genomic Characterization of Squamous Cell Lung Cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the Hallmarks of Lung Adenocarcinoma with Massively Parallel Sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef]
- Stephens, P.J.; McBride, D.J.; Lin, M.L.; Varela, I.; Pleasance, E.D.; Simpson, J.T.; Stebbings, L.A.; Leroy, C.; Edkins, S.; Mudie, L.J.; et al. Complex Landscapes of Somatic Rearrangement in Human Breast Cancer Genomes. Nature 2009, 462, 1005–1010. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.; Rajesh, Y.; Mitra, P.; Mandal, M. ETV6 Gene Aberrations in Non-Haematological Malignancies: A Review Highlighting ETV6 Associated Fusion Genes in Solid Tumors. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188389. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.; Cotzia, P.; Hyman, D.M.; Drilon, A.; Tap, W.D.; Zhang, L.; Hechtman, J.F.; Frosina, D.; Jungbluth, A.A.; Murali, R.; et al. NTRK Fusions Define a Novel Uterine Sarcoma Subtype With Features of Fibrosarcoma. Am. J. Surg. Pathol. 2018, 42, 791–798. [Google Scholar] [CrossRef]
- Costigan, D.C.; Nucci, M.R.; Dickson, B.C.; Chang, M.C.; Song, S.; Sholl, L.M.; Hornick, J.L.; Fletcher, C.D.M.; Kolin, D.L. NTRK -Rearranged Uterine Sarcomas: Clinicopathologic Features of 15 Cases, Literature Review, and Risk Stratification. Am. J. Surg. Pathol. 2022, 46, 1415–1429. [Google Scholar] [CrossRef]
- Dang, X.; Xiang, T.; Zhao, C.; Tang, H.; Cui, P. EML4-NTRK3 Fusion Cervical Sarcoma: A Case Report and Literature Review. Front. Med. 2022, 9, 832376. [Google Scholar] [CrossRef]
- Devereaux, K.A.; Weiel, J.J.; Mills, A.M.; Kunder, C.A.; Longacre, T.A. Neurofibrosarcoma Revisited: An Institutional Case Series of Uterine Sarcomas Harboring Kinase-Related Fusions With Report of a Novel FGFR1-TACC1 Fusion. Am. J. Surg. Pathol. 2021, 45, 638–652. [Google Scholar] [CrossRef]
- Hodgson, A.; Pun, C.; Djordjevic, B.; Turashvili, G. NTRK-Rearranged Cervical Sarcoma: Expanding the Clinicopathologic Spectrum. Int. J. Gynecol. Pathol. 2021, 40, 73–77. [Google Scholar] [CrossRef]
- Iyama, K.; Matsuse, M.; Mitsutake, N.; Rogounovitch, T.; Saenko, V.; Suzuki, K.; Ashizawa, M.; Ookouchi, C.; Suzuki, S.; Mizunuma, H.; et al. Identification of Three Novel Fusion Oncogenes, SQSTM1/NTRK3, AFAP1L2/RET, and PPFIBP2/RET, in Thyroid Cancers of Young Patients in Fukushima. Thyroid 2017, 27, 811–818. [Google Scholar] [CrossRef]
- Lanman, T.; Hayden Gephart, M.; Bui, N.; Toland, A.; Nagpal, S. Isolated Leptomeningeal Progression in a Patient with NTRK Fusion+ Uterine Sarcoma: A Case Report. Case Rep. Oncol. 2021, 14, 1841–1846. [Google Scholar] [CrossRef]
- Manea, C.A.; Badiu, D.C.; Ploscaru, I.C.; Zgura, A.; Bacinschi, X.; Smarandache, C.G.; Serban, D.; Popescu, C.G.; Grigorean, V.T.; Botnarciuc, V. A Review of NTRK Fusions in Cancer. Ann. Med. Surg. 2022, 79, 103893. [Google Scholar] [CrossRef] [PubMed]
- Nilforoushan, N.; Wethington, S.L.; Nonogaki, H.; Gross, J.; Vang, R.; Xing, D. NTRK-Fusion Sarcoma of the Uterine Cervix: Report of 2 Cases With Comparative Clinicopathologic Features. Int. J. Gynecol. Pathol. 2022, 41, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Rabban, J.T.; Devine, W.P.; Sangoi, A.R.; Poder, L.; Alvarez, E.; Davis, J.L.; Rudzinski, E.; Garg, K.; Bean, G.R. NTRK Fusion Cervical Sarcoma: A Report of Three Cases, Emphasising Morphological and Immunohistochemical Distinction from Other Uterine Sarcomas, Including Adenosarcoma. Histopathology 2020, 77, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Busam, K.J.; Benayed, R.; Cimera, R.; Wang, J.; Denley, R.; Rao, M.; Aryeequaye, R.; Mullaney, K.; Cao, L.; et al. Identification of NTRK3 Fusions in Childhood Melanocytic Neoplasms. J. Mol. Diagn. 2017, 19, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, F.; Nakatani, F.; Asano, N.; Wakai, S.; Sekimizu, M.; Mitani, S.; Kubo, T.; Kawai, A.; Ichikawa, H.; Yoshida, A. Novel NTRK3 Fusions in Fibrosarcomas of Adults. Am. J. Surg. Pathol. 2019, 43, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Tognon, C.E.; Godinho, F.J.; Yasaitis, L.; Hock, H.; Herschkowitz, J.I.; Lannon, C.L.; Cho, E.; Kim, S.J.; Bronson, R.T.; et al. ETV6-NTRK3 Fusion Oncogene Initiates Breast Cancer from Committed Mammary Progenitors via Activation of AP1 Complex. Cancer Cell 2007, 12, 542–558. [Google Scholar] [CrossRef] [PubMed]
- Tognon, C.; Garnett, M.; Kenward, E.; Kay, R.; Morrison, K.; Sorensen, P.H. The Chimeric Protein Tyrosine Kinase ETV6-NTRK3 Requires Both Ras-Erk1/2 and PI3-Kinase-Akt Signaling for Fibroblast Transformation. Cancer Res. 2001, 61, 8909–8916. [Google Scholar]
- Tognon, C.E.; Martin, M.J.; Moradian, A.; Trigo, G.; Rotblat, B.; Cheng, S.W.; Pollard, M.; Uy, E.; Chow, C.; Carboni, J.M.; et al. A Tripartite Complex Composed of ETV6-NTRK3, IRS1 and IGF1R Is Required for ETV6-NTRK3-Mediated Membrane Localization and Transformation. Oncogene 2012, 31, 1334–1340. [Google Scholar] [CrossRef]
- Eguchi, M.; Eguchi-Ishimae, M.; Tojo, A.; Morishita, K.; Suzuki, K.; Sato, Y.; Kudoh, S.; Tanaka, K.; Setoyama, M.; Nagamura, F.; et al. Fusion of ETV6 to Neurotrophin-3 Receptor TRKC in Acute Myeloid Leukemia with t(12;15)(P13;Q25). Blood 1999, 93, 1355–1363. [Google Scholar] [CrossRef]
- Leeman-Neill, R.J.; Kelly, L.M.; Liu, P.; Brenner, A.V.; Little, M.P.; Bogdanova, T.I.; Evdokimova, V.N.; Hatch, M.; Zurnadzy, L.Y.; Nikiforova, M.N.; et al. ETV6-NTRK3 Is a Common Chromosomal Rearrangement in Radiation-Associated Thyroid Cancer. Cancer 2014, 120, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.R.; Nucifora, G. The Leukemia-Associated Gene TEL Encodes a Transcription Repressor Which Associates with SMRT and MSin3A. Biochem. Biophys. Res. Commun. 1999, 264, 871–877. [Google Scholar] [CrossRef]
- Guidez, F.; Petrie, K.; Ford, A.M.; Lu, H.; Bennett, C.A.; MacGregor, A.; Hannemann, J.; Ito, Y.; Ghysdael, J.; Greaves, M.; et al. Recruitment of the Nuclear Receptor Corepressor N-CoR by the TEL Moiety of the Childhood Leukemia-Associated TEL-AML1 Oncoprotein. Blood 2000, 96, 2557–2561. [Google Scholar] [CrossRef]
- Guiton, M.; Gunn-Moore, F.J.; Glass, D.J.; Geis, D.R.; Yancopoulos, G.D.; Tavare, J.M. Naturally Occurring Tyrosine Kinase Inserts Block High Affinity Binding of Phospholipase C Gamma and Shc to TrkC and Neurotrophin-3 Signaling. J. Biol. Chem. 1995, 270, 20384–20390. [Google Scholar] [CrossRef]
- Wai, D.H.; Knezevich, S.R.; Lucas, T.; Jansen, B.; Kay, R.J.; Sorensen, P.H. The ETV6-NTRK3 Gene Fusion Encodes a Chimeric Protein Tyrosine Kinase That Transforms NIH3T3 Cells. Oncogene 2000, 19, 906–915. [Google Scholar] [CrossRef]
- Morrison, K.B.; Tognon, C.E.; Garnett, M.J.; Deal, C.; Sorensen, P.H. ETV6-NTRK3 Transformation Requires Insulin-like Growth Factor 1 Receptor Signaling and Is Associated with Constitutive IRS-1 Tyrosine Phosphorylation. Oncogene 2002, 21, 5684–5695. [Google Scholar] [CrossRef] [PubMed]
- Hanke, S.; Mann, M. The Phosphotyrosine Interactome of the Insulin Receptor Family and Its Substrates IRS-1 and IRS-2. Mol. Cell. Proteomics. 2009, 8, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.G., Jr.; Backer, J.M.; Sun, X.J.; Shoelson, S.; Hu, P.; Schlessinger, J.; Yoakim, M.; Schaffhausen, B.; White, M.F. IRS-1 Activates Phosphatidylinositol 3′-Kinase by Associating with Src Homology 2 Domains of P85. Proc. Natl. Acad. Sci. USA 1992, 89, 10350–10354. [Google Scholar] [CrossRef]
- Myers, M.G., Jr.; Wang, L.M.; Sun, X.J.; Zhang, Y.; Yenush, L.; Schlessinger, J.; Pierce, J.H.; White, M.F. Role of IRS-1-GRB-2 Complexes in Insulin Signaling. Mol. Cell Biol. 1994, 14, 3577–3587. [Google Scholar] [CrossRef]
- Lamballe, F.; Tapley, P.; Barbacid, M. TrkC Encodes Multiple Neurotrophin-3 Receptors with Distinct Biological Properties and Substrate Specificities. EMBO J. 1993, 12, 3083–3094. [Google Scholar] [CrossRef] [PubMed]
- Lamballe, F.; Klein, R.; Barbacid, M. The Trk Family of Oncogenes and Neurotrophin Receptors. Princess Takamatsu Symp. 1991, 22, 153–170. [Google Scholar]
- Liu, Q.; Schwaller, J.; Kutok, J.; Cain, D.; Aster, J.C.; Williams, I.R.; Gilliland, D.G. Signal Transduction and Transforming Properties of the TEL-TRKC Fusions Associated with t(12;15)(P13;Q25) in Congenital Fibrosarcoma and Acute Myelogenous Leukemia. EMBO J. 2000, 19, 1827–1838. [Google Scholar] [CrossRef]
- Dubus, P.; Coindre, J.M.; Groppi, A.; Jouan, H.; Ferrer, J.; Cohen, C.; Rivel, J.; Copin, M.C.; Leroy, J.P.; de Muret, A.; et al. The Detection of Tel-TrkC Chimeric Transcripts Is More Specific than TrkC Immunoreactivity for the Diagnosis of Congenital Fibrosarcoma. J. Pathol. 2001, 193, 88–94. [Google Scholar] [CrossRef]
- Henno, S.; Loeuillet, L.; Henry, C.; D’Herve, D.; Azzis, O.; Ferrer, J.; Poulain, P.; Babut, J.M.; Merlio, J.P.; Jouan, H.; et al. Cellular Mesoblastic Nephroma: Morphologic, Cytogenetic and Molecular Links with Congenital Fibrosarcoma. Pathol. Res. Pract. 2003, 199, 35–40. [Google Scholar] [CrossRef]
- Choi, H.; Larsen, B.; Lin, Z.-Y.; Breitkreutz, A.; Mellacheruvu, D.; Fermin, D.; Qin, Z.S.; Tyers, M.; Gingras, A.-C.; Nesvizhskii, A.I. SAINT: Probabilistic Scoring of Affinity Purification-Mass Spectrometry Data. Nat. Methods 2011, 8, 70–73. [Google Scholar] [CrossRef]
- Mellacheruvu, D.; Wright, Z.; Couzens, A.L.; Lambert, J.-P.; St-Denis, N.A.; Li, T.; Miteva, Y.V.; Hauri, S.; Sardiu, M.E.; Low, T.Y.; et al. The CRAPome: A Contaminant Repository for Affinity Purification-Mass Spectrometry Data. Nat. Methods 2013, 10, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-Enabled Heat Mapping for All. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef]
- Knight, J.D.R.; Choi, H.; Gupta, G.D.; Pelletier, L.; Raught, B.; Nesvizhskii, A.I.; Gingras, A.-C. ProHits-Viz: A Suite of Web Tools for Visualizing Interaction Proteomics Data. Nat. Methods 2017, 14, 645–646. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A Web Server for Functional Enrichment Analysis and Functional Annotation of Gene Lists (2021 Update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating Viruses and Cellular Organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The Reactome Pathway Knowledgebase 2022. Nucleic Acids Res. 2022, 50, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Tognon, C.; Knezevich, S.R.; Huntsman, D.; Roskelley, C.D.; Melnyk, N.; Mathers, J.A.; Becker, L.; Carneiro, F.; MacPherson, N.; Horsman, D.; et al. Expression of the ETV6-NTRK3 Gene Fusion as a Primary Event in Human Secretory Breast Carcinoma. Cancer Cell 2002, 2, 367–376. [Google Scholar] [CrossRef]
- Skalova, A.; Vanecek, T.; Sima, R.; Laco, J.; Weinreb, I.; Perez-Ordonez, B.; Starek, I.; Geierova, M.; Simpson, R.H.; Passador-Santos, F.; et al. Mammary Analogue Secretory Carcinoma of Salivary Glands, Containing the ETV6-NTRK3 Fusion Gene: A Hitherto Undescribed Salivary Gland Tumor Entity. Am. J. Surg. Pathol. 2010, 34, 599–608. [Google Scholar] [CrossRef]
- Kralik, J.M.; Kranewitter, W.; Boesmueller, H.; Marschon, R.; Tschurtschenthaler, G.; Rumpold, H.; Wiesinger, K.; Erdel, M.; Petzer, A.L.; Webersinke, G. Characterization of a Newly Identified ETV6-NTRK3 Fusion Transcript in Acute Myeloid Leukemia. Diagn. Pathol. 2011, 6, 19. [Google Scholar] [CrossRef]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable Kinase-Activating Lesions in Ph-like Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef]
- Wang, Y.; Miller, S.; Roulston, D.; Bixby, D.; Shao, L. Genome-Wide Single-Nucleotide Polymorphism Array Analysis Improves Prognostication of Acute Lymphoblastic Leukemia/Lymphoma. J. Mol. Diagn. 2016, 18, 595–603. [Google Scholar] [CrossRef]
- Forghieri, F.; Morselli, M.; Potenza, L.; Maccaferri, M.; Pedrazzi, L.; Paolini, A.; Bonacorsi, G.; Artusi, T.; Giacobbi, F.; Corradini, G.; et al. Chronic Eosinophilic Leukaemia with ETV6-NTRK3 Fusion Transcript in an Elderly Patient Affected with Pancreatic Carcinoma. Eur. J. Haematol. 2011, 86, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, S.; Skalova, A.; Agaimy, A.; Bishop, J.A.; Laco, J.; Leivo, I.; Franchi, A.; Larsen, S.R.; Erentaite, D.; Ulhoi, B.P.; et al. ETV6 Gene Rearrangements Characterize a Morphologically Distinct Subset of Sinonasal Low-Grade Non-Intestinal-Type Adenocarcinoma: A Novel Translocation-Associated Carcinoma Restricted to the Sinonasal Tract. Am. J. Surg. Pathol. 2017, 41, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The Genomic Landscape of Diffuse Intrinsic Pontine Glioma and Pediatric Non-Brainstem High-Grade Glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar] [CrossRef]
- Brenca, M.; Rossi, S.; Polano, M.; Gasparotto, D.; Zanatta, L.; Racanelli, D.; Valori, L.; Lamon, S.; Dei Tos, A.P.; Maestro, R. Transcriptome Sequencing Identifies ETV6-NTRK3 as a Gene Fusion Involved in GIST. J. Pathol. 2016, 238, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Shi, E.; Chmielecki, J.; Tang, C.M.; Wang, K.; Heinrich, M.C.; Kang, G.; Corless, C.L.; Hong, D.; Fero, K.E.; Murphy, J.D.; et al. FGFR1 and NTRK3 Actionable Alterations in “Wild-Type” Gastrointestinal Stromal Tumors. J. Transl. Med. 2016, 14, 339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wu, G.; Miller, C.P.; Tatevossian, R.G.; Dalton, J.D.; Tang, B.; Orisme, W.; Punchihewa, C.; Parker, M.; Qaddoumi, I.; et al. Whole-Genome Sequencing Identifies Genetic Alterations in Pediatric Low-Grade Gliomas. Nat. Genet. 2013, 45, 602–612. [Google Scholar] [CrossRef]
- Dogan, S.; Wang, L.; Ptashkin, R.N.; Dawson, R.R.; Shah, J.P.; Sherman, E.J.; Michael Tuttle, R.; Fagin, J.A.; Klimstra, D.S.; Katabi, N.; et al. Mammary Analog Secretory Carcinoma of the Thyroid Gland: A Primary Thyroid Adenocarcinoma Harboring ETV6-NTRK3 Fusion. Mod. Pathol. 2016, 29, 985–995. [Google Scholar] [CrossRef]
- Wu, B.; Loh, T.K.S.; Vanecek, T.; Michal, M.; Petersson, F. (Mammary Analogue) Secretory Carcinoma of the Nasal Cavity: Report of a Rare Case with P63 and DOG1 Expression and Uncommon Exon 4-Exon 14 ETV6-NTRK3 Fusion Diagnosed with Next Generation Sequencing. Head Neck Pathol. 2020, 14, 542–549. [Google Scholar] [CrossRef]
- Pemovska, T.; Kontro, M.; Yadav, B.; Edgren, H.; Eldfors, S.; Szwajda, A.; Almusa, H.; Bespalov, M.M.; Ellonen, P.; Elonen, E.; et al. Individualized Systems Medicine Strategy to Tailor Treatments for Patients with Chemorefractory Acute Myeloid Leukemia. Cancer Discov. 2013, 3, 1416–1429. [Google Scholar] [CrossRef]
- Chen, S.; Nagel, S.; Schneider, B.; Dai, H.; Geffers, R.; Kaufmann, M.; Meyer, C.; Pommerenke, C.; Thress, K.S.; Li, J.; et al. A New ETV6-NTRK3 Cell Line Model Reveals MALAT1 as a Novel Therapeutic Target—A Short Report. Cell. Oncol. 2018, 41, 93–101. [Google Scholar] [CrossRef]
- Gu, T.L.; Popova, L.; Reeves, C.; Nardone, J.; Macneill, J.; Rush, J.; Nimer, S.D.; Polakiewicz, R.D. Phosphoproteomic Analysis Identifies the M0-91 Cell Line as a Cellular Model for the Study of TEL-TRKC Fusion-Associated Leukemia. Leukemia 2007, 21, 563–566. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, X.; Salokas, K.; Tamene, F.; Jiu, Y.; Weldatsadik, R.G.; Öhman, T.; Varjosalo, M. An AP-MS- and BioID-Compatible MAC-Tag Enables Comprehensive Mapping of Protein Interactions and Subcellular Localizations. Nat. Commun. 2018, 9, 1188. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Salokas, K.; Weldatsadik, R.G.; Gawriyski, L.; Varjosalo, M. Combined Proximity Labeling and Affinity Purification-Mass Spectrometry Workflow for Mapping and Visualizing Protein Interaction Networks. Nat. Protoc. 2020, 15, 3182–3211. [Google Scholar] [CrossRef] [PubMed]
- Budenholzer, L.; Cheng, C.L.; Li, Y.; Hochstrasser, M. Proteasome Structure and Assembly. J. Mol. Biol. 2017, 429, 3500–3524. [Google Scholar] [CrossRef]
- Park, J.; Kim, J.; Park, B.; Yang, K.M.; Sun, E.J.; Tognon, C.E.; Sorensen, P.H.; Kim, S.J. Novel Identification of STAT1 as a Crucial Mediator of ETV6-NTRK3-Induced Tumorigenesis. Oncogene 2018, 37, 2270–2284. [Google Scholar] [CrossRef]
- Gu, H.; Maeda, H.; Moon, J.J.; Lord, J.D.; Yoakim, M.; Nelson, B.H.; Neel, B.G. New Role for Shc in Activation of the Phosphatidylinositol 3-Kinase/Akt Pathway. Mol. Cell. Biol. 2000, 20, 7109–7120. [Google Scholar] [CrossRef]
- Ravichandran, K.S. Signaling via Shc Family Adapter Proteins. Oncogene 2001, 20, 6322–6330. [Google Scholar] [CrossRef]
- Xu, Y.R.; Lei, C.Q. TAK1-TABs Complex: A Central Signalosome in Inflammatory Responses. Front. Immunol. 2020, 11, 608976. [Google Scholar] [CrossRef]
- Watanabe, S.; Take, H.; Takeda, K.; Yu, Z.X.; Iwata, N.; Kajigaya, S. Characterization of the CIN85 Adaptor Protein and Identification of Components Involved in CIN85 Complexes. Biochem. Biophys. Res. Commun. 2000, 278, 167–174. [Google Scholar] [CrossRef]
- Szymkiewicz, I.; Kowanetz, K.; Soubeyran, P.; Dinarina, A.; Lipkowitz, S.; Dikic, I. CIN85 Participates in Cbl-b-Mediated down-Regulation of Receptor Tyrosine Kinases. J. Biol. Chem. 2002, 277, 39666–39672. [Google Scholar] [CrossRef]
- Feng, L.; Wang, J.T.; Jin, H.; Qian, K.; Geng, J.G. SH3KBP1-Binding Protein 1 Prevents Epidermal Growth Factor Receptor Degradation by the Interruption of c-Cbl-CIN85 Complex. Cell Biochem. Funct. 2011, 29, 589–596. [Google Scholar] [CrossRef]
- Kowanetz, K.; Crosetto, N.; Haglund, K.; Schmidt, M.H.H.; Heldin, C.H.; Dikic, I. Suppressors of T-Cell Receptor Signaling Sts-1 and Sts-2 Bind to Cbl and Inhibit Endocytosis of Receptor Tyrosine Kinases. J. Biol. Chem. 2004, 279, 32786–32795. [Google Scholar] [CrossRef]
- Wakioka, T.; Sasaki, A.; Mitsui, K.; Yokouchi, M.; Inoue, A.; Komiya, S.; Yoshimura, A. APS, an Adaptor Protein Containing Pleckstrin Homology (PH) and Src Homology-2 (SH2) Domains Inhibits the JAK-STAT Pathway in Collaboration with c-Cbl. Leukemia 1999, 13, 760–767. [Google Scholar] [CrossRef]
- Gavet, O.; Pines, J. Progressive Activation of CyclinB1-Cdk1 Coordinates Entry to Mitosis. Dev. Cell 2010, 18, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Nantajit, D.; Fan, M.; Duru, N.; Wen, Y.; Reed, J.C.; Li, J.J. Cyclin B1/Cdk1 Phosphorylation of Mitochondrial P53 Induces Anti-Apoptotic Response. PLoS ONE 2010, 5, e12341. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cleary, R.A.; Wang, R.; Tang, D.D. Role of the Adapter Protein Abi1 in Actin-Associated Signaling and Smooth Muscle Contraction. J. Biol. Chem. 2013, 288, 20713–20722. [Google Scholar] [CrossRef] [PubMed]
- Lamorte, L.; Rodrigues, S.; Sangwan, V.; Turner, C.E.; Park, M. Crk Associates with a Multimolecular Paxillin/GIT2/Beta-PIX Complex and Promotes Rac-Dependent Relocalization of Paxillin to Focal Contacts. Mol. Biol. Cell 2003, 14, 2818–2831. [Google Scholar] [CrossRef]
- Gangisetty, O.; Lauffart, B.; Sondarva, G.V.; Chelsea, D.M.; Still, I.H. The Transforming Acidic Coiled Coil Proteins Interact with Nuclear Histone Acetyltransferases. Oncogene 2004, 23, 2559–2563. [Google Scholar] [CrossRef]
- Hart, M.J.; de los Santos, R.; Albert, I.N.; Rubinfeld, B.; Polakis, P. Downregulation of Beta-Catenin by Human Axin and Its Association with the APC Tumor Suppressor, Beta-Catenin and GSK3 Beta. Curr. Biol. 1998, 8, 573–581. [Google Scholar] [CrossRef]
- Lemmon, M.A. Pleckstrin Homology (PH) Domains and Phosphoinositides. Biochem. Soc. Symp. 2007, 74, 81–93. [Google Scholar] [CrossRef]
- Anderson, C.A.; Kovar, D.R.; Gardel, M.L.; Winkelman, J.D. LIM Domain Proteins in Cell Mechanobiology. Cytoskeleton 2021, 78, 303–311. [Google Scholar] [CrossRef]
- Drilon, A.; Nagasubramanian, R.; Blake, J.F.; Ku, N.; Tuch, B.B.; Ebata, K.; Smith, S.; Lauriault, V.; Kolakowski, G.R.; Brandhuber, B.J.; et al. A Next-Generation TRK Kinase Inhibitor Overcomes Acquired Resistance to Prior TRK Kinase Inhibition in Patients with TRK Fusion-Positive Solid Tumors. Cancer Discov. 2017, 7, 963–972. [Google Scholar] [CrossRef]
- Galic, S.; Hauser, C.; Kahn, B.B.; Haj, F.G.; Neel, B.G.; Tonks, N.K.; Tiganis, T. Coordinated Regulation of Insulin Signaling by the Protein Tyrosine Phosphatases PTP1B and TCPTP. Mol. Cell. Biol. 2005, 25, 819–829. [Google Scholar] [CrossRef]
- Knauf, U.; Tschopp, C.; Gram, H. Negative Regulation of Protein Translation by Mitogen-Activated Protein Kinase-Interacting Kinases 1 and 2. Mol. Cell. Biol. 2001, 21, 5500–5511. [Google Scholar] [CrossRef]
- Ueda, T.; Watanabe-Fukunaga, R.; Fukuyama, H.; Nagata, S.; Fukunaga, R. Mnk2 and Mnk1 Are Essential for Constitutive and Inducible Phosphorylation of Eukaryotic Initiation Factor 4E but Not for Cell Growth or Development. Mol. Cell. Biol. 2004, 24, 6539–6549. [Google Scholar] [CrossRef]
- Uniacke, J.; Holterman, C.E.; Lachance, G.; Franovic, A.; Jacob, M.D.; Fabian, M.R.; Payette, J.; Holcik, M.; Pause, A.; Lee, S. An Oxygen-Regulated Switch in the Protein Synthesis Machinery. Nature 2012, 486, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Uniacke, J.; Perera, J.K.; Lachance, G.; Francisco, C.B.; Lee, S. Cancer Cells Exploit EIF4E2-Directed Synthesis of Hypoxia Response Proteins to Drive Tumor Progression. Cancer Res. 2014, 74, 1379–1389. [Google Scholar] [CrossRef] [PubMed]
- Vomastek, T.; Schaeffer, H.J.; Tarcsafalvi, A.; Smolkin, M.E.; Bissonette, E.A.; Weber, M.J. Modular Construction of a Signaling Scaffold: MORG1 Interacts with Components of the ERK Cascade and Links ERK Signaling to Specific Agonists. Proc. Natl. Acad. Sci. USA 2004, 101, 6981–6986. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Q.; Tsiaras, W.G.; Araki, T.; Wen, G.; Minichiello, L.; Klein, R.; Neel, B.G. Receptor-Specific Regulation of Phosphatidylinositol 3′-Kinase Activation by the Protein Tyrosine Phosphatase Shp2. Mol. Cell Biol. 2002, 22, 4062–4072. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of Glycogen Synthase Kinase-3 by Insulin Mediated by Protein Kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. MTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Xu, D.; Qu, C.K. Protein Tyrosine Phosphatases in the JAK/STAT Pathway. Front. Biosci. 2008, 13, 4925–4932. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Janke, L.J.; Zhao, Y.; Seth, A.; Ma, J.; Finkelstein, D.; Smith, S.; Ebata, K.; Tuch, B.B.; Hunger, S.P.; et al. ETV6-NTRK3 Induces Aggressive Acute Lymphoblastic Leukemia Highly Sensitive to Selective TRK Inhibition. Blood 2018, 132, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Roovers, K.; Assoian, R.K. Integrating the MAP Kinase Signal into the G1 Phase Cell Cycle Machinery. Bioessays 2000, 22, 818–826. [Google Scholar] [CrossRef]
- Xie, X.; Chang, S.W.; Tatsumoto, T.; Chan, A.M.; Miki, T. TIM, a Dbl-Related Protein, Regulates Cell Shape and Cytoskeletal Organization in a Rho-Dependent Manner. Cell. Signal. 2005, 17, 461–471. [Google Scholar] [CrossRef]
- Fernandez-Madrid, F.; Tang, N.; Alansari, H.; Granda, J.L.; Tait, L.; Amirikia, K.C.; Moroianu, M.; Wang, X.; Karvonen, R.L. Autoantibodies to Annexin XI-A and Other Autoantigens in the Diagnosis of Breast Cancer. Cancer Res. 2004, 64, 5089–5096. [Google Scholar] [CrossRef]
- Huang, B.; Trujillo, M.A.; Fujikura, K.; Qiu, M.; Chen, F.; Felsenstein, M.; Zhou, C.; Skaro, M.; Gauthier, C.; Macgregor-Das, A.; et al. Molecular Characterization of Organoids Derived from Pancreatic Intraductal Papillary Mucinous Neoplasms. J. Pathol. 2020, 252, 252–262. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Lukyanova, E.N.; Fedorova, M.S.; Kalinin, D.V.; Melnikova, N.V.; Stepanov, O.A.; Kiseleva, M.V.; Kaprin, A.D.; Pudova, E.A.; Kudryavtseva, A.V. Novel Genes Associated with the Development of Carotid Paragangliomas. Mol. Biol. 2019, 53, 613–626. [Google Scholar] [CrossRef]
- Igci, Y.Z.; Arslan, A.; Akarsu, E.; Erkilic, S.; Igci, M.; Oztuzcu, S.; Cengiz, B.; Gogebakan, B.; Cakmak, E.A.; Demiryurek, A.T. Differential Expression of a Set of Genes in Follicular and Classic Variants of Papillary Thyroid Carcinoma. Endocr. Pathol. 2011, 22, 86–96. [Google Scholar] [CrossRef]
- Majewska, H.; Skalova, A.; Stodulski, D.; Klimkova, A.; Steiner, P.; Stankiewicz, C.; Biernat, W. Mammary Analogue Secretory Carcinoma of Salivary Glands: A New Entity Associated with ETV6 Gene Rearrangement. Virchows Arch. 2015, 466, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Strauss, B.L.; Bratthauer, G.L.; Tavassoli, F.A. STAT 5a Expression in the Breast Is Maintained in Secretory Carcinoma, in Contrast to Other Histologic Types. Hum. Pathol. 2006, 37, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, A.; Taira, T.; Abe, H.; Takase, Y.; Kurita, T.; Sadashima, E.; Hattori, S.; Imamura, I.; Matsumoto, S.; Fujisaki, H.; et al. Diagnostic Utility of Phosphorylated Signal Transducer and Activator of Transcription 5 Immunostaining in the Diagnosis of Mammary Analogue Secretory Carcinoma of the Salivary Gland: A Comparative Study of Salivary Gland Cancers. Cancer Cytopathol. 2015, 123, 603–611. [Google Scholar] [CrossRef]
- Horita, M.; Andreu, E.J.; Benito, A.; Arbona, C.; Sanz, C.; Benet, I.; Prosper, F.; Fernandez-Luna, J.L. Blockade of the Bcr-Abl Kinase Activity Induces Apoptosis of Chronic Myelogenous Leukemia Cells by Suppressing Signal Transducer and Activator of Transcription 5-Dependent Expression of Bcl-XL. J. Exp. Med. 2000, 191, 977–984. [Google Scholar] [CrossRef]
- Taipale, M.; Krykbaeva, I.; Whitesell, L.; Santagata, S.; Zhang, J.; Liu, Q.; Gray, N.S.; Lindquist, S. Chaperones as Thermodynamic Sensors of Drug-Target Interactions Reveal Kinase Inhibitor Specificities in Living Cells. Nat. Biotechnol. 2013, 31, 630–637. [Google Scholar] [CrossRef]
- Gotoh, T.; Cai, D.; Tian, X.; Feig, L.A.; Lerner, A. P130Cas Regulates the Activity of AND-34, a Novel Ral, Rap1, and R-Ras Guanine Nucleotide Exchange Factor. J. Biol. Chem. 2000, 275, 30118–30123. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Canaff, L.; Rajadurai, C.V.; Fils-Aime, N.; Tian, J.; Dai, M.; Korah, J.; Villatoro, M.; Park, M.; Ali, S.; et al. Breast Cancer Anti-Estrogen Resistance 3 Inhibits Transforming Growth Factor Beta/Smad Signaling and Associates with Favorable Breast Cancer Disease Outcomes. Breast Cancer Res. 2014, 16, 476. [Google Scholar] [CrossRef]
- Jin, W.; Kim, B.C.; Tognon, C.; Lee, H.J.; Patel, S.; Lannon, C.L.; Maris, J.M.; Triche, T.J.; Sorensen, P.H.; Kim, S.J. The ETV6-NTRK3 Chimeric Tyrosine Kinase Suppresses TGF-Beta Signaling by Inactivating the TGF-Beta Type II Receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 16239–16244. [Google Scholar] [CrossRef]
- Zhang, L.; Shay, J.W. Multiple Roles of APC and Its Therapeutic Implications in Colorectal Cancer. J. Natl. Cancer Inst. 2017, 109, djw332. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Heinen, C.D.; Goss, K.H.; Cornelius, J.R.; Babcock, G.F.; Knudsen, E.S.; Kowalik, T.; Groden, J. The APC Tumor Suppressor Controls Entry into S-Phase through Its Ability to Regulate the Cyclin D/RB Pathway. Gastroenterology 2002, 123, 751–763. [Google Scholar] [CrossRef]
- Wingelhofer, B.; Neubauer, H.A.; Valent, P.; Han, X.; Constantinescu, S.N.; Gunning, P.T.; Muller, M.; Moriggl, R. Implications of STAT3 and STAT5 Signaling on Gene Regulation and Chromatin Remodeling in Hematopoietic Cancer. Leukemia 2018, 32, 1713–1726. [Google Scholar] [CrossRef] [PubMed]
- Asmamaw, M.D.; Shi, X.J.; Zhang, L.R.; Liu, H.M. A Comprehensive Review of SHP2 and Its Role in Cancer. Cell. Oncol. 2022, 45, 729–753. [Google Scholar] [CrossRef] [PubMed]
- Nyga, R.; Pecquet, C.; Harir, N.; Gu, H.; Dhennin-Duthille, I.; Regnier, A.; Gouilleux-Gruart, V.; Lassoued, K.; Gouilleux, F. Activated STAT5 Proteins Induce Activation of the PI 3-Kinase/Akt and Ras/MAPK Pathways via the Gab2 Scaffolding Adapter. Biochem. J. 2005, 390, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, I.; Kitamura, T.; Wakao, H.; Tanaka, H.; Hashimoto, K.; Albanese, C.; Downward, J.; Pestell, R.G.; Kanakura, Y. Transcriptional Regulation of the Cyclin D1 Promoter by STAT5: Its Involvement in Cytokine-Dependent Growth of Hematopoietic Cells. EMBO J. 1999, 18, 1367–1377. [Google Scholar] [CrossRef]
- Canoy, R.J.; Shmakova, A.; Karpukhina, A.; Shepelev, M.; Germini, D.; Vassetzky, Y. Factors That Affect the Formation of Chromosomal Translocations in Cells. Cancers 2022, 14, 5110. [Google Scholar] [CrossRef]
- Tognon, C.E.; Rafn, B.; Cetinbas, N.M.; Kamura, T.; Trigo, G.; Rotblat, B.; Okumura, F.; Matsumoto, M.; Chow, C.; Davare, M.; et al. Insulin-like Growth Factor 1 Receptor Stabilizes the ETV6-NTRK3 Chimeric Oncoprotein by Blocking Its KPC1/Rnf123-Mediated Proteasomal Degradation. J. Biol. Chem. 2018, 293, 12502–12515. [Google Scholar] [CrossRef]
- Jadhav, T.; Geetha, T.; Jiang, J.; Wooten, M.W. Identification of a Consensus Site for TRAF6/P62 Polyubiquitination. Biochem. Biophys. Res. Commun. 2008, 371, 521–524. [Google Scholar] [CrossRef]
- Jadhav, T.S.; Wooten, M.W.; Wooten, M.C. Mining the TRAF6/P62 Interactome for a Selective Ubiquitination Motif. BMC Proc. 2011, 5 (Suppl. 2), S4. [Google Scholar] [CrossRef]
- Wooten, M.W.; Geetha, T.; Babu, J.R.; Seibenhener, M.L.; Peng, J.; Cox, N.; Diaz-Meco, M.T.; Moscat, J. Essential Role of Sequestosome 1/P62 in Regulating Accumulation of Lys63-Ubiquitinated Proteins. J. Biol. Chem. 2008, 283, 6783–6789. [Google Scholar] [CrossRef]
- Takaesu, G.; Kobayashi, T.; Yoshimura, A. TGFβ-Activated Kinase 1 (TAK1)-Binding Proteins (TAB) 2 and 3 Negatively Regulate Autophagy. J. Biochem. 2012, 151, 157–166. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinnunen, M.; Liu, X.; Niemelä, E.; Öhman, T.; Gawriyski, L.; Salokas, K.; Keskitalo, S.; Varjosalo, M. The Impact of ETV6-NTRK3 Oncogenic Gene Fusions on Molecular and Signaling Pathway Alterations. Cancers 2023, 15, 4246. https://doi.org/10.3390/cancers15174246
Kinnunen M, Liu X, Niemelä E, Öhman T, Gawriyski L, Salokas K, Keskitalo S, Varjosalo M. The Impact of ETV6-NTRK3 Oncogenic Gene Fusions on Molecular and Signaling Pathway Alterations. Cancers. 2023; 15(17):4246. https://doi.org/10.3390/cancers15174246
Chicago/Turabian StyleKinnunen, Matias, Xiaonan Liu, Elina Niemelä, Tiina Öhman, Lisa Gawriyski, Kari Salokas, Salla Keskitalo, and Markku Varjosalo. 2023. "The Impact of ETV6-NTRK3 Oncogenic Gene Fusions on Molecular and Signaling Pathway Alterations" Cancers 15, no. 17: 4246. https://doi.org/10.3390/cancers15174246
APA StyleKinnunen, M., Liu, X., Niemelä, E., Öhman, T., Gawriyski, L., Salokas, K., Keskitalo, S., & Varjosalo, M. (2023). The Impact of ETV6-NTRK3 Oncogenic Gene Fusions on Molecular and Signaling Pathway Alterations. Cancers, 15(17), 4246. https://doi.org/10.3390/cancers15174246