
Innovative Breakthroughs for the Treatment of Advanced and Metastatic Synovial Sarcoma

 ,
,  , , and
, , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. SyS Molecular Biology
3. SyS Current Therapies
3.1. SyS Standard of Care in the Front-Line Setting
3.2. SyS Second Line Setting and Beyond
3.2.1. Trabectedin
3.2.2. Pazopanib and Tyrosine Kinase Inhibitors
4. SyS Innovative Therapies and Ongoing Clinical Trials
4.1. Epigenetic Modifiers
4.2. BRD9 Degraders
5. Immunotherapy in Advanced Metastatic SyS: Selected Use of Immune Check Point Inhibitors and Adoptive Transfer of Engineered Immune Effectors
5.1. Immune Checkpoint Inhibitors in SyS
Biomarkers of the Immune Tumor Microenvironment
5.2. Adoptive T-Cell-Based Cancer Immunotherapy Targeting Cancer/Testis Antigens in SyS
5.2.1. NY-ESO-1
5.2.2. MAGE-A4
5.2.3. PRAME
6. Conclusions
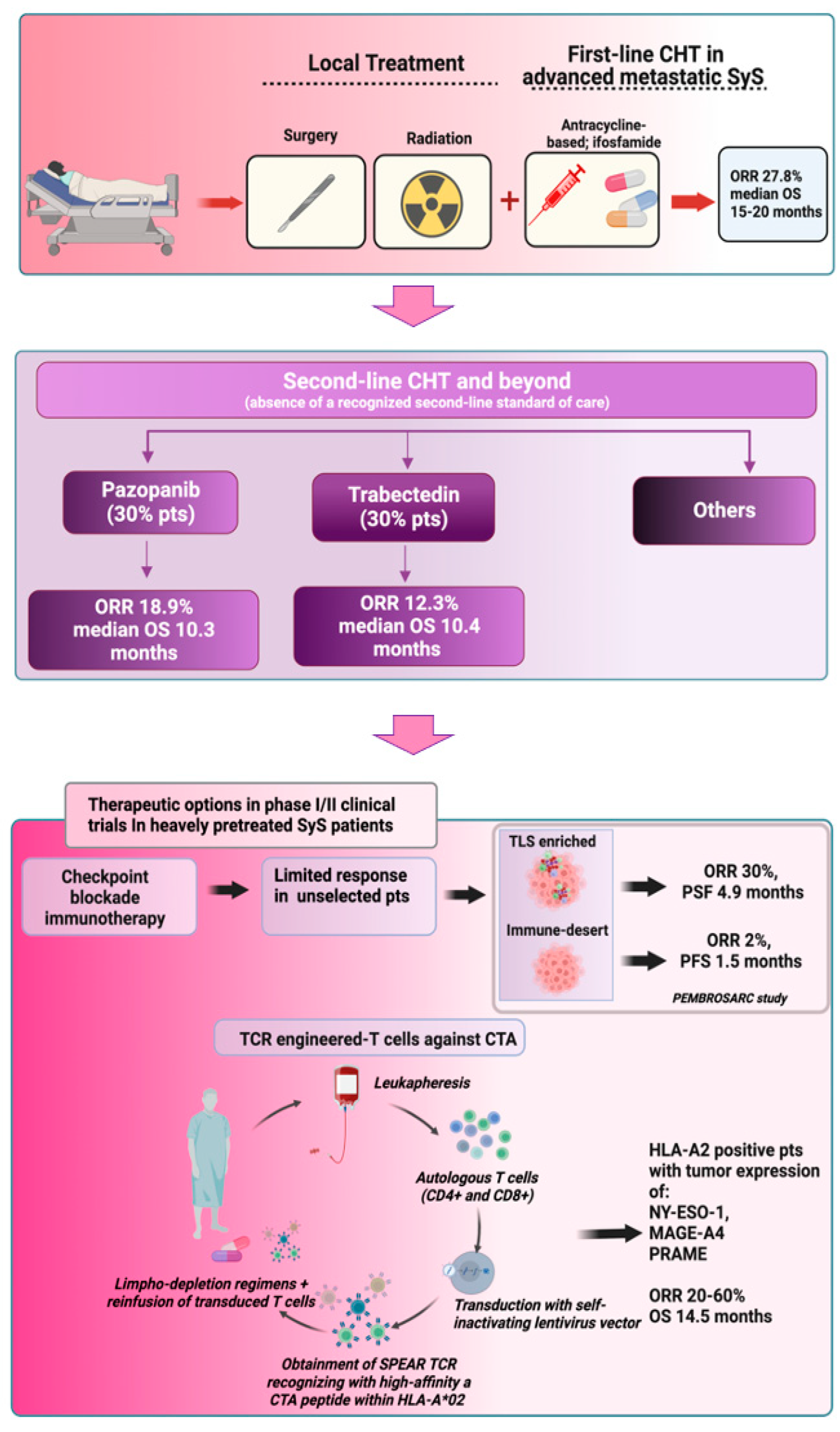
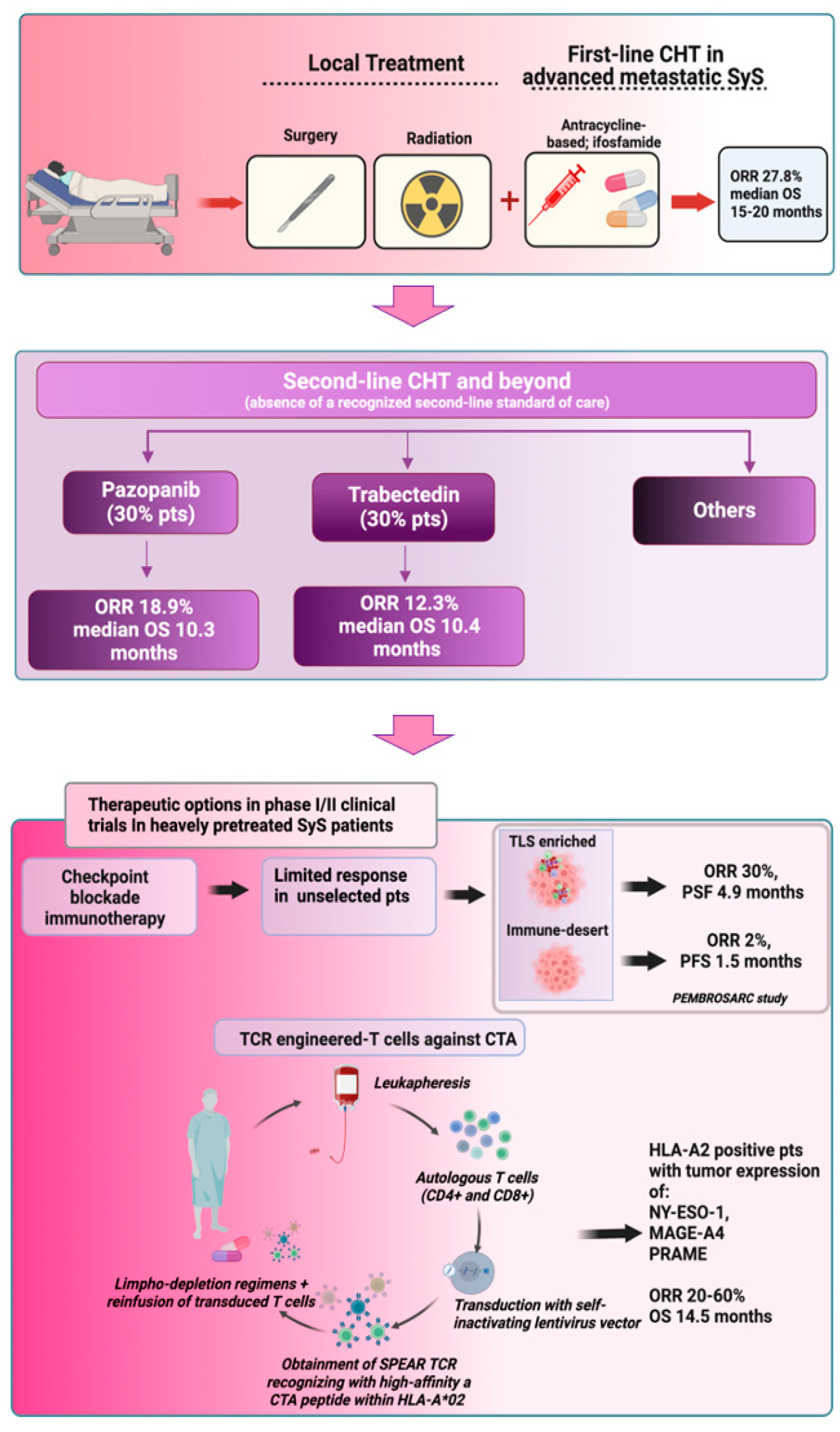
- Current front line treatments, represented by anthracycline and ifosfamide-based chemotherapy, can offer a limited response rate in advanced metastatic SyS, which is below 30%. Response rates further decrease in second line and beyond settings, where in the absence of a recognized standard of care, pazopanib and trabectedin are the most used drugs.
- Epigenetic modifiers, such as HDAC inhibitors and EZH2 inhibitors, have not shown anti-tumor efficacy in SyS in early clinical trials. Other epigenetic drugs, such as BRD9 degraders based on PROTAC technology, are now entering clinical evaluation in SyS.
- Clinical trial results indicate that genetic signatures and biomarkers of the tumor immune microenvironment are highly relevant and predictive of response to both chemotherapy and immunological approaches.
- The use of ICIs in SyS is still challenging. SyS did not emerge as an ICI-sensitive tumor. To improve the response rate, future studies should evaluate ICI-based approaches in selected patients based on tumor immune microenvironment markers.
- Adoptive transfer of TCR-engineered T-cells targeting cancer/testis antigens highly expressed in SyS (NY-ESO-1, MAGE-A4, PRAME) has achieved remarkable ORR in early clinical trials in heavily pretreated advanced metastatic SyS patients with CTA-positive tumors and expressing HLA-A2 aplotype.
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Gambarotti, M. Synovial Sarcoma (SS). In Atlas of Musculoskeletal Tumors and Tumorlike Lesions: The Rizzoli Case Archive; Picci, P., Manfrini, M., Fabbri, N., Gambarotti, M., Vanel, D., Eds.; Springer International Publishing: Cham, Switzerland, 2014; pp. 359–364. [Google Scholar] [CrossRef]
- Gazendam, A.M.; Popovic, S.; Munir, S.; Parasu, N.; Wilson, D.; Ghert, M. Synovial Sarcoma: A Clinical Review. Curr. Oncol. 2021, 28, 1909–1920. [Google Scholar] [CrossRef] [PubMed]
- Hale, R.; Sandakly, S.; Shipley, J.; Walters, Z. Epigenetic Targets in Synovial Sarcoma: A Mini-Review. Front. Oncol. 2019, 9, 1078. [Google Scholar] [CrossRef]
- Vlenterie, M.; Litiere, S.; Rizzo, E.; Marreaud, S.; Judson, I.; Gelderblom, H.; Le Cesne, A.; Wardelmann, E.; Messiou, C.; Gronchi, A.; et al. Outcome of chemotherapy in advanced synovial sarcoma patients: Review of 15 clinical trials from the European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group; setting a new landmark for studies in this entity. Eur. J. Cancer 2016, 58, 62–72. [Google Scholar] [CrossRef]
- Guillou, L.; Benhattar, J.; Bonichon, F.; Gallagher, G.; Terrier, P.; Stauffer, E.; Somerhausen Nde, S.; Michels, J.J.; Jundt, G.; Vince, D.R.; et al. Histologic grade, but not SYT-SSX fusion type, is an important prognostic factor in patients with synovial sarcoma: A multicenter, retrospective analysis. J. Clin. Oncol. 2004, 22, 4040–4050. [Google Scholar] [CrossRef] [PubMed]
- Baranov, E.; McBride, M.J.; Bellizzi, A.M.; Ligon, A.H.; Fletcher, C.D.M.; Kadoch, C.; Hornick, J.L. A Novel SS18-SSX Fusion-specific Antibody for the Diagnosis of Synovial Sarcoma. Am. J. Surg. Pathol. 2020, 44, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Landuzzi, L.; Ruzzi, F.; Lollini, P.L.; Scotlandi, K. Synovial Sarcoma Preclinical Modeling: Integrating Transgenic Mouse Models and Patient-Derived Models for Translational Research. Cancers 2023, 15, 588. [Google Scholar] [CrossRef]
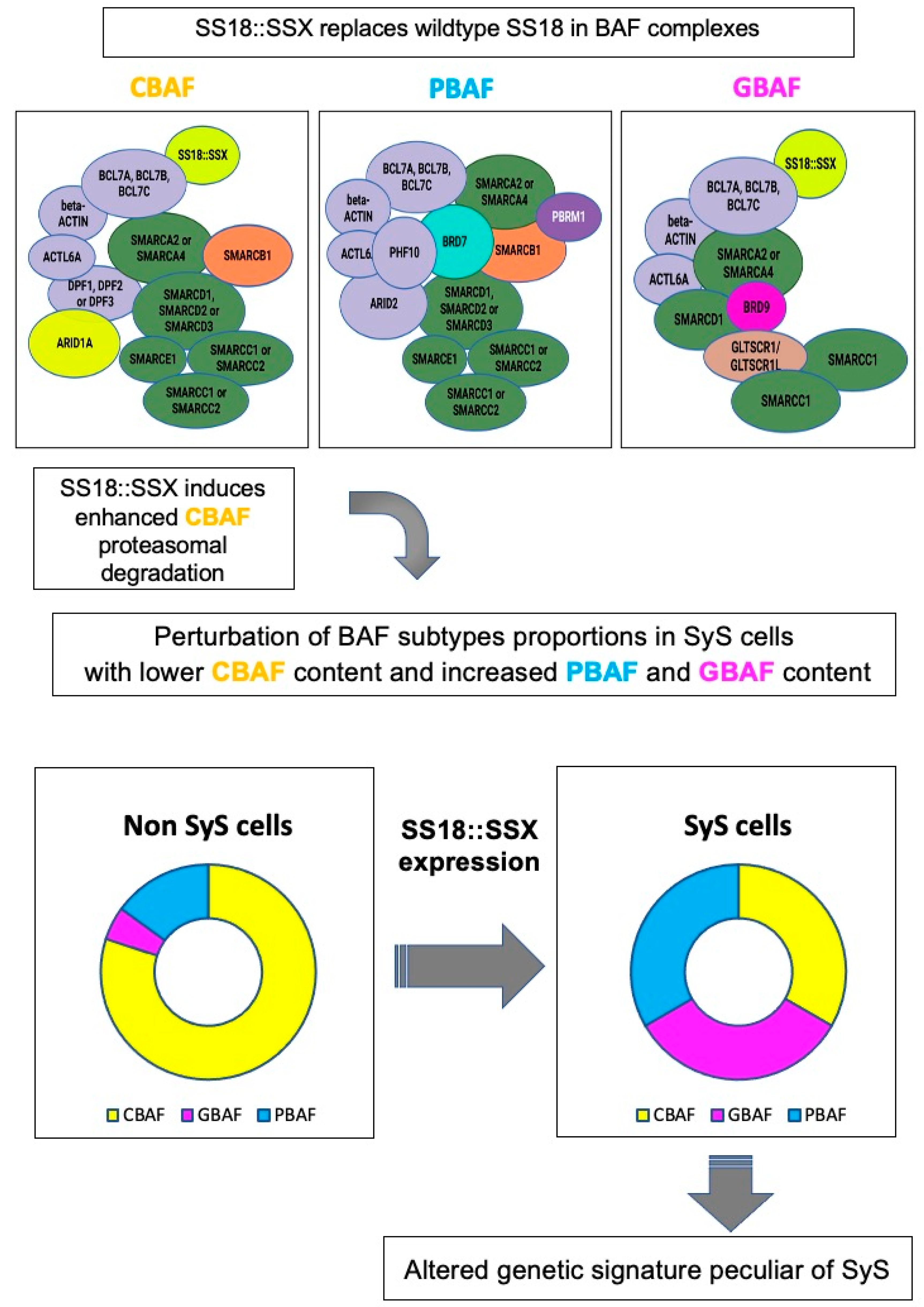
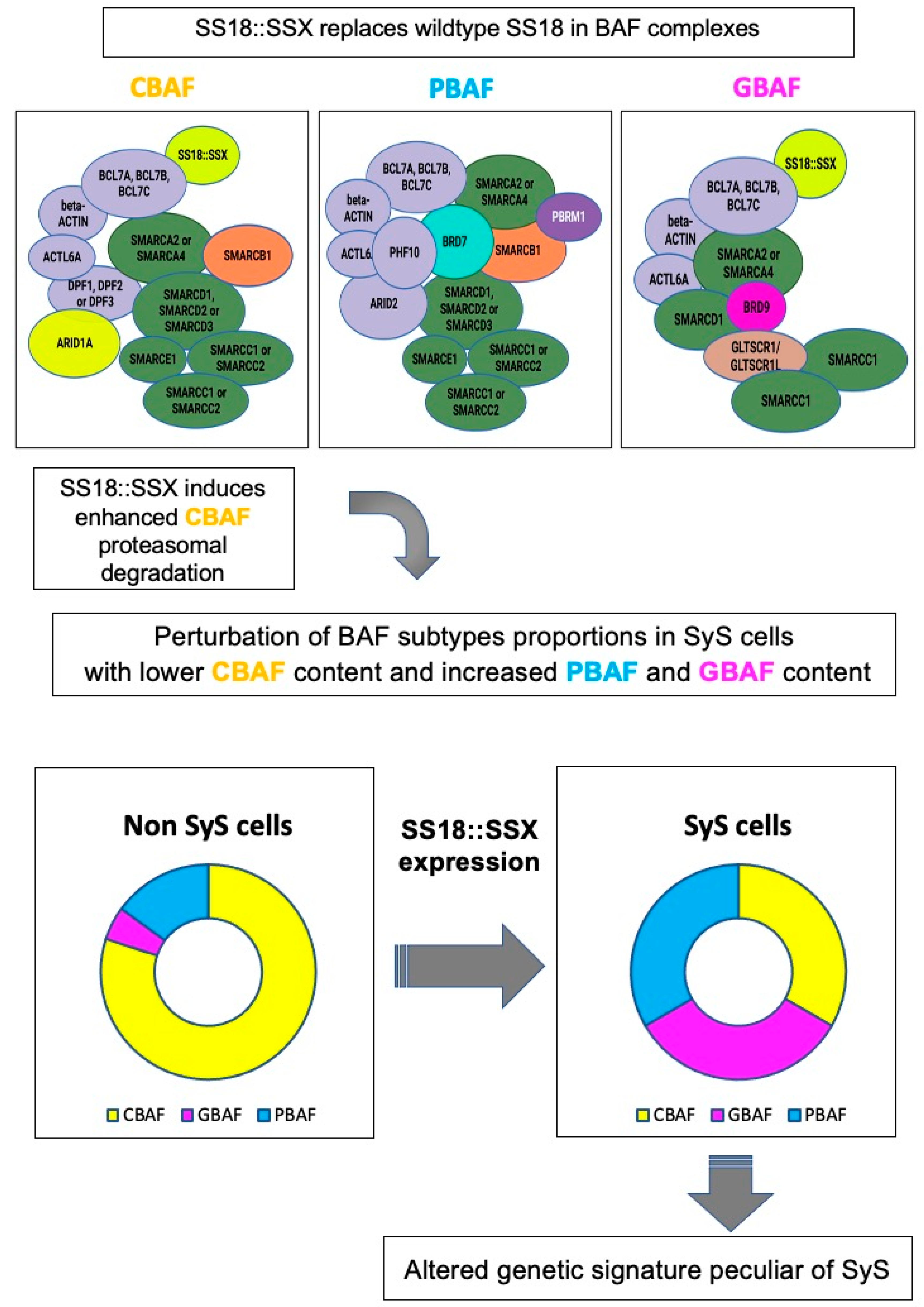
- Li, J.; Mulvihill, T.S.; Li, L.; Barrott, J.J.; Nelson, M.L.; Wagner, L.; Lock, I.C.; Pozner, A.; Lambert, S.L.; Ozenberger, B.B.; et al. A Role for SMARCB1 in Synovial Sarcomagenesis Reveals That SS18-SSX Induces Canonical BAF Destruction. Cancer Discov. 2021, 11, 2620–2637. [Google Scholar] [CrossRef]
- El Beaino, M.; Rassy, E.; Hadid, B.; Araujo, D.M.; Pavlidis, N.; Lin, P.P. Synovial Sarcoma: A Complex Disease with Multifaceted Signaling and Epigenetic Landscapes. Curr. Oncol. Rep. 2020, 22, 124. [Google Scholar] [CrossRef]
- Nacev, B.A.; Jones, K.B.; Intlekofer, A.M.; Yu, J.S.E.; Allis, C.D.; Tap, W.D.; Ladanyi, M.; Nielsen, T.O. The epigenomics of sarcoma. Nat. Rev. Cancer 2020, 20, 608–623. [Google Scholar] [CrossRef]
- Michel, B.C.; D’Avino, A.R.; Cassel, S.H.; Mashtalir, N.; McKenzie, Z.M.; McBride, M.J.; Valencia, A.M.; Zhou, Q.; Bocker, M.; Soares, L.M.M.; et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat. Cell Biol. 2018, 20, 1410–1420. [Google Scholar] [CrossRef]
- Piunti, A.; Shilatifard, A. The roles of Polycomb repressive complexes in mammalian development and cancer. Nat. Rev. Mol. Cell Biol. 2021, 22, 326–345. [Google Scholar] [CrossRef]
- McBride, M.J.; Pulice, J.L.; Beird, H.C.; Ingram, D.R.; D’Avino, A.R.; Shern, J.F.; Charville, G.W.; Hornick, J.L.; Nakayama, R.T.; Garcia-Rivera, E.M.; et al. The SS18-SSX Fusion Oncoprotein Hijacks BAF Complex Targeting and Function to Drive Synovial Sarcoma. Cancer Cell 2018, 33, 1128–1141.e127. [Google Scholar] [CrossRef] [Green Version]
- Boulay, G.; Cironi, L.; Garcia, S.P.; Rengarajan, S.; Xing, Y.H.; Lee, L.; Awad, M.E.; Naigles, B.; Iyer, S.; Broye, L.C.; et al. The chromatin landscape of primary synovial sarcoma organoids is linked to specific epigenetic mechanisms and dependencies. Life Sci. Alliance 2021, 4, e202000808. [Google Scholar] [CrossRef]
- Cooley, C.; Su, L. HDAC2 links ubiquitination to tumor suppression in synovial sarcoma. Mol. Cell. Oncol. 2021, 8, 1914291. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Sampaio, A.V.; Jones, K.B.; Pacheco, M.; Goytain, A.; Lin, S.; Poulin, N.; Yi, L.; Rossi, F.M.; Kast, J.; et al. Deconstruction of the SS18-SSX fusion oncoprotein complex: Insights into disease etiology and therapeutics. Cancer Cell 2012, 21, 333–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, M.; Barys, L.; O’Reilly, T.; Young, S.; Gorbatcheva, B.; Monahan, J.; Zumstein-Mecker, S.; Choong, P.F.; Dickinson, I.; Crowe, P.; et al. Comprehensive mapping of p53 pathway alterations reveals an apparent role for both SNP309 and MDM2 amplification in sarcomagenesis. Clin. Cancer Res. 2011, 17, 416–426. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, K.; Ikeya, M.; Fukuta, M.; Woltjen, K.; Tamaki, S.; Takahara, N.; Kato, T., Jr.; Sato, S.; Otsuka, T.; Toguchida, J. Identification of target genes of synovial sarcoma-associated fusion oncoprotein using human pluripotent stem cells. Biochem. Biophys. Res. Commun. 2013, 432, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.O.; Poulin, N.M.; Ladanyi, M. Synovial sarcoma: Recent discoveries as a roadmap to new avenues for therapy. Cancer Discov. 2015, 5, 124–134. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Oda, Y.; Kawaguchi, K.; Takahira, T.; Yamamoto, H.; Tanaka, K.; Matsuda, S.; Sakamoto, A.; Iwamoto, Y.; Tsuneyoshi, M. PTEN and other tumor suppressor gene mutations as secondary genetic alterations in synovial sarcoma. Oncol. Rep. 2004, 11, 1011–1015. [Google Scholar] [CrossRef]
- Saito, T.; Oda, Y.; Sakamoto, A.; Kawaguchi, K.; Tanaka, K.; Matsuda, S.; Tamiya, S.; Iwamoto, Y.; Tsuneyoshi, M. APC mutations in synovial sarcoma. J. Pathol. 2002, 196, 445–449. [Google Scholar] [CrossRef]
- Subramaniam, M.M.; Calabuig-Farinas, S.; Pellin, A.; Llombart-Bosch, A. Mutational analysis of E-cadherin, beta-catenin and APC genes in synovial sarcomas. Histopathology 2010, 57, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Naka, N.; Takenaka, S.; Araki, N.; Miwa, T.; Hashimoto, N.; Yoshioka, K.; Joyama, S.; Hamada, K.; Tsukamoto, Y.; Tomita, Y.; et al. Synovial sarcoma is a stem cell malignancy. Stem Cells 2010, 28, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Moreau-Bachelard, C.; Campion, L.; Toulmonde, M.; Le Cesne, A.; Brahmi, M.; Italiano, A.; Mir, O.; Piperno-Neumann, S.; Laurence, V.; Firmin, N.; et al. Patterns of care and outcomes of 417 patients with METAstatic SYNovial sarcoma (METASYN): Real-life data from the French Sarcoma Group (FSG). ESMO Open 2022, 7, 100402. [Google Scholar] [CrossRef]
- Carroll, C.; Patel, N.; Gunsoy, N.B.; Stirnadel-Farrant, H.A.; Pokras, S. Meta-analysis of pazopanib and trabectedin effectiveness in previously treated metastatic synovial sarcoma (second-line setting and beyond). Future Oncol. 2022, 18, 3651–3665. [Google Scholar] [CrossRef] [PubMed]
- Pokras, S.; Tseng, W.Y.; Espirito, J.L.; Beeks, A.; Culver, K.; Nadler, E. Treatment patterns and outcomes in metastatic synovial sarcoma: A real-world study in the US oncology network. Future Oncol. 2022, 18, 3637–3650. [Google Scholar] [CrossRef]
- Schoffski, P.; Ray-Coquard, I.L.; Cioffi, A.; Bui, N.B.; Bauer, S.; Hartmann, J.T.; Krarup-Hansen, A.; Grunwald, V.; Sciot, R.; Dumez, H.; et al. Activity of eribulin mesylate in patients with soft-tissue sarcoma: A phase 2 study in four independent histological subtypes. Lancet Oncol. 2011, 12, 1045–1052. [Google Scholar] [CrossRef]
- Kantidakis, G.; Litiere, S.; Neven, A.; Vinches, M.; Judson, I.; Blay, J.Y.; Wardelmann, E.; Stacchiotti, S.; D’Ambrosio, L.; Marreaud, S.; et al. New benchmarks to design clinical trials with advanced or metastatic liposarcoma or synovial sarcoma patients: An EORTC—Soft Tissue and Bone Sarcoma Group (STBSG) meta-analysis based on a literature review for soft-tissue sarcomas. Eur. J. Cancer 2022, 174, 261–276. [Google Scholar] [CrossRef]
- Garcia-Carbonero, R.; Supko, J.G.; Maki, R.G.; Manola, J.; Ryan, D.P.; Harmon, D.; Puchalski, T.A.; Goss, G.; Seiden, M.V.; Waxman, A.; et al. Ecteinascidin-743 (ET-743) for chemotherapy-naive patients with advanced soft tissue sarcomas: Multicenter phase II and pharmacokinetic study. J. Clin. Oncol. 2005, 23, 5484–5492. [Google Scholar] [CrossRef]
- Le Cesne, A.; Cresta, S.; Maki, R.G.; Blay, J.Y.; Verweij, J.; Poveda, A.; Casali, P.G.; Balana, C.; Schoffski, P.; Grosso, F.; et al. A retrospective analysis of antitumour activity with trabectedin in translocation-related sarcomas. Eur. J. Cancer 2012, 48, 3036–3044. [Google Scholar] [CrossRef]
- Sanfilippo, R.; Dileo, P.; Blay, J.Y.; Constantinidou, A.; Le Cesne, A.; Benson, C.; Vizzini, L.; Contu, M.; Baldi, G.G.; Dei Tos, A.P.; et al. Trabectedin in advanced synovial sarcomas: A multicenter retrospective study from four European institutions and the Italian Rare Cancer Network. Anticancer Drugs 2015, 26, 678–681. [Google Scholar] [CrossRef] [Green Version]
- Kawai, A.; Araki, N.; Sugiura, H.; Ueda, T.; Yonemoto, T.; Takahashi, M.; Morioka, H.; Hiraga, H.; Hiruma, T.; Kunisada, T.; et al. Trabectedin monotherapy after standard chemotherapy versus best supportive care in patients with advanced, translocation-related sarcoma: A randomised, open-label, phase 2 study. Lancet Oncol. 2015, 16, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.C.; Van Tine, B.A. Relapsed Synovial Sarcoma: Treatment Options. Curr. Treat. Options Oncol. 2023, 24, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Mo, F.; Pu, L.; Li, Q.; Ma, X. Pretreatment Inflammatory Indexes as Prognostic Predictors of Survival in Patients Suffering From Synovial Sarcoma. Front. Oncol. 2019, 9, 955. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ortega, D.Y.; Alvarez-Cano, A.; Sanchez-Llamas, L.A.; Caro-Sanchez, C.H.S.; Martinez-Said, H.; Luna-Ortiz, K.; Cuellar-Hubbe, M. Neutrophil/lymphocyte ratio is associated with survival in synovial sarcoma. Surg. Oncol. 2018, 27, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Fausti, V.; De Vita, A.; Vanni, S.; Ghini, V.; Gurrieri, L.; Riva, N.; Casadei, R.; Maraldi, M.; Ercolani, G.; Cavaliere, D.; et al. Systemic Inflammatory Indices in Second-Line Soft Tissue Sarcoma Patients: Focus on Lymphocyte/Monocyte Ratio and Trabectedin. Cancers 2023, 15, 80. [Google Scholar] [CrossRef]
- Mir, O.; Brodowicz, T.; Italiano, A.; Wallet, J.; Blay, J.Y.; Bertucci, F.; Chevreau, C.; Piperno-Neumann, S.; Bompas, E.; Salas, S.; et al. Safety and efficacy of regorafenib in patients with advanced soft tissue sarcoma (REGOSARC): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016, 17, 1732–1742. [Google Scholar] [CrossRef]
- Brodowicz, T.; Mir, O.; Wallet, J.; Italiano, A.; Blay, J.Y.; Bertucci, F.; Eisterer, W.; Chevreau, C.; Piperno-Neumann, S.; Bompas, E.; et al. Efficacy and safety of regorafenib compared to placebo and to post-cross-over regorafenib in advanced non-adipocytic soft tissue sarcoma. Eur. J. Cancer 2018, 99, 28–36. [Google Scholar] [CrossRef]
- Wanior, M.; Kramer, A.; Knapp, S.; Joerger, A.C. Exploiting vulnerabilities of SWI/SNF chromatin remodelling complexes for cancer therapy. Oncogene 2021, 40, 3637–3654. [Google Scholar] [CrossRef]
- Damerell, V.; Pepper, M.S.; Prince, S. Molecular mechanisms underpinning sarcomas and implications for current and future therapy. Signal Transduct. Target. Ther. 2021, 6, 246. [Google Scholar] [CrossRef]
- Schmitt, T.; Mayer-Steinacker, R.; Mayer, F.; Grunwald, V.; Schutte, J.; Hartmann, J.T.; Kasper, B.; Husing, J.; Hajda, J.; Ottawa, G.; et al. Vorinostat in refractory soft tissue sarcomas—Results of a multi-centre phase II trial of the German Soft Tissue Sarcoma and Bone Tumour Working Group (AIO). Eur. J. Cancer 2016, 64, 74–82. [Google Scholar] [CrossRef]
- Cassier, P.A.; Lefranc, A.; Amela, E.Y.; Chevreau, C.; Bui, B.N.; Lecesne, A.; Ray-Coquard, I.; Chabaud, S.; Penel, N.; Berge, Y.; et al. A phase II trial of panobinostat in patients with advanced pretreated soft tissue sarcoma. A study from the French Sarcoma Group. Br. J. Cancer 2013, 109, 909–914. [Google Scholar] [CrossRef]
- Gounder, M.; Schoffski, P.; Jones, R.L.; Agulnik, M.; Cote, G.M.; Villalobos, V.M.; Attia, S.; Chugh, R.; Chen, T.W.; Jahan, T.; et al. Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: An international, open-label, phase 2 basket study. Lancet Oncol. 2020, 21, 1423–1432. [Google Scholar] [CrossRef] [PubMed]
- Bekes, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Fuchs, J.R.; Schulte, B.C.; Fuchs, J.W.; Agulnik, M. Emerging targeted and cellular therapies in the treatment of advanced and metastatic synovial sarcoma. Front. Oncol. 2023, 13, 1123464. [Google Scholar] [CrossRef] [PubMed]
- Jerby-Arnon, L.; Neftel, C.; Shore, M.E.; Weisman, H.R.; Mathewson, N.D.; McBride, M.J.; Haas, B.; Izar, B.; Volorio, A.; Boulay, G.; et al. Opposing immune and genetic mechanisms shape oncogenic programs in synovial sarcoma. Nat. Med. 2021, 27, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Korman, A.J.; Garrett-Thomson, S.C.; Lonberg, N. The foundations of immune checkpoint blockade and the ipilimumab approval decennial. Nat. Rev. Drug Discov. 2022, 21, 509–528. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Pardoll, D.M. Neoadjuvant checkpoint blockade for cancer immunotherapy. Science 2020, 367, eaax0182. [Google Scholar] [CrossRef] [PubMed]
- Versluis, J.M.; Long, G.V.; Blank, C.U. Learning from clinical trials of neoadjuvant checkpoint blockade. Nat. Med. 2020, 26, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Bellera, C.; D’Angelo, S. PD1/PD-L1 targeting in advanced soft-tissue sarcomas: A pooled analysis of phase II trials. J. Hematol. Oncol. 2020, 13, 55. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Burgess, M.; Bolejack, V.; Schuetze, S.M.; Tine, B.A.V.; Attia, S.; Riedel, R.F.; Hu, J.; Davis, L.E.; Okuno, S.H.; Priebat, D.A.; et al. Clinical activity of pembrolizumab (P) in undifferentiated pleomorphic sarcoma (UPS) and dedifferentiated/pleomorphic liposarcoma (LPS): Final results of SARC028 expansion cohorts. J. Clin. Oncol. 2019, 37, 11015. [Google Scholar] [CrossRef]
- Maki, R.G.; Jungbluth, A.A.; Gnjatic, S.; Schwartz, G.K.; D’Adamo, D.R.; Keohan, M.L.; Wagner, M.J.; Scheu, K.; Chiu, R.; Ritter, E.; et al. A Pilot Study of Anti-CTLA4 Antibody Ipilimumab in Patients with Synovial Sarcoma. Sarcoma 2013, 2013, 168145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somaiah, N.; Conley, A.P.; Parra, E.R.; Lin, H.; Amini, B.; Solis Soto, L.; Salazar, R.; Barreto, C.; Chen, H.; Gite, S.; et al. Durvalumab plus tremelimumab in advanced or metastatic soft tissue and bone sarcomas: A single-centre phase 2 trial. Lancet Oncol. 2022, 23, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Chawla, S.P.; Van Tine, B.A.; Pollack, S.M.; Ganjoo, K.N.; Elias, A.D.; Riedel, R.F.; Attia, S.; Choy, E.; Okuno, S.H.; Agulnik, M.; et al. Phase II Randomized Study of CMB305 and Atezolizumab Compared With Atezolizumab Alone in Soft-Tissue Sarcomas Expressing NY-ESO-1. J. Clin. Oncol. 2022, 40, 1291–1300. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Richards, A.L.; Conley, A.P.; Woo, H.J.; Dickson, M.A.; Gounder, M.; Kelly, C.; Keohan, M.L.; Movva, S.; Thornton, K.; et al. Pilot study of bempegaldesleukin in combination with nivolumab in patients with metastatic sarcoma. Nat. Commun. 2022, 13, 3477. [Google Scholar] [CrossRef] [PubMed]
- Champiat, S.; Dercle, L.; Ammari, S.; Massard, C.; Hollebecque, A.; Postel-Vinay, S.; Chaput, N.; Eggermont, A.; Marabelle, A.; Soria, J.C.; et al. Hyperprogressive Disease Is a New Pattern of Progression in Cancer Patients Treated by Anti-PD-1/PD-L1. Clin. Cancer Res. 2017, 23, 1920–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klemen, N.D.; Hwang, S.; Bradic, M.; Rosenbaum, E.; Dickson, M.A.; Gounder, M.M.; Kelly, C.M.; Keohan, M.L.; Movva, S.; Thornton, K.A.; et al. Long-term Follow-up and Patterns of Response, Progression, and Hyperprogression in Patients after PD-1 Blockade in Advanced Sarcoma. Clin. Cancer Res. 2022, 28, 939–947. [Google Scholar] [CrossRef]
- Gordon, E.M.; Chawla, S.P.; Tellez, W.A.; Younesi, E.; Thomas, S.; Chua-Alcala, V.S.; Chomoyan, H.; Valencia, C.; Brigham, D.A.; Moradkhani, A.; et al. SAINT: A Phase I/Expanded Phase II Study Using Safe Amounts of Ipilimumab, Nivolumab and Trabectedin as First-Line Treatment of Advanced Soft Tissue Sarcoma. Cancers 2023, 15, 906. [Google Scholar] [CrossRef]
- Seong, G.; D’Angelo, S.P. New therapeutics for soft tissue sarcomas: Overview of current immunotherapy and future directions of soft tissue sarcomas. Front. Oncol. 2023, 13, 1150765. [Google Scholar] [CrossRef]
- Petitprez, F.; de Reynies, A.; Keung, E.Z.; Chen, T.W.; Sun, C.M.; Calderaro, J.; Jeng, Y.M.; Hsiao, L.P.; Lacroix, L.; Bougouin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef]
- Italiano, A.; Bessede, A.; Pulido, M.; Bompas, E.; Piperno-Neumann, S.; Chevreau, C.; Penel, N.; Bertucci, F.; Toulmonde, M.; Bellera, C.; et al. Pembrolizumab in soft-tissue sarcomas with tertiary lymphoid structures: A phase 2 PEMBROSARC trial cohort. Nat. Med. 2022, 28, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Bazhin, A.V.; Pretzsch, E.; Jacob, S.; Yu, H.; Zhu, J.; Albertsmeier, M.; Lindner, L.H.; Knosel, T.; Werner, J.; et al. A novel immune-related gene signature predicting survival in sarcoma patients. Mol. Ther. Oncolytics 2022, 24, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, E.; Seier, K.; Bandlamudi, C.; Dickson, M.; Gounder, M.; Keohan, M.L.; Chi, P.; Kelly, C.; Movva, S.; Nacev, B.; et al. HLA Genotyping in Synovial Sarcoma: Identifying HLA-A*02 and Its Association with Clinical Outcome. Clin. Cancer Res. 2020, 26, 5448–5455. [Google Scholar] [CrossRef] [PubMed]
- Iura, K.; Maekawa, A.; Kohashi, K.; Ishii, T.; Bekki, H.; Otsuka, H.; Yamada, Y.; Yamamoto, H.; Harimaya, K.; Iwamoto, Y.; et al. Cancer-testis antigen expression in synovial sarcoma: NY-ESO-1, PRAME, MAGEA4, and MAGEA1. Hum. Pathol. 2017, 61, 130–139. [Google Scholar] [CrossRef]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: Long-term follow-up and correlates with response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef] [Green Version]
- Jungbluth, A.A.; Antonescu, C.R.; Busam, K.J.; Iversen, K.; Kolb, D.; Coplan, K.; Chen, Y.T.; Stockert, E.; Ladanyi, M.; Old, L.J. Monophasic and biphasic synovial sarcomas abundantly express cancer/testis antigen NY-ESO-1 but not MAGE-A1 or CT7. Int. J. Cancer 2001, 94, 252–256. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, S.P.; Melchiori, L.; Merchant, M.S.; Bernstein, D.; Glod, J.; Kaplan, R.; Grupp, S.; Tap, W.D.; Chagin, K.; Binder, G.K.; et al. Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 (c259)T Cells in Synovial Sarcoma. Cancer Discov. 2018, 8, 944–957. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, I.; Lowther, D.E.; Dryer-Minnerly, R.; Wang, R.; Fayngerts, S.; Nunez, D.; Betts, G.; Bath, N.; Tipping, A.J.; Melchiori, L.; et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J. Immunother. Cancer 2019, 7, 276. [Google Scholar] [CrossRef]
- Gyurdieva, A.; Zajic, S.; Chang, Y.F.; Houseman, E.A.; Zhong, S.; Kim, J.; Nathenson, M.; Faitg, T.; Woessner, M.; Turner, D.C.; et al. Biomarker correlates with response to NY-ESO-1 TCR T cells in patients with synovial sarcoma. Nat. Commun. 2022, 13, 5296. [Google Scholar] [CrossRef]
- Ishihara, M.; Nishida, Y.; Kitano, S.; Kawai, A.; Muraoka, D.; Momose, F.; Harada, N.; Miyahara, Y.; Seo, N.; Hattori, H.; et al. A phase 1 trial of NY-ESO-1-specific TCR-engineered T-cell therapy combined with a lymph node-targeting nanoparticulate peptide vaccine for the treatment of advanced soft tissue sarcoma. Int. J. Cancer 2023, 152, 2554–2566. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Busam, K.J.; Iversen, K.; Kolb, D.; Coplan, K.; Spagnoli, G.C.; Ladanyi, M.; Old, L.J.; Jungbluth, A.A. MAGE antigen expression in monophasic and biphasic synovial sarcoma. Hum. Pathol. 2002, 33, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, J.P.; Crowley, D.J.; Wiedermann, G.E.; Quinn, L.L.; Crossland, K.L.; Tunbridge, H.M.; Cornforth, T.V.; Barnes, C.S.; Ahmed, T.; Howe, K.; et al. Preclinical evaluation of an affinity-enhanced MAGE-A4-specific T-cell receptor for adoptive T-cell therapy. Oncoimmunology 2020, 9, 1682381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, D.S.; Van Tine, B.A.; Biswas, S.; McAlpine, C.; Johnson, M.L.; Olszanski, A.J.; Clarke, J.M.; Araujo, D.; Blumenschein, G.R., Jr.; Kebriaei, P.; et al. Autologous T cell therapy for MAGE-A4(+) solid cancers in HLA-A*02(+) patients: A phase 1 trial. Nat. Med. 2023, 29, 104–114. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Attia, S.; Blay, J.-Y.; Strauss, S.J.; Morales, C.M.V.; Razak, A.R.A.; Winkle, E.V.; Annareddy, T.; Sattigari, C.; Diamantopoulos, E.; et al. Identification of response stratification factors from pooled efficacy analyses of afamitresgene autoleucel (“Afami-cel” [Formerly ADP-A2M4]) in metastatic synovial sarcoma and myxoid/round cell liposarcoma phase 1 and phase 2 trials. J. Clin. Oncol. 2022, 40, 11562. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Tine, B.A.V.; Attia, S.; Blay, J.-Y.; Strauss, S.J.; Morales, C.M.V.; Razak, A.R.A.; Winkle, E.V.; Trivedi, T.; Biswas, S.; et al. SPEARHEAD-1: A phase 2 trial of afamitresgene autoleucel (Formerly ADP-A2M4) in patients with advanced synovial sarcoma or myxoid/round cell liposarcoma. J. Clin. Oncol. 2021, 39, 11504. [Google Scholar] [CrossRef]
- Roszik, J.; Wang, W.L.; Livingston, J.A.; Roland, C.L.; Ravi, V.; Yee, C.; Hwu, P.; Futreal, A.; Lazar, A.J.; Patel, S.R.; et al. Overexpressed PRAME is a potential immunotherapy target in sarcoma subtypes. Clin. Sarcoma Res. 2017, 7, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luk, S.J.; van der Steen, D.M.; Hagedoorn, R.S.; Jordanova, E.S.; Schilham, M.W.; Bovee, J.V.; Cleven, A.H.; Falkenburg, J.F.; Szuhai, K.; Heemskerk, M.H. PRAME and HLA Class I expression patterns make synovial sarcoma a suitable target for PRAME specific T-cell receptor gene therapy. Oncoimmunology 2018, 7, e1507600. [Google Scholar] [CrossRef] [PubMed]
- Wermke, M.; Tsimberidou, A.-M.; Mohamed, A.; Mayer-Mokler, A.; Satelli, A.; Reinhardt, C.; Araujo, D.; Maurer, D.; Blumenschein, G.J.; Singh, H.; et al. 959 Safety and anti-tumor activity of TCR-engineered autologous, PRAME-directed T cells across multiple advanced solid cancers at low doses—Clinical update on the ACTengine® IMA203 trial. J. ImmunoTher. Cancer 2021, 9, A1009. [Google Scholar] [CrossRef]
- D’Angelo, S.; Demetri, G.; Tine, B.; Druta, M.; Glod, J.; Chow, W.; Pandya, N.; Hasan, A.; Chiou, V.L.; Tress, J.; et al. 298 Final analysis of the phase 1 trial of NY-ESO-1–specific T-cell receptor (TCR) T-cell therapy (letetresgene autoleucel; GSK3377794) in patients with advanced synovial sarcoma (SS). J. ImmunoTher. Cancer 2020, 8, A325. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Target Antigen | Trial Number | Study Description | Main Results and References |
|---|---|---|---|
| NY-ESO-1 | NCT00670748 | Pilot phase I study: NY-ESO-1 TCR-T-cells plus IL2 in heavily pretreated metastatic HLA-A*02-positive SyS patients with >50% expression of NY-ESO-1 | ORR 61% objective clinical responses in 11/18 SyS patients [66] |
| NCT01343043 | Pilot phase I study: NY-ESO-1 TCR-T-cells plus IL2 in heavily pretreated metastatic HLA-A*02-positive SyS patients among four cohorts allocated between different levels of NY-ESO-1 expression and lympho-depletion regimens | ORR 20–50% [68,69,70,80] | |
| NCT03967223 | Phase II study: First generation of NY-ESO-1 TCR-T-cells as a first line treatment in advanced metastatic, previously untreated HLA-A*02-positive patients with NY-ESO-1-positive SyS or MRCLS and as a second line treatment after first line anthracycline-based chemotherapy | Active, not recruiting [70] | |
| NCT04526509 | Phase I master protocol of three different next generation NY-ESO-1 TCR-T-cell co-expressing CD8 alpha cell surface receptor, or co-expressing the dominant-negative TGF-beta receptor type II, or engineered using the epigenetically reprogrammed (Epi-R) manufacturing process | Active, not recruiting [70] | |
| NCT04939701 | Phase I/II trial: Human artificial adjuvant vector cells (aAVC) loaded with the CD1d ligand alpha-galactosylceramide and modified to express NY-ESO-1 in combination with pembrolizumab for SyS, MRCLS, ovarian carcinoma, non-small cell lung cancer, and esophageal squamous cell carcinoma | Active, not recruiting [45] | |
| MAGE-A4 | NCT03132922 | Phase I multi-tumor trial: Afami-cel (carrying TCR specific for a MAGE-A4230−239 peptide, GVYDGREHTV, presented by HLA-A*02) in HLA-A*02+ patients with advanced metastatic MAGE-A4-expressing solid tumors, across nine tumor types including SyS | ORR 44% for SyS and 9% for all other tumors [74] |
| NCT04044768 | Phase II SPEARHEAD-1 trial: A single arm open-label clinical trial on ADP-A2M4 SPEAR™ T-Cells in HLA-A*02 eligible and MAGE-A4 positive subjects with metastatic or inoperable SyS or MRCLS | ORR 40.7% in SyS [75,76] | |
| PRAME | NCT03686124 | The ACTENGINE IMA203/IMA203CD8 trial: TCR-T-cells directed against an HLA-A*02-restricted peptide derived from PRAME after lymphodepletion, with or without nivolumab in patients with advanced solid tumors | ORR 60% in SyS Objective clinical responses in 3/5 SyS patients [79] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Landuzzi, L.; Manara, M.C.; Pazzaglia, L.; Lollini, P.-L.; Scotlandi, K. Innovative Breakthroughs for the Treatment of Advanced and Metastatic Synovial Sarcoma. Cancers 2023, 15, 3887. https://doi.org/10.3390/cancers15153887
Landuzzi L, Manara MC, Pazzaglia L, Lollini P-L, Scotlandi K. Innovative Breakthroughs for the Treatment of Advanced and Metastatic Synovial Sarcoma. Cancers. 2023; 15(15):3887. https://doi.org/10.3390/cancers15153887
Chicago/Turabian StyleLanduzzi, Lorena, Maria Cristina Manara, Laura Pazzaglia, Pier-Luigi Lollini, and Katia Scotlandi. 2023. "Innovative Breakthroughs for the Treatment of Advanced and Metastatic Synovial Sarcoma" Cancers 15, no. 15: 3887. https://doi.org/10.3390/cancers15153887
APA StyleLanduzzi, L., Manara, M. C., Pazzaglia, L., Lollini, P.-L., & Scotlandi, K. (2023). Innovative Breakthroughs for the Treatment of Advanced and Metastatic Synovial Sarcoma. Cancers, 15(15), 3887. https://doi.org/10.3390/cancers15153887