
Models in Pancreatic Neuroendocrine Neoplasms: Current Perspectives and Future Directions

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Cell Lines of pNENs
2.1. Animal-Derived Cell Lines
2.2. Established Human Cell Lines
2.3. Primary Human 2D Cultures
3. Animal Models of PNENs
3.1. Patient-Derived Xenografts of PNENs
3.2. Genetically Engineered Mouse Models (GEMMs) of PNENs
3.3. SV40 Tag
3.4. Conventional Germline Heterozygous Knockouts
3.5. Conventional Germline Homozygous Knockouts
3.6. Induced Activation Models
3.7. Homozygous Knock-In
3.8. Other Animal Genetic Models in PNEN Research
4. 3D Models of pNENs
4.1. Spheroids
4.2. ECM-Supported Constructs
4.3. Bioengineered Hybrid Models
5. Discussion
6. Future Directions
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients With Neuroendocrine Tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef]
- Das, S.; Dasari, A. Epidemiology, Incidence, and Prevalence of Neuroendocrine Neoplasms: Are There Global Differences? Curr. Oncol. Rep. 2021, 23, 43. [Google Scholar] [CrossRef]
- Nagtegaal, I.D.; Odze, R.D.; Klimstra, D.; Paradis, V.; Rugge, M.; Schirmacher, P.; Washington, K.M.; Carneiro, F.; Cree, I.A. The 2019 WHO classification of tumours of the digestive system. Histopathology 2020, 76, 182–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Zeng, L.; Ke, N.W.; Tan, C.L.; Tian, B.L.; Liu, X.B.; Xiang, B.; Zhang, Y. World Health Organization grading classification for pancreatic neuroendocrine neoplasms: A comprehensive analysis from a large Chinese institution. BMC Cancer 2020, 20, 906. [Google Scholar] [CrossRef]
- Yachida, S.; Vakiani, E.; White, C.M.; Zhong, Y.; Saunders, T.; Morgan, R.; de Wilde, R.F.; Maitra, A.; Hicks, J.; Demarzo, A.M.; et al. Small cell and large cell neuroendocrine carcinomas of the pancreas are genetically similar and distinct from well-differentiated pancreatic neuroendocrine tumors. Am. J. Surg. Pathol. 2012, 36, 173–184. [Google Scholar] [CrossRef]
- Buicko, J.L.; Finnerty, B.M.; Zhang, T.; Kim, B.J.; Fahey, T.J., III; Du, Y.-C.N. Insights into the biology and treatment strategies of pancreatic neuroendocrine tumors. Ann. Pancreat. Cancer 2019, 2, 12. [Google Scholar] [CrossRef]
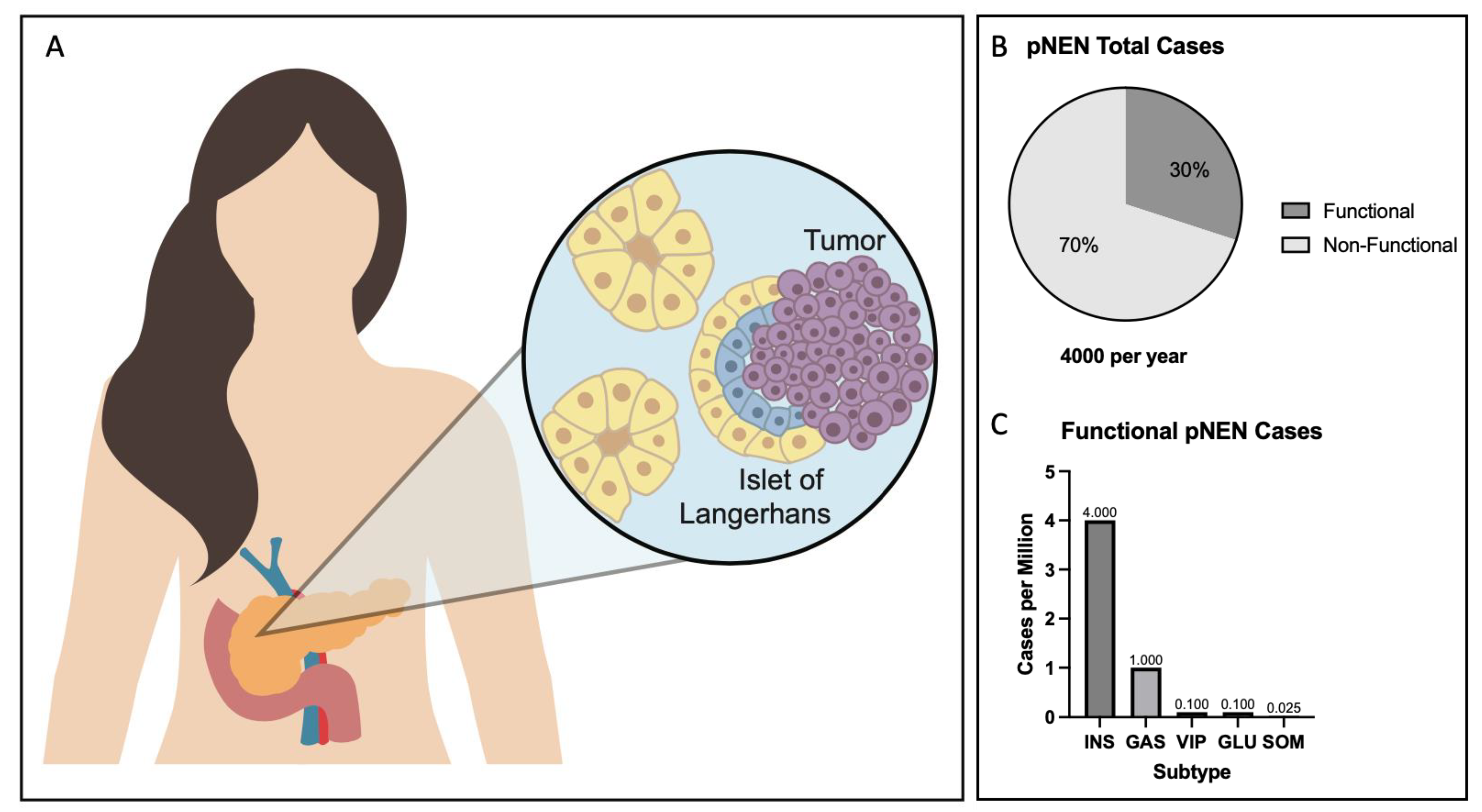
- Da Silva Xavier, G. The Cells of the Islets of Langerhans. J. Clin. Med. 2018, 7, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnavion, R.; Teinturier, R.; Jaafar, R.; Ripoche, D.; Leteurtre, E.; Chen, Y.J.; Rehfeld, J.F.; Lepinasse, F.; Hervieu, V.; Pattou, F.; et al. Islet Cells Serve as Cells of Origin of Pancreatic Gastrin-Positive Endocrine Tumors. Mol. Cell Biol. 2015, 35, 3274–3283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Igarashi, H.; Jensen, R.T. Pancreatic neuroendocrine tumors: Clinical features, diagnosis and medical treatment: Advances. Best. Pract. Res. Clin. Gastroenterol. 2012, 26, 737–753. [Google Scholar] [CrossRef] [Green Version]
- Gorelik, M.; Ahmad, M.; Grossman, D.; Grossman, M.; Cooperman, A.M. Nonfunctioning Incidental Pancreatic Neuroendocrine Tumors: Who, When, and How to Treat? Surg. Clin. N. Am. 2018, 98, 157–167. [Google Scholar] [CrossRef]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef] [Green Version]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Brandi, M.L.; Agarwal, S.K.; Perrier, N.D.; Lines, K.E.; Valk, G.D.; Thakker, R.V. Multiple Endocrine Neoplasia Type 1: Latest Insights. Endocr. Rev. 2021, 42, 133–170. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.S.; Laddha, S.V.; Lewis, P.W.; Koletsky, M.S.; Robzyk, K.; Da Silva, E.; Torres, P.J.; Untch, B.R.; Li, J.; Bose, P.; et al. ATRX, DAXX or MEN1 mutant pancreatic neuroendocrine tumors are a distinct alpha-cell signature subgroup. Nat. Commun. 2018, 9, 4158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulve, N.; Su, C.; Deng, Z.; Soldan, S.S.; Vladimirova, O.; Wickramasinghe, J.; Zheng, H.; Kossenkov, A.V.; Lieberman, P.M. DAXX-ATRX regulation of p53 chromatin binding and DNA damage response. Nat. Commun. 2022, 13, 5033. [Google Scholar] [CrossRef] [PubMed]
- Greidinger, A.; Miller-Samuel, S.; Giri, V.N.; Woo, M.S.-A.; Akumalla, S.; Zeigler-Johnson, C.; Keith, S.W.; Silver, D.P. Neuroendocrine Tumors Are Enriched in Cowden Syndrome. JCO Precis. Oncol. 2020, 4, 551–556. [Google Scholar] [CrossRef]
- Serrano, J.; Goebel, S.U.; Peghini, P.L.; Lubensky, I.A.; Gibril, F.; Jensen, R.T. Alterations in the p16INK4a/CDKN2A tumor suppressor gene in gastrinomas. J. Clin. Endocrinol. Metab. 2000, 85, 4146–4156. [Google Scholar] [CrossRef]
- Muscarella, P.; Melvin, W.S.; Fisher, W.E.; Foor, J.; Ellison, E.C.; Herman, J.G.; Schirmer, W.J.; Hitchcock, C.L.; DeYoung, B.R.; Weghorst, C.M. Genetic alterations in gastrinomas and nonfunctioning pancreatic neuroendocrine tumors: An analysis of p16/MTS1 tumor suppressor gene inactivation. Cancer Res. 1998, 58, 237–240. [Google Scholar]
- Alvarez, M.J.; Subramaniam, P.S.; Tang, L.H.; Grunn, A.; Aburi, M.; Rieckhof, G.; Komissarova, E.V.; Hagan, E.A.; Bodei, L.; Clemons, P.A.; et al. A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat. Genet. 2018, 50, 979–989. [Google Scholar] [CrossRef]
- Dreijerink, K.M.; Hackeng, W.M.; Singhi, A.D.; Heaphy, C.M.; Brosens, L.A. Clinical implications of cell-of-origin epigenetic characteristics in non-functional pancreatic neuroendocrine tumors. J. Pathol. 2022, 256, 143–148. [Google Scholar] [CrossRef]
- Vijayvergia, N.; Boland, P.M.; Handorf, E.; Gustafson, K.S.; Gong, Y.; Cooper, H.S.; Sheriff, F.; Astsaturov, I.; Cohen, S.J.; Engstrom, P.F. Molecular profiling of neuroendocrine malignancies to identify prognostic and therapeutic markers: A Fox Chase Cancer Center Pilot Study. Br. J. Cancer 2016, 115, 564–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hijioka, S.; Hosoda, W.; Mizuno, N.; Hara, K.; Imaoka, H.; Bhatia, V.; Mekky, M.A.; Tajika, M.; Tanaka, T.; Ishihara, M.; et al. Does the WHO 2010 classification of pancreatic neuroendocrine neoplasms accurately characterize pancreatic neuroendocrine carcinomas? J. Gastroenterol. 2015, 50, 564–572. [Google Scholar] [CrossRef] [Green Version]
- Akirov, A.; Larouche, V.; Alshehri, S.; Asa, S.L.; Ezzat, S. Treatment Options for Pancreatic Neuroendocrine Tumors. Cancers 2019, 11, 828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogoza, O.; Megnis, K.; Kudrjavceva, M.; Gerina-Berzina, A.; Rovite, V. Role of Somatostatin Signalling in Neuroendocrine Tumours. Int. J. Mol. Sci. 2022, 23, 1447. [Google Scholar] [CrossRef]
- Caplin, M.E.; Pavel, M.; Ćwikła, J.B.; Phan, A.T.; Raderer, M.; Sedláčková, E.; Cadiot, G.; Wolin, E.M.; Capdevila, J.; Wall, L.; et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N. Engl. J. Med. 2014, 371, 224–233. [Google Scholar] [CrossRef]
- Qian, Z.R.; Li, T.; Ter-Minassian, M.; Yang, J.; Chan, J.A.; Brais, L.K.; Masugi, Y.; Thiaglingam, A.; Brooks, N.; Nishihara, R.; et al. Association Between Somatostatin Receptor Expression and Clinical Outcomes in Neuroendocrine Tumors. Pancreas 2016, 45, 1386–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.; Chan, D.L.; Wong, M.H.; Li, B.T.; Lumba, S.; Clarke, S.J.; Samra, J.; Pavlakis, N. Systematic Review of the Role of Targeted Therapy in Metastatic Neuroendocrine Tumors. Neuroendocrinology 2017, 104, 209–222. [Google Scholar] [CrossRef] [Green Version]
- Roquin, G.; Baudin, E.; Lombard-Bohas, C.; Cadiot, G.; Dominguez, S.; Guimbaud, R.; Niccoli, P.; Legoux, J.L.; Mitry, E.; Rohmer, V.; et al. Chemotherapy for Well-Differentiated Pancreatic Neuroendocrine Tumours with a Ki-67 Index ≥10%: Is There a More Effective Antitumour Regimen? A Retrospective Multicentre Study of the French Group of Endocrine Tumours (GTE). Neuroendocrinology 2018, 106, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Dumont, R.A.; Seiler, D.; Marincek, N.; Brunner, P.; Radojewski, P.; Rochlitz, C.; Müller-Brand, J.; Maecke, H.R.; Briel, M.; Walter, M.A. Survival after somatostatin based radiopeptide therapy with (90)Y-DOTATOC vs. (90)Y-DOTATOC plus (177)Lu-DOTATOC in metastasized gastrinoma. Am. J. Nucl. Med. Mol. Imaging 2015, 5, 46–55. [Google Scholar]
- van Schaik, E.; van Vliet, E.I.; Feelders, R.A.; Krenning, E.P.; Khan, S.; Kamp, K.; Valkema, R.; van Nederveen, F.H.; Teunissen, J.J.M.; Kwekkeboom, D.J.; et al. Improved Control of Severe Hypoglycemia in Patients with Malignant Insulinomas by Peptide Receptor Radionuclide Therapy. J. Clin. Endocrinol. Metab. 2011, 96, 3381–3389. [Google Scholar] [CrossRef]
- Egal, E.S.A.; Jacenik, D.; Soares, H.P.; Beswick, E.J. Translational challenges in pancreatic neuroendocrine tumor immunotherapy. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188640. [Google Scholar] [CrossRef]
- Palmieri, L.J.; Dermine, S.; Barré, A.; Dhooge, M.; Brezault, C.; Cottereau, A.S.; Coriat, R. Medical Treatment of Advanced Pancreatic Neuroendocrine Neoplasms. J. Clin. Med. 2020, 9, 1860. [Google Scholar] [CrossRef]
- Hahn, W.C.; Meyerson, M. Telomerase activation, cellular immortalization and cancer. Ann. Med. 2001, 33, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Wright, W.E. Hayflick, his limit, and cellular ageing. Nat. Rev. Mol. Cell Biol. 2000, 1, 72–76. [Google Scholar] [CrossRef]
- Kim, T.K.; Eberwine, J.H. Mammalian cell transfection: The present and the future. Anal. Bioanal. Chem. 2010, 397, 3173–3178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monazzam, A.; Li, S.-C.; Wargelius, H.; Razmara, M.; Bajic, D.; Mi, J.; Bergquist, J.; Crona, J.; Skogseid, B. Generation and characterization of CRISPR/Cas9-mediated MEN1 knockout BON1 cells: A human pancreatic neuroendocrine cell line. Sci. Rep. 2020, 10, 14572. [Google Scholar] [CrossRef]
- Efrat, S.; Linde, S.; Kofod, H.; Spector, D.; Delannoy, M.; Grant, S.; Hanahan, D.; Baekkeskov, S. Beta-cell lines derived from transgenic mice expressing a hybrid insulin gene-oncogene. Proc. Natl. Acad. Sci. USA 1988, 85, 9037–9041. [Google Scholar] [CrossRef]
- Miyazaki, J.; Araki, K.; Yamato, E.; Ikegami, H.; Asano, T.; Shibasaki, Y.; Oka, Y.; Yamamura, K. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: Special reference to expression of glucose transporter isoforms. Endocrinology 1990, 127, 126–132. [Google Scholar] [CrossRef]
- Powers, A.C.; Efrat, S.; Mojsov, S.; Spector, D.; Habener, J.F.; Hanahan, D. Proglucagon processing similar to normal islets in pancreatic alpha-like cell line derived from transgenic mouse tumor. Diabetes 1990, 39, 406–414. [Google Scholar] [CrossRef]
- Rindi, G.; Grant, S.G.; Yiangou, Y.; Ghatei, M.A.; Bloom, S.R.; Bautch, V.L.; Solcia, E.; Polak, J.M. Development of neuroendocrine tumors in the gastrointestinal tract of transgenic mice. Heterogeneity of hormone expression. Am. J. Pathol. 1990, 136, 1349–1363. [Google Scholar] [PubMed]
- Grant, S.G.; Seidman, I.; Hanahan, D.; Bautch, V.L. Early invasiveness characterizes metastatic carcinoid tumors in transgenic mice. Cancer Res. 1991, 51, 4917–4923. [Google Scholar]
- Hamaguchi, K.; Gaskins, H.R.; Leiter, E.H. NIT-1, a pancreatic beta-cell line established from a transgenic NOD/Lt mouse. Diabetes 1991, 40, 842–849. [Google Scholar] [CrossRef]
- Pettengill, O.S.; Memoli, V.A.; Brinck-Johnsen, T.; Longnecker, D.S. Cell lines derived from pancreatic tumors of Tg(Ela-1-SV40E)Bri18 transgenic mice express somatostatin and T antigen. Carcinogenesis 1994, 15, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Babu, V.; Paul, N.; Yu, R. Animal models and cell lines of pancreatic neuroendocrine tumors. Pancreas 2013, 42, 912–923. [Google Scholar] [CrossRef] [PubMed]
- Gazdar, A.F.; Chick, W.L.; Oie, H.K.; Sims, H.L.; King, D.L.; Weir, G.C.; Lauris, V. Continuous, clonal, insulin- and somatostatin-secreting cell lines established from a transplantable rat islet cell tumor. Proc. Natl. Acad. Sci. USA 1980, 77, 3519–3523. [Google Scholar] [CrossRef]
- Asfari, M.; Janjic, D.; Meda, P.; Li, G.; Halban, P.A.; Wollheim, C.B. Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology 1992, 130, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Santerre, R.F.; Cook, R.A.; Crisel, R.M.; Sharp, J.D.; Schmidt, R.J.; Williams, D.C.; Wilson, C.P. Insulin synthesis in a clonal cell line of simian virus 40-transformed hamster pancreatic beta cells. Proc. Natl. Acad. Sci. USA 1981, 78, 4339–4343. [Google Scholar] [CrossRef]
- Capodanno, Y.; Buishand, F.O.; Pang, L.Y.; Kirpensteijn, J.; Mol, J.A.; Argyle, D.J. Notch pathway inhibition targets chemoresistant insulinoma cancer stem cells. Endocr.-Relat. Cancer 2018, 25, 131–144. [Google Scholar] [CrossRef] [Green Version]
- Kaku, M.; Nishiyama, T.; Yagawa, K.; Abe, M. Establishment of a carcinoembryonic antigen-producing cell line from human pancreatic carcinoma. Gan 1980, 71, 596–601. [Google Scholar]
- Gueli, N.; Toto, A.; Palmieri, G.; Carmenini, G.; Delpino, A.; Ferrini, U. In vitro growth of a cell line originated from a human insulinoma. J. Exp. Clin. Cancer Res. 1987, 6, 281–285. [Google Scholar]
- Evers, B.M.; Townsend, C.M., Jr.; Upp, J.R.; Allen, E.; Hurlbut, S.C.; Kim, S.W.; Rajaraman, S.; Singh, P.; Reubi, J.C.; Thompson, J.C. Establishment and characterization of a human carcinoid in nude mice and effect of various agents on tumor growth. Gastroenterology 1991, 101, 303–311. [Google Scholar] [CrossRef]
- Tillotson, L.G.; Lodestro, C.; Höcker, M.; Wiedenmann, B.; Newcomer, C.E.; Reid, L.M. Isolation, maintenance, and characterization of human pancreatic islet tumor cells expressing vasoactive intestinal peptide. Pancreas 2001, 22, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Zhong, Y.; Patrascu, R.; Davis, M.B.; Morsberger, L.A.; Griffin, C.A.; Hruban, R.H.; Laheru, D.; Iacobuzio-Donahue, C.A. Establishment and characterization of a new cell line, A99, from a primary small cell carcinoma of the pancreas. Pancreas 2011, 40, 905–910. [Google Scholar] [CrossRef] [Green Version]
- Krampitz, G.W.; George, B.M.; Willingham, S.B.; Volkmer, J.P.; Weiskopf, K.; Jahchan, N.; Newman, A.M.; Sahoo, D.; Zemek, A.J.; Yanovsky, R.L.; et al. Identification of tumorigenic cells and therapeutic targets in pancreatic neuroendocrine tumors. Proc. Natl. Acad. Sci. USA 2016, 113, 4464–4469. [Google Scholar] [CrossRef] [PubMed]
- Benten, D.; Behrang, Y.; Unrau, L.; Weissmann, V.; Wolters-Eisfeld, G.; Burdak-Rothkamm, S.; Stahl, F.R.; Anlauf, M.; Grabowski, P.; Möbs, M.; et al. Establishment of the First Well-differentiated Human Pancreatic Neuroendocrine Tumor Model. Mol. Cancer Res. 2018, 16, 496–507. [Google Scholar] [CrossRef] [Green Version]
- Lou, X.; Ye, Z.; Xu, X.; Jiang, M.; Lu, R.; Jing, D.; Zhang, W.; Gao, H.; Wang, F.; Zhang, Y.; et al. Establishment and characterization of the third non-functional human pancreatic neuroendocrine tumor cell line. Hum. Cell 2022, 35, 1248–1261. [Google Scholar] [CrossRef]
- Viol, F.; Sipos, B.; Fahl, M.; Clauditz, T.S.; Amin, T.; Kriegs, M.; Nieser, M.; Izbicki, J.R.; Huber, S.; Lohse, A.W.; et al. Novel preclinical gastroenteropancreatic neuroendocrine neoplasia models demonstrate the feasibility of mutation-based targeted therapy. Cell Oncol. (Dordr.) 2022, 45, 1401–1419. [Google Scholar] [CrossRef]
- Poitout, V.; Olson, L.K.; Robertson, R.P. Insulin-secreting cell lines: Classification, characteristics and potential applications. Diabetes Metab. 1996, 22, 7–14. [Google Scholar]
- Sullivan, C.S.; Pipas, J.M. T antigens of simian virus 40: Molecular chaperones for viral replication and tumorigenesis. Microbiol. Mol. Biol. Rev. 2002, 66, 179–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawlor, N.; Youn, A.; Kursawe, R.; Ucar, D.; Stitzel, M.L. Alpha TC1 and Beta-TC-6 genomic profiling uncovers both shared and distinct transcriptional regulatory features with their primary islet counterparts. Sci. Rep. 2017, 7, 11959. [Google Scholar] [CrossRef] [Green Version]
- Chick, W.L.; Warren, S.; Chute, R.N.; Like, A.A.; Lauris, V.; Kitchen, K.C. A transplantable insulinoma in the rat. Proc. Natl. Acad. Sci. USA 1977, 74, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Hohmeier, H.E.; Newgard, C.B. Cell lines derived from pancreatic islets. Mol. Cell Endocrinol. 2004, 228, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Praz, G.A.; Halban, P.A.; Wollheim, C.B.; Blondel, B.; Strauss, A.J.; Renold, A.E. Regulation of immunoreactive-insulin release from a rat cell line (RINm5F). Biochem. J. 1983, 210, 345–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acosta-Montalvo, A.; Saponaro, C.; Kerr-Conte, J.; Prehn, J.H.M.; Pattou, F.; Bonner, C. Proglucagon-Derived Peptides Expression and Secretion in Rat Insulinoma INS-1 Cells. Front. Cell Dev. Biol. 2020, 8, 590763. [Google Scholar] [CrossRef]
- Gleason, C.E.; Gonzalez, M.; Harmon, J.S.; Robertson, R.P. Determinants of glucose toxicity and its reversibility in the pancreatic islet β-cell line, HIT-T15. Am. J. Physiol.-Endocrinol. Metab. 2000, 279, E997–E1002. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Gleason, C.E.; Tran, P.O.; Harmon, J.S.; Robertson, R.P. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc. Natl. Acad. Sci. USA 1999, 96, 10857–10862. [Google Scholar] [CrossRef]
- Gragnoli, C. The CM cell line derived from liver metastasis of malignant human insulinoma is not a valid beta cell model for in vitro studies. J. Cell. Physiol. 2008, 216, 569–570. [Google Scholar] [CrossRef]
- Luley, K.B.; Biedermann, S.B.; Kunstner, A.; Busch, H.; Franzenburg, S.; Schrader, J.; Grabowski, P.; Wellner, U.F.; Keck, T.; Brabant, G.; et al. A Comprehensive Molecular Characterization of the Pancreatic Neuroendocrine Tumor Cell Lines BON-1 and QGP-1. Cancers 2020, 12, 691. [Google Scholar] [CrossRef] [Green Version]
- Vandamme, T.; Peeters, M.; Dogan, F.; Pauwels, P.; Van Assche, E.; Beyens, M.; Mortier, G.; Vandeweyer, G.; de Herder, W.; Van Camp, G.; et al. Whole-exome characterization of pancreatic neuroendocrine tumor cell lines BON-1 and QGP-1. J. Mol. Endocrinol. 2015, 54, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Vandamme, T.; Beyens, M.; Peeters, M.; Van Camp, G.; de Beeck, K.O. Next generation exome sequencing of pancreatic neuroendocrine tumor cell lines BON-1 and QGP-1 reveals different lineages. Cancer Genet. 2015, 208, 523. [Google Scholar] [CrossRef]
- Baroni, M.G.; Cavallo, M.G.; Mark, M.; Monetini, L.; Stoehrer, B.; Pozzilli, P. Beta-cell gene expression and functional characterisation of the human insulinoma cell line CM. J. Endocrinol. 1999, 161, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Künstner, A.; Busch, H.; Franzenburg, S.; Luley, K.; Viol, F.; Schrader, J.; Konukiewitz, B.; Wellner, U.F.; Meyhöfer, S.M.; et al. Differential Effects of Somatostatin, Octreotide, and Lanreotide on Neuroendocrine Differentiation and Proliferation in Established and Primary NET Cell Lines: Possible Crosstalk with TGF-β Signaling. Int. J. Mol. Sci. 2022, 23, 15868. [Google Scholar] [CrossRef]
- Ohmoto, A.; Suzuki, M.; Takai, E.; Rokutan, H.; Fujiwara, Y.; Morizane, C.; Yanagihara, K.; Shibata, T.; Yachida, S. Establishment of preclinical chemotherapy models for gastroenteropancreatic neuroendocrine carcinoma. Oncotarget 2018, 9, 21086–21099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, A.; Romano, D.; Saveanu, A.; Roche, C.; Albertelli, M.; Barbieri, F.; Brue, T.; Niccoli, P.; Delpero, J.R.; Garcia, S.; et al. Anti-proliferative and anti-secretory effects of everolimus on human pancreatic neuroendocrine tumors primary cultures: Is there any benefit from combination with somatostatin analogs? Oncotarget 2017, 8, 41044–41063. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, A.; Blanchard, M.P.; Albertelli, M.; Barbieri, F.; Brue, T.; Niccoli, P.; Delpero, J.R.; Monges, G.; Garcia, S.; Ferone, D.; et al. Pasireotide and octreotide antiproliferative effects and sst2 trafficking in human pancreatic neuroendocrine tumor cultures. Endocr. Relat. Cancer 2014, 21, 691–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falletta, S.; Partelli, S.; Rubini, C.; Nann, D.; Doria, A.; Marinoni, I.; Polenta, V.; Di Pasquale, C.; Degli Uberti, E.; Perren, A.; et al. mTOR inhibitors response and mTOR pathway in pancreatic neuroendocrine tumors. Endocr. Relat. Cancer 2016, 23, 883–891. [Google Scholar] [CrossRef] [Green Version]
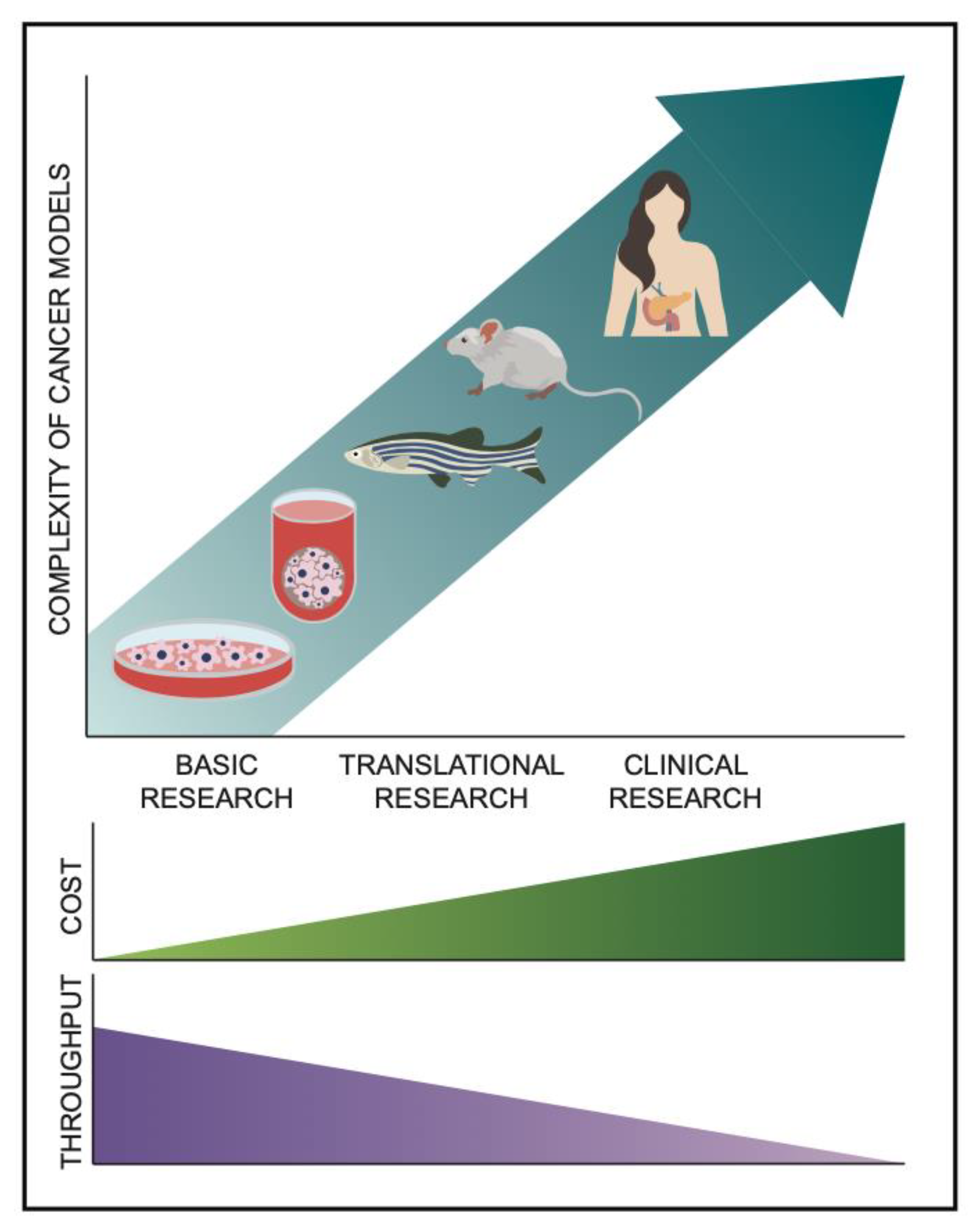
- Mukherjee, P.; Roy, S.; Ghosh, D.; Nandi, S.K. Role of animal models in biomedical research: A review. Lab. Anim. Res. 2022, 38, 18. [Google Scholar] [CrossRef]
- Goto, T. Patient-Derived Tumor Xenograft Models: Toward the Establishment of Precision Cancer Medicine. J. Pers. Med. 2020, 10, 64. [Google Scholar] [CrossRef]
- Lai, Y.; Wei, X.; Lin, S.; Qin, L.; Cheng, L.; Li, P. Current status and perspectives of patient-derived xenograft models in cancer research. J. Hematol. Oncol. 2017, 10, 106. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Zhang, L.; Serra, S.; Law, C.; Wei, A.; Stockley, T.L.; Ezzat, S.; Asa, S.L. Establishment and Characterization of a Human Neuroendocrine Tumor Xenograft. Endocr. Pathol. 2016, 27, 97–103. [Google Scholar] [CrossRef]
- Chamberlain, C.E.; German, M.S.; Yang, K.; Wang, J.; VanBrocklin, H.; Regan, M.; Shokat, K.M.; Ducker, G.S.; Kim, G.E.; Hann, B.; et al. A Patient-derived Xenograft Model of Pancreatic Neuroendocrine Tumors Identifies Sapanisertib as a Possible New Treatment for Everolimus-resistant Tumors. Mol. Cancer Ther. 2018, 17, 2702–2709. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, K.; Toshimitsu, K.; Matano, M.; Fujita, M.; Fujii, M.; Togasaki, K.; Ebisudani, T.; Shimokawa, M.; Takano, A.; Takahashi, S.; et al. An Organoid Biobank of Neuroendocrine Neoplasms Enables Genotype-Phenotype Mapping. Cell 2020, 183, 1420–1435.e1421. [Google Scholar] [CrossRef] [PubMed]
- Pham, N.-A.; Radulovich, N.; Ibrahimov, E.; Martins-Filho, S.N.; Li, Q.; Pintilie, M.; Weiss, J.; Raghavan, V.; Cabanero, M.; Denroche, R.E.; et al. Patient-derived tumor xenograft and organoid models established from resected pancreatic, duodenal and biliary cancers. Sci. Rep. 2021, 11, 10619. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.G.; Borbon, L.C.; Mudd, J.L.; Abusada, E.; AghaAmiri, S.; Ghosh, S.C.; Vargas, S.H.; Li, G.; Beyer, G.V.; McDonough, M.; et al. Establishment of Novel Neuroendocrine Carcinoma Patient-Derived Xenograft Models for Receptor Peptide-Targeted Therapy. Cancers 2022, 14, 1910. [Google Scholar] [CrossRef]
- Gaudenzi, G.; Albertelli, M.; Dicitore, A.; Würth, R.; Gatto, F.; Barbieri, F.; Cotelli, F.; Florio, T.; Ferone, D.; Persani, L.; et al. Patient-derived xenograft in zebrafish embryos: A new platform for translational research in neuroendocrine tumors. Endocrine 2017, 57, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Kersten, K.; de Visser, K.E.; van Miltenburg, M.H.; Jonkers, J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol. Med. 2017, 9, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature 1985, 315, 115–122. [Google Scholar] [CrossRef]
- Zumsteg, A.; Strittmatter, K.; Klewe-Nebenius, D.; Antoniadis, H.; Christofori, G. A bioluminescent mouse model of pancreatic beta-cell carcinogenesis. Carcinogenesis 2010, 31, 1465–1474. [Google Scholar] [CrossRef] [Green Version]
- Mandriota, S.J.; Jussila, L.; Jeltsch, M.; Compagni, A.; Baetens, D.; Prevo, R.; Banerji, S.; Huarte, J.; Montesano, R.; Jackson, D.G.; et al. Vascular endothelial growth factor-C-mediated lymphangiogenesis promotes tumour metastasis. Embo J. 2001, 20, 672–682. [Google Scholar] [CrossRef] [Green Version]
- Gannon, G.; Mandriota, S.J.; Cui, L.; Baetens, D.; Pepper, M.S.; Christofori, G. Overexpression of vascular endothelial growth factor-A165 enhances tumor angiogenesis but not metastasis during beta-cell carcinogenesis. Cancer Res. 2002, 62, 603–608. [Google Scholar]
- Kopfstein, L.; Veikkola, T.; Djonov, V.G.; Baeriswyl, V.; Schomber, T.; Strittmatter, K.; Stacker, S.A.; Achen, M.G.; Alitalo, K.; Christofori, G. Distinct roles of vascular endothelial growth factor-D in lymphangiogenesis and metastasis. Am. J. Pathol. 2007, 170, 1348–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, T.; Hanahan, D. Elevated levels of IGF-1 receptor convey invasive and metastatic capability in a mouse model of pancreatic islet tumorigenesis. Cancer Cell 2002, 1, 339–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saupe, F.; Schwenzer, A.; Jia, Y.; Gasser, I.; Spenlé, C.; Langlois, B.; Kammerer, M.; Lefebvre, O.; Hlushchuk, R.; Rupp, T.; et al. Tenascin-C Downregulates Wnt Inhibitor Dickkopf-1, Promoting Tumorigenesis in a Neuroendocrine Tumor Model. Cell Rep. 2013, 5, 482–492. [Google Scholar] [CrossRef] [Green Version]
- Hunter, K.E.; Palermo, C.; Kester, J.C.; Simpson, K.; Li, J.P.; Tang, L.H.; Klimstra, D.S.; Vlodavsky, I.; Joyce, J.A. Heparanase promotes lymphangiogenesis and tumor invasion in pancreatic neuroendocrine tumors. Oncogene 2014, 33, 1799–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Perl, A.; Wilgenbus, P.; Dahl, U.; Semb, H.; Christofori, G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 1998, 392, 190–193. [Google Scholar] [CrossRef]
- Zhang, Y.; Manouchehri Doulabi, E.; Herre, M.; Cedervall, J.; Qiao, Q.; Miao, Z.; Hamidi, A.; Hellman, L.; Kamali-Moghaddam, M.; Olsson, A.K. Platelet-Derived PDGFB Promotes Recruitment of Cancer-Associated Fibroblasts, Deposition of Extracellular Matrix and Tgfβ Signaling in the Tumor Microenvironment. Cancers 2022, 14, 1947. [Google Scholar] [CrossRef]
- Kobayashi, S.; Contractor, T.; Vosburgh, E.; Du, Y.N.; Tang, L.H.; Clausen, R.; Harris, C.R. Alleles of Insm1 determine whether RIP1-Tag2 mice produce insulinomas or nonfunctioning pancreatic neuroendocrine tumors. Oncogenesis 2019, 8, 16. [Google Scholar] [CrossRef] [Green Version]
- Contractor, T.; Clausen, R.; Harris, G.R.; Rosenfeld, J.A.; Carpizo, D.R.; Tang, L.; Harris, C.R. IGF2 drives formation of ileal neuroendocrine tumors in patients and mice. Endocr. Relat. Cancer 2020, 27, 175–186. [Google Scholar] [CrossRef]
- Adams, T.E.; Alpert, S.; Hanahan, D. Non-tolerance and autoantibodies to a transgenic self antigen expressed in pancreatic beta cells. Nature 1987, 325, 223–228. [Google Scholar] [CrossRef]
- Onrust, S.V.; Hartl, P.M.; Rosen, S.D.; Hanahan, D. Modulation of L-selectin ligand expression during an immune response accompanying tumorigenesis in transgenic mice. J. Clin. Investig. 1996, 97, 54–64. [Google Scholar] [CrossRef] [Green Version]
- Chun, M.G.; Mao, J.H.; Chiu, C.W.; Balmain, A.; Hanahan, D. Polymorphic genetic control of tumor invasion in a mouse model of pancreatic neuroendocrine carcinogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 17268–17273. [Google Scholar] [CrossRef] [PubMed]
- Hunter, K.; Quick, M.; Sadanandam, A.; Hanahan, D.; Joyce, J. Identification and characterization of poorly differentiated invasive carcinomas in a mouse model of pancreatic neuroendocrine tumorigenesis. PLoS ONE 2013, 8, e64472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, R.H., Jr.; Memoli, V.A.; Longnecker, D.S. Hyperplasia and tumors of the islets of Langerhans in mice bearing an elastase I-SV40 T-antigen fusion gene. Carcinogenesis 1990, 11, 1393–1398. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Hammer, R.E.; Messing, A.; Palmiter, R.D.; Brinster, R.L. Pancreatic neoplasia induced by SV40 T-antigen expression in acinar cells of transgenic mice. Science 1987, 238, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Götz, W.; Schucht, C.; Roth, J.; Theuring, F.; Herken, R. Endocrine pancreatic tumors in MSV-SV40 large T transgenic mice. Am. J. Pathol. 1993, 142, 1493–1503. [Google Scholar]
- Cartier, N.; Miquerol, L.; Tulliez, M.; Lepetit, N.; Levrat, F.; Grimber, G.; Briand, P.; Kahn, A. Diet-dependent carcinogenesis of pancreatic islets and liver in transgenic mice expressing oncogenes under the control of the L-type pyruvate kinase gene promoter. Oncogene 1992, 7, 1413–1422. [Google Scholar]
- Montag, A.G.; Oka, T.; Baek, K.H.; Choi, C.S.; Jay, G.; Agarwal, K. Tumors in hepatobiliary tract and pancreatic islet tissues of transgenic mice harboring gastrin simian virus 40 large tumor antigen fusion gene. Proc. Natl. Acad. Sci. USA 1993, 90, 6696–6700. [Google Scholar] [CrossRef]
- Efrat, S.; Teitelman, G.; Anwar, M.; Ruggiero, D.; Hanahan, D. Glucagon gene regulatory region directs oncoprotein expression to neurons and pancreatic alpha cells. Neuron 1988, 1, 605–613. [Google Scholar] [CrossRef]
- Asa, S.L.; Lee, Y.C.; Drucker, D.J. Development of colonic and pancreatic endocrine tumours in mice expressing a glucagon-SV40 T antigen transgene. Virchows Arch. 1996, 427, 595–606. [Google Scholar] [CrossRef]
- Murphy, D.; Bishop, A.; Rindi, G.; Murphy, M.N.; Stamp, G.W.; Hanson, J.; Polak, J.M.; Hogan, B. Mice transgenic for a vasopressin-SV40 hybrid oncogene develop tumors of the endocrine pancreas and the anterior pituitary. A possible model for human multiple endocrine neoplasia type 1. Am. J. Pathol. 1987, 129, 552–566. [Google Scholar] [PubMed]
- Dyer, K.R.; Messing, A. Peripheral neuropathy associated with functional islet cell adenomas in SV40 transgenic mice. J. Neuropathol. Exp. Neurol. 1989, 48, 399–412. [Google Scholar] [CrossRef]
- Dyer, K.R.; Messing, A. Metal-inducible pathology in the liver, pancreas, and kidney of transgenic mice expressing SV40 early region genes. Am. J. Pathol. 1989, 135, 401–410. [Google Scholar] [PubMed]
- Lee, Y.C.; Asa, S.L.; Drucker, D.J. Glucagon gene 5′-flanking sequences direct expression of simian virus 40 large T antigen to the intestine, producing carcinoma of the large bowel in transgenic mice. J. Biol. Chem. 1992, 267, 10705–10708. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.J.; Upchurch, B.H.; Rindi, G.; Leiter, A.B. Studies in transgenic mice reveal potential relationships between secretin-producing cells and other endocrine cell types. J. Biol. Chem. 1995, 270, 885–891. [Google Scholar] [CrossRef] [Green Version]
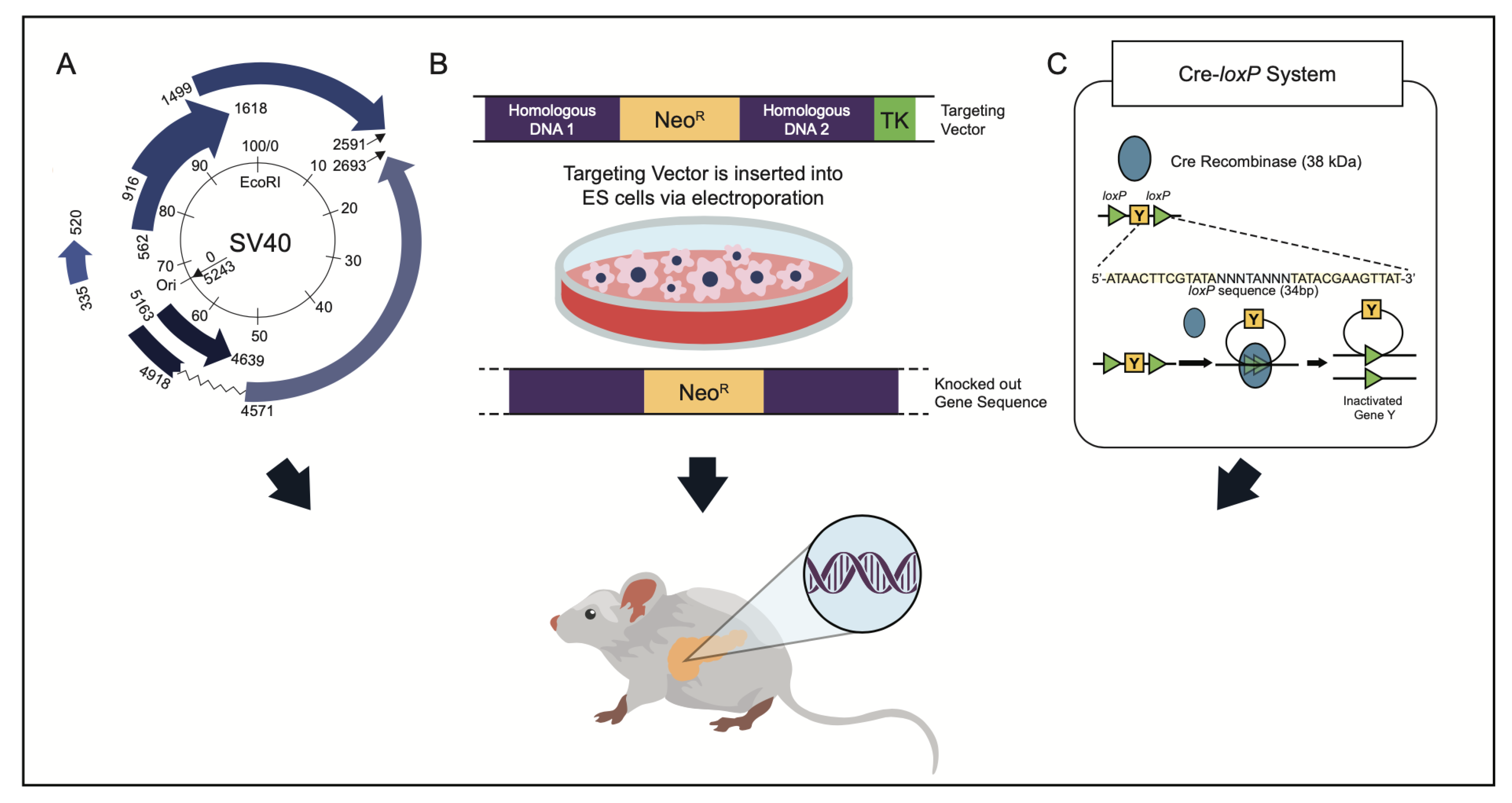
- Jonkers, J.; Berns, A. Conditional mouse models of sporadic cancer. Nat. Rev. Cancer 2002, 2, 251–265. [Google Scholar] [CrossRef]
- Crabtree, J.S.; Scacheri, P.C.; Ward, J.M.; Garrett-Beal, L.; Emmert-Buck, M.R.; Edgemon, K.A.; Lorang, D.; Libutti, S.K.; Chandrasekharappa, S.C.; Marx, S.J.; et al. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 1118–1123. [Google Scholar] [CrossRef]
- Loffler, K.A.; Biondi, C.A.; Gartside, M.G.; Serewko-Auret, M.M.; Duncan, R.; Tonks, I.D.; Mould, A.W.; Waring, P.; Muller, H.K.; Kay, G.F.; et al. Lack of augmentation of tumor spectrum or severity in dual heterozygous Men1 and Rb1 knockout mice. Oncogene 2007, 26, 4009–4017. [Google Scholar] [CrossRef] [Green Version]
- Williams, B.O.; Remington, L.; Albert, D.M.; Mukai, S.; Bronson, R.T.; Jacks, T. Cooperative tumorigenic effects of germline mutations in Rb and p53. Nat. Genet. 1994, 7, 480–484. [Google Scholar] [CrossRef]
- Pei, X.H.; Bai, F.; Li, Z.; Smith, M.D.; Whitewolf, G.; Jin, R.; Xiong, Y. Cytoplasmic CUL9/PARC ubiquitin ligase is a tumor suppressor and promotes p53-dependent apoptosis. Cancer Res. 2011, 71, 2969–2977. [Google Scholar] [CrossRef] [Green Version]
- Harvey, M.; Vogel, H.; Lee, E.Y.; Bradley, A.; Donehower, L.A. Mice deficient in both p53 and Rb develop tumors primarily of endocrine origin. Cancer Res. 1995, 55, 1146–1151. [Google Scholar] [PubMed]
- Harding, B.; Lemos, M.C.; Reed, A.A.; Walls, G.V.; Jeyabalan, J.; Bowl, M.R.; Tateossian, H.; Sullivan, N.; Hough, T.; Fraser, W.D.; et al. Multiple endocrine neoplasia type 1 knockout mice develop parathyroid, pancreatic, pituitary and adrenal tumours with hypercalcaemia, hypophosphataemia and hypercorticosteronaemia. Endocr. Relat. Cancer 2009, 16, 1313–1327. [Google Scholar] [CrossRef]
- Furuta, M.; Yano, H.; Zhou, A.; Rouillé, Y.; Holst, J.J.; Carroll, R.; Ravazzola, M.; Orci, L.; Furuta, H.; Steiner, D.F. Defective prohormone processing and altered pancreatic islet morphology in mice lacking active SPC2. Proc. Natl. Acad. Sci. USA 1997, 94, 6646–6651. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.B.; Reens, J.; Brocklehurst, S.R.; Betts, C.J.; Bickerton, S.; Bigley, A.L.; Jenkins, R.P.; Whalley, N.M.; Morgan, D.; Smith, D.M. Islets of Langerhans from prohormone convertase-2 knockout mice show α-cell hyperplasia and tumorigenesis with elevated α-cell neogenesis. Int. J. Exp. Pathol. 2014, 95, 29–48. [Google Scholar] [CrossRef]
- Gelling, R.W.; Du, X.Q.; Dichmann, D.S.; Romer, J.; Huang, H.; Cui, L.; Obici, S.; Tang, B.; Holst, J.J.; Fledelius, C.; et al. Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc. Natl. Acad. Sci. USA 2003, 100, 1438–1443. [Google Scholar] [CrossRef]
- Yu, R.; Dhall, D.; Nissen, N.N.; Zhou, C.; Ren, S.G. Pancreatic neuroendocrine tumors in glucagon receptor-deficient mice. PLoS ONE 2011, 6, e23397. [Google Scholar] [CrossRef] [Green Version]
- Neumann, C.A.; Krause, D.S.; Carman, C.V.; Das, S.; Dubey, D.P.; Abraham, J.L.; Bronson, R.T.; Fujiwara, Y.; Orkin, S.H.; Van Etten, R.A. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature 2003, 424, 561–565. [Google Scholar] [CrossRef] [Green Version]
- Tuttle, R.L.; Gill, N.S.; Pugh, W.; Lee, J.P.; Koeberlein, B.; Furth, E.E.; Polonsky, K.S.; Naji, A.; Birnbaum, M.J. Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nat. Med. 2001, 7, 1133–1137. [Google Scholar] [CrossRef]
- Pelengaris, S.; Khan, M.; Evan, G.I. Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell 2002, 109, 321–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, B.C.; Klimstra, D.S.; Varmus, H.E. The c-myc and PyMT oncogenes induce different tumor types in a somatic mouse model for pancreatic cancer. Genes. Dev. 2003, 17, 3127–3138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Rahman, L.; Voeller, D.; Kastanos, E.; Yang, S.X.; Feigenbaum, L.; Allegra, C.; Kaye, F.J.; Steeg, P.; Zajac-Kaye, M. Transgenic expression of human thymidylate synthase accelerates the development of hyperplasia and tumors in the endocrine pancreas. Oncogene 2007, 26, 4817–4824. [Google Scholar] [CrossRef] [Green Version]
- Carter, A.M.; Kumar, N.; Herring, B.; Tan, C.; Guenter, R.; Telange, R.; Howse, W.; Viol, F.; McCaw, T.R.; Bickerton, H.H.; et al. Cdk5 drives formation of heterogeneous pancreatic neuroendocrine tumors. Oncogenesis 2021, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Vijayakurup, V.; Maeng, K.; Lee, H.S.; Meyer, B.; Burkett, S.; Nawab, A.; Dougherty, M.W.; Jobin, C.; Mahmud, I.; Garrett, T.J.; et al. Thymidylate synthase accelerates Men1-mediated pancreatic tumor progression and reduces survival. JCI Insight 2022, 7, e147417. [Google Scholar] [CrossRef] [PubMed]
- Alliouachene, S.; Tuttle, R.L.; Boumard, S.; Lapointe, T.; Berissi, S.; Germain, S.; Jaubert, F.; Tosh, D.; Birnbaum, M.J.; Pende, M. Constitutively active Akt1 expression in mouse pancreas requires S6 kinase 1 for insulinoma formation. J. Clin. Investig. 2008, 118, 3629–3638. [Google Scholar] [CrossRef]
- Sotillo, R.; Dubus, P.; Martin, J.; de la Cueva, E.; Ortega, S.; Malumbres, M.; Barbacid, M. Wide spectrum of tumors in knock-in mice carrying a Cdk4 protein insensitive to INK4 inhibitors. EMBO J. 2001, 20, 6637–6647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, Y.; Yamamoto, M.; Mizoguchi, H.; Watanabe, C.; Ito, R.; Yamamoto, S.; Sun, X.Y.; Murata, Y. Mice deficient for glucagon gene-derived peptides display normoglycemia and hyperplasia of islet {alpha}-cells but not of intestinal L-cells. Mol. Endocrinol. 2009, 23, 1990–1999. [Google Scholar] [CrossRef]
- Kim, H.; Kim, M.; Im, S.K.; Fang, S. Mouse Cre-LoxP system: General principles to determine tissue-specific roles of target genes. Lab. Anim. Res. 2018, 34, 147–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnuson, M.A.; Osipovich, A.B. Pancreas-specific Cre driver lines and considerations for their prudent use. Cell Metab. 2013, 18, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Bertolino, P.; Radovanovic, I.; Casse, H.; Aguzzi, A.; Wang, Z.Q.; Zhang, C.X. Genetic ablation of the tumor suppressor menin causes lethality at mid-gestation with defects in multiple organs. Mech. Dev. 2003, 120, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Biondi, C.A.; Gartside, M.G.; Waring, P.; Loffler, K.A.; Stark, M.S.; Magnuson, M.A.; Kay, G.F.; Hayward, N.K. Conditional inactivation of the MEN1 gene leads to pancreatic and pituitary tumorigenesis but does not affect normal development of these tissues. Mol. Cell Biol. 2004, 24, 3125–3131. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.C.; He, M.; Powell, A.; Adem, A.; Lorang, D.; Heller, C.; Grover, A.C.; Ylaya, K.; Hewitt, S.M.; Marx, S.J.; et al. Recapitulation of pancreatic neuroendocrine tumors in human multiple endocrine neoplasia type I syndrome via Pdx1-directed inactivation of Men1. Cancer Res. 2009, 69, 1858–1866. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Herrera, P.L.; Carreira, C.; Bonnavion, R.; Seigne, C.; Calender, A.; Bertolino, P.; Zhang, C.X. Alpha cell-specific Men1 ablation triggers the transdifferentiation of glucagon-expressing cells and insulinoma development. Gastroenterology 2010, 138, 1954–1965. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.C.; Ylaya, K.; Pechhold, K.; Wilson, A.; Adem, A.; Hewitt, S.M.; Libutti, S.K. Multiple endocrine neoplasia type 1 deletion in pancreatic alpha-cells leads to development of insulinomas in mice. Endocrinology 2010, 151, 4024–4030. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Cao, Y.; Li, F.; Su, Y.; Li, Y.; Peng, Y.; Cheng, Y.; Zhang, C.; Wang, W.; Ning, G. Targeting beta-catenin signaling for therapeutic intervention in MEN1-deficient pancreatic neuroendocrine tumours. Nat. Commun. 2014, 5, 5809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lines, K.E.; Vas Nunes, R.P.; Frost, M.; Yates, C.J.; Stevenson, M.; Thakker, R.V. A MEN1 pancreatic neuroendocrine tumour mouse model under temporal control. Endocr. Connect. 2017, 6, 232–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.; Tang, L.H.; Davidson, C.; Vosburgh, E.; Chen, W.; Foran, D.J.; Notterman, D.A.; Levine, A.J.; Xu, E.Y. Two well-differentiated pancreatic neuroendocrine tumor mouse models. Cell Death Differ. 2020, 27, 269–283. [Google Scholar] [CrossRef] [Green Version]
- Duan, S.; Sawyer, T.W.; Sontz, R.A.; Wieland, B.A.; Diaz, A.F.; Merchant, J.L. GFAP-directed Inactivation of Men1 Exploits Glial Cell Plasticity in Favor of Neuroendocrine Reprogramming. Cell Mol. Gastroenterol. Hepatol. 2022, 14, 1025–1051. [Google Scholar] [CrossRef]
- Bertolino, P.; Tong, W.M.; Galendo, D.; Wang, Z.Q.; Zhang, C.X. Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Mol. Endocrinol. 2003, 17, 1880–1892. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.C.; Adem, A.; Ylaya, K.; Wilson, A.; He, M.; Lorang, D.; Hewitt, S.M.; Pechhold, K.; Harlan, D.M.; Lubensky, I.A.; et al. Deciphering von Hippel-Lindau (VHL/Vhl)-associated pancreatic manifestations by inactivating Vhl in specific pancreatic cell populations. PLoS ONE 2009, 4, e4897. [Google Scholar] [CrossRef] [Green Version]
- Glenn, S.T.; Jones, C.A.; Sexton, S.; LeVea, C.M.; Caraker, S.M.; Hajduczok, G.; Gross, K.W. Conditional deletion of p53 and Rb in the renin-expressing compartment of the pancreas leads to a highly penetrant metastatic pancreatic neuroendocrine carcinoma. Oncogene 2014, 33, 5706–5715. [Google Scholar] [CrossRef] [Green Version]
- Murphy, P.A.; Begum, S.; Hynes, R.O. Tumor angiogenesis in the absence of fibronectin or its cognate integrin receptors. PLoS ONE 2015, 10, e0120872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzopardi, S.; Pang, S.; Klimstra, D.S.; Du, Y.N. p53 and p16(Ink4a)/p19(Arf) Loss Promotes Different Pancreatic Tumor Types from PyMT-Expressing Progenitor Cells. Neoplasia 2016, 18, 610–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, Y.; Kodama, Y.; Shiokawa, M.; Kakiuchi, N.; Marui, S.; Kuwada, T.; Sogabe, Y.; Tomono, T.; Mima, A.; Morita, T.; et al. Rb and p53 Execute Distinct Roles in the Development of Pancreatic Neuroendocrine Tumors. Cancer Res. 2020, 80, 3620–3630. [Google Scholar] [CrossRef] [PubMed]
- Masiello, P.; Wollheim, C.B.; Gori, Z.; Blondel, B.; Bergamini, E. Streptozotocin-induced functioning islet cell tumor in the rat: High frequency of induction and biological properties of the tumor cells. Toxicol. Pathol. 1984, 12, 274–280. [Google Scholar] [CrossRef]
- Hully, J.R.; Su, Y.; Lohse, J.K.; Griep, A.E.; Sattler, C.A.; Haas, M.J.; Dragan, Y.; Peterson, J.; Neveu, M.; Pitot, H.C. Transgenic hepatocarcinogenesis in the rat. Am. J. Pathol. 1994, 145, 384–397. [Google Scholar]
- Haas, M.J.; Dragan, Y.P.; Hikita, H.; Shimel, R.; Takimoto, K.; Heath, S.; Vaughan, J.; Pitot, H.C. Transgene expression and repression in transgenic rats bearing the phosphoenolpyruvate carboxykinase-simian virus 40 T antigen or the phosphoenolpyruvate carboxykinase-transforming growth factor-alpha constructs. Am. J. Pathol. 1999, 155, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Gaudenzi, G.; Carra, S.; Dicitore, A.; Cantone, M.; Persani, L.; Vitale, G. Fishing for neuroendocrine tumors. Endocr.-Relat. Cancer 2020, 27, R163–R176. [Google Scholar] [CrossRef]
- Vitale, G.; Gaudenzi, G.; Dicitore, A.; Cotelli, F.; Ferone, D.; Persani, L. Zebrafish as an innovative model for neuroendocrine tumors. Endocr.-Relat. Cancer 2014, 21, R67–R83. [Google Scholar] [CrossRef]
- Yang, H.W.; Kutok, J.L.; Lee, N.H.; Piao, H.Y.; Fletcher, C.D.; Kanki, J.P.; Look, A.T. Targeted expression of human MYCN selectively causes pancreatic neuroendocrine tumors in transgenic zebrafish. Cancer Res. 2004, 64, 7256–7262. [Google Scholar] [CrossRef] [Green Version]
- Salanga, M.; Horb, M. Xenopus as a model for GI/pancreas disease. Curr. Pathobiol. Rep. 2015, 3, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Naert, T.; Dimitrakopoulou, D.; Tulkens, D.; Demuynck, S.; Carron, M.; Noelanders, R.; Eeckhout, L.; Van Isterdael, G.; Deforce, D.; Vanhove, C. RBL1 (p107) functions as tumor suppressor in glioblastoma and small-cell pancreatic neuroendocrine carcinoma in Xenopus tropicalis. Oncogene 2020, 39, 2692–2706. [Google Scholar] [CrossRef]
- Capodanno, Y.; Altieri, B.; Elders, R.; Colao, A.; Faggiano, A.; Schrader, J. Canine insulinoma as a model for human malignant insulinoma research: Novel perspectives for translational clinical studies. Transl. Oncol. 2022, 15, 101269. [Google Scholar] [CrossRef]
- de Vries, C.; Konukiewitz, B.; Weichert, W.; Klöppel, G.; Aupperle-Lellbach, H.; Steiger, K. Do Canine Pancreatic Neuroendocrine Neoplasms Resemble Human Pancreatic Neuroendocrine Tumours? A Comparative Morphological and Immunohistochemical Investigation. J. Comp. Pathol. 2020, 181, 73–85. [Google Scholar] [CrossRef]
- Wong, C.; Vosburgh, E.; Levine, A.J.; Cong, L.; Xu, E.Y. Human neuroendocrine tumor cell lines as a three-dimensional model for the study of human neuroendocrine tumor therapy. J. Vis. Exp. 2012, 66, e4218. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.; Laddha, S.V.; Tang, L.; Vosburgh, E.; Levine, A.J.; Normant, E.; Sandy, P.; Harris, C.R.; Chan, C.S.; Xu, E.Y. The bromodomain and extra-terminal inhibitor CPI203 enhances the antiproliferative effects of rapamycin on human neuroendocrine tumors. Cell Death Dis. 2014, 5, e1450. [Google Scholar] [CrossRef] [Green Version]
- Al-Ramadan, A.; Mortensen, A.C.; Carlsson, J.; Nestor, M.V. Analysis of radiation effects in two irradiated tumor spheroid models. Oncol. Lett. 2018, 15, 3008–3016. [Google Scholar] [CrossRef] [Green Version]
- Bresciani, G.; Hofland, L.J.; Dogan, F.; Giamas, G.; Gagliano, T.; Zatelli, M.C. Evaluation of Spheroid 3D Culture Methods to Study a Pancreatic Neuroendocrine Neoplasm Cell Line. Front. Endocrinol. 2019, 10, 682. [Google Scholar] [CrossRef]
- Lundsten, S.; Spiegelberg, D.; Stenerlöw, B.; Nestor, M. The HSP90 inhibitor onalespib potentiates 177Lu-DOTATATE therapy in neuroendocrine tumor cells. Int. J. Oncol. 2019, 55, 1287–1295. [Google Scholar] [CrossRef]
- Matrood, S.; de Prisco, N.; Wissniowski, T.T.; Wiese, D.; Jabari, S.; Griesmann, H.; Wanzel, M.; Stiewe, T.; Neureiter, D.; Klieser, E.; et al. Modulation of Pancreatic Neuroendocrine Neoplastic Cell Fate by Autophagy-Mediated Death. Neuroendocrinology 2020, 111, 965–985. [Google Scholar] [CrossRef]
- Herrera-Martínez, A.D.; Feelders, R.A.; Van den Dungen, R.; Dogan-Oruc, F.; van Koetsveld, P.M.; Castaño, J.P.; de Herder, W.W.; Hofland, L.J. Effect of the Tryptophan Hydroxylase Inhibitor Telotristat on Growth and Serotonin Secretion in 2D and 3D Cultured Pancreatic Neuroendocrine Tumor Cells. Neuroendocrinology 2020, 110, 351–363. [Google Scholar] [CrossRef]
- Wagener, N.; Buchholz, M.; Bertolino, P.; Zhang, C.X.; Di Fazio, P. Exploring the MEN1 dependent modulation of caspase 8 and caspase 3 in human pancreatic and murine embryo fibroblast cells. Apoptosis 2022, 27, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Gulde, S.; Foscarini, A.; April-Monn, S.L.; Genio, E.; Marangelo, A.; Satam, S.; Helbling, D.; Falconi, M.; Toledo, R.A.; Schrader, J.; et al. Combined Targeting of Pathogenetic Mechanisms in Pancreatic Neuroendocrine Tumors Elicits Synergistic Antitumor Effects. Cancers 2022, 14, 5481. [Google Scholar] [CrossRef]
- April-Monn, S.L.; Wiedmer, T.; Skowronska, M.; Maire, R.; Schiavo Lena, M.; Trippel, M.; Di Domenico, A.; Muffatti, F.; Andreasi, V.; Capurso, G.; et al. Three-Dimensional Primary Cell Culture: A Novel Preclinical Model for Pancreatic Neuroendocrine Tumors. Neuroendocrinology 2021, 111, 273–287. [Google Scholar] [CrossRef]
- Shi, X.; Li, Y.; Yuan, Q.; Tang, S.; Guo, S.; Zhang, Y.; He, J.; Zhang, X.; Han, M.; Liu, Z.; et al. Integrated profiling of human pancreatic cancer organoids reveals chromatin accessibility features associated with drug sensitivity. Nat. Commun. 2022, 13, 2169. [Google Scholar] [CrossRef] [PubMed]
- Hogenson, T.L.; Xie, H.; Phillips, W.J.; Toruner, M.D.; Li, J.J.; Horn, I.P.; Kennedy, D.J.; Almada, L.L.; Marks, D.L.; Carr, R.M.; et al. Culture media composition influences patient-derived organoid ability to predict therapeutic responses in gastrointestinal cancers. JCI Insight 2022, 7, e158060. [Google Scholar] [CrossRef] [PubMed]
- Dayton, T.L.; Alcala, N.; Moonen, L.; den Hartigh, L.; Mangiante, L.; Lap, L.; Dost, A.F.M.; Beumer, J.; Levy, S.; van Leeuwaarde, R.S.; et al. Druggable Growth Dependencies and Tumor Evolution Analysis in Patient-Derived Organoids of Neuroendocrine Cancer. bioRxiv 2022. [Google Scholar] [CrossRef]
- April-Monn, S.L.; Detjen, K.; Kirchner, P.; Bräutigam, K.; Trippel, M.A.; Marques, I.J.; Grob, T.; Statzer, C.; Maire, R.S.; Kollàr, A.; et al. Patient-derived tumoroids of advanced high-grade neuroendocrine neoplasms mimic patient chemotherapy responses and guide the design of personalized combination therapies. bioRxiv 2022. [Google Scholar] [CrossRef]
- Herring, B.; Whitt, J.; Aweda, T.; Ou, J.; Guenter, R.; Lapi, S.; Berry, J.; Chen, H.; Liu, X.; Rose, J.B.; et al. A growth model of neuroendocrine tumor surrogates and the efficacy of a novel somatostatin-receptor–guided antibody-drug conjugate: Perspectives on clinical response? Surgery 2020, 167, 197–203. [Google Scholar] [CrossRef]
- Herring, B.; Jang, S.; Whitt, J.; Goliwas, K.; Aburjania, Z.; Dudeja, V.; Ren, B.; Berry, J.; Bibb, J.; Frost, A.; et al. Ex Vivo Modeling of Human Neuroendocrine Tumors in Tissue Surrogates. Front. Endocrinol. 2021, 12, 710009. [Google Scholar] [CrossRef]
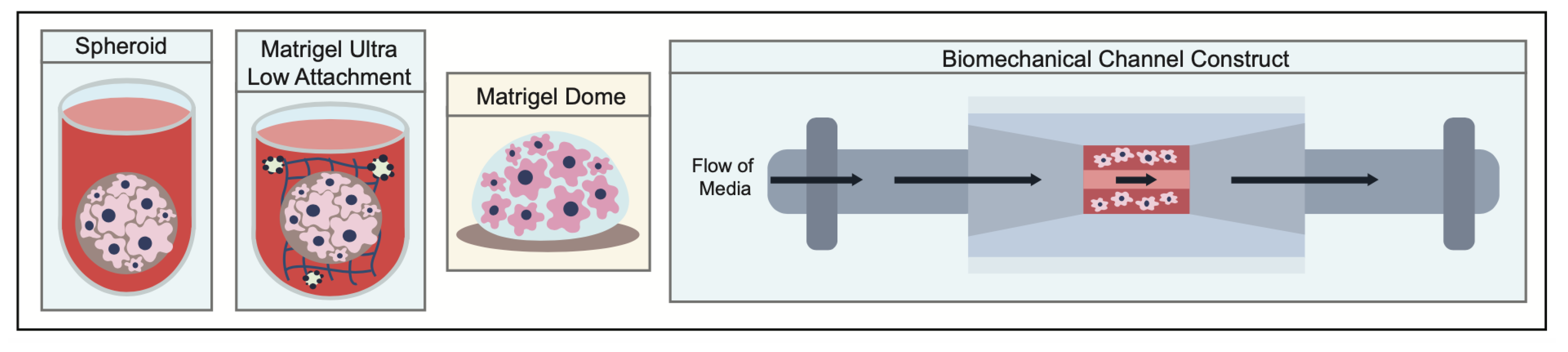
- Białkowska, K.; Komorowski, P.; Bryszewska, M.; Miłowska, K. Spheroids as a Type of Three-Dimensional Cell Cultures-Examples of Methods of Preparation and the Most Important Application. Int. J. Mol. Sci. 2020, 21, 6225. [Google Scholar] [CrossRef]
- Skardal, A.; Aleman, J.; Forsythe, S.; Rajan, S.; Murphy, S.; Devarasetty, M.; Pourhabibi Zarandi, N.; Nzou, G.; Wicks, R.; Sadri-Ardekani, H.; et al. Drug compound screening in single and integrated multi-organoid body-on-a-chip systems. Biofabrication 2020, 12, 025017. [Google Scholar] [CrossRef] [PubMed]
- Ear, P.H.; Li, G.; Wu, M.; Abusada, E.; Bellizzi, A.M.; Howe, J.R. Establishment and Characterization of Small Bowel Neuroendocrine Tumor Spheroids. J. Vis. Exp. 2019, 152, e60303. [Google Scholar] [CrossRef]
- Ishiguro, T.; Ohata, H.; Sato, A.; Yamawaki, K.; Enomoto, T.; Okamoto, K. Tumor-derived spheroids: Relevance to cancer stem cells and clinical applications. Cancer Sci. 2017, 108, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Dominijanni, A.; Mazzocchi, A.; Shelkey, E.; Forsythe, S.; Devarsetty, M.; Soker, S. Bioengineered Tumor Organoids. Curr. Opin. Biomed. Eng. 2020, 13, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.S.; Postovit, L.M.; Lajoie, G.A. Matrigel: A complex protein mixture required for optimal growth of cell culture. Proteomics 2010, 10, 1886–1890. [Google Scholar] [CrossRef] [PubMed]
- Devarasetty, M.; Forsythe, S.D.; Shelkey, E.; Soker, S. In Vitro Modeling of the Tumor Microenvironment in Tumor Organoids. Tissue Eng. Regen. Med. 2020, 17, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, K.K.; van den Berg, J.G.; Weeber, F.; van de Haar, J.; Velds, A.; Kaing, S.; Peters, D.; Eskens, F.; de Groot, D.A.; Tesselaar, M.E.T.; et al. Patient-Derived Organoid Models of Human Neuroendocrine Carcinoma. Front. Endocrinol. 2021, 12, 627819. [Google Scholar] [CrossRef]
- Votanopoulos, K.I.; Mazzocchi, A.; Sivakumar, H.; Forsythe, S.; Aleman, J.; Levine, E.A.; Skardal, A. Appendiceal Cancer Patient-Specific Tumor Organoid Model for Predicting Chemotherapy Efficacy Prior to Initiation of Treatment: A Feasibility Study. Ann. Surg. Oncol. 2019, 26, 139–147. [Google Scholar] [CrossRef]
- Forsythe, S.D.; Sivakumar, H.; Erali, R.A.; Wajih, N.; Li, W.; Shen, P.; Levine, E.A.; Miller, K.E.; Skardal, A.; Votanopoulos, K.I. Patient-Specific Sarcoma Organoids for Personalized Translational Research: Unification of the Operating Room with Rare Cancer Research and Clinical Implications. Ann. Surg. Oncol. 2022, 29, 7354–7367. [Google Scholar] [CrossRef]
- Forsythe, S.D.; Erali, R.A.; Laney, P.; Sivakumar, H.; Li, W.; Skardal, A.; Soker, S.; Votanopoulos, K.I. Application of immune enhanced organoids in modeling personalized Merkel cell carcinoma research. Sci. Rep. 2022, 12, 13865. [Google Scholar] [CrossRef]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschenes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsythe, S.D.; Erali, R.A.; Edenhoffer, N.; Meeker, W.; Wajih, N.; Schaaf, C.R.; Laney, P.; Vanezuela, C.D.; Li, W.; Levine, E.A.; et al. Cisplatin exhibits superiority over MMC as a perfusion agent in a peritoneal mesothelioma patient specific organoid HIPEC platform. Sci. Rep. 2023, 13, 11640. [Google Scholar] [CrossRef]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernandez-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsythe, S.D.; Sasikumar, S.; Moaven, O.; Sivakumar, H.; Shen, P.; Levine, E.A.; Soker, S.; Skardal, A.; Votanopoulos, K.I. Personalized Identification of Optimal HIPEC Perfusion Protocol in Patient-Derived Tumor Organoid Platform. Ann. Surg. Oncol. 2020, 27, 4950–4960. [Google Scholar] [CrossRef]
- Forsythe, S.D.; Erali, R.A.; Sasikumar, S.; Laney, P.; Shelkey, E.; D’Agostino, R.; Miller, L.D.; Shen, P.; Levine, E.A.; Soker, S.; et al. Organoid Platform in Preclinical Investigation of Personalized Immunotherapy Efficacy in Appendiceal Cancer: Feasibility Study. Clin. Cancer Res. 2021, 27, 5141–5150. [Google Scholar] [CrossRef]
- Aisenbrey, E.A.; Murphy, W.L. Synthetic alternatives to Matrigel. Nat. Rev. Mater. 2020, 5, 539–551. [Google Scholar] [CrossRef]
- Aleman, J.; Skardal, A. A multi-site metastasis-on-a-chip microphysiological system for assessing metastatic preference of cancer cells. Biotechnol. Bioeng. 2019, 116, 936–944. [Google Scholar] [CrossRef]
- Caddeo, S.; Boffito, M.; Sartori, S. Tissue Engineering Approaches in the Design of Healthy and Pathological In Vitro Tissue Models. Front. Bioeng. Biotechnol. 2017, 5, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skardal, A.; Devarasetty, M.; Forsythe, S.; Atala, A.; Soker, S. A reductionist metastasis-on-a-chip platform for in vitro tumor progression modeling and drug screening. Biotechnol. Bioeng. 2016, 113, 2020–2032. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Li, Z.-X.; Zhu, Y.-J.; Fu, J.; Zhao, X.-F.; Zhang, Y.-N.; Wang, S.; Wu, J.-M.; Wang, K.-T.; Wu, R.; et al. Single-Cell Transcriptome Analysis Uncovers Intratumoral Heterogeneity and Underlying Mechanisms for Drug Resistance in Hepatobiliary Tumor Organoids. Adv. Sci. 2021, 8, 2003897. [Google Scholar] [CrossRef]
- van Riet, J.; van de Werken, H.J.G.; Cuppen, E.; Eskens, F.; Tesselaar, M.; van Veenendaal, L.M.; Klumpen, H.J.; Dercksen, M.W.; Valk, G.D.; Lolkema, M.P.; et al. The genomic landscape of 85 advanced neuroendocrine neoplasms reveals subtype-heterogeneity and potential therapeutic targets. Nat. Commun. 2021, 12, 4612. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Lu, J.; Li, Q.; Xiong, F.; Zhang, Y.; Zhu, J.; Peng, G.; Yang, J. Single-cell heterogeneity analysis and CRISPR screens in MIN6 cell line reveal transcriptional regulators of insulin. Cell Cycle 2021, 20, 2053–2065. [Google Scholar] [CrossRef] [PubMed]
- Al Shihabi, A.; Davarifar, A.; Nguyen, H.T.L.; Tavanaie, N.; Nelson, S.D.; Yanagawa, J.; Federman, N.; Bernthal, N.; Hornicek, F.; Soragni, A. Personalized chordoma organoids for drug discovery studies. Sci. Adv. 2022, 8, eabl3674. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Wells, J.M.; Helmrath, M.A.; Zorn, A.M. Organoid Center Strategies for Accelerating Clinical Translation. Cell Stem Cell 2018, 22, 806–809. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Name | Year | Species | Tumor Subtype | Primary/ Metastasis | Notable Features or Mutations | References |
|---|---|---|---|---|---|---|
| βTC | 1988 | Mouse | Insulinoma | Primary | SV40 T-Antigen in RIP-Tag Mice | [37] |
| MIN6 | 1990 | Mouse | Insulinoma | Primary | SV40 T-Antigen in transgenic mice | [38] |
| Alpha TC1 | 1990 | Mouse | Glucagonoma | Primary (Alpha Cell) | PR proglucagon promoter-driven SV40 T antigen from C57BL/6 x DBA/2 mouse | [39] |
| BTC | 1990 | Mouse | Insulinoma | Primary | Polyoma small T antigen and SV40 from C57BL/6J double transgenic RIP1Tag2/ Rip2pyST1 | [40,41] |
| NIT-1 | 1991 | Mouse | β-Cell Adenoma | Primary | NOD/Lt mice insulin-promoter/SV40 T-antigen | [42] |
| TGP61 | 1994 | Mouse | Insulinoma | Primary | Tg(Ela-1-SV40E)Bri18 transgenic mice with SV40 T antigen | [43] |
| Mu Islet (E6/E7) | 2009 | Mouse | NA | Primary (Alpha Cells) | C57BLKS/J Mouse Islet cells transduced by HPV E6 and E7 antigens | [44] |
| RIN | 1980 | Rat | Insulinoma | Primary | X-ray induced Inbred NEDH rat strain | [45] |
| INS1 | 1992 | Rat | Insulinoma | Primary | X-ray induced Inbred NEDH rat strain | [46] |
| HIT | 1981 | Syrian Hamster | Primary Islet Culture | Primary | Islets transformed from SV40 | [47] |
| canINS | 2017 | Dog | Insulinoma | Primary | NA | [48] |
| QGP-1 | 1980 | Human | Somatostatinoma Delta Cells | Primary | ATRX, KRAS, TP53, APC | [49] |
| CM | 1987 | Human | Insulinoma | Met (Ascites) | Severe Chromosomal Aberrations | [50] |
| BON-1 | 1991 | Human | NF pNEC | Met (LN) | ATRX, TSC2, NRAS, TP53, BRCA2, APC, CDK2A/B | [51] |
| HuNET | 2001 | Human | VIP | Primary | NA | [52] |
| A99 | 2011 | Human | SCC | Met (Liver) | p53, KRAS | [53] |
| APL1 | 2016 | Human | NF, G1 | Met (Liver) | CD47+, HGF/MET | [54] |
| NT-3 | 2018 | Human | WD Insulinoma | Met (LN) | SSTR+, MEN1, VEGF+ | [55] |
| SPNE1 | 2022 | Human | pNET | Primary | CD44+, SSTR+ | [56] |
| NT-18P | 2022 | Human | G3 | Primary | DAXX, MEN1, p53, MSH6 | [57] |
| NT-18LM | 2022 | Human | G3 | Met (Liver) | DAXX, MEN1, MSH6 | [57] |
| NT-36 | 2022 | Human | G3 | Primary (recurrence) | DAXX, RAD50, MEN1, MSH6 | [57] |
| NT-32 | 2022 | Human | LC pNEC | Primary | BRAF, RB1, p53 | [57] |
| Author | Year | Host Species | Strain | Tumor Type | Xenograft Source | PDX Method/Site | Success Rate | References |
|---|---|---|---|---|---|---|---|---|
| Yang | 2016 | Mouse | NOD-SCID | NET | Tumor | Subcu flank Tissue | 3/58 (5.2%) * | [80] |
| Chamberlain | 2018 | Mouse | Athymic Nude | Insulinoma (liver met) | Tumor | Subcu Tissue | 1/1 (100%) | [81] |
| Kawasaki | 2020 | Mouse | NOG | NETG3, NEC (LC) | Organoid | Renal/spleen | 2/2 (100%) | [82] |
| Pham | 2020 | Mouse | NOD SCID | pNET | Tumor | Subcu, Orthotopic | 1/5 (20%) | [83] |
| Tran | 2022 | Mouse | NSG | NETG2 | Tumor | Subcu Cells | 0/3 (0%) | [84] |
| Gaudenzi | 2017 | Zebrafish | Tg(fli1a:EGFP) y1 | NETG1 (LM) | Tumor | Embryo injection (cells) | 1/1 (100%) | [85] |
| Model Name | Year | pNEN Subtype | Target Gene | Strain | Model Type | Tumor Development | % of Mice w pNENs | Other Tumors | References |
|---|---|---|---|---|---|---|---|---|---|
| RIP-Tag | 1985 | Beta Cell Tumors, pNEC | SV40 Tag (RIP) | B6D2Fl (C57BL/6J × DBA/21 | Transgenic | 10–20 weeks | 100% | None | [87,102,103] |
| RIP1Tag2/RIP2PyST1 | 1990 | Beta Cell Tumors | SV40 Tag and polyoma small T-antigen (PyST) | RIP-Tag Mice | Transgenic | 6–14 weeks | 100% | Colon | [40,41] |
| RIP-Tag5 | 1996 | Beta Cell Tumor | SV40 Tag (RIP) | C3Heb/FeJ (CH3) | Transgenic | 17–22 Weeks | 100% | None | [101] |
| Rip1Tag2xRip1E-Cad, Rip1Tag2xRip1dnE-cad | 1998 | Beta Cell Tumors | SV40 Tag (E-Cad) | B6D2Fl (C57BL/6J × DBA/21 | Transgenic | 10–16 Weeks | 7.8%, 50.6% | None | [96] |
| RipVEGF-C × Rip1Tag2 | 2001 | Insulinomas | SV40 Tag (RIP) and VEGF-C (RIP) | B6D2Fl (C57BL/6J × DBA/21 | Transgenic | 10 Weeks | 100% | None | [89] |
| Rip1Tag2/Rip1VEGF-A | 2002 | pNETs | SV40 Tag (RIP) and VEGF-A (RIP) | B6D2Fl (C57BL/6J × DBA/21 | Transgenic | 10 weeks | 100% | None | [90] |
| RIP7-Igf-1R, RIP1-Tag2 | 2002 | Invasive Carcinoma, Beta Cell Hyperlplasia | SV40 Tag (RIP) and IGF1R (RIP) | B6D2Fl (C57BL/6J × DBA/21 | Transgenic | 5–15 weeks | 100% | None | [92] |
| RIP-Tag2,Rag−/− | 2005 | Beta Cell Hyperplasia | Rag1 | C57-B16-J | Transgenic, Homozygous KO | 13.5 Weeks | 100% | None | [95] |
| Rip1Tag2;Rip1VEGF-D | 2007 | Beta Cell Tumors | SV40 Tag (RIP) and VEGF-D (RIP) | C57BL/6 | Transgenic | 12–14 Weeks | 100% | None | [91] |
| RipTag-IRES-Luciferase (RTL) 1 | 2010 | Insulinoma | SV40 Tag (RIP-IRES) | C57Bl/6 | Transgenic | 7–10 weeks | 100% | None | [88] |
| RT2/TNC, RT2/TNCKO | 2013 | Insulinoma | TNC | C57Bl6, C57Bl6 × 129/Sv-C57Bl6 | Transgenic | 8–12 weeks | NA | None | [93] |
| Hpa-Tg RT2, Hpse−/− RT2 | 2013 | Islet Cell Carcinoma | Heparanase | C57BL/6 | Transgenic, Homozygous KO | 13.5 weeks | 100% | None | [94] |
| RIP-Tag2 | 2019 | NF pNETs | SV40 Tag (RIP), Insm1 low | RT2 AB6F1 | Transgenic | 17 weeks | 100% | None | [98] |
| RIP-TAG | 2020 | pNET | RIP-Tag | RT2 B6A(F1) | Transgenic | 8–12 Weeks | 100% | siNET | [99] |
| RIP-TAG2, pl-PDGFB KO | 2022 | Islet Cell Carcinoma | RIP-Tag2, PDGFB | C57BL/6 | Transgenic, Selective KO | 8–14 Weeks | 100% | None | [97] |
| Model Name | Year | pNEN Subtype | Target Gene | Strain | Model Type | Tumor Development | % of Mice w pNENs | Other Tumors | References |
|---|---|---|---|---|---|---|---|---|---|
| VT-C (AVP-Tag) | 1987 | Islet Dysplasia | SV40-Tag (Vasopressin) | C57B1/KJ X SJL F1 | Transgenic | 90–140 Days Hyp | 7/8 | Pituitary | [111] |
| ESLV Tg (Ela-1,SV40E)Bri18 | 1987 | D-Cell Hyperplasia, Insulinomas | SV40-Tag (Elastase) | C57/SJL F2 | Transgenic | 8 weeks Hyp 20 Weeks Tumors | 100% | Pancreatic Exocrine Tumor | [104,105] |
| Glu2-Tag | 1988 | Alpha Cell Hyperplasia | SV40-Tag (Glu2) | C57BL/6J × DBA/2J | Transgenic | 5 months Hyp, 9–12-months tumors | 100% | None | [109] |
| SV-202 | 1989 | Islet Cell Adenoma | SV40-Tag (MT) | C57BL/6JXSJL F1 | Transgenic | 15 Weeks tumors, 20 Weeks (Death) | 100% | Liver | [112,113] |
| MSV125 | 1990 | Insulinoma | MSV-SV40 | NA | Transgenic | 2–12 Months | 100% | Brain, Eye, Kidney, Sarcoma | [106] |
| L-PK/Tag | 1992 | Islet Cell Carcinoma GLUTag-Ytg | SV40 Tag (L-Type Pyruvate-Kinase) | (C57BL/6 × DBA)F1 | Transgenic | NA | 80% | Liver | [107] |
| GLUTag-Ytg | 1992 | Islet Cell Carcinoma | SV40 Tag (RG) | CD1 | Transgenic | 11–12 weeks | 100% | Colon | [110,114] |
| GP1.5 Tag, GP10.5 Tag | 1993 | Pancreatic Islet Cell Tumors | SV40 Tag (Gastrin) | CD1 | Transgenic | 80–100 Days (Death) | 100% | Hepatobiliary Tract | [108] |
| Secretin-Tag | 1995 | Insulinoma | SV40 Tag (Secretin) | B6D2F1× B6D2F1 embryos (CD6) | Transgenic | 12 Weeks | >80% | siNET, Colon | [115] |
| Model Name | Year | pNEN Subtype | Gene | Strain | Model Type | Tumor Development | % of Mice w pNENs | Other Tumors | References |
|---|---|---|---|---|---|---|---|---|---|
| Rb1+/p53+/− | 1994 | Islet Cell Tumors (pNEC) | Rb1, p53 | (C57BLx CBA) × C57BL/6 | Global Heterozygous KO | 9 months, 3–6 months | 14%, 23% | Many | [119,121] |
| MEN1+/−Rb1+/− | 2007 | Islet Cell Tumors | MEN1, Rb1 | C57BL/6j:129 × FVB/N:129 | Global Heterozygous KO | 402 days | 55% | Many | [118] |
| Men1+/1 | 2009 | Insulinoma, Glucagonoma, NF | MEN1 | C57BL/6 | Global Heterozygous KO | 9–12 months | 60% | Many | [122] |
| Cul9+/− | 2011 | Insulinoma | CUL9 | BL/6 | Global Heterozygous KO | 21 months | 1/23 (4.3%) | Many | [120] |
| Model Name | Year | pNEN Subtype | Target Gene | Strain | Model Type | Tumor Development | % of Mice w pNENs | Other Tumors | References |
|---|---|---|---|---|---|---|---|---|---|
| Rb1+/−p53−/− | 1994 | Islet Cell Tumors (pNEC) | Rb1, p53 | (C57BLx CBA) × C57BL/6 | Global Heterozygous KO | 3–6 months | 14%, 23% | Many | [119,121] |
| SPC−/− | 1997 | Alpha and Delta Cell Hyperplasia | SPC | C57BL/6J | Homozygous KO | 3 Months | 100% | None | [123] |
| Gcgr−/− | 2003 | Glucagonoma | Gcgr | C57BL/6J | Homozygous KO | 8 weeks (hyperplasia) | 100% | None | [125] |
| Prdx−/− | 2003 | Pancreatic Islet Cell Adenoma | PrDX1 | B6 | Homozygous KO | 9 months | 9% | Many | [127] |
| Gcgr−/− | 2011 | Glucagonoma, NF | Gcgr2 | C57BL/6 × DBA1/lacJ | Homozygous KO | 5–7 months, 10–12 months | 100% | None | [126] |
| PC2−/− | 2014 | Glucagonoma | PCKS2 | C57Bl6 | Homozygous KO | 3 months (hyper) 6–8 months | 100% | None | [124] |
| Name | Year | pNEN Subtype | Target Gene | Strain | Model Type | Tumor Development | % of Mice w pNENs | Other Tumors | References |
|---|---|---|---|---|---|---|---|---|---|
| RIP-MyrAkt1 | 2001 | NET | MyrAKT (pS473) (RIP) | B6SJLF1/J | Transgenic, IA | 8–12 weeks (hyperplasia) | 100% | NA | [128] |
| pIns-c-MycERTAM/BCL-XL | 2002 | Islet Cell Carcinoma | MYC, BCL-xl (RIP) | (CBA × C57BL/6)F1 | Transgenic, IA | 2 Weeks (post activation) | 100% | None | [129] |
| Elastase-tv-a;RCAS-c-myc;p16−/−p19-/- | 2003 | Insulinoma | c-Myc | FVB (lnk4a/Arf null) | Transgenic, IA | 7 months | 4/14 | Sarcoma, Lymphoma | [130] |
| TS-T1 | 2007 | Islet Cell Tumors | Thymidylate Synthase | FVB | Transgenic, IA | 9–24 months | 23% hyper, 6% adenoma | None | [131] |
| RIP-MyrAkt1 (SK61−/−) | 2008 | Insulinoma | Akt1 and (S6K1) | B6SJLF1/J × (C57Bl/6xDBA/2) | Transgenic, IA | 1 year (death) | 19/23 | Lung, Pancreatic Carcinoma | [134] |
| INS-p25OE | 2021 | Beta Cell (WD) | CDK5R1 | Ins2-rtTA × tetOp-p25GFP | Transgenic, IA | 10–15 weeks | 100% | None | [132] |
| hTS/Men1–/– | 2022 | Islet Cell Carcinoma | Thymidylate Synthase, MEN1 | FVB × (C57BL/6J × NIH Black Swiss females or 129/SvEvTacFBR) × (129/Ola × 129/Sv) | Transgenic, IA, Inducible homozygous KO | 10 months | 100% | Pituitary | [133] |
| hTS/MEN1+/− | 2022 | Islet Cell Carcinomas | Thymidylate Synthase, MEN1 | FVB × (C57BL/6J × NIH Black Swiss females or 129/SvEvTacFBR) × (129/Ola × 129/Sv) | Transgenic, IA, Inducible heterozygous KO | 22 months | 100% | Pituitary | [133] |
| Name | Year | pNEN Subtype | Target Gene | Strain | Model Type | Tumor Development | % of Mice w pNENs | Other Tumors | References |
|---|---|---|---|---|---|---|---|---|---|
| Cdk4R24C/R24C | 2001 | Beta Cell Tumor, PP, Glucagonoma | Cdk4 (R24C) | mixed 129/Sv CD-1 | Homozygous KI | 8 months detectable, dead at 16 | 34% | Many | [135] |
| Gcggfp/gfp | 2009 | Alpha Cell Hyperplasia | Gcg | C57/BL6J | Homozygous KI | 2 months | 100% | None | [136] |
| Name | Year | pNEN Subtype | Target Gene | Strain | Model Type | Tumor Development | % of Mice w pNENs | Other Tumors | References |
|---|---|---|---|---|---|---|---|---|---|
| Men1TSM/+, Men1ΔN3–8/+ | 2001 | Pancreatic Islet Tumors | MEN1 | C57BL/6J × NIH Black Swiss females or 129/SvEvTacFBR | Cre-LoxP Heterozygous KO | 9 months (hyperplasia) | 28% | Many | [117] |
| Men1+/T | 2003 | Insulinoma, Glucagonoma | MEN1 | (129/Ola × 129/Sv) | Cre-LoxP Heterozygous KO | 8+ months | >60% | Many | [148] |
| Men1loxP/loxP Rip-cre+ | 2004 | Insulinoma | MEN1 | C57BL/6J | Cre-LoxP Homozygous KO | 4 months (hyperplasia), 9 months tumors | 12/12 hyperplasia, 7/12 tumors | Pituitary, Prolactinomas | [140] |
| MEN1+/−;Rb1ΔX2/+ | 2007 | Insulinoma, Glucagonoma | MEN1, Rb1 | C57/129 | Cre-LoxP Heterozygous KO | 210–360 Days | 10/18 hyperplasia, 1/18 | Many | [118] |
| Pdx1-Cre, MEN1 f/f,Pdx1-Cre MEN1f/+ | 2009 | Insulinoma | MEN1 (PDX1) | FVB;129Sv | Cre-LoxP Homozygous KO | 5–6 months hyperplasia, 10–12 tumors | >80% | None | [141] |
| MEN1F/F-GluCre+ | 2010 | Glucagonoma, Insulinoma, Mixed | MEN1 (alpha cell only) | R26R | Cre-LoxP Homozygous KO | 2–3 months (hyperplasia), 7 months | 100% | None | [142] |
| Glu-Cre;Men1 f/+ | 2010 | Glucagonoma, Insulinoma | MEN1 (alpha cell only) | Glu-Cre;Z/AP | Cre-LoxP Homozygous KO | 13–14 months | 100% | None | [143] |
| βMen1/Bcat-KO | 2014 | Insulinoma | MEN1, β-Catenin | 129/SvJ × C57BL/6J | Cre-LoxP Homozygous KO | 8 months | 33% | None | [144] |
| (Men1L/L/RIP2-CreER) | 2017 | Insulinoma | MEN1 | C57Bl/6 × 129S | Cre-LoxP Homozygous KO | 2–3 months | 100% | None | [145] |
| MPR (Men1flox/floxPtenflox/flox RIP-Cre) | 2020 | PNETG1/G2 (WD) | MEN1, PTEN | Mixed | Cre-LoxP Homozygous KO | 7 weeks | 100% | Pituitary | [146] |
| (Men1flox/flox Ptenflox/flox MIP-Cre) | 2020 | PNETG1/G2 (WD) | MEN1, PTEN | Mixed | Cre-LoxP Homozygous KO | 7 weeks | 100% | Pituitary | [146] |
| GFAPΔMen1, GFAPΔMen1, Ss−/−, GFAPΔMen1;ΔKif3a; Sst−/− | 2022 | Islet Hyperplasia | MEN1, Sst, Kif3a (GFAP Expressing) | C57BL/6J | Cre-LoxP Homozygous KO | 15–24 months | 50% | Pituitary Prolactinomas, Gastric | [147] |
| Sox10ΔMen1 | 2022 | Islet Hyperplasia | MEN1 (Sox10 expressing cells) | C57BL/6J | Cre-LoxP Transgenic, Induced Homozygous KO | 10–12 months | 57% | Gastric | [147] |
| Study Name | Year | pNEN Subtype | Target Gene | Strain | Model Type | Tumor Development | % of Mice w pNENs | Other Tumors | References |
|---|---|---|---|---|---|---|---|---|---|
| Pdx1-Cre, VHLf/f | 2009 | Adenomas (VHL) | VHL (PDX1) | A/J and C57BL/6 | Cre-LoxP Homozygous KO | 16–18 months | NA | Pancreatic | [149] |
| RenCre x floxed p53/Rb1 | 2014 | Glucagonoma | p53, Rb | C57BL/6J | Cre-LoxP Homozygous KO | 22 weeks | 100% | Sarcoma | [150] |
| Rosa-CreER; FN f/f ; RIP-Tag | 2015 | Islet Cell Tumors | Fibronectin | Many | Cre-LoxP Homozygous KO, Transgenic | 7–11 weeks | 100% | None | [151] |
| Pdx1-tTA; tet-o-MT; p48-cre p16/p19lox/lox | 2016 | pNET (Beta Cell Hyperplasia) | PyMT, INK4A/ARF (PDX1, PTF1A) | ICR, C57BL/6, FVB/N | Cre-LoxP Transgenic | 400–600 days (survival) | 3/35 (8.6%) | Pancreatic Acinar Ductal Carcinoma | [152] |
| RIP7-rtTA; tet-o-MT; p48-cre p53lox/lo | 2016 | pNET (Beta Cell Hyperplasia | PyMT, p53 (RIP, PTF1A | ICR, C57BL/6, FVB/N | Cre-LoxP Transgenic | 400–600 days (survival) | 2/12 (16.7%) | None | [152] |
| RIP7-rtTA; tet-o-MT; p48-cre p16/p19lox/lox | 2016 | pNET (Beta Cell Hyperplasia | PyMT, INK4A/ARF (RIP, PTF1A) | ICR, C57BL/6, FVB/N | Cre-LoxP Transgenic | 400–600 days (survival) | 12/60 (20%) | None | [152] |
| RIP7-rtTA; tet-o-MT; p48-cre p53lox/lox; p16/p19lox/lox | 2016 | pNET (Beta Cell Hyperplasia | PyMT, p53(PDX1, PTF1A | ICR, C57BL/6, FVB/N | Cre-LoxP Transgenic | 400–600 days (survival) | 12/30 (40%) | None | [152] |
| Pdx1-Cre;Rb f/ | 2020 | pNET (WD) | Rb (PDX1) | Pdx1-Cre, Rosa26R, Rb flox | Cre-LoxP Homozygous KO | 18–20 months | 80% | None | [153] |
| Pdx1-Cre;Trp53R172H;Rb f/f | 2020 | pNET | p53, Rb (PDX1) | Pdx1-Cre, Rosa26R, Rb flox, LSL-Trp53R172H | Cre-LoxP Homozygous and Heterozygous KO | 6 months | 100% | None | [153] |
| Study Name | Year | Host Species | Subspecies Model | Tumor Type | Gene | Establishment Method | Tumor Development | Success Rate | References |
|---|---|---|---|---|---|---|---|---|---|
| Chick | 1977 | Rat | NEDH inbred albino | Insulinoma | NA | X-ray-Induced | 4 months | 92% | [61] |
| Maisello | 1984 | Rat | Wistar | Islet Cell Tumor | NA | Chemical | 11–24 months | 81–100% | [154] |
| a/b-SV40 Tag | 1994 | Rat | Transgenic- Sprague Dawley | Islet Cell Tumors | a/b-SV40 Tag | Transgenic | 3–5 months | 33% | [155] |
| PEPCK-TAg | 1999 | Rat | Transgenic Sprague-Dawley | Islet Cell Carcinomas | SV40 Tag (PEPCK Promoter) | Transgenic | 5–8 months | 100% | [156] |
| pPEPCK-TGFAlpha | 1999 | Rat | Transgenic Sprague-Dawley | Islet Cell Carcinomas | TGFAlpha (PEPCK Promoter) | Transgenic | 5–8 months | 100% | [156] |
| z-myod–MYCN, core-z-myod-MYCN | 2004 | Zebrafish | NA | pNET | MYCN | Transgenic | 4–6 months | 4/250 (1.6%) | [159] |
| rb1/rbl1 | 2020 | Frog | X. tropicalis | SC pNEC | Rb1, rbl1 | CRISPR/Cas9 Homozygous KO | 70 days | 86% | [161] |
| rb1/rbl1/tp53cr2 | 2020 | Frog | X. tropicalis | SC pNEC | Rb1, rbl1, tp53 | CRISPR/Cas9 Homozygous KO | 70 days | 77% | [161] |
| Model Type | Cell Source | Success Rate | Year | Studies Performed | References |
|---|---|---|---|---|---|
| Spheroid | BON-1, QGP-1 | 100% | 2012, 2014 | Formation, Drug Screen, IHC, Model Comparison | [164,165] |
| Spheroid | BON-1 | NA | 2018 | Formation, Radiation, IHC | [166] |
| Spheroid | BON-11 | 100% | 2019 | Formation, Drug Screen, IHC | [167] |
| Spheroid | BON-1 | NA | 2019 | Formation, Drug Screen | [168] |
| Spheroid | BON-1, HMEG2725 | NA | 2020 | Formation, Drug Screen | [169] |
| Spheroid | BON-1, QGP-1 | NA | 2020 | Drug Screen | [170] |
| Spheroid | BON-1, QGP-1 | NA | 2022 | Formation, Drug Screen | [171] |
| Spheroid | BON-1, INS-1E, NT-3, Primary pNET | 4/4 (100%) (Primary Drug Screen) | 2022 | Formation, IHC, Drug Screen | [172] |
| Organoid | pNETG3, pNEC | 3/8 (37.5%) | 2020 | IHC, Genomic, RNAseq, Methylation Array, Drug Screen, PDX Formation, Transcriptomic, CRISPR/Cas9 | [82] |
| Organoid | pNET (INS, NF) | 8/11 (72.7%) Culture 6/7 (86%) Drug Testing | 2021 | IHC, Drug Screen | [173] |
| Organoid | pNETG1, G2, pNEC | 4/39 (10.3%) | 2022 | IHC, Genomic, Transcriptomic, Drug Screen | [174] |
| Organoid | pNET | 4/13 (30.7%) | 2022 | IHC, Media Comparison | [175] |
| Organoid | pNET, pNEC (LC) | pNET 0%, pNEC 1/1 (100%) | 2022 | IHC, Drug Screen, Genomic, RNAseq | [176] |
| Organoid, Spheroid | pNETG3, pNEC (LC), QGP-1, NT-3 | 3/3 (100%) | 2022 | IHC, Drug Screen, Genomic, RNAseq, Transcriptomic, Model Comparison | [177] |
| Bioreactor | BON-1, QGP-1, PDX, Primary pNET | 100% (Both) | 2020, 2021 | IHC, Drug Screen, Flow Cytometry, Invasion | [178,179] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forsythe, S.D.; Pu, T.; Andrews, S.G.; Madigan, J.P.; Sadowski, S.M. Models in Pancreatic Neuroendocrine Neoplasms: Current Perspectives and Future Directions. Cancers 2023, 15, 3756. https://doi.org/10.3390/cancers15153756
Forsythe SD, Pu T, Andrews SG, Madigan JP, Sadowski SM. Models in Pancreatic Neuroendocrine Neoplasms: Current Perspectives and Future Directions. Cancers. 2023; 15(15):3756. https://doi.org/10.3390/cancers15153756
Chicago/Turabian StyleForsythe, Steven D., Tracey Pu, Stephen G. Andrews, James P. Madigan, and Samira M. Sadowski. 2023. "Models in Pancreatic Neuroendocrine Neoplasms: Current Perspectives and Future Directions" Cancers 15, no. 15: 3756. https://doi.org/10.3390/cancers15153756
APA StyleForsythe, S. D., Pu, T., Andrews, S. G., Madigan, J. P., & Sadowski, S. M. (2023). Models in Pancreatic Neuroendocrine Neoplasms: Current Perspectives and Future Directions. Cancers, 15(15), 3756. https://doi.org/10.3390/cancers15153756