

Environmental Determinants of Ferroptosis in Cancer


{kind=link}
{kind=link}
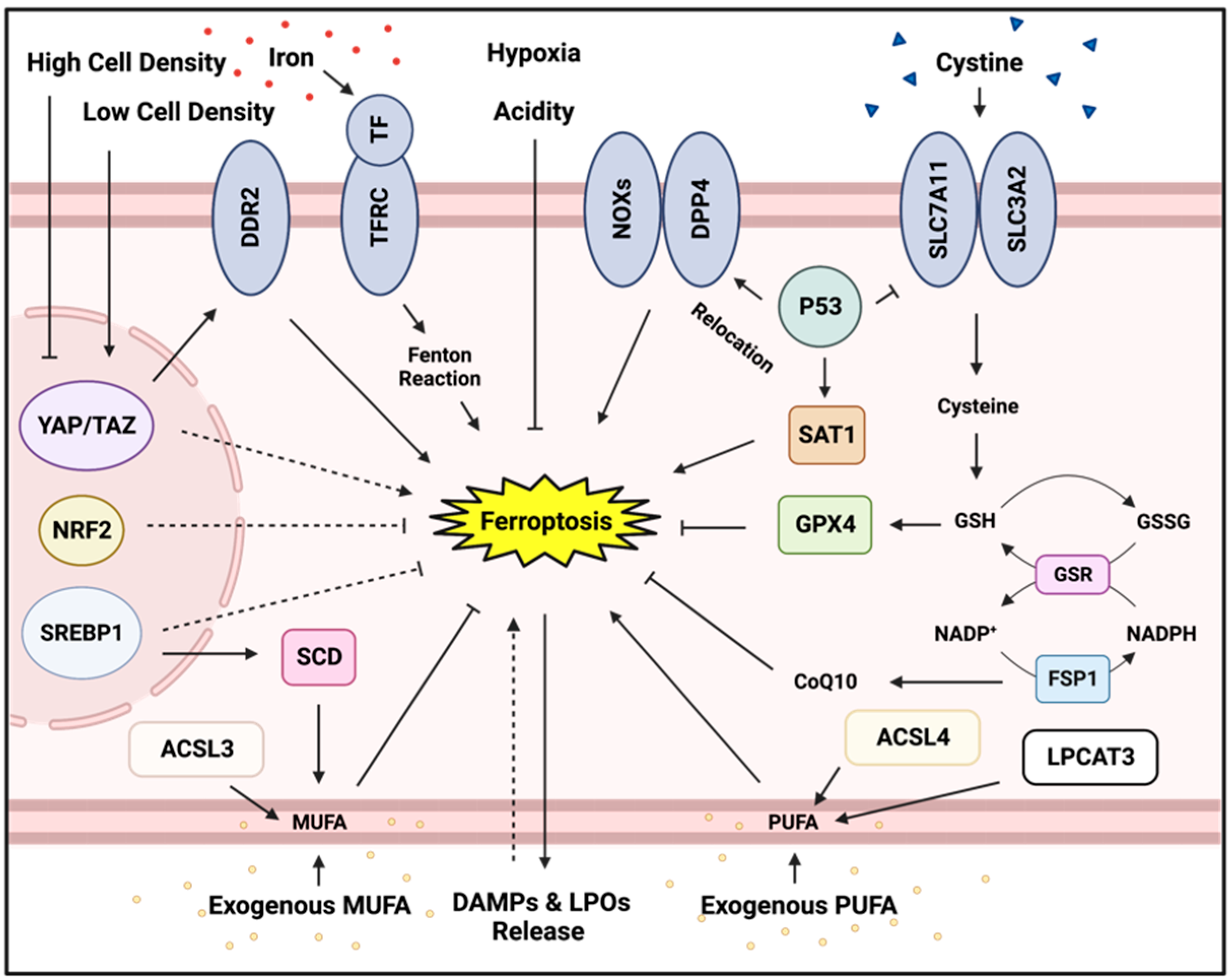
Abstract
:Simple Summary
Abstract
1. Introduction
2. Ferroptosis, an Iron-Dependent Form of Regulated Cell Death
3. Genetic Regulators and Protectors of Ferroptosis
3.1. The GPX4-Dependent and -Independent Antioxidant Defense Pathways
3.2. NADPH
3.3. DDR2
3.4. p53
3.5. SREBP1
4. Microenvironmental Regulators and Protectors of Ferroptosis
4.1. Cellular Contact
4.2. Epithelial-Mesenchymal Transition (EMT)
4.3. Mono- and Polyunsaturated Fatty Acids
4.4. Adjacent Cells and Exosomes
4.5. Bioenergetic Depletions
4.6. Lactate
4.7. Hypoxia and Acidity
4.8. Immune Cells
4.9. Broader Influences
5. Conclusions
6. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A. Hypoxia and drug resistance. Cancer Metastasis Rev. 1994, 13, 139–168. [Google Scholar] [CrossRef]
- Chen, J.L.; Lucas, J.E.; Schroeder, T.; Mori, S.; Wu, J.; Nevins, J.; Dewhirst, M.; West, M.; Chi, J.T. The genomic analysis of lactic acidosis and acidosis response in human cancers. PLoS Genet. 2008, 4, e1000293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.L.; Merl, D.; Peterson, C.W.; Wu, J.; Liu, P.Y.; Yin, H.; Muoio, D.M.; Ayer, D.E.; West, M.; Chi, J.T. Lactic acidosis triggers starvation response with paradoxical induction of TXNIP through MondoA. PLoS Genet. 2010, 6, e1001093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Wang, J.; Zhang, T.; Xu, D.; Hu, W.; Feng, Z. The interplay between tumor suppressor p53 and hypoxia signaling pathways in cancer. Front. Cell Dev. Biol. 2021, 9, 648808. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.H.; Zhang, X.P.; Liu, F.; Wang, W. Modeling the interplay between the HIF-1 and p53 pathways in hypoxia. Sci. Rep. 2015, 5, 13834. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef] [Green Version]
- Lucas, J.E.; Kung, H.N.; Chi, J.T. Latent factor analysis to discover pathway-associated putative segmental aneuploidies in human cancers. PLoS Comput. Biol. 2010, 6, e1000920. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Lucas, J.E.; Chen, J.L.; LaMonte, G.; Wu, J.; Wang, M.C.; Koumenis, C.; Chi, J.T. Functional interaction between responses to lactic acidosis and hypoxia regulates genomic transcriptional outputs. Cancer Res. 2012, 72, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Song, C.W.; Park, H.J.; Lee, C.K.; Griffin, R. Implications of increased tumor blood flow and oxygenation caused by mild temperature hyperthermia in tumor treatment. Int. J. Hyperthermia 2005, 21, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Tie, Y.; Tang, F.; Wei, Y.Q.; Wei, X.W. Immunosuppressive cells in cancer: Mechanisms and potential therapeutic targets. J. Hematol. Oncol. 2022, 15, 61. [Google Scholar] [CrossRef] [PubMed]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, G.; Zhuang, L.; Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef]
- Setayeshpour, Y.; Chi, J.T. Editorial: Novel insights into ferroptosis. Front. Cell Dev. Biol. 2021, 9, 754160. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.T.; Zhou, T.T.; Liu, B.; Bao, J.K. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef]
- Labi, V.; Erlacher, M. How cell death shapes cancer. Cell Death Dis. 2015, 6, e1675. [Google Scholar] [CrossRef] [Green Version]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef] [PubMed]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An overview from discovery, molecular mechanisms to physiology and pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021, 593, 586–590. [Google Scholar] [CrossRef]
- Mishima, E.; Nakamura, T.; Zheng, J.; Zhang, W.; Mourão, A.S.D.; Sennhenn, P.; Conrad, M. DHODH inhibitors sensitize to ferroptosis by FSP1 inhibition. Nature 2023, 619, E9–E18. [Google Scholar] [CrossRef]
- Lachaier, E.; Louandre, C.; Godin, C.; Saidak, Z.; Baert, M.; Diouf, M.; Chauffert, B.; Galmiche, A. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014, 34, 6417–6422. [Google Scholar] [PubMed]
- Eaton, J.K.; Furst, L.; Ruberto, R.A.; Moosmayer, D.; Hilpmann, A.; Ryan, M.J.; Zimmermann, K.; Cai, L.L.; Niehues, M.; Badock, V.; et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat. Chem. Biol. 2020, 16, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; You, E.; Jeong, J.; Ko, P.; Kim, J.W.; Rhee, S. DDR2 controls the epithelial-mesenchymal-transition-related gene expression via c-Myb acetylation upon matrix stiffening. Sci. Rep. 2017, 7, 6847. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, M.E.; Martin, E.E.; Anwar, T.; Arellano-Garcia, C.; Medhora, N.; Lama, A.; Chen, Y.C.; Tanager, K.S.; Yoon, E.; Kidwell, K.M.; et al. Mesenchymal stem cell-induced DDR2 mediates stromal-breast cancer interactions and metastasis Growth. Cell Rep. 2017, 18, 1215–1228. [Google Scholar] [CrossRef]
- Lin, C.C.; Yang, W.H.; Lin, Y.T.; Tang, X.; Chen, P.H.; Ding, C.C.; Qu, D.C.; Alvarez, J.V.; Chi, J.T. DDR2 upregulation confers ferroptosis susceptibility of recurrent breast tumors through the Hippo pathway. Oncogene 2021, 40, 2018–2034. [Google Scholar] [CrossRef]
- Ozaki, T.; Nakagawara, A. Role of p53 in cell death and human cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yang, L.; Zhang, X.; Cui, W.; Liu, Y.; Sun, Q.R.; He, Q.; Zhao, S.; Zhang, G.A.; Wang, Y.; et al. Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep. 2019, 20, e47563. [Google Scholar] [CrossRef]
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 2019, 21, 579–591. [Google Scholar] [CrossRef]
- Ou, Y.; Wang, S.J.; Li, D.; Chu, B.; Gu, W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–E6812. [Google Scholar] [CrossRef] [PubMed]
- Lisek, K.; Campaner, E.; Ciani, Y.; Walerych, D.; Del Sal, G. Mutant p53 tunes the NRF2-dependent antioxidant response to support survival of cancer cells. Oncotarget 2018, 9, 20508–20523. [Google Scholar] [CrossRef] [Green Version]
- Leu, J.I.; Murphy, M.E.; George, D.L. Mechanistic basis for impaired ferroptosis in cells expressing the African-centric S47 variant of p53. Proc. Natl. Acad. Sci. USA 2019, 116, 8390–8396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, K.S.; Leu, J.I.; Barnoud, T.; Vonteddu, P.; Gnanapradeepan, K.; Lin, C.; Liu, Q.; Barton, J.C.; Kossenkov, A.V.; George, D.L.; et al. African-centric TP53 variant increases iron accumulation and bacterial pathogenesis but improves response to malaria toxin. Nat. Commun. 2020, 11, 473. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: Convergent physiology—Divergent pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Yi, J.; Zhu, J.; Wu, J.; Thompson, C.B.; Jiang, X. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 31189–31197. [Google Scholar] [CrossRef]
- Hong, X.; Roh, W.; Sullivan, R.J.; Wong, K.H.K.; Wittner, B.S.; Guo, H.; Dubash, T.D.; Sade-Feldman, M.; Wesley, B.; Horwitz, E.; et al. The lipogenic regulator SREBP2 induces transferrin in circulating melanoma cells and suppresses ferroptosis. Cancer Discov. 2021, 11, 678–695. [Google Scholar] [CrossRef]
- Paton, C.M.; Ntambi, J.M. Biochemical and physiological function of stearoyl-CoA desaturase. Am. J. Physiol. Endocrinol. Metab. 2009, 297, e28–e37. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, e4966–e4975. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging biological principles of metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Gupta, G.P.; Massagué, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [Green Version]
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hapach, L.A.; Mosier, J.A.; Wang, W.; Reinhart-King, C.A. Engineered models to parse apart the metastatic cascade. NPJ. Precis. Oncol. 2019, 3, 20. [Google Scholar] [CrossRef] [Green Version]
- Mittal, V. Epithelial mesenchymal transition in tumor metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.H.; Ding, C.C.; Sun, T.; Rupprecht, G.; Lin, C.C.; Hsu, D.; Chi, J.T. The Hippo pathway effector TAZ regulates ferroptosis in renal cell carcinoma. Cell Rep. 2019, 28, 2501–2508.e4. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.H.; Chi, J.T. Hippo pathway effectors YAP/TAZ as novel determinants of ferroptosis. Mol. Cell Oncol. 2019, 7, 1699375. [Google Scholar] [CrossRef]
- Magesh, S.; Cai, D. Roles of YAP/TAZ in ferroptosis. Trends Cell Biol. 2022, 32, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Chi, J.T. Regulation of ferroptosis in cancer cells by YAP/TAZ and Hippo pathways: The therapeutic implications. Genes Dis. 2020, 8, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Cho, P.; Selfors, L.M.; Kuiken, H.J.; Kaul, R.; Fujiwara, T.; Harris, I.S.; Zhang, T.; Gygi, S.P.; Brugge, J.S. 3D culture models with CRISPR screens reveal hyperactive NRF2 as a prerequisite for spheroid formation via regulation of proliferation and ferroptosis. Mol. Cell 2020, 80, 828–844.e6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Peng, L.; Hu, K.; Gu, N. Stress relaxation-induced colon tumor multicellular spheroid culture based on biomimetic hydrogel for nanoenzyme ferroptosis sensitization evaluation. Adv. Healthc. Mater. 2023, 12, e2202009. [Google Scholar] [CrossRef]
- Wu, J.; Minikes, A.M.; Gao, M.; Bian, H.; Li, Y.; Stockwell, B.R.; Chen, Z.N.; Jiang, X. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 2019, 572, 402–406. [Google Scholar] [CrossRef]
- Yang, W.H.; Lin, C.C.; Wu, J.; Chao, P.Y.; Chen, K.; Chen, P.H.; Chi, J.T. The Hippo Pathway effector YAP promotes ferroptosis via the E3 ligase SKP2. Mol. Cancer Res. 2021, 19, 1005–1014. [Google Scholar] [CrossRef]
- Du, K.; Maeso-Díaz, R.; Oh, S.H.; Wang, E.; Chen, T.; Pan, C.; Xiang, K.; Dutta, R.K.; Wang, X.F.; Chi, J.T.; et al. Targeting YAP-mediated HSC death susceptibility and senescence for treatment of liver fibrosis. Hepatology 2023, 77, 1998–2015. [Google Scholar] [CrossRef]
- Du, K.; Oh, S.H.; Dutta, R.K.; Sun, T.; Yang, W.H.; Chi, J.T.; Diehl, A.M. Inhibiting xCT/SLC7A11 induces ferroptosis of myofibroblastic hepatic stellate cells but exacerbates chronic liver injury. Liver Int. 2021, 41, 2214–2227. [Google Scholar] [CrossRef]
- Tang, X.; Ding, C.K.; Wu, J.; Sjol, J.; Wardell, S.; Spasojevic, I.; George, D.; McDonnell, D.P.; Hsu, D.S.; Chang, J.T.; et al. Cystine addiction of triple-negative breast cancer associated with EMT augmented death signaling. Oncogene 2017, 36, 4235–4242. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; You, J.H.; Kim, M.S.; Roh, J.L. Epigenetic reprogramming of epithelial-mesenchymal transition promotes ferroptosis of head and neck cancer. Redox Biol. 2020, 37, 101697. [Google Scholar] [CrossRef]
- Wang, X.; Liu, M.; Chu, Y.; Liu, Y.; Cao, X.; Zhang, H.; Huang, Y.; Gong, A.; Liao, X.; Wang, D.; et al. O-GlcNAcylation of ZEB1 facilitated mesenchymal pancreatic cancer cell ferroptosis. Int. J. Biol. Sci. 2022, 18, 4135–4150. [Google Scholar] [CrossRef]
- Schuster, E.; Taftaf, R.; Reduzzi, C.; Albert, M.K.; Romero-Calvo, I.; Liu, H. Better together: Circulating tumor cell clustering in metastatic cancer. Trends Cancer 2021, 7, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Chin, V.L.; Lim, C.L. Epithelial-mesenchymal plasticity-engaging stemness in an interplay of phenotypes. Stem Cell Investig. 2019, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Roh, J.L. Epithelial-mesenchymal plasticity: Implications for ferroptosis vulnerability and cancer therapy. Crit. Rev. Oncol. Hematol. 2023, 185, 103964. [Google Scholar] [CrossRef] [PubMed]
- Ubellacker, J.M.; Tasdogan, A.; Ramesh, V.; Shen, B.; Mitchell, E.C.; Martin-Sandoval, M.S.; Gu, Z.; McCormick, M.L.; Durham, A.B.; Spitz, D.R.; et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 2020, 585, 113–118. [Google Scholar] [CrossRef]
- Magtanong, L.; Ko, P.J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem. Biol. 2019, 26, 420–432.e9. [Google Scholar] [CrossRef]
- Mashima, T.; Seimiya, H.; Tsuruo, T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br. J. Cancer 2009, 100, 1369–1372. [Google Scholar] [CrossRef] [Green Version]
- Glatz, J.F.; Luiken, J.J.; Bonen, A. Membrane fatty acid transporters as regulators of lipid metabolism: Implications for metabolic disease. Physiol. Rev. 2010, 90, 367–417. [Google Scholar] [CrossRef] [Green Version]
- Schwenk, R.W.; Holloway, G.P.; Luiken, J.J.; Bonen, A.; Glatz, J.F. Fatty acid transport across the cell membrane: Regulation by fatty acid transporters. Prostaglandins Leukot. Essent. Fatty Acids 2010, 82, 149–154. [Google Scholar] [CrossRef]
- Liang, D.; Feng, Y.; Zandkarimi, F.; Wang, H.; Zhang, Z.; Kim, J.; Cai, Y.; Gu, W.; Stockwell, B.R.; Jiang, X. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell 2023, 186, 2748–2764.e22. [Google Scholar] [CrossRef]
- Zhang, Q.; Deng, T.; Zhang, H.; Zuo, D.; Zhu, Q.; Bai, M.; Liu, R.; Ning, T.; Zhang, L.; Yu, Z.; et al. Adipocyte-derived exosomal MTTP suppresses ferroptosis and promotes chemoresistance in colorectal cancer. Adv. Sci. 2022, 9, e2203357. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wang, B.; Zhao, Y.; Tao, Z.; Wang, Y.; Chen, G.; Hu, X. Mammary adipocytes protect triple-negative breast cancer cells from ferroptosis. J. Hematol. Oncol. 2022, 15, 72. [Google Scholar] [CrossRef] [PubMed]
- Ping, Q.; Yan, R.; Cheng, X.; Wang, W.; Zhong, Y.; Hou, Z.; Shi, Y.; Wang, C.; Li, R. Cancer-associated fibroblasts: Overview, progress, challenges, and directions. Cancer Gene Ther. 2021, 28, 984–999. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Encarnación-Rosado, J.; Lin, E.Y.; Sohn, A.S.W.; Zhang, H.; Mancias, J.D.; Kimmelman, A.C. Autophagy supports mitochondrial metabolism through the regulation of iron homeostasis in pancreatic cancer. Sci. Adv. 2023, 9, eadf9284. [Google Scholar] [CrossRef]
- Yang, W.H.; Huang, Z.; Wu, J.; Ding, C.C.; Murphy, S.K.; Chi, J.T. A TAZ-ANGPTL4-NOX2 axis regulates ferroptotic cell death and chemoresistance in epithelial ovarian cancer. Mol. Cancer Res. 2020, 18, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Wu, J.; Ding, C.K.; Lu, M.; Keenan, M.M.; Lin, C.C.; Lin, C.A.; Wang, C.C.; George, D.; Hsu, D.S.; et al. Cystine deprivation triggers programmed necrosis in VHL-deficient renal cell carcinomas. Cancer Res. 2016, 76, 1892–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badgley, M.A.; Kremer, D.M.; Maurer, H.C.; DelGiorno, K.E.; Lee, H.J.; Purohit, V.; Sagalovskiy, I.R.; Ma, A.; Kapilian, J.; Firl, C.E.M. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020, 368, 85–89. [Google Scholar] [CrossRef]
- Zalyte, E.; Cicenas, J. Starvation mediates pancreatic cancer cell sensitivity to ferroptosis via ERK1/2, JNK and changes in the cell mesenchymal state. Int. J. Mol. Med. 2022, 49, 84. [Google Scholar] [CrossRef]
- Song, X.; Liu, J.; Kuang, F.; Chen, X.; Zeh, H.J., 3rd; Kang, R.; Kroemer, G.; Xie, Y.; Tang, D. PDK4 dictates metabolic resistance to ferroptosis by suppressing pyruvate oxidation and fatty acid synthesis. Cell Rep. 2021, 34, 108767. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Qiu, Q.; Wang, L.; Mao, H.; Hu, J.; Chen, Z.; Du, Y.; Liu, X. Energy-stress-mediated AMPK activation promotes GPX4-dependent ferroptosis through the JAK2/STAT3/P53 axis in renal cancer. Oxid. Med. Cell Longev. 2022, 2022, 2353115. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, M.; Yao, X.; Fei, Y.; Lin, Z.; Li, Z.; Cai, K.; Zhao, Y.; Luo, Z. HCAR1/MCT1 regulates tumor ferroptosis through the lactate-mediated AMPK-SCD1 activity and its therapeutic implications. Cell Rep. 2020, 33, 108487. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Dou, J.; Yang, Y.; Sun, S.; Chen, R.; Zhang, Z.; Wei, H.; Li, J.; Wu, Z. Drug-induced lactate confers ferroptosis resistance via p38-SGK1-NEDD4L-dependent upregulation of GPX4 in NSCLC cells. Cell Death Discov. 2023, 9, 165. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1 and mechanisms of hypoxia sensing. Curr. Opin. Cell Biol. 2001, 13, 167–171. [Google Scholar] [CrossRef]
- Chi, J.T.; Wang, Z.; Nuyten, D.S.; Rodriguez, E.H.; Schaner, M.E.; Salim, A.; Wang, Y.; Kristensen, G.B.; Helland, A.; Børresen-Dale, A.L.; et al. Gene expression programs in response to hypoxia: Cell type specificity and prognostic significance in human cancers. PLoS Med. 2006, 3, e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Z.; Yang, G.; Zhang, W.; Liu, Q.; Liu, G.; Liu, P.; Xu, L.; Wang, J.; Yan, Z.; Han, H.; et al. Hypoxia blocks ferroptosis of hepatocellular carcinoma via suppression of METTL14 triggered YTHDF2-dependent silencing of SLC7A11. J. Cell Mol. Med. 2021, 25, 10197–10212. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Chen, P.; Liu, J.; Zhu, S.; Kroemer, G.; Klionsky, D.J.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci. Adv. 2019, 5, eaaw2238. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Samanta, D.; Xiang, L.; Zhang, H.; Hu, H.; Chen, I.; Bullen, J.W.; Semenza, G.L. Chemotherapy triggers HIF-1-dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proc. Natl. Acad. Sci. USA 2015, 112, e4600–e4609. [Google Scholar] [CrossRef]
- Ding, C.C.; Rose, J.; Sun, T.; Wu, J.; Chen, P.H.; Lin, C.C.; Yang, W.H.; Chen, K.Y.; Lee, H.; Xu, E.; et al. MESH1 is a cytosolic NADPH phosphatase that regulates ferroptosis. Nat. Metab. 2020, 2, 270–277. [Google Scholar] [CrossRef]
- Ye, J.; Fan, J.; Venneti, S.; Wan, Y.W.; Pawel, B.R.; Zhang, J.; Finley, L.W.; Lu, C.; Lindsten, T.; Cross, J.R.; et al. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 2014, 4, 1406–1417. [Google Scholar] [CrossRef] [Green Version]
- Samanta, D.; Park, Y.; Andrabi, S.A.; Shelton, L.M.; Gilkes, D.M.; Semenza, G.L. PHGDH expression is required for mitochondrial redox homeostasis, breast cancer stem cell maintenance, and lung metastasis. Cancer Res. 2016, 76, 4430–4442. [Google Scholar] [CrossRef] [Green Version]
- Ding, M.; Zhang, S.; Guo, Y.; Yao, J.; Shen, Q.; Huang, M.; Chen, W.; Yu, S.; Zheng, Y.; Lin, Y.; et al. Tumor microenvironment acidity triggers lipid accumulation in liver cancer via SCD1 activation. Mol. Cancer Res. 2022, 20, 810–822. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, P.; Zhang, Y.N.; Lu, P.; Huang, X.; Wang, Y.; Ran, L.; Xin, H.; Xu, X.; Gao, W.; et al. Biodegradable nanoplatform upregulates tumor microenvironment acidity for enhanced cancer therapy via synergistic induction of apoptosis, ferroptosis, and anti-angiogenesis. J. Nanobiotechnology 2023, 21, 59. [Google Scholar] [CrossRef] [PubMed]
- Dang, Q.; Sun, Z.; Wang, Y.; Wang, L.; Liu, Z.; Han, X. Ferroptosis: A double-edged sword mediating immune tolerance of cancer. Cell Death Dis. 2022, 13, 925. [Google Scholar] [CrossRef]
- Wen, Q.; Liu, J.; Kang, R.; Zhou, B.; Tang, D. The release and activity of HMGB1 in ferroptosis. Biochem. Biophys. Res. Commun. 2019, 510, 278–283. [Google Scholar] [CrossRef]
- Demuynck, R.; Efimova, I.; Naessens, F.; Krysko, D.V. Immunogenic ferroptosis and where to find it? J. Immunother. Cancer 2021, 9, e003430. [Google Scholar] [CrossRef]
- Ye, F.; Chai, W.; Xie, M.; Yang, M.; Yu, Y.; Cao, L.; Yang, L. HMGB1 regulates erastin-induced ferroptosis via RAS-JNK/p38 signaling in HL-60/NRASQ61L cells. Am. J. Cancer Res. 2019, 9, 730–739. [Google Scholar]
- Hayashi, K.; Nikolos, F.; Lee, Y.C.; Jain, A.; Tsouko, E.; Gao, H.; Kasabyan, A.; Leung, H.E.; Osipov, A.; Jung, S.Y.; et al. Tipping the immunostimulatory and inhibitory DAMP balance to harness immunogenic cell death. Nat. Commun. 2020, 11, 6299. [Google Scholar] [CrossRef]
- Perez, M.A.; Magtanong, L.; Dixon, S.J.; Watts, J.L. Dietary lipids induce ferroptosis in Caenorhabditis elegans and human cancer cells. Dev. Cell 2020, 54, 447–454.e4. [Google Scholar] [CrossRef]
- Ferrer, M.; Mourikis, N.; Davidson, E.E.; Kleeman, S.O.; Zaccaria, M.; Habel, J.; Rubino, R.; Gao, Q.; Flint, T.R.; Young, L.; et al. Ketogenic diet promotes tumor ferroptosis but induces relative corticosterone deficiency that accelerates cachexia. Cell Metab. 2023, 00185–00187. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Pratt, D.A. Ferroptosis: A flexible constellation of related biochemical mechanisms. Mol. Cell 2023, 83, 1030–1042. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Riegman, M.; Sagie, L.; Galed, C.; Levin, T.; Steinberg, N.; Dixon, S.J.; Wiesner, U.; Bradbury, M.S.; Niethammer, P.; Zaritsky, A. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat. Cell Biol. 2020, 22, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Katikaneni, A.; Jelcic, M.; Gerlach, G.F.; Ma, Y.; Overholtzer, M.; Niethammer, P. Lipid peroxidation regulates long-range wound detection through 5-lipoxygenase in zebrafish. Nat. Cell Biol. 2020, 22, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.H.; Tseng, W.H.; Chi, J.T. The Intersection of DNA Damage Response and Ferroptosis-A Rationale for Combination Therapeutics. Biology 2020, 9, 187. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, G.; Condello, S.; Huang, H.; Cardenas, H.; Tanner, E.J.; Wei, J.; Ji, Y.; Li, J.; Tan, Y.; et al. Frizzled-7 identifies platinum-tolerant ovarian cancer cells susceptible to ferroptosis. Cancer Res. 2021, 81, 384–399. [Google Scholar] [CrossRef]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Nie, Q.; Hu, Y.; Yu, X.; Li, X.; Fang, X. Induction and application of ferroptosis in cancer therapy. Cancer Cell Int. 2022, 22, 12. [Google Scholar] [CrossRef]
- Hendricks, J.M.; Doubravsky, C.E.; Wehri, E.; Li, Z.; Roberts, M.A.; Deol, K.K.; Lange, M.; Lasheras-Otero, I.; Momper, J.D.; Dixon, S.J.; et al. Identification of structurally diverse FSP1 inhibitors that sensitize cancer cells to ferroptosis. Cell Chem. Biol. 2023, 00114–00119. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Setayeshpour, Y.; Lee, Y.; Chi, J.-T. Environmental Determinants of Ferroptosis in Cancer. Cancers 2023, 15, 3861. https://doi.org/10.3390/cancers15153861
Setayeshpour Y, Lee Y, Chi J-T. Environmental Determinants of Ferroptosis in Cancer. Cancers. 2023; 15(15):3861. https://doi.org/10.3390/cancers15153861
Chicago/Turabian StyleSetayeshpour, Yasaman, Yunji Lee, and Jen-Tsan Chi. 2023. "Environmental Determinants of Ferroptosis in Cancer" Cancers 15, no. 15: 3861. https://doi.org/10.3390/cancers15153861
APA StyleSetayeshpour, Y., Lee, Y., & Chi, J.-T. (2023). Environmental Determinants of Ferroptosis in Cancer. Cancers, 15(15), 3861. https://doi.org/10.3390/cancers15153861