
Abrogating Metastatic Properties of Triple-Negative Breast Cancer Cells by EGFR and PI3K Dual Inhibitors
 ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Line and Culture Conditions
2.2. Small Molecules Preparation
2.3. Cell Viability Assay
2.4. IC50 Determination
2.5. Wound-Healing Assay
2.6. Immunofluorescence and Immunocytochemistry
2.7. Image Acquisition and Analysis
2.8. Statistical Analysis
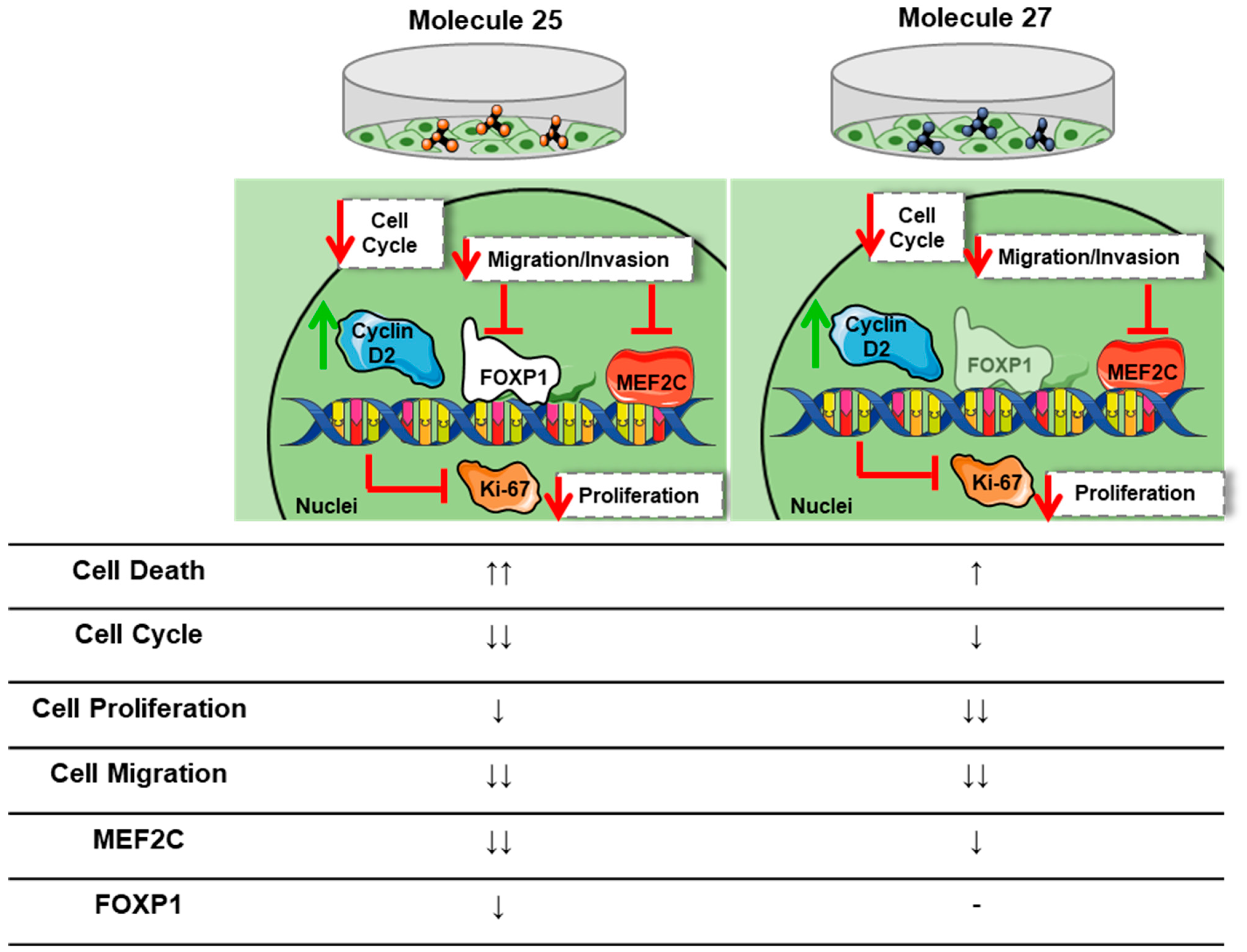
3. Results
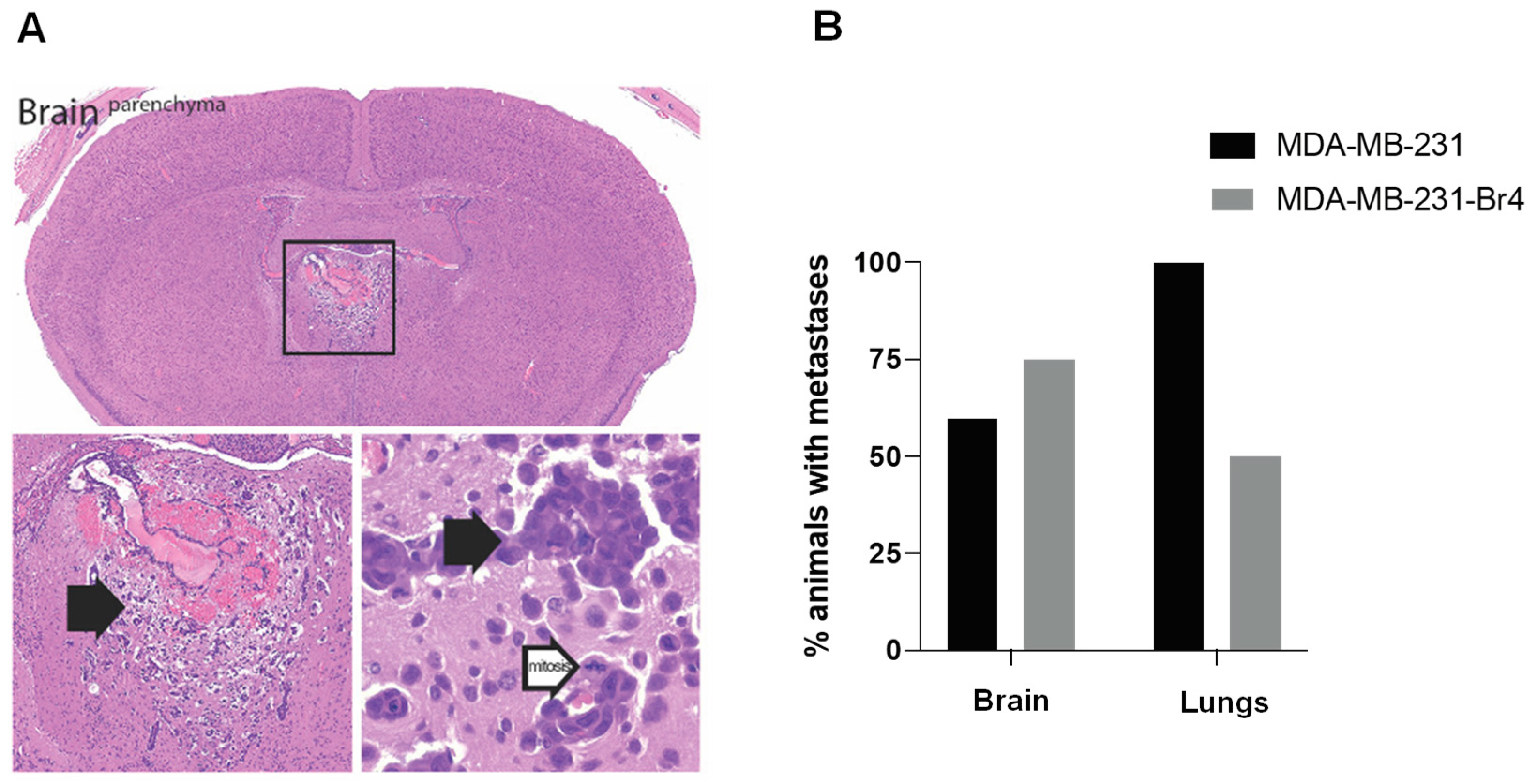
3.1. Characterization of the Developed Cell Line MDA-MB-231 Br4
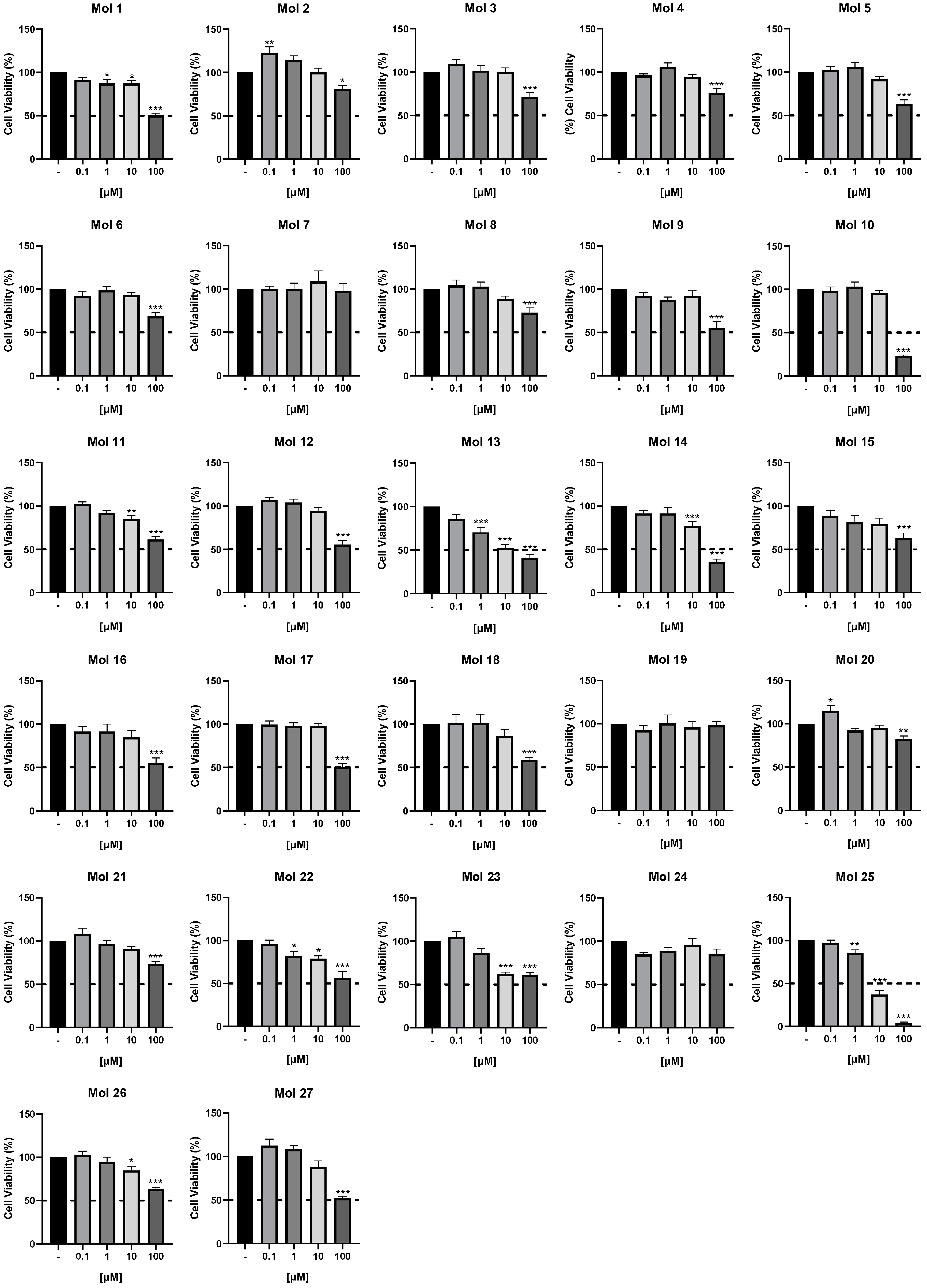
3.2. Small Molecules Cytotoxic Effects in TNBC Cells
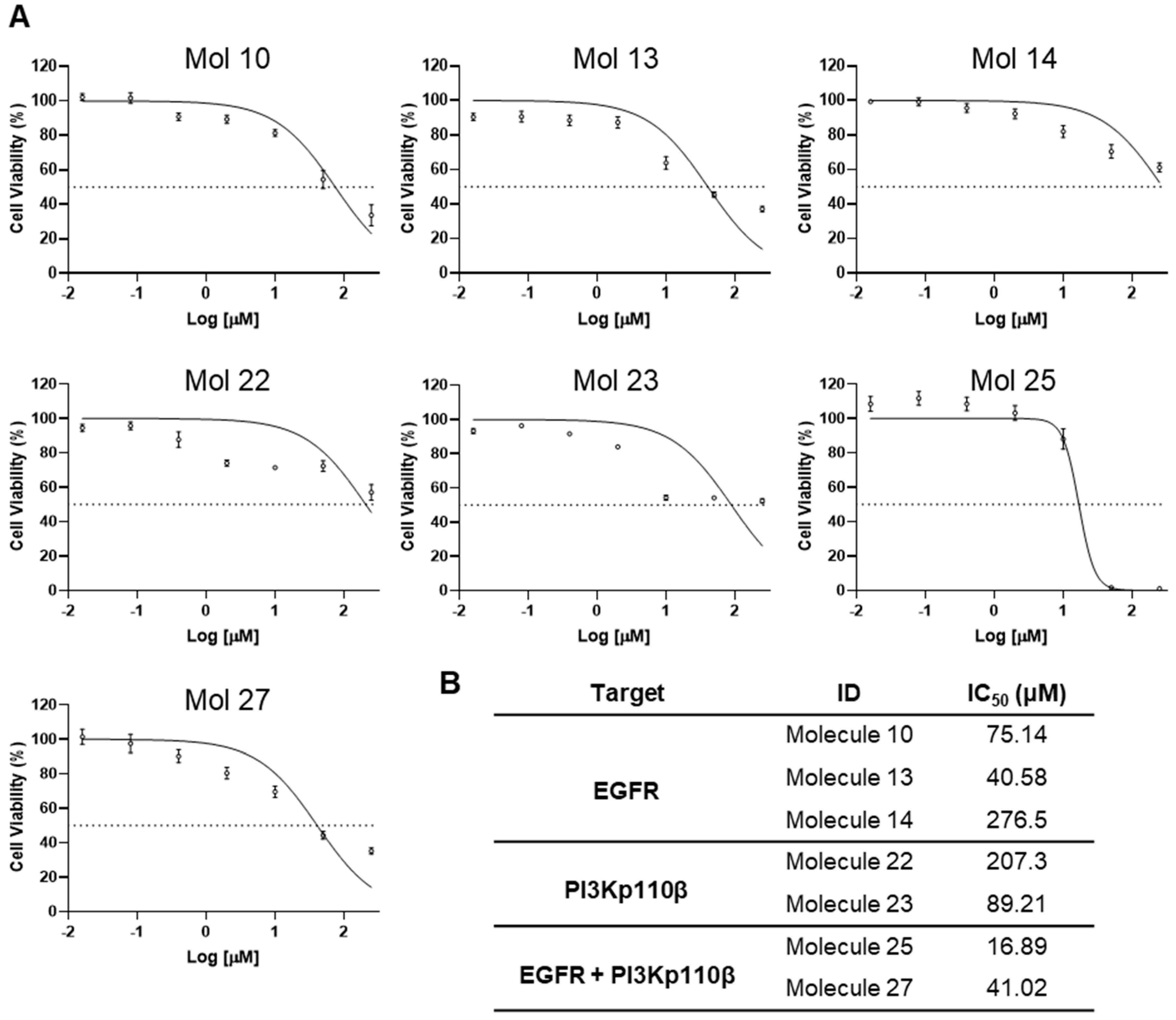
3.3. Determination of Half-Maximal Inhibitory Concentration of Inhibitors
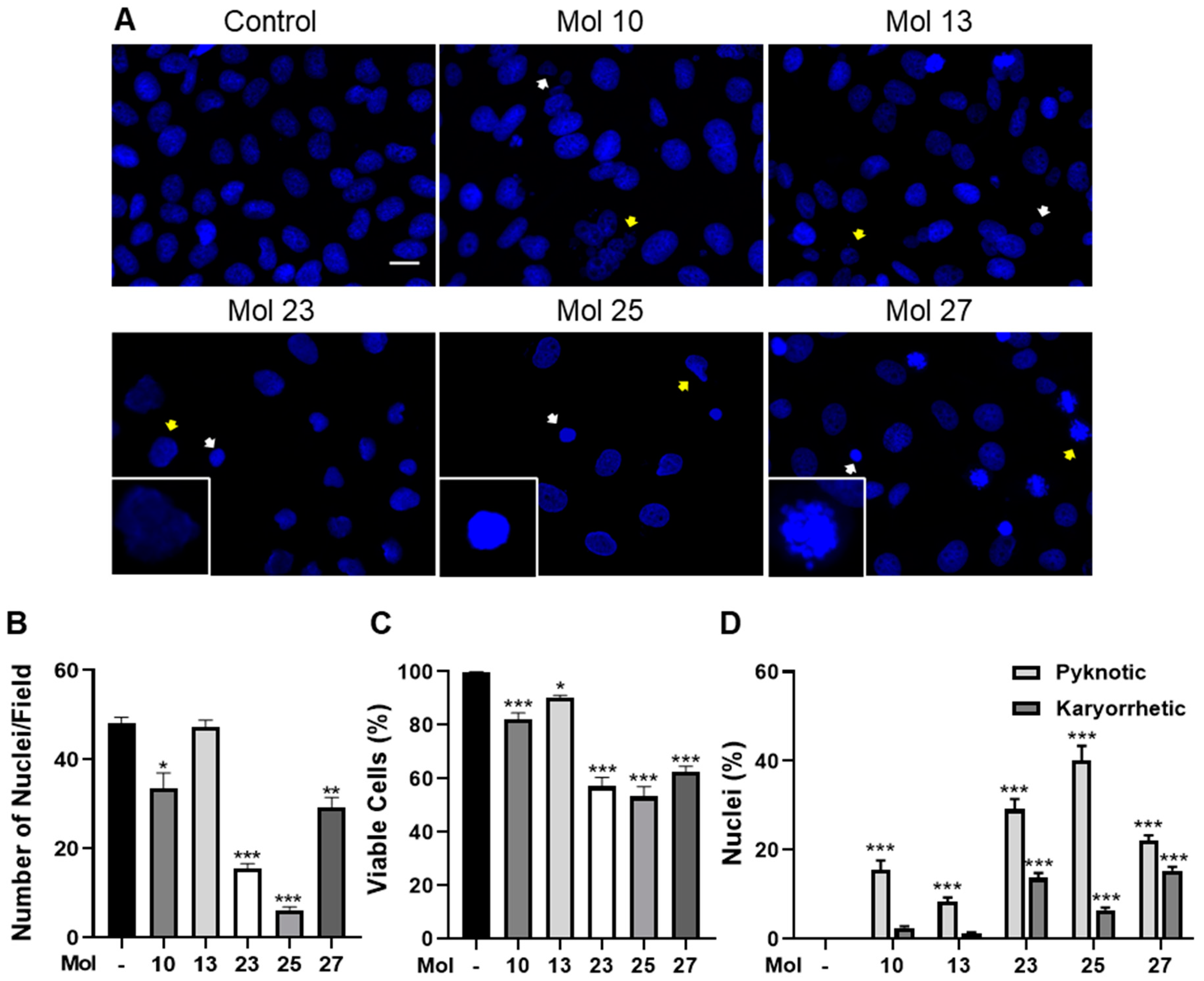
3.4. Small Molecule Inhibitors Induce TNBC Cell Death
3.5. Small Molecule Inhibitors Affect TNBC Cells Migration
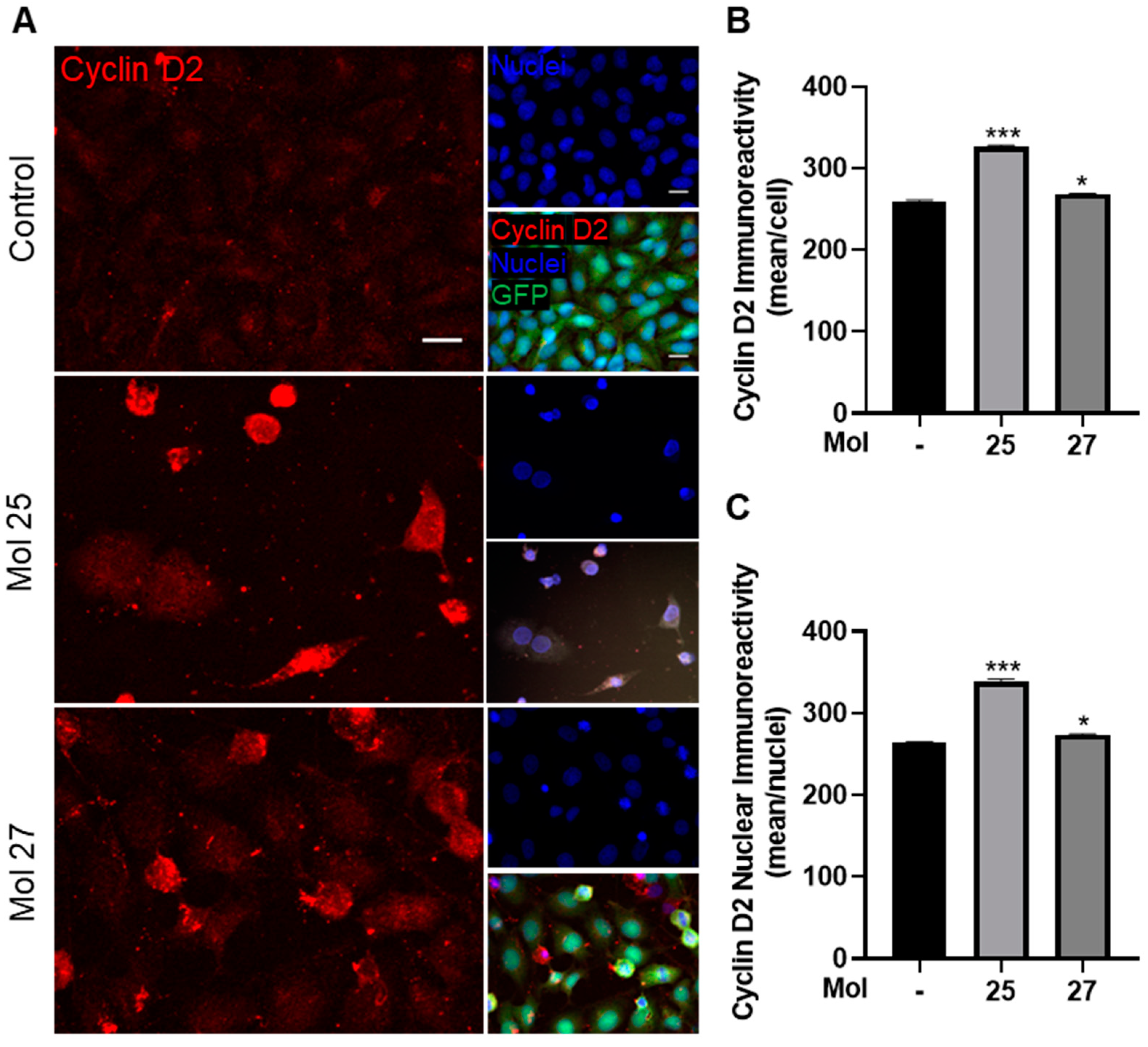
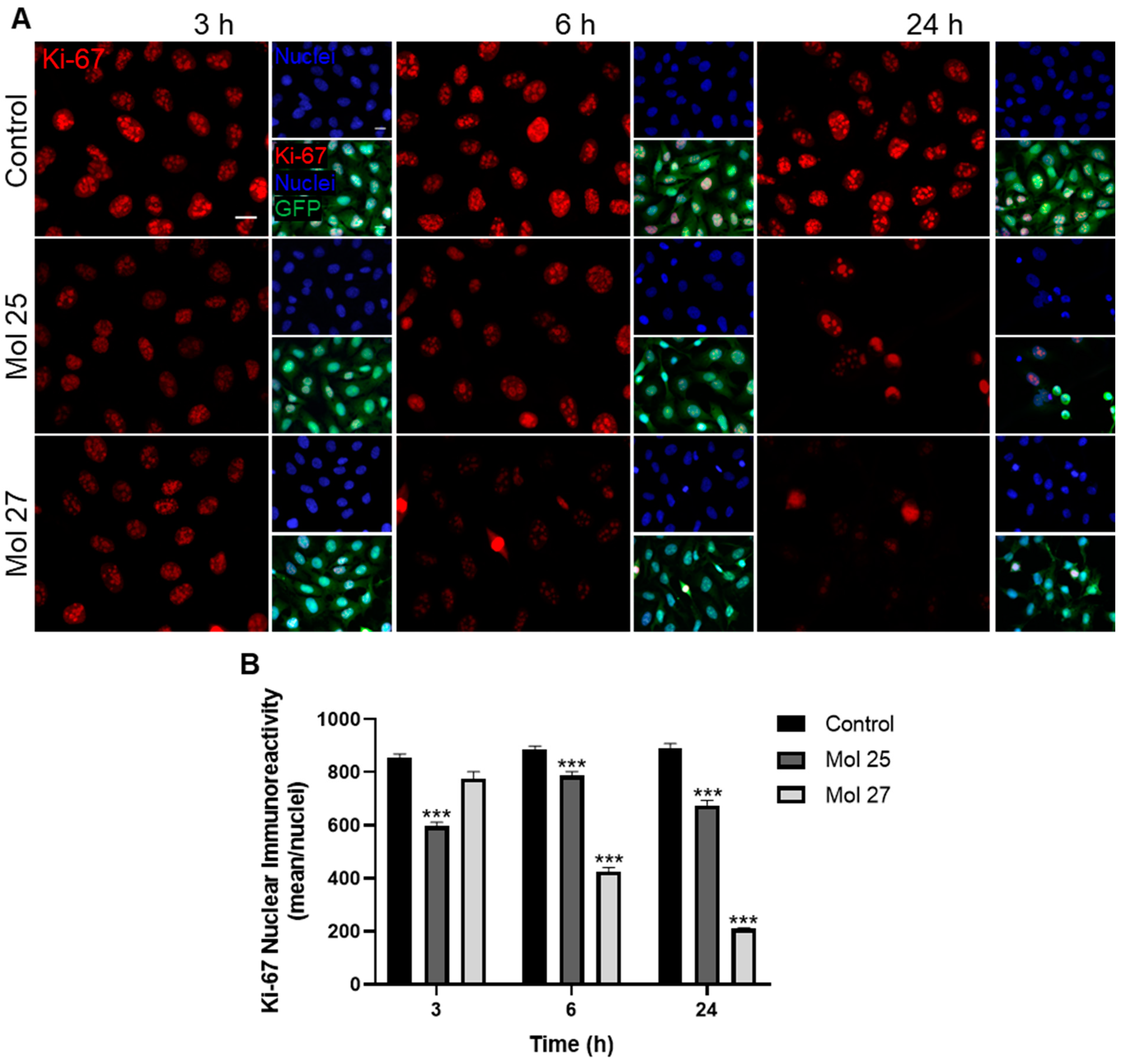
3.6. Dual-Inhibitor Molecules Modulate Cell Cycle and TNBC Cells Proliferation
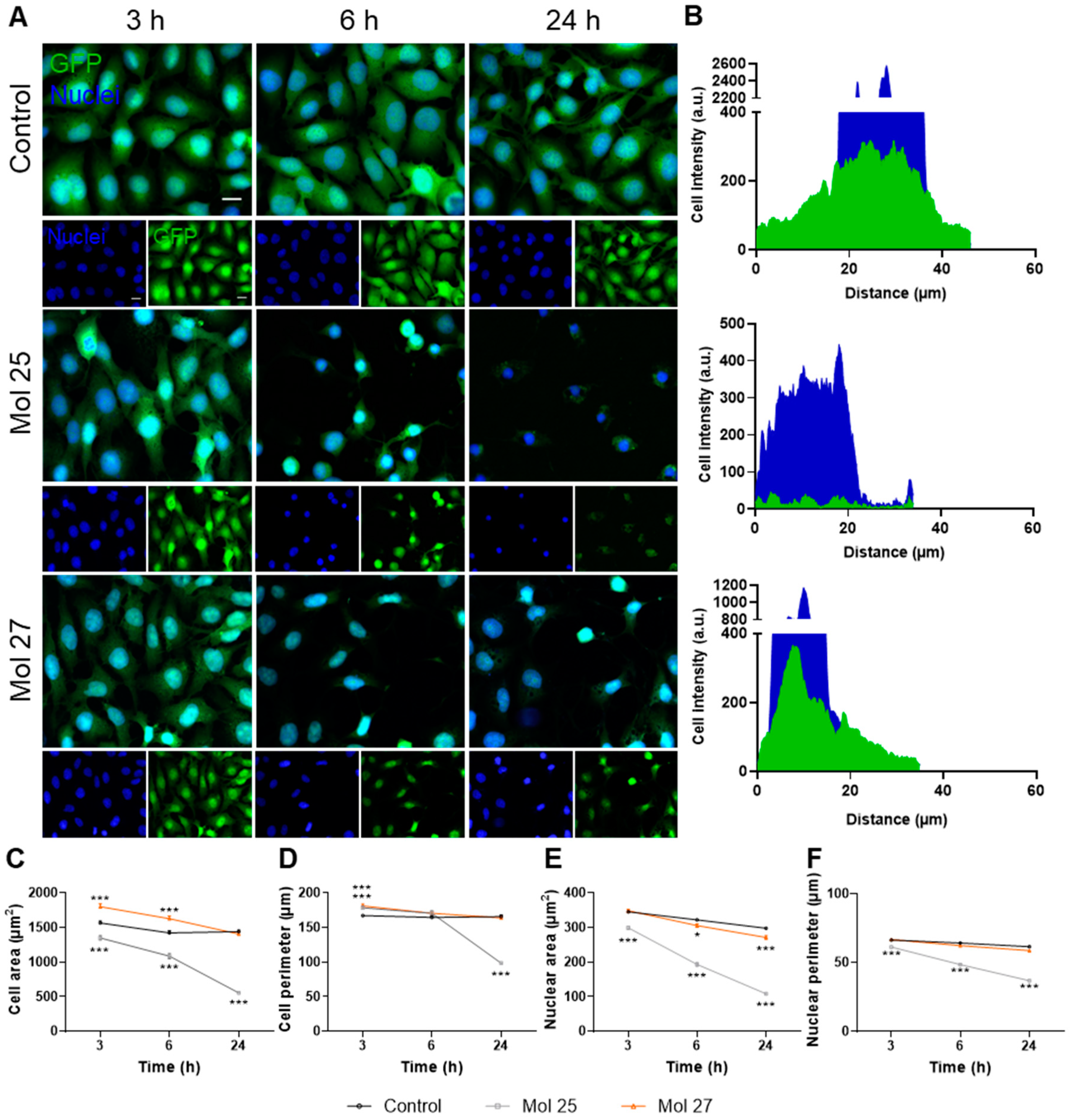
3.7. Dual-Inhibitor Molecules Alter TNBC Cells Morphology
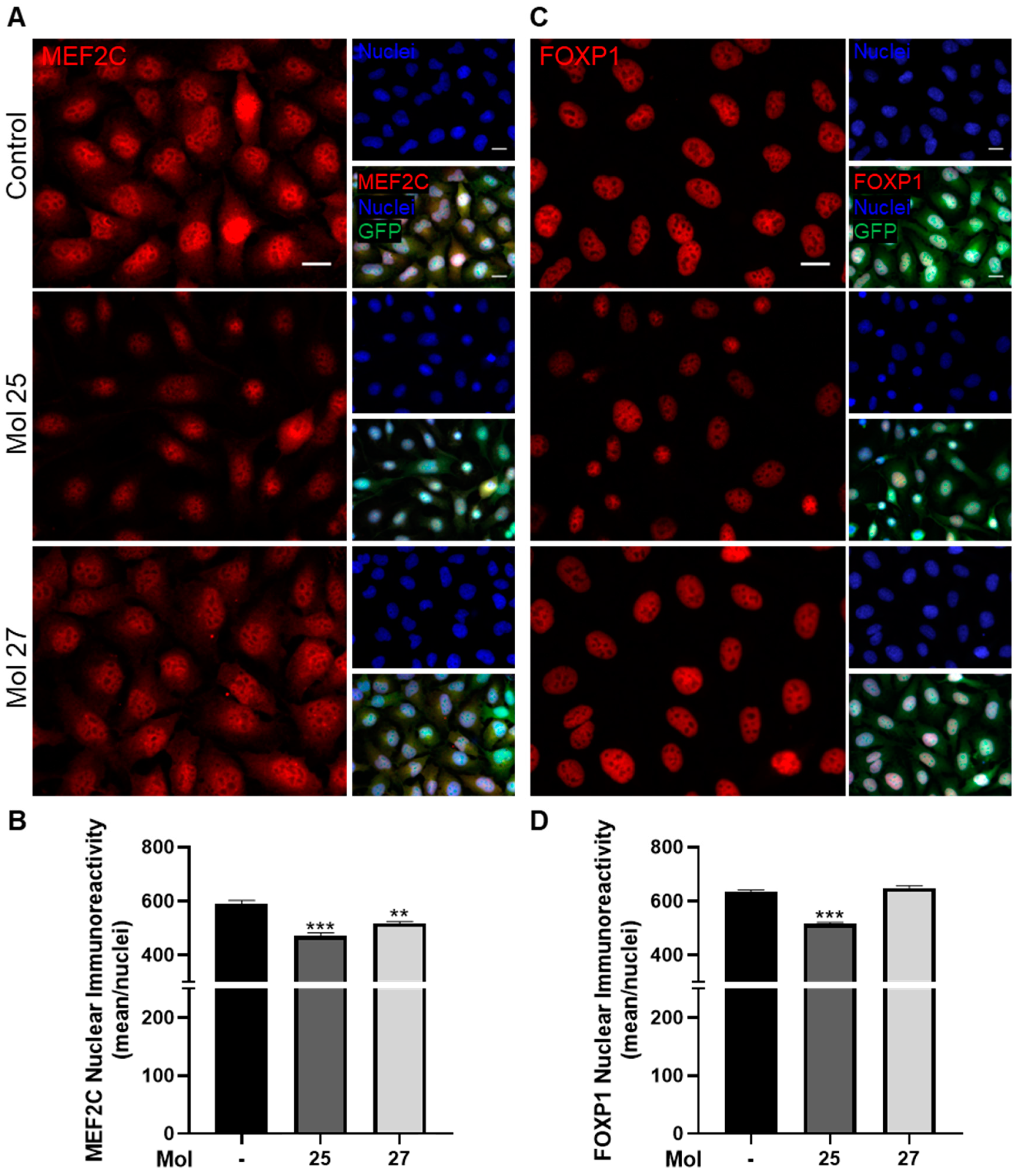
3.8. Dual-Inhibitor Molecules Have Downstream Effects in EGFR/PI3K Pathway
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Shi, A.; Lu, C.; Song, T.; Zhang, Z.; Zhao, J. Breast Cancer: Epidemiology and Etiology. Cell Biochem. Biophys. 2015, 72, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Uscanga-Perales, G.; Santuario-Facio, S.; Ortiz-López, R. Triple negative breast cancer: Deciphering the biology and heterogeneity. Med. Univ. 2016, 18, 105–114. [Google Scholar] [CrossRef]
- Derakhshan, F.; Reis-Filho, J.S. Pathogenesis of Triple-Negative Breast Cancer. Annu. Rev. Pathol. Mech. Dis. 2022, 17, 181–204. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Ma, X.; Du, Y.; Feng, J. Understanding Patterns of Brain Metastasis in Triple-Negative Breast Cancer and Exploring Potential Therapeutic Targets. OncoTargets Ther. 2021, 14, 589–607. [Google Scholar] [CrossRef]
- Branco, V.; Pimentel, J.; Brito, M.A.; Carvalho, C. Thioredoxin, Glutathione and Related Molecules in Tumors of the Nervous System. Curr. Med. Chem. 2020, 27, 1878–1900. [Google Scholar] [CrossRef]
- Custódio-Santos, T.; Videira, M.A.; Brito, M.A. Brain metastasization of breast cancer. BBA-Rev. Cancer 2017, 1868, 132–147. [Google Scholar] [CrossRef]
- Jerusalem, G.; Collignon, J.; Schroeder, H.; Lousberg, L. Triple-negative breast cancer: Treatment challenges and solutions. Breast Cancer Targets Ther. 2016, 8, 93–107. [Google Scholar] [CrossRef]
- Park, H.S.; Jang, M.H.; Kim, E.J.; Kim, H.J.; Lee, H.J.; Kim, Y.J.; Kim, J.H.; Kang, E.; Kim, S.-W.; Kim, I.A.; et al. High EGFR gene copy number predicts poor outcome in triple-negative breast cancer. Mod. Pathol. 2014, 27, 1212–1222. [Google Scholar] [CrossRef]
- Song, X.; Liu, Z.; Yu, Z. EGFR Promotes the Development of Triple Negative Breast Cancer Through JAK/STAT3 Signaling. Cancer Manag. Res. 2020, 12, 703–717. [Google Scholar] [CrossRef]
- Maennling, A.E.; Tur, M.K.; Niebert, M.; Klockenbring, T.; Zeppernick, F.; Gattenlöhner, S.; Meinhold-Heerlein, I.; Hussain, A.F. Molecular Targeting Therapy against EGFR Family in Breast Cancer: Progress and Future Potentials. Cancers 2019, 11, 1826. [Google Scholar] [CrossRef]
- Si, Y.; Xu, Y.; Guan, J.; Chen, K.; Kim, S.; Yang, E.S.; Zhou, L.; Liu, X.M. Anti-EGFR antibody-drug conjugate for triple-negative breast cancer therapy. Eng. Life Sci. 2021, 21, 37–44. [Google Scholar] [CrossRef]
- Sabatier, R.; Lopez, M.; Guille, A.; Billon, E.; Carbuccia, N.; Garnier, S.; Adelaide, J.; Extra, J.-M.; Cappiello, M.-A.; Charafe-Jauffret, E.; et al. High Response to Cetuximab in a Patient With EGFR-Amplified Heavily Pretreated Metastatic Triple-Negative Breast Cancer. JCO Precis. Oncol. 2019, 3, 1–8. [Google Scholar] [CrossRef]
- Ali, R.; Wendt, M.K. The paradoxical functions of EGFR during breast cancer progression. Signal Transduct. Target. Ther. 2017, 2, 16042. [Google Scholar] [CrossRef]
- Lev, S. Targeted therapy and drug resistance in triple-negative breast cancer: The EGFR axis. Biochem. Soc. Trans. 2020, 48, 657–665. [Google Scholar] [CrossRef] [PubMed]
- López-Knowles, E.; O’Toole, S.A.; McNeil, C.M.; Millar, E.K.; Qiu, M.R.; Crea, P.; Daly, R.J.; Musgrove, E.A.; Sutherland, R.L. PI3K pathway activation in breast cancer is associated with the basal-like phenotype and cancer-specific mortality. Int. J. Cancer 2010, 126, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Pascual, J.; Turner, N.C. Targeting the PI3-kinase pathway in triple-negative breast cancer. Ann. Oncol. 2019, 30, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M. Inhibition of the PI3K/AKT/mTOR Pathway in Solid Tumors. J. Clin. Oncol. 2016, 34, 3803–3815. [Google Scholar] [CrossRef]
- Ortega, M.A.; Fraile-Martínez, O.; Asúnsolo, Á.; Buján, J.; García-Honduvilla, N.; Coca, S. Signal Transduction Pathways in Breast Cancer: The Important Role of PI3K/Akt/mTOR. J. Oncol. 2020, 2020, 9258396. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Lou, Y.; Zhong, R.; Han, B. MMP9 activation triggered by epidermal growth factor induced FoxO1 nuclear exclusion in non-small cell lung cancer. Tumor Biol. 2014, 35, 6673–6678. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, E.; Clocchiatti, A.; Piccinin, S.; Sgorbissa, A.; Viviani, G.; Peruzzo, P.; Romeo, S.; Rossi, S.; Tos, A.P.D.; Maestro, R.; et al. MEF2 Is a Converging Hub for Histone Deacetylase 4 and Phosphatidylinositol 3-Kinase/Akt-Induced Transformation. Mol. Cell. Biol. 2013, 33, 4473–4491. [Google Scholar] [CrossRef] [PubMed]
- Pridham, K.J.; Varghese, R.T.; Sheng, Z. The Role of Class IA Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunits in Glioblastoma. Front. Oncol. 2017, 7, 312. [Google Scholar] [CrossRef] [PubMed]
- Pazarentzos, E.; Giannikopoulos, P.; Hrustanovic, G.; St John, J.; Olivas, V.R.; Gubens, M.A.; Balassanian, R.; Weissman, J.; Polkinghorn, W.; Bivona, T.G. Oncogenic activation of the PI3-kinase p110β isoform via the tumor-derived PIK3CβD1067V kinase domain mutation. Oncogene 2016, 35, 1198–1205. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Y.; Wang, Q.; Chen, Z.; Li, X.; Wu, Z.; Hu, C.; Liao, D.; Zhang, W.; Chen, Z.-S. The PI3K subunits, P110α and P110β are potential targets for overcoming P-gp and BCRP-mediated MDR in cancer. Mol. Cancer 2020, 19, 10. [Google Scholar] [CrossRef]
- Halacli, S.O.; Dogan, A.L. FOXP1 regulation via the PI3K/Akt/p70S6K signaling pathway in breast cancer cells. Oncol. Lett. 2015, 9, 1482–1488. [Google Scholar] [CrossRef]
- Banham, A.; Beasley, N.; Campo, E.; Fernandez, P.L.; Fidler, C.; Gatter, K.; Jones, M.; Mason, D.Y.; Prime, J.E.; Trougouboff, P.; et al. The FOXP1 winged helix transcription factor is a novel candidate tumor suppressor gene on chromosome 3p. Cancer Res. 2001, 61, 8820–8829. [Google Scholar]
- Brown, P.J.; Ashe, S.L.; Leich, E.; Burek, C.; Barrans, S.; Fenton, J.A.; Jack, A.S.; Pulford, K.; Rosenwald, A.; Banham, A.H. Potentially oncogenic B-cell activation–induced smaller isoforms of FOXP1 are highly expressed in the activated B cell–like subtype of DLBCL. Blood 2008, 111, 2816–2824. [Google Scholar] [CrossRef]
- De Silva, P.; Garaud, S.; Solinas, C.; de Wind, A.; Eyden, G.V.D.; Jose, V.; Gu-Trantien, C.; Migliori, E.; Boisson, A.; Naveaux, C.; et al. FOXP1 negatively regulates tumor infiltrating lymphocyte migration in human breast cancer. Ebiomedicine 2019, 39, 226–238. [Google Scholar] [CrossRef]
- Evron, E.; Umbricht, C.B.; Korz, D.; Raman, V.; Loeb, D.M.; Niranjan, B.; Buluwela, L.; Weitzman, S.A.; Marks, J.; Sukumar, S. Loss of cyclin D2 expression in the majority of breast cancers is associated with promoter hypermethylation. Cancer Res. 2001, 61, 2782. [Google Scholar] [PubMed]
- Russo, L.C.; Araujo, C.B.; Iwai, L.K.; Ferro, E.S.; Forti, F.L. A Cyclin D2-derived peptide acts on specific cell cycle phases by activating ERK1/2 to cause the death of breast cancer cells. J. Proteom. 2017, 151, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-S.; Wang, S.-C.; Yen, Y.-T.; Lee, T.-H.; Wen, W.-C.; Lin, R.-K. Hypermethylation of CCND2 in Lung and Breast Cancer Is a Potential Biomarker and Drug Target. Int. J. Mol. Sci. 2018, 19, 3096. [Google Scholar] [CrossRef] [PubMed]
- Ostrander, J.H.; Daniel, A.R.; Lofgren, K.; Kleer, C.G.; Lange, C.A. Breast Tumor Kinase (Protein Tyrosine Kinase 6) Regulates Heregulin-Induced Activation of ERK5 and p38 MAP Kinases in Breast Cancer Cells. Cancer Res. 2007, 67, 4199–4209. [Google Scholar] [CrossRef]
- Chen, X.; Gao, B.; Ponnusamy, M.; Lin, Z.; Liu, J. MEF2 signaling and human diseases. Oncotarget 2017, 8, 112152–112165. [Google Scholar] [CrossRef]
- Di Giorgio, E.; Hancock, W.W.; Brancolini, C. MEF2 and the tumorigenic process, hic sunt leones. Biochim. Biophys. Acta (BBA) Rev. Cancer 2018, 1870, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Sereno, M.; Haskó, J.; Molnár, K.; Medina, S.J.; Reisz, Z.; Malhó, R.; Videira, M.; Tiszlavicz, L.; Booth, S.A.; Wilhelm, I.; et al. Downregulation of circulating miR 802-5p and miR 194-5p and upregulation of brain MEF2C along breast cancer brain metastasization. Mol. Oncol. 2020, 14, 520–538. [Google Scholar] [CrossRef]
- Galego, S.; Kauppila, L.A.; Malhó, R.; Pimentel, J.; Brito, M.A. Myocyte Enhancer Factor 2C as a New Player in Human Breast Cancer Brain Metastases. Cells 2021, 10, 378. [Google Scholar] [CrossRef]
- Franco, C.; Kausar, S.; Silva, M.F.B.; Guedes, R.C.; Falcao, A.O.; Brito, M.A. Multi-Targeting Approach in Glioblastoma Using Computer-Assisted Drug Discovery Tools to Overcome the Blood–Brain Barrier and Target EGFR/PI3Kp110β Signaling. Cancers 2022, 14, 3506. [Google Scholar] [CrossRef]
- Godinho-Pereira, J.; Lopes, M.D.; Garcia, A.R.; Botelho, H.M.; Malhó, R.; Figueira, I.; Brito, M.A. A Drug Screening Reveals Minocycline Hydrochloride as a Therapeutic Option to Prevent Breast Cancer Cells Extravasation across the Blood–Brain Barrier. Biomedicines 2022, 10, 1988. [Google Scholar] [CrossRef]
- Godinho-Pereira, J.; Garcia, A.R.; Figueira, I.; Malhó, R.; Brito, M.A. Behind Brain Metastases Formation: Cellular and Molecular Alterations and Blood–Brain Barrier Disruption. Int. J. Mol. Sci. 2021, 22, 7057. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef]
- Goel, S.; Hidalgo, M.; Perez-Soler, R. EGFR inhibitor-mediated apoptosis in solid tumors. J. Exp. Ther. Oncol. 2007, 6, 305–320. [Google Scholar]
- Price, J.; Tiganis, T.; Agarwal, A.; Djakiew, D.; Thompson, E.W. Epidermal growth factor promotes MDA-MB-231 breast cancer cell migration through a phosphatidylinositol 3′-kinase and phospholipase C-dependent mechanism. Cancer Res. 1999, 59, 5475–5478. [Google Scholar] [PubMed]
- Cantley, L.C. The Phosphoinositide 3-Kinase Pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.; Chen, J.; Skoog, L.; Lindblom, A. Cyclin D2 expression in familial and sporadic breast cancer. Oncol. Rep. 2002, 9, 1157–1161. [Google Scholar] [CrossRef]
- Chierico, L.; Rizzello, L.; Guan, L.; Joseph, A.S.; Lewis, A.; Battaglia, G. The role of the two splice variants and extranuclear pathway on Ki-67 regulation in non-cancer and cancer cells. PLoS ONE 2017, 12, e0171815. [Google Scholar] [CrossRef]
- Halacli, S.O. FOXP1 enhances tumor cell migration by repression of NFAT1 transcriptional activity in MDA-MB-231 cells. Cell Biol. Int. 2017, 41, 102–110. [Google Scholar] [CrossRef]
- Grupka, N.L.; Lear-Kaul, K.C.; Kleinschmidt-DeMasters, B.K.; Singh, M. Epidermal Growth Factor Receptor Status in Breast Cancer Metastases to the Central Nervous System. Comparison with HER-2/neu status. Arch. Pathol. Lab. Med. 2004, 128, 974–979. [Google Scholar] [CrossRef]
- Shen, T.; Guo, Q. EGFR signaling pathway occupies an important position in cancer-related downstream signaling pathways of Pyk2. Cell Biol. Int. 2020, 44, 2–13. [Google Scholar] [CrossRef]
- Nakai, K.; Hung, M.-C.; Yamaguchi, H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am. J. Cancer Res. 2016, 6, 1609–1623. [Google Scholar]
- Miller, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Jemal, A.; Kramer, J.L.; Siegel, R.L. Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 2019, 69, 363–385. [Google Scholar] [CrossRef]
- Nyati, M.K.; Maheshwari, D.; Hanasoge, S.; Sreekumar, A.; Rynkiewicz, S.D.; Chinnaiyan, A.M.; Leopold, W.R.; Ethier, S.P.; Lawrence, T.S. Radiosensitization by Pan ErbB Inhibitor CI-1033 in Vitro and in Vivo. Clin. Cancer Res. 2004, 10, 691–700. [Google Scholar] [CrossRef][Green Version]
- Gomaa, H.A.M.; Ali, A.T.; Gabbar, M.A.; Kandeil, M.A. The Effect of Canertinib on Sensitivity of Cytotoxic Drugs in Tamoxifen-Resistant Breast Cancer Cells In Vitro. Int. J. Genom. 2018, 2018, 7628734. [Google Scholar] [CrossRef]
- Skvortsov, S.; Skvortsova, I.; Sarg, B.; Loeffler-Ragg, J.; Lindner, H.; Lukas, P.; Tabernero, J.; Zwierzina, H. Irreversible pan-ErbB tyrosine kinase inhibitor CI-1033 induces caspase-independent apoptosis in colorectal cancer DiFi cell line. Apoptosis 2005, 10, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Allen, L. CI-1033, an irreversible pan-erbB receptor inhibitor and its potential application for the treatment of breast cancer. Semin. Oncol. 2003, 30 (Suppl. S16), 65–78. [Google Scholar] [CrossRef] [PubMed]
- Rixe, O.; Franco, S.X.; Yardley, D.A.; Johnston, S.R.; Martín, M.; Arun, B.K.; Letrent, S.P.; Rugo, H.S. A randomized, phase II, dose-finding study of the pan-ErbB receptor tyrosine-kinase inhibitor CI-1033 in patients with pretreated metastatic breast cancer. Cancer Chemother. Pharmacol. 2009, 64, 1139–1148. [Google Scholar] [CrossRef] [PubMed]
- Zinner, R.G.; Nemunaitis, J.; Eiseman, I.; Shin, H.J.C.; Olson, S.C.; Christensen, J.; Huang, X.; Lenehan, P.F.; Donato, N.J.; Shin, D.M. Phase I Clinical and Pharmacodynamic Evaluation of Oral CI-1033 in Patients with Refractory Cancer. Clin. Cancer Res. 2007, 13, 3006–3014. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jänne, P.A.; von Pawel, J.; Cohen, R.B.; Crino, L.; Butts, C.A.; Olson, S.S.; Eiseman, I.A.; Chiappori, A.A.; Yeap, B.Y.; Lenehan, P.F.; et al. Multicenter, Randomized, Phase II Trial of CI-1033, an Irreversible Pan-ERBB Inhibitor, for Previously Treated Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2007, 25, 3936–3944. [Google Scholar] [CrossRef]
- Roberts, M.S.; Anstine, L.J.; Finke, V.S.; Bryson, B.L.; Webb, B.M.; Weber-Bonk, K.L.; Seachrist, D.D.; Majmudar, P.R.; Keri, R.A. KLF4 defines the efficacy of the epidermal growth factor receptor inhibitor, erlotinib, in triple-negative breast cancer cells by repressing the EGFR gene. Breast Cancer Res. 2020, 22, 66. [Google Scholar] [CrossRef]
- Xu, H.-Y.; Chen, Z.-W.; Hou, J.-C.; DU, F.-X.; Liu, J.-C. Jolkinolide B induces apoptosis in MCF-7 cells through inhibition of the PI3K/Akt/mTOR signaling pathway. Oncol. Rep. 2013, 29, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Henkels, K.M.; Mallets, E.R.; Dennis, P.B.; Gomez-Cambronero, J. S6K is a morphogenic protein with a mechanism involving Filamin-A phosphorylation and phosphatidic acid binding. FASEB J. 2015, 29, 1299–1313. [Google Scholar] [CrossRef] [PubMed]
- Stefanello, S.T.; Luchtefeld, I.; Liashkovich, I.; Pethö, Z.; Azzam, I.; Bulk, E.; Rosso, G.; Döhlinger, L.; Hesse, B.; Oeckinghaus, A.; et al. Impact of the Nuclear Envelope on Malignant Transformation, Motility, and Survival of Lung Cancer Cells. Adv. Sci. 2021, 8, e2102757. [Google Scholar] [CrossRef]
- Smith, E.R.; Leal, J.; Amaya, C.; Li, B.; Xu, X.-X. Nuclear Lamin A/C Expression Is a Key Determinant of Paclitaxel Sensitivity. Mol. Cell. Biol. 2021, 41, e0064820. [Google Scholar] [CrossRef] [PubMed]
- Muscarella, A.M.; Dai, W.; Mitchell, P.G.; Zhang, W.; Wang, H.; Jia, L.; Stossi, F.; Mancini, M.A.; Chiu, W.; Zhang, X.H.-F. Unique cellular protrusions mediate breast cancer cell migration by tethering to osteogenic cells. NPJ Breast Cancer 2020, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Hume, R.D.; Pensa, S.; Brown, E.J.; Kreuzaler, P.A.; Hitchcock, J.; Husmann, A.; Campbell, J.J.; Lloyd-Thomas, A.O.; Cameron, R.E.; Watson, C.J. Tumour cell invasiveness and response to chemotherapeutics in adipocyte invested 3D engineered anisotropic collagen scaffolds. Sci. Rep. 2018, 8, 12658. [Google Scholar] [CrossRef]
- Kang, D.Y.; Sp, N.; Kim, D.H.; Joung, Y.H.; Lee, H.G.; Park, Y.M.; Yang, Y.M. Salidroside inhibits migration, invasion and angiogenesis of MDA-MB 231 TNBC cells by regulating EGFR/Jak2/STAT3 signaling via MMP2. Int. J. Oncol. 2018, 53, 877–885. [Google Scholar] [CrossRef]
- Chun, J.; Kim, Y.S. Platycodin D inhibits migration, invasion, and growth of MDA-MB-231 human breast cancer cells via suppression of EGFR-mediated Akt and MAPK pathways. Chem. Biol. Interact. 2013, 205, 212–221. [Google Scholar] [CrossRef]
- Hsieh, C.-Y.; Tsai, P.-C.; Tseng, C.-H.; Chen, Y.-L.; Chang, L.-S.; Lin, S.-R. Inhibition of EGF/EGFR activation with naphtho[1,2-b]furan-4,5-dione blocks migration and invasion of MDA-MB-231 cells. Toxicol. Vitr. 2013, 27, 1–10. [Google Scholar] [CrossRef]
- Cui, W.; Zhang, S.; Shan, C.; Zhou, L.; Zhou, Z. microRNA-133a regulates the cell cycle and proliferation of breast cancer cells by targeting epidermal growth factor receptor through the EGFR/Akt signaling pathway. FEBS J. 2013, 280, 3962–3974. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Min, K.-N.; Joung, K.-E.; Kim, D.-K.; Sheen, Y.-Y. Anti-Cancer Effect of IN-2001 in MDA-MB-231 Human Breast Cancer. Biomol. Ther. 2012, 20, 313–319. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Keam, B.; Im, S.-A.; Lee, K.-H.; Han, S.-W.; Oh, D.-Y.; Kim, J.H.; Lee, S.-H.; Han, W.; Kim, D.-W.; Kim, T.-Y.; et al. Ki-67 can be used for further classification of triple negative breast cancer into two subtypes with different response and prognosis. Breast Cancer Res. 2011, 13, R22. [Google Scholar] [CrossRef]
- de Azambuja, E.; Cardoso, F.; de Castro, G.; Colozza, M.; Mano, M.S.; Durbecq, V.; Sotiriou, C.; Larsimont, D.; Piccart-Gebhart, M.J.; Paesmans, M. Ki-67 as prognostic marker in early breast cancer: A meta-analysis of published studies involving 12 155 patients. Br. J. Cancer 2007, 96, 1504–1513. [Google Scholar] [CrossRef]
- Martin, J.L.; Julovi, S.M.; Lin, M.Z.; de Silva, H.C.; Boyle, F.M.; Baxter, R.C. Inhibition of basal-like breast cancer growth by FTY720 in combination with epidermal growth factor receptor kinase blockade. Breast Cancer Res. 2017, 19, 90. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.A.; Biggs, W.H., III; Treiber, D.K.; Atteridge, C.E.; Azimioara, M.D.; Benedetti, M.G.; Carter, T.A.; Ciceri, P.; Edeen, P.T.; Floyd, M.; et al. A small molecule–kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [Google Scholar] [CrossRef]
- Shigekawa, T.; Ijichi, N.; Ikeda, K.; Horie-Inoue, K.; Shimizu, C.; Saji, S.; Aogi, K.; Tsuda, H.; Osaki, A.; Saeki, T.; et al. FOXP1, an Estrogen-Inducible Transcription Factor, Modulates Cell Proliferation in Breast Cancer Cells and 5-Year Recurrence-Free Survival of Patients with Tamoxifen-Treated Breast Cancer. Horm. Cancer 2011, 2, 286–297. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Molecule (Mol) ID | Chemical Formula | ZINC ID | Molecular Weight (g/mol) |
|---|---|---|---|---|
| EGFR | 1 | C14H10BrN3 | ZINC132618 | 290.153 |
| 2 | C14H8BrClFN3 | ZINC955717 | 352.594 | |
| 3 | C14H9BrFN3 | ZINC99087 | 318.149 | |
| 4 | C14H11N3O | ZINC65031 | 237.262 | |
| 5 | C18H12BrN3 | ZINC94936 | 350.219 | |
| 6 | C13H12N6 | ZINC4710712 | 252.281 | |
| 7 | C20H17N3OS | ZINC2664933 | 347.443 | |
| 8 | C17H18N4O2S | ZINC71920558 | 342.424 | |
| 9 | C20H17N3 | ZINC13863969 | 299.377 | |
| 10 | C22H21FN6 | ZINC9074069 | 388.45 | |
| 11 | C18H20FN7O | ZINC13010674 | 369.404 | |
| 12 | C18H21N7O | ZINC22735958 | 351.414 | |
| 13 | C19H15N3OS | ZINC117048 | 333.416 | |
| 14 | C14H9ClIN3 | ZINC955103 | 381.604 | |
| 15 | C15H12BrN3 | ZINC122234 | 314.186 | |
| 16 | C14H9Cl2N3 | ZINC140100 | 300.156 | |
| 17 | C14H10BrN3 | ZINC144105 | 300.159 | |
| 18 | C16H14ClN3 | ZINC48331888 | 283.762 | |
| PI3Kp110β | 19 | C27H36F2N4OS | ZINC20729292 | 502.675 |
| 20 | C28H30FN3O3 | ZINC36307506 | 475.564 | |
| 21 | C27H31N3O3S | ZINC9873787 | 477.63 | |
| 22 | C22H23NO4 | ZINC977288 | 365.429 | |
| 23 | C30H42N2O9 | ZINC68202727 | 574.671 | |
| 24 | C21H23ClFNO5 | ZINC218287337 | 423.868 | |
| EGFR + PI3K | 25 | C24H25ClFN5O3 | ZINC27439698 | 485.947 |
| 26 | C28H21Cl2N3O3 | ZINC20615563 | 518.4 | |
| 27 | C17H13BrN4O | ZINC1488208 | 369.222 |
| Target Protein | Primary Antibody | Secondary Antibody |
|---|---|---|
| Cyclin D2 | Cyclin D2 (1:100) | Alexa Fluor® 555 (1:500) |
| Thermo Fisher Scientific, | Thermo Fisher Scientific, | |
| #AHF0112, Mouse | #A31570, Donkey Anti-Mouse | |
| FOXP1 | FOXP1 (1:200) | Alexa Fluor® 555 (1:500) |
| Thermo Fisher Scientific, | Thermo Fisher Scientific, | |
| #PA5-52006, Rabbit | #A21428 Goat Anti-Rabbit | |
| Ki-67 | Ki-67 (1:100) | Alexa Fluor® 555 (1:500) |
| Thermo Fisher Scientific, | Thermo Fisher Scientific, | |
| #PA5-19462, Rabbit | #A21428 Goat Anti-Rabbit | |
| MEF2C | MEF2C (1:50) | Alexa Fluor® 555 (1:500) |
| Thermo Fisher Scientific, | Thermo Fisher Scientific, | |
| #PA5-28247, Rabbit | #A21428 Goat Anti-Rabbit |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia, A.R.; Mendes, A.; Custódia, C.; Faria, C.C.; Barata, J.T.; Malhó, R.; Figueira, I.; Brito, M.A. Abrogating Metastatic Properties of Triple-Negative Breast Cancer Cells by EGFR and PI3K Dual Inhibitors. Cancers 2023, 15, 3973. https://doi.org/10.3390/cancers15153973
Garcia AR, Mendes A, Custódia C, Faria CC, Barata JT, Malhó R, Figueira I, Brito MA. Abrogating Metastatic Properties of Triple-Negative Breast Cancer Cells by EGFR and PI3K Dual Inhibitors. Cancers. 2023; 15(15):3973. https://doi.org/10.3390/cancers15153973
Chicago/Turabian StyleGarcia, Ana Rita, Avilson Mendes, Carlos Custódia, Cláudia C. Faria, João T. Barata, Rui Malhó, Inês Figueira, and Maria Alexandra Brito. 2023. "Abrogating Metastatic Properties of Triple-Negative Breast Cancer Cells by EGFR and PI3K Dual Inhibitors" Cancers 15, no. 15: 3973. https://doi.org/10.3390/cancers15153973
APA StyleGarcia, A. R., Mendes, A., Custódia, C., Faria, C. C., Barata, J. T., Malhó, R., Figueira, I., & Brito, M. A. (2023). Abrogating Metastatic Properties of Triple-Negative Breast Cancer Cells by EGFR and PI3K Dual Inhibitors. Cancers, 15(15), 3973. https://doi.org/10.3390/cancers15153973