
Minimal Residual Disease in Multiple Myeloma: Past, Present, and Future
 ,
,  ,
, 

Abstract
Simple Summary
Abstract
1. Prognostic and Clinical Relevance of Minimal Residual Disease in Multiple Myeloma
1.1. Definition of Minimal Residual Disease
1.2. Baseline and Post-Treatment Outcome Predictors
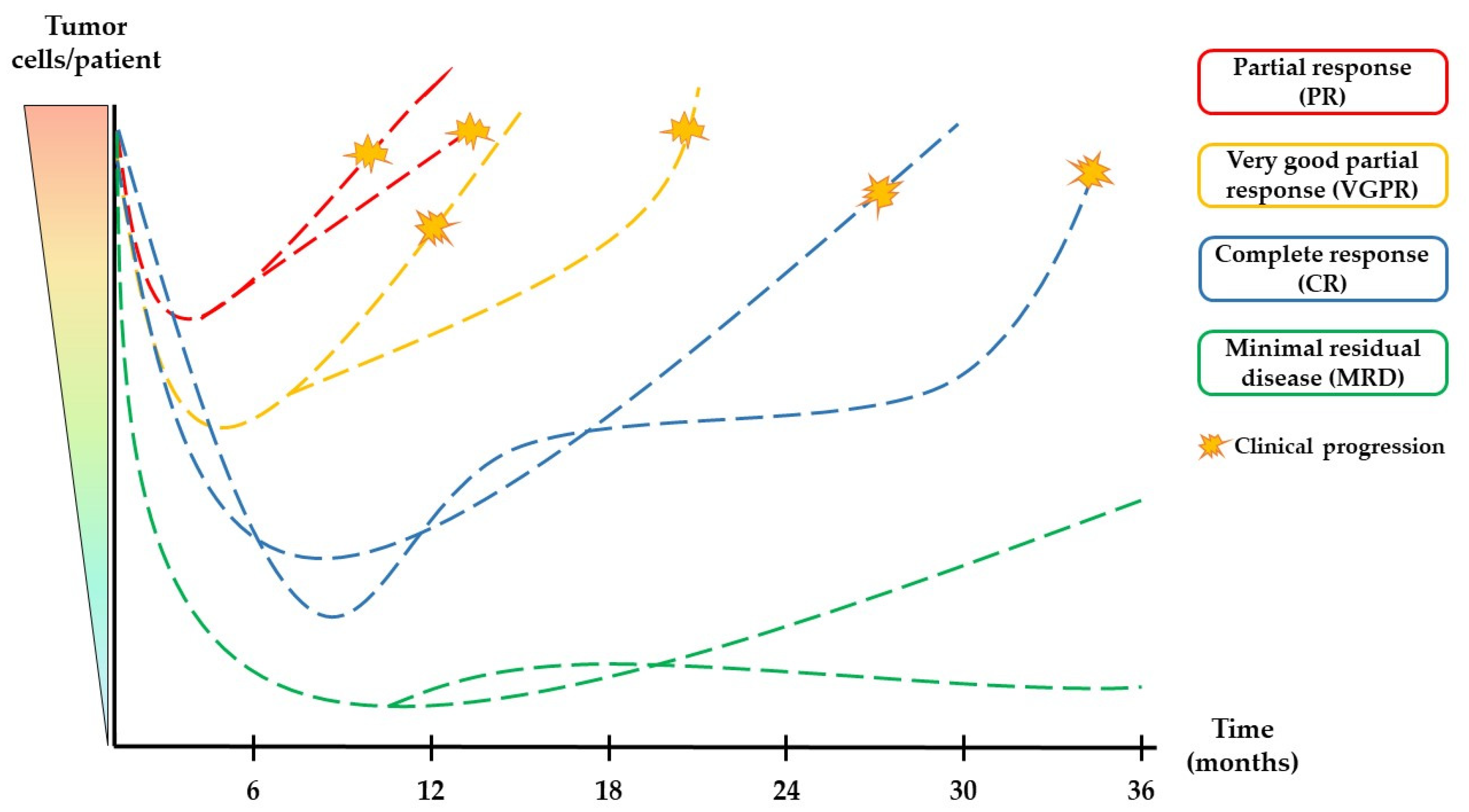
1.3. Assessment of Treatment Responses in Multiple Myeloma Patients
2. Alternatives for the Detection of Minimal Residual Disease
2.1. Immunophenotypic Approaches
2.1.1. Molecular Basis, Potential Targets, and Technical Considerations
2.1.2. Traditional and Next-Generation Flow Cytometry Approaches
2.1.3. Evidence
2.2. Molecular Approaches
2.2.1. Molecular Basis, Potential Targets, and Technical Considerations
2.2.2. Allele-Specific Oligonucleotide Quantitative PCR
2.2.3. Evidence
2.2.4. Next-Generation Sequencing
2.2.5. Evidences
2.2.6. Digital PCR
2.2.7. Evidence
2.3. Overall Comparison of Contemporary MRD Strategies
3. Future Perspectives
3.1. Liquid Biopsy: Circulating Tumor DNA and Circulating Tumor Cells
3.2. Imaging Techniques
3.3. Mass Spectrometry
4. Open Questions in the Field of MRD
- (a)
- Which patients should be evaluated?
- (b)
- What is the optimal time point for MRD evaluation?
- (c)
- How can we use MRD information?
- (d)
- Which strategy is the best?
5. Conclusions
- (1)
- MRD studies should be performed in the bone marrow, using validated and standardized procedures capable of assessing high sensitivity thresholds, ideally 10−6, which currently includes only NGF and NGS. Each institution may choose the most appropriate based on availability, expertise, and other aspects.
- (2)
- Bone-marrow-based MRD analyses should be performed using the first pull of the aspirate to prevent hemodilution. Evaluating MRD outside the bone marrow is an appealing and complementing option; incorporation of liquid biopsies and imaging techniques in prospective clinical trials would be very helpful to avoid invasive procedures, although more evidence is needed.
- (3)
- The evaluation of MRD should be performed in parallel with other clinical routine assessments at relevant time points, at least including post induction, post transplantation in candidate patients, post consolidation, post maintenance, and periodically during the subsequent follow-up. Single-time-point MRD may be prognostic and informative, but consecutive assessments in order to characterize MRD kinetics should be the goal.
- (4)
- If available, we recommend performing MRD studies for all patients diagnosed with multiple myeloma, in and outside of clinical trials. Results must be interpreted in the particular clinical–biological context of each patient and used for prognostic purposes. Interventional strategies based on MRD should be limited to clinical trials designed with that aim.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumar, S.K.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Pandey, S.; Kapoor, P.; Dingli, D.; Hayman, S.R.; Leung, N.; et al. Continued Improvement in Survival in Multiple Myeloma: Changes in Early Mortality and Outcomes in Older Patients. Leukemia 2014, 28, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, S.; Marcheselli, L.; Bari, A.; Liardo, E.V.; Marcheselli, R.; Luminari, S.; Quaresima, M.; Cirilli, C.; Ferri, P.; Federico, M.; et al. Survival of Multiple Myeloma Patients in the Era of Novel Therapies Confirms the Improvement in Patients Younger than 75 Years: A Population-Based Analysis. Br. J. Haematol. 2013, 163, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Avet-Loiseau, H.; Rawstron, A.C.; Owen, R.G.; Child, J.A.; Thakurta, A.; Sherrington, P.; Samur, M.K.; Georgieva, A.; Anderson, K.C.; et al. Association of Minimal Residual Disease with Superior Survival Outcomes in Patients With Multiple Myeloma: A Meta-Analysis. JAMA Oncol. 2017, 3, 28. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Kumar, S. Multiple Myeloma Current Treatment Algorithms. Blood Cancer J. 2020, 10, 94. [Google Scholar] [CrossRef] [PubMed]
- Goswami, M.; McGowan, K.S.; Lu, K.; Jain, N.; Candia, J.; Hensel, N.F.; Tang, J.; Calvo, K.R.; Battiwalla, M.; Barrett, A.J.; et al. A Multigene Array for Measurable Residual Disease Detection in AML Patients Undergoing SCT. Bone Marrow Transplant. 2015, 50, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Paiva, B.; San-Miguel, J.; Avet-Loiseau, H. MRD in Multiple Myeloma: Does CR Really Matter? Blood 2022, 140, 2423–2428. [Google Scholar] [CrossRef]
- Munshi, N.C.; Avet-Loiseau, H.; Anderson, K.C.; Neri, P.; Paiva, B.; Samur, M.; Dimopoulos, M.; Kulakova, M.; Lam, A.; Hashim, M.; et al. A Large Meta-Analysis Establishes the Role of MRD Negativity in Long-Term Survival Outcomes in Patients with Multiple Myeloma. Blood Adv. 2020, 4, 5988–5999. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Gregory, W.M.; De Tute, R.M.; Davies, F.E.; Bell, S.E.; Drayson, M.T.; Cook, G.; Jackson, G.H.; Morgan, G.J.; Child, J.A.; et al. Minimal Residual Disease in Myeloma by Flow Cytometry: Independent Prediction of Survival Benefit per Log Reduction. Blood 2015, 125, 1932–1935. [Google Scholar] [CrossRef]
- Corre, J.; Munshi, N.C.; Avet-Loiseau, H. Risk Factors in Multiple Myeloma: Is It Time for a Revision? Blood 2021, 137, 16–19. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple Myeloma: 2022 Update on Diagnosis, Risk Stratification, and Management. Am. J. Hematol. 2022, 97, 1086–1107. [Google Scholar] [CrossRef]
- Chng, W.J.; Dispenzieri, A.; Chim, C.-S.; Fonseca, R.; Goldschmidt, H.; Lentzsch, S.; Munshi, N.; Palumbo, A.; Miguel, J.S.; Sonneveld, P.; et al. IMWG Consensus on Risk Stratification in Multiple Myeloma. Leukemia 2014, 28, 269–277. [Google Scholar] [CrossRef]
- Ludwig, H.; Bolejack, V.; Crowley, J.; Bladé, J.; Miguel, J.S.; Kyle, R.A.; Rajkumar, S.V.; Shimizu, K.; Turesson, I.; Westin, J.; et al. Survival and Years of Life Lost in Different Age Cohorts of Patients with Multiple Myeloma. J. Clin. Oncol. 2010, 28, 1599–1605. [Google Scholar] [CrossRef]
- Sonneveld, P.; Avet-Loiseau, H.; Lonial, S.; Usmani, S.; Siegel, D.; Anderson, K.C.; Chng, W.-J.; Moreau, P.; Attal, M.; Kyle, R.A.; et al. Treatment of Multiple Myeloma with High-Risk Cytogenetics: A Consensus of the International Myeloma Working Group. Blood 2016, 127, 2955–2962. [Google Scholar] [CrossRef]
- Greipp, P.R.; Miguel, J.S.; Durie, B.G.M.; Crowley, J.J.; Barlogie, B.; Bladé, J.; Boccadoro, M.; Child, J.A.; Avet-Loiseau, H.; Kyle, R.A.; et al. International Staging System for Multiple Myeloma. J. Clin. Oncol. 2005, 23, 3412–3420. [Google Scholar] [CrossRef]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report from International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef]
- D’Agostino, M.; Cairns, D.A.; Lahuerta, J.J.; Wester, R.; Bertsch, U.; Waage, A.; Zamagni, E.; Mateos, M.-V.; Dall’Olio, D.; Van De Donk, N.W.C.J.; et al. Second Revision of the International Staging System (R2-ISS) for Overall Survival in Multiple Myeloma: A European Myeloma Network (EMN) Report Within the HARMONY Project. J. Clin. Oncol. 2022, 40, 3406–3418. [Google Scholar] [CrossRef]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. A High-Risk, Double-Hit, Group of Newly Diagnosed Myeloma Identified by Genomic Analysis. Leukemia 2019, 33, 159–170. [Google Scholar] [CrossRef]
- Fonseca, R.; Bergsagel, P.L.; Drach, J.; Shaughnessy, J.; Gutierrez, N.; Stewart, A.K.; Morgan, G.; Van Ness, B.; Chesi, M.; Minvielle, S.; et al. International Myeloma Working Group Molecular Classification of Multiple Myeloma: Spotlight Review. Leukemia 2009, 23, 2210–2221. [Google Scholar] [CrossRef]
- Boyd, K.D.; Ross, F.M.; Chiecchio, L.; Dagrada, G.P.; Konn, Z.J.; Tapper, W.J.; Walker, B.A.; Wardell, C.P.; Gregory, W.M.; Szubert, A.J.; et al. A Novel Prognostic Model in Myeloma Based on Co-Segregating Adverse FISH Lesions and the ISS: Analysis of Patients Treated in the MRC Myeloma IX Trial. Leukemia 2012, 26, 349–355. [Google Scholar] [CrossRef]
- Perrot, A.; Lauwers-Cances, V.; Tournay, E.; Hulin, C.; Chretien, M.-L.; Royer, B.; Dib, M.; Decaux, O.; Jaccard, A.; Belhadj, K.; et al. Development and Validation of a Cytogenetic Prognostic Index Predicting Survival in Multiple Myeloma. J. Clin. Oncol. 2019, 37, 1657–1665. [Google Scholar] [CrossRef]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of Novel Mutational Drivers Reveals Oncogene Dependencies in Multiple Myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Proposed Guidelines for Protocol Studies. IV. New Agents in the Treatment of Chronic Granulocytic Leukemia. Prepared by a Committee of the Chronic Leukemia--Myeloma Task Force, National Cancer Institute. Cancer Chemother. Rep. 3 1968, 1, 53–62.
- Alexanian, R.; Bonnet, J.; Gehan, E.; Haut, A.; Hewlett, J.; Lane, M.; Monto, R.; Wilson, H. Combination Chemotherapy for Multiple Myeloma. Cancer 1972, 30, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Bladé, J.; Samson, D.; Reece, D.; Apperley, J.; BJÖrkstrand, B.; Gahrton, G.; Gertz, M.; Giralt, S.; Jagannath, S.; Vesole, D.; et al. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high-dose therapy and haemopoietic stem cell transplantation: Annotation. Br. J. Haematol. 1998, 102, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Van De Velde, H.J.K.; Liu, X.; Chen, G.; Cakana, A.; Deraedt, W.; Bayssas, M. Complete Response Correlates with Long-Term Survival and Progression-Free Survival in High-Dose Therapy in Multiple Myeloma. Haematologica 2007, 92, 1399–1406. [Google Scholar] [CrossRef]
- Sonneveld, P.; Goldschmidt, H.; Rosiñol, L.; Bladé, J.; Lahuerta, J.J.; Cavo, M.; Tacchetti, P.; Zamagni, E.; Attal, M.; Lokhorst, H.M.; et al. Bortezomib-Based Versus Nonbortezomib-Based Induction Treatment Before Autologous Stem-Cell Transplantation in Patients with Previously Untreated Multiple Myeloma: A Meta-Analysis of Phase III Randomized, Controlled Trials. J. Clin. Oncol. 2013, 31, 3279–3287. [Google Scholar] [CrossRef]
- Van De Velde, H.; Londhe, A.; Ataman, O.; Johns, H.L.; Hill, S.; Landers, E.; Berlin, J.A. Association between Complete Response and Outcomes in Transplant-Eligible Myeloma Patients in the Era of Novel Agents. Eur. J. Haematol. 2017, 98, 269–279. [Google Scholar] [CrossRef]
- Durie, B.G.M.; Harousseau, J.-L.; Miguel, J.S.; Bladé, J.; Barlogie, B.; Anderson, K.; Gertz, M.; Dimopoulos, M.; Westin, J.; Sonneveld, P.; et al. International Uniform Response Criteria for Multiple Myeloma. Leukemia 2006, 20, 1467–1473. [Google Scholar] [CrossRef]
- Kapoor, P.; Kumar, S.K.; Dispenzieri, A.; Lacy, M.Q.; Buadi, F.; Dingli, D.; Russell, S.J.; Hayman, S.R.; Witzig, T.E.; Lust, J.A.; et al. Importance of Achieving Stringent Complete Response After Autologous Stem-Cell Transplantation in Multiple Myeloma. J. Clin. Oncol. 2013, 31, 4529–4535. [Google Scholar] [CrossRef]
- Martínez-López, J.; Paiva, B.; López-Anglada, L.; Mateos, M.-V.; Cedena, T.; Vidríales, M.-B.; Sáez-Gómez, M.A.; Contreras, T.; Oriol, A.; Rapado, I.; et al. Critical Analysis of the Stringent Complete Response in Multiple Myeloma: Contribution of SFLC and Bone Marrow Clonality. Blood 2015, 126, 858–862. [Google Scholar] [CrossRef]
- Lehners, N.; Becker, N.; Benner, A.; Pritsch, M.; Löpprich, M.; Mai, E.K.; Hillengass, J.; Goldschmidt, H.; Raab, M.-S. Analysis of Long-Term Survival in Multiple Myeloma after First-Line Autologous Stem Cell Transplantation: Impact of Clinical Risk Factors and Sustained Response. Cancer Med. 2018, 7, 307–316. [Google Scholar] [CrossRef]
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.; Lonial, S.; Bladé, J.; Mateos, M.-V.; et al. International Myeloma Working Group Consensus Criteria for Response and Minimal Residual Disease Assessment in Multiple Myeloma. Lancet Oncol. 2016, 17, e328–e346. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Orfao, A.; Beksac, M.; Bezdickova, L.; Brooimans, R.A.; Bumbea, H.; Dalva, K.; Fuhler, G.; Gratama, J.; Hose, D.; et al. Report of the European Myeloma Network on Multiparametric Flow Cytometry in Multiple Myeloma and Related Disorders. Haematologica 2008, 93, 431–438. [Google Scholar] [CrossRef]
- Flores-Montero, J.; De Tute, R.; Paiva, B.; Perez, J.J.; Böttcher, S.; Wind, H.; Sanoja, L.; Puig, N.; Lecrevisse, Q.; Vidriales, M.B.; et al. Immunophenotype of Normal vs. Myeloma Plasma Cells: Toward Antibody Panel Specifications for MRD Detection in Multiple Myeloma. Cytometry B Clin. Cytom. 2016, 90, 61–72. [Google Scholar] [CrossRef]
- on behalf of the EuroFlow Consortium (EU-FP6, LSHB-CT-2006-018708); Van Dongen, J.J.M.; Lhermitte, L.; Böttcher, S.; Almeida, J.; Van Der Velden, V.H.J.; Flores-Montero, J.; Rawstron, A.; Asnafi, V.; Lécrevisse, Q.; et al. EuroFlow Antibody Panels for Standardized N-Dimensional Flow Cytometric Immunophenotyping of Normal, Reactive and Malignant Leukocytes. Leukemia 2012, 26, 1908–1975. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Paiva, B.; Stetler-Stevenson, M. Assessment of Minimal Residual Disease in Myeloma and the Need for a Consensus Approach. Cytometry B Clin. Cytom. 2016, 90, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Pedreira, C.E.; Costa, E.S.D.; Lecrevise, Q.; Grigore, G.; Fluxa, R.; Verde, J.; Hernandez, J.; Van Dongen, J.J.M.; Orfao, A. From Big Flow Cytometry Datasets to Smart Diagnostic Strategies: The EuroFlow Approach. J. Immunol. Methods 2019, 475, 112631. [Google Scholar] [CrossRef]
- San Miguel, J.F.; Almeida, J.; Mateo, G.; Bladé, J.; López-Berges, C.; Caballero, D.; Hernández, J.; Moro, M.J.; Fernández-Calvo, J.; Díaz-Mediavilla, J.; et al. Immunophenotypic Evaluation of the Plasma Cell Compartment in Multiple Myeloma: A Tool for Comparing the Efficacy of Different Treatment Strategies and Predicting Outcome. Blood 2002, 99, 1853–1856. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Davies, F.E.; DasGupta, R.; Ashcroft, A.J.; Patmore, R.; Drayson, M.T.; Owen, R.G.; Jack, A.S.; Child, J.A.; Morgan, G.J. Flow Cytometric Disease Monitoring in Multiple Myeloma: The Relationship between Normal and Neoplastic Plasma Cells Predicts Outcome after Transplantation. Blood 2002, 100, 3095–3100. [Google Scholar] [CrossRef]
- Paiva, B.; Vidriales, M.-B.; Cerveró, J.; Mateo, G.; Pérez, J.J.; Montalbán, M.A.; Sureda, A.; Montejano, L.; Gutiérrez, N.C.; De Coca, A.G.; et al. Multiparameter Flow Cytometric Remission Is the Most Relevant Prognostic Factor for Multiple Myeloma Patients Who Undergo Autologous Stem Cell Transplantation. Blood 2008, 112, 4017–4023. [Google Scholar] [CrossRef]
- Paiva, B.; Martinez-Lopez, J.; Vidriales, M.-B.; Mateos, M.-V.; Montalban, M.-A.; Fernandez-Redondo, E.; Alonso, L.; Oriol, A.; Teruel, A.-I.; De Paz, R.; et al. Comparison of Immunofixation, Serum Free Light Chain, and Immunophenotyping for Response Evaluation and Prognostication in Multiple Myeloma. J. Clin. Oncol. 2011, 29, 1627–1633. [Google Scholar] [CrossRef] [PubMed]
- Rawstron, A.C.; Child, J.A.; De Tute, R.M.; Davies, F.E.; Gregory, W.M.; Bell, S.E.; Szubert, A.J.; Navarro-Coy, N.; Drayson, M.T.; Feyler, S.; et al. Minimal Residual Disease Assessed by Multiparameter Flow Cytometry in Multiple Myeloma: Impact on Outcome in the Medical Research Council Myeloma IX Study. J. Clin. Oncol. 2013, 31, 2540–2547. [Google Scholar] [CrossRef] [PubMed]
- Paiva, B.; Cedena, M.-T.; Puig, N.; Arana, P.; Vidriales, M.-B.; Cordon, L.; Flores-Montero, J.; Gutierrez, N.C.; Martín-Ramos, M.-L.; Martinez-Lopez, J.; et al. Minimal Residual Disease Monitoring and Immune Profiling in Multiple Myeloma in Elderly Patients. Blood 2016, 127, 3165–3174. [Google Scholar] [CrossRef] [PubMed]
- Rawstron, A.C.; De Tute, R.M.; Haughton, J.; Owen, R.G. Measuring Disease Levels in Myeloma Using Flow Cytometry in Combination with Other Laboratory Techniques: Lessons from the Past 20 Years at the Leeds Haematological Malignancy Diagnostic Service. Cytometry B Clin. Cytom. 2016, 90, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Flores-Montero, J.; Sanoja-Flores, L.; Paiva, B.; Puig, N.; García-Sánchez, O.; Böttcher, S.; Van Der Velden, V.H.J.; Pérez-Morán, J.-J.; Vidriales, M.-B.; García-Sanz, R.; et al. Next Generation Flow for Highly Sensitive and Standardized Detection of Minimal Residual Disease in Multiple Myeloma. Leukemia 2017, 31, 2094–2103. [Google Scholar] [CrossRef]
- Romano, A.; Palumbo, G.A.; Parrinello, N.L.; Conticello, C.; Martello, M.; Terragna, C. Minimal Residual Disease Assessment Within the Bone Marrow of Multiple Myeloma: A Review of Caveats, Clinical Significance and Future Perspectives. Front. Oncol. 2019, 9, 699. [Google Scholar] [CrossRef]
- Puig, N.; Flores-Montero, J.; Burgos, L.; Cedena, M.-T.; Cordón, L.; Pérez, J.-J.; Sanoja-Flores, L.; Manrique, I.; Rodríguez-Otero, P.; Rosiñol, L.; et al. Reference Values to Assess Hemodilution and Warn of Potential False-Negative Minimal Residual Disease Results in Myeloma. Cancers 2021, 13, 4924. [Google Scholar] [CrossRef]
- Roshal, M.; Flores-Montero, J.A.; Gao, Q.; Koeber, M.; Wardrope, J.; Durie, B.G.M.; Dogan, A.; Orfao, A.; Landgren, O. MRD Detection in Multiple Myeloma: Comparison between MSKCC 10-Color Single-Tube and EuroFlow 8-Color 2-Tube Methods. Blood Adv. 2017, 1, 728–732. [Google Scholar] [CrossRef]
- Takamatsu, H.; Yoroidaka, T.; Fujisawa, M.; Kobori, K.; Hanawa, M.; Yamashita, T.; Murata, R.; Ueda, M.; Nakao, S.; Matsue, K. Comparison of Minimal Residual Disease Detection in Multiple Myeloma by SRL 8-Color Single-Tube and EuroFlow 8-Color 2-Tube Multiparameter Flow Cytometry. Int. J. Hematol. 2019, 109, 377–381. [Google Scholar] [CrossRef]
- Bayly, E.; Nguyen, V.; Binek, A.; Piggin, A.; Baldwin, K.; Westerman, D.; Came, N. Validation of a Modified Pre-lysis Sample Preparation Technique for Flow Cytometric Minimal Residual Disease Assessment in Multiple Myeloma, Chronic Lymphocytic Leukemia, and B-Non Hodgkin Lymphoma. Cytometry B Clin. Cytom. 2020, 98, 385–398. [Google Scholar] [CrossRef]
- Riebl, V.; Dold, S.M.; Wider, D.; Follo, M.; Ihorst, G.; Waldschmidt, J.M.; Jung, J.; Rassner, M.; Greil, C.; Wäsch, R.; et al. Ten Color Multiparameter Flow Cytometry in Bone Marrow and Apheresis Products for Assessment and Outcome Prediction in Multiple Myeloma Patients. Front. Oncol. 2021, 11, 708231. [Google Scholar] [CrossRef]
- Rosiñol, L.; Oriol, A.; Rios, R.; Sureda, A.; Blanchard, M.J.; Hernández, M.T.; Martínez-Martínez, R.; Moraleda, J.M.; Jarque, I.; Bargay, J.; et al. Bortezomib, Lenalidomide, and Dexamethasone as Induction Therapy Prior to Autologous Transplant in Multiple Myeloma. Blood 2019, 134, 1337–1345. [Google Scholar] [CrossRef]
- Paiva, B.; Puig, N.; Cedena, M.-T.; Rosiñol, L.; Cordón, L.; Vidriales, M.-B.; Burgos, L.; Flores-Montero, J.; Sanoja-Flores, L.; Lopez-Anglada, L.; et al. Measurable Residual Disease by Next-Generation Flow Cytometry in Multiple Myeloma. J. Clin. Oncol. 2020, 38, 784–792. [Google Scholar] [CrossRef]
- Oliva, S.; Bruinink, D.H.O.; Rihova, L.; D’Agostino, M.; Pantani, L.; Capra, A.; Van Der Holt, B.; Troia, R.; Petrucci, M.T.; Villanova, T.; et al. Minimal Residual Disease Assessment by Multiparameter Flow Cytometry in Transplant-Eligible Myeloma in the EMN02/HOVON 95 MM Trial. Blood Cancer J. 2021, 11, 106. [Google Scholar] [CrossRef]
- De Tute, R.M.; Pawlyn, C.; Cairns, D.A.; Davies, F.E.; Menzies, T.; Rawstron, A.; Jones, J.R.; Hockaday, A.; Henderson, R.; Cook, G.; et al. Minimal Residual Disease After Autologous Stem-Cell Transplant for Patients with Myeloma: Prognostic Significance and the Impact of Lenalidomide Maintenance and Molecular Risk. J. Clin. Oncol. 2022, 40, 2889–2900. [Google Scholar] [CrossRef]
- Diamond, B.; Korde, N.; Lesokhin, A.M.; Smith, E.L.; Shah, U.; Mailankody, S.; Hultcrantz, M.; Hassoun, H.; Lu, S.X.; Tan, C.; et al. Dynamics of Minimal Residual Disease in Patients with Multiple Myeloma on Continuous Lenalidomide Maintenance: A Single-Arm, Single-Centre, Phase 2 Trial. Lancet Haematol. 2021, 8, e422–e432. [Google Scholar] [CrossRef]
- Moreau, P.; Attal, M.; Hulin, C.; Arnulf, B.; Belhadj, K.; Benboubker, L.; Béné, M.C.; Broijl, A.; Caillon, H.; Caillot, D.; et al. Bortezomib, Thalidomide, and Dexamethasone with or without Daratumumab before and after Autologous Stem-Cell Transplantation for Newly Diagnosed Multiple Myeloma (CASSIOPEIA): A Randomised, Open-Label, Phase 3 Study. The Lancet 2019, 394, 29–38. [Google Scholar] [CrossRef]
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic Complexity of Multiple Myeloma and Its Clinical Implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113. [Google Scholar] [CrossRef]
- Maura, F.; Bolli, N.; Angelopoulos, N.; Dawson, K.J.; Leongamornlert, D.; Martincorena, I.; Mitchell, T.J.; Fullam, A.; Gonzalez, S.; Szalat, R.; et al. Genomic Landscape and Chronological Reconstruction of Driver Events in Multiple Myeloma. Nat. Commun. 2019, 10, 3835. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread Genetic Heterogeneity in Multiple Myeloma: Implications for Targeted Therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef]
- Puig, N.; Conde, I.; Jiménez, C.; Sarasquete, M.E.; Balanzategui, A.; Alcoceba, M.; Quintero, J.; Chillón, M.C.; Sebastián, E.; Corral, R.; et al. The Predominant Myeloma Clone at Diagnosis, CDR3 Defined, Is Constantly Detectable across All Stages of Disease Evolution. Leukemia 2015, 29, 1435–1437. [Google Scholar] [CrossRef]
- Rustad, E.H.; Misund, K.; Bernard, E.; Coward, E.; Yellapantula, V.D.; Hultcrantz, M.; Ho, C.; Kazandjian, D.; Korde, N.; Mailankody, S.; et al. Stability and Uniqueness of Clonal Immunoglobulin CDR3 Sequences for MRD Tracking in Multiple Myeloma. Am. J. Hematol. 2019, 94, 1364–1373. [Google Scholar] [CrossRef]
- Medina, A.; Puig, N.; Flores-Montero, J.; Jimenez, C.; Sarasquete, M.-E.; Garcia-Alvarez, M.; Prieto-Conde, I.; Chillon, C.; Alcoceba, M.; Gutierrez, N.C.; et al. Comparison of Next-Generation Sequencing (NGS) and next-Generation Flow (NGF) for Minimal Residual Disease (MRD) Assessment in Multiple Myeloma. Blood Cancer J. 2020, 10, 108. [Google Scholar] [CrossRef] [PubMed]
- Medina, A.; Jiménez, C.; Sarasquete, M.E.; González, M.; Chillón, M.C.; Balanzategui, A.; Prieto-Conde, I.; García-Álvarez, M.; Puig, N.; González-Calle, V.; et al. Molecular Profiling of Immunoglobulin Heavy-Chain Gene Rearrangements Unveils New Potential Prognostic Markers for Multiple Myeloma Patients. Blood Cancer J. 2020, 10, 14. [Google Scholar] [CrossRef] [PubMed]
- Van Der Velden, V.H.J.; Hochhaus, A.; Cazzaniga, G.; Szczepanski, T.; Gabert, J.; Van Dongen, J.J.M. Detection of Minimal Residual Disease in Hematologic Malignancies by Real-Time Quantitative PCR: Principles, Approaches, and Laboratory Aspects. Leukemia 2003, 17, 1013–1034. [Google Scholar] [CrossRef]
- Sarasquete, M.E.; García-Sanz, R.; González, D.; Martínez, J.; Mateo, G.; Martínez, P.; Ribera, J.M.; Hernández, J.M.; Lahuerta, J.J.; Orfão, A.; et al. Minimal Residual Disease Monitoring in Multiple Myeloma: A Comparison between Allelic-Specific Oligonucleotide Real-Time Quantitative Polymerase Chain Reaction and Flow Cytometry. Haematologica 2005, 90, 1365–1372. [Google Scholar] [PubMed]
- Pott, C.; Brüggemann, M.; Ritgen, M.; Van Der Velden, V.H.J.; Van Dongen, J.J.M.; Kneba, M. MRD Detection in B-Cell Non-Hodgkin Lymphomas Using Ig Gene Rearrangements and Chromosomal Translocations as Targets for Real-Time Quantitative PCR. In Lymphoma; Küppers, R., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; Volume 1956, pp. 199–228. ISBN 978-1-4939-9150-1. [Google Scholar]
- Van Dongen, J.J.M.; Langerak, A.W.; Brüggemann, M.; Evans, P.A.S.; Hummel, M.; Lavender, F.L.; Delabesse, E.; Davi, F.; Schuuring, E.; García-Sanz, R.; et al. Design and Standardization of PCR Primers and Protocols for Detection of Clonal Immunoglobulin and T-Cell Receptor Gene Recombinations in Suspect Lymphoproliferations: Report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003, 17, 2257–2317. [Google Scholar] [CrossRef]
- Puig, N.; Sarasquete, M.E.; Balanzategui, A.; Martínez, J.; Paiva, B.; García, H.; Fumero, S.; Jiménez, C.; Alcoceba, M.; Chillón, M.C.; et al. Critical Evaluation of ASO RQ-PCR for Minimal Residual Disease Evaluation in Multiple Myeloma. A Comparative Analysis with Flow Cytometry. Leukemia 2014, 28, 391–397. [Google Scholar] [CrossRef]
- Puig, N.; Sarasquete, M.E.; Alcoceba, M.; Balanzategui, A.; Chillón, M.C.; Sebastián, E.; Díaz, M.G.; San Miguel, J.F.; García-Sanz, R. Kappa Deleting Element as an Alternative Molecular Target for Minimal Residual Disease Assessment by Real-Time Quantitative PCR in Patients with Multiple Myeloma. Eur. J. Haematol. 2012, 89, 328–335. [Google Scholar] [CrossRef]
- Van Der Velden, V.H.J.; Cazzaniga, G.; Schrauder, A.; Hancock, J.; Bader, P.; Panzer-Grumayer, E.R.; Flohr, T.; Sutton, R.; Cave, H.; Madsen, H.O.; et al. Analysis of Minimal Residual Disease by Ig/TCR Gene Rearrangements: Guidelines for Interpretation of Real-Time Quantitative PCR Data. Leukemia 2007, 21, 604–611. [Google Scholar] [CrossRef]
- Pott, C.; Sehn, L.H.; Belada, D.; Gribben, J.; Hoster, E.; Kahl, B.; Kehden, B.; Nicolas-Virelizier, E.; Spielewoy, N.; Fingerle-Rowson, G.; et al. MRD Response in Relapsed/Refractory FL after Obinutuzumab plus Bendamustine or Bendamustine Alone in the GADOLIN Trial. Leukemia 2020, 34, 522–532. [Google Scholar] [CrossRef]
- O’Connor, D.; Moorman, A.V.; Wade, R.; Hancock, J.; Tan, R.M.R.; Bartram, J.; Moppett, J.; Schwab, C.; Patrick, K.; Harrison, C.J.; et al. Use of Minimal Residual Disease Assessment to Redefine Induction Failure in Pediatric Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2017, 35, 660–667. [Google Scholar] [CrossRef]
- Pott, C.; Hoster, E.; Delfau-Larue, M.-H.; Beldjord, K.; Böttcher, S.; Asnafi, V.; Plonquet, A.; Siebert, R.; Callet-Bauchu, E.; Andersen, N.; et al. Molecular Remission Is an Independent Predictor of Clinical Outcome in Patients with Mantle Cell Lymphoma after Combined Immunochemotherapy: A European MCL Intergroup Study. Blood 2010, 115, 3215–3223. [Google Scholar] [CrossRef]
- Bakkus, M.H.C.; Bouko, Y.; Samson, D.; Apperley, J.F.; Thielemans, K.; Camp, B.V.; Benner, A.; Goldschmidt, H.; Moos, M.; Cremer, F.W. Post-Transplantation Tumour Load in Bone Marrow, as Assessed by Quantitative ASO-PCR, Is a Prognostic Parameter in Multiple Myeloma: Post-PBSCT Tumour Load Is Prognostic in Myeloma. Br. J. Haematol. 2004, 126, 665–674. [Google Scholar] [CrossRef]
- González, D.; Garcia-Sanz, R. Incomplete DJH Rearrangements. In Multiple Myeloma; Humana Press: Totowa, NJ, USA, 2005; Volume 113, pp. 165–174. ISBN 978-1-59259-916-5. [Google Scholar]
- Puig, N.; Sarasquete, M.E.; Alcoceba, M.; Balanzategui, A.; Chillón, M.C.; Sebastián, E.; Marín, L.A.; Díaz, M.G.; San Miguel, J.F.; Sanz, R.G. The Use of CD138 Positively Selected Marrow Samples Increases the Applicability of Minimal Residual Disease Assessment by PCR in Patients with Multiple Myeloma. Ann. Hematol. 2013, 92, 97–100. [Google Scholar] [CrossRef]
- Korthals, M.; Sehnke, N.; Kronenwett, R.; Bruns, I.; Mau, J.; Zohren, F.; Haas, R.; Kobbe, G.; Fenk, R. The Level of Minimal Residual Disease in the Bone Marrow of Patients with Multiple Myeloma before High-Dose Therapy and Autologous Blood Stem Cell Transplantation Is an Independent Predictive Parameter. Biol. Blood Marrow Transplant. 2012, 18, 423–431.e3. [Google Scholar] [CrossRef]
- Ladetto, M.; Pagliano, G.; Ferrero, S.; Cavallo, F.; Drandi, D.; Santo, L.; Crippa, C.; De Rosa, L.; Pregno, P.; Grasso, M.; et al. Major Tumor Shrinking and Persistent Molecular Remissions After Consolidation With Bortezomib, Thalidomide, and Dexamethasone in Patients With Autografted Myeloma. J. Clin. Oncol. 2010, 28, 2077–2084. [Google Scholar] [CrossRef]
- Martinez-Lopez, J.; Sanchez-Vega, B.; Barrio, S.; Cuenca, I.; Ruiz-Heredia, Y.; Alonso, R.; Rapado, I.; Marin, C.; Cedena, M.-T.; Paiva, B.; et al. Analytical and Clinical Validation of a Novel In-House Deep-Sequencing Method for Minimal Residual Disease Monitoring in a Phase II Trial for Multiple Myeloma. Leukemia 2017, 31, 1446–1449. [Google Scholar] [CrossRef]
- Miller, J.E. Immunoglobulin and T Cell Receptor Gene Rearrangement: Principle. In Molecular Genetic Pathology; Cheng, L., Zhang, D.Y., Eble, J.N., Eds.; Springer: New York, NY, USA, 2013; pp. 825–856. ISBN 978-1-4614-4799-3. [Google Scholar]
- EuroClonality-NGS Working Group; Brüggemann, M.; Kotrová, M.; Knecht, H.; Bartram, J.; Boudjogrha, M.; Bystry, V.; Fazio, G.; Froňková, E.; Giraud, M.; et al. Standardized Next-Generation Sequencing of Immunoglobulin and T-Cell Receptor Gene Recombinations for MRD Marker Identification in Acute Lymphoblastic Leukaemia; a EuroClonality-NGS Validation Study. Leukemia 2019, 33, 2241–2253. [Google Scholar] [CrossRef]
- EuroMRD Working Group; Cazzaniga, G.; Songia, S.; Biondi, A. PCR Technology to Identify Minimal Residual Disease. In Leukemia Stem Cells; Cobaleda, C., Sánchez-García, I., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2021; Volume 2185, pp. 77–94. ISBN 978-1-07-160809-8. [Google Scholar]
- Faham, M.; Zheng, J.; Moorhead, M.; Carlton, V.E.H.; Stow, P.; Coustan-Smith, E.; Pui, C.-H.; Campana, D. Deep-Sequencing Approach for Minimal Residual Disease Detection in Acute Lymphoblastic Leukemia. Blood 2012, 120, 5173–5180. [Google Scholar] [CrossRef]
- Van Den Brand, M.; Rijntjes, J.; Möbs, M.; Steinhilber, J.; Van Der Klift, M.Y.; Heezen, K.C.; Kroeze, L.I.; Reigl, T.; Porc, J.; Darzentas, N.; et al. Next-Generation Sequencing–Based Clonality Assessment of Ig Gene Rearrangements. J. Mol. Diagn. 2021, 23, 1105–1115. [Google Scholar] [CrossRef]
- Kotrova, M.; Trka, J.; Kneba, M.; Brüggemann, M. Is Next-Generation Sequencing the Way to Go for Residual Disease Monitoring in Acute Lymphoblastic Leukemia? Mol. Diagn. Ther. 2017, 21, 481–492. [Google Scholar] [CrossRef]
- Yao, Q.; Bai, Y.; Orfao, A.; Chim, C.S. Standardized Minimal Residual Disease Detection by Next-Generation Sequencing in Multiple Myeloma. Front. Oncol. 2019, 9, 449. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lopez, J.; Lahuerta, J.J.; Pepin, F.; González, M.; Barrio, S.; Ayala, R.; Puig, N.; Montalban, M.A.; Paiva, B.; Weng, L.; et al. Prognostic Value of Deep Sequencing Method for Minimal Residual Disease Detection in Multiple Myeloma. Blood 2014, 123, 3073–3079. [Google Scholar] [CrossRef] [PubMed]
- Perrot, A.; Lauwers-Cances, V.; Corre, J.; Robillard, N.; Hulin, C.; Chretien, M.-L.; Dejoie, T.; Maheo, S.; Stoppa, A.-M.; Pegourie, B.; et al. Minimal Residual Disease Negativity Using Deep Sequencing Is a Major Prognostic Factor in Multiple Myeloma. Blood 2018, 132, 2456–2464. [Google Scholar] [CrossRef]
- Goicoechea, I.; Puig, N.; Cedena, M.-T.; Burgos, L.; Cordón, L.; Vidriales, M.-B.; Flores-Montero, J.; Gutierrez, N.C.; Calasanz, M.-J.; Ramos, M.-L.M.; et al. Deep MRD Profiling Defines Outcome and Unveils Different Modes of Treatment Resistance in Standard- and High-Risk Myeloma. Blood 2021, 137, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, F.; Zhou, X.; Mei, J.; Song, P.; An, Z.; Zhao, Q.; Guo, X.; Wang, X.; Zhai, Y. Achieving Minimal Residual Disease-Negative by Multiparameter Flow Cytometry May Ameliorate a Poor Prognosis in MM Patients with High-Risk Cytogenetics: A Retrospective Single-Center Analysis. Ann. Hematol. 2019, 98, 1185–1195. [Google Scholar] [CrossRef]
- Costa, L.J.; Derman, B.A.; Bal, S.; Sidana, S.; Chhabra, S.; Silbermann, R.; Ye, J.C.; Cook, G.; Cornell, R.F.; Holstein, S.A.; et al. International Harmonization in Performing and Reporting Minimal Residual Disease Assessment in Multiple Myeloma Trials. Leukemia 2021, 35, 18–30. [Google Scholar] [CrossRef]
- Lahuerta, J.-J.; Paiva, B.; Vidriales, M.-B.; Cordón, L.; Cedena, M.-T.; Puig, N.; Martinez-Lopez, J.; Rosiñol, L.; Gutierrez, N.C.; Martín-Ramos, M.-L.; et al. Depth of Response in Multiple Myeloma: A Pooled Analysis of Three PETHEMA/GEM Clinical Trials. J. Clin. Oncol. 2017, 35, 2900–2910. [Google Scholar] [CrossRef]
- Martin, T.; Usmani, S.Z.; Berdeja, J.G.; Agha, M.; Cohen, A.D.; Hari, P.; Avigan, D.; Deol, A.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene Autoleucel, an Anti–B-Cell Maturation Antigen Chimeric Antigen Receptor T-Cell Therapy, for Relapsed/Refractory Multiple Myeloma: CARTITUDE-1 2-Year Follow-Up. J. Clin. Oncol. 2023, 41, 1265–1274. [Google Scholar] [CrossRef]
- Moreau, P.; Garfall, A.L.; Van De Donk, N.W.C.J.; Nahi, H.; San-Miguel, J.F.; Oriol, A.; Nooka, A.K.; Martin, T.; Rosinol, L.; Chari, A.; et al. Teclistamab in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2022, 387, 495–505. [Google Scholar] [CrossRef]
- Voorhees, P.M.; Kaufman, J.L.; Laubach, J.; Sborov, D.W.; Reeves, B.; Rodriguez, C.; Chari, A.; Silbermann, R.; Costa, L.J.; Anderson, L.D.; et al. Daratumumab, Lenalidomide, Bortezomib, and Dexamethasone for Transplant-Eligible Newly Diagnosed Multiple Myeloma: The GRIFFIN Trial. Blood 2020, 136, 936–945. [Google Scholar] [CrossRef]
- Mateos, M.-V.; Cavo, M.; Blade, J.; Dimopoulos, M.A.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lucio, P.; Nagy, Z.; et al. Overall Survival with Daratumumab, Bortezomib, Melphalan, and Prednisone in Newly Diagnosed Multiple Myeloma (ALCYONE): A Randomised, Open-Label, Phase 3 Trial. Lancet 2020, 395, 132–141. [Google Scholar] [CrossRef]
- San-Miguel, J.; Avet-Loiseau, H.; Paiva, B.; Kumar, S.; Dimopoulos, M.A.; Facon, T.; Mateos, M.-V.; Touzeau, C.; Jakubowiak, A.; Usmani, S.Z.; et al. Sustained Minimal Residual Disease Negativity in Newly Diagnosed Multiple Myeloma and the Impact of Daratumumab in MAIA and ALCYONE. Blood 2022, 139, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Facon, T.; Kumar, S.K.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab, Lenalidomide, and Dexamethasone versus Lenalidomide and Dexamethasone Alone in Newly Diagnosed Multiple Myeloma (MAIA): Overall Survival Results from a Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2021, 22, 1582–1596. [Google Scholar] [CrossRef]
- Sonneveld, P.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.-V.; et al. Overall Survival with Daratumumab, Bortezomib, and Dexamethasone in Previously Treated Multiple Myeloma (CASTOR): A Randomized, Open-Label, Phase III Trial. J. Clin. Oncol. 2023, 41, 1600–1609. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Suzuki, K.; Plesner, T.; et al. Overall Survival with Daratumumab, Lenalidomide, and Dexamethasone in Previously Treated Multiple Myeloma (POLLUX): A Randomized, Open-Label, Phase III Trial. J. Clin. Oncol. 2023, 41, 1590–1599. [Google Scholar] [CrossRef]
- Martin, T.; Dimopoulos, M.-A.; Mikhael, J.; Yong, K.; Capra, M.; Facon, T.; Hajek, R.; Špička, I.; Baker, R.; Kim, K.; et al. Isatuximab, Carfilzomib, and Dexamethasone in Patients with Relapsed Multiple Myeloma: Updated Results from IKEMA, a Randomized Phase 3 Study. Blood Cancer J. 2023, 13, 72. [Google Scholar] [CrossRef]
- Del Giudice, I.; Raponi, S.; Della Starza, I.; De Propris, M.S.; Cavalli, M.; De Novi, L.A.; Cappelli, L.V.; Ilari, C.; Cafforio, L.; Guarini, A.; et al. Minimal Residual Disease in Chronic Lymphocytic Leukemia: A New Goal? Front. Oncol. 2019, 9, 689. [Google Scholar] [CrossRef]
- Drandi, D.; Kubiczkova-Besse, L.; Ferrero, S.; Dani, N.; Passera, R.; Mantoan, B.; Gambella, M.; Monitillo, L.; Saraci, E.; Ghione, P.; et al. Minimal Residual Disease Detection by Droplet Digital PCR in Multiple Myeloma, Mantle Cell Lymphoma, and Follicular Lymphoma. J. Mol. Diagn. 2015, 17, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Emerson, R.O.; Sherwood, A.; Loh, M.L.; Angiolillo, A.; Howie, B.; Vogt, J.; Rieder, M.; Kirsch, I.; Carlson, C.; et al. Detection of Minimal Residual Disease in B Lymphoblastic Leukemia by High-Throughput Sequencing of IGH. Clin. Cancer Res. 2014, 20, 4540–4548. [Google Scholar] [CrossRef]
- ERIC (European Research Initiative on CLL); Rawstron, A.C.; Fazi, C.; Agathangelidis, A.; Villamor, N.; Letestu, R.; Nomdedeu, J.; Palacio, C.; Stehlikova, O.; Kreuzer, K.-A.; et al. A Complementary Role of Multiparameter Flow Cytometry and High-Throughput Sequencing for Minimal Residual Disease Detection in Chronic Lymphocytic Leukemia: An European Research Initiative on CLL Study. Leukemia 2016, 30, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Ladetto, M.; Brüggemann, M.; Monitillo, L.; Ferrero, S.; Pepin, F.; Drandi, D.; Barbero, D.; Palumbo, A.; Passera, R.; Boccadoro, M.; et al. Next-Generation Sequencing and Real-Time Quantitative PCR for Minimal Residual Disease Detection in B-Cell Disorders. Leukemia 2014, 28, 1299–1307. [Google Scholar] [CrossRef]
- Takamatsu, H.; Takezako, N.; Zheng, J.; Moorhead, M.; Carlton, V.E.H.; Kong, K.A.; Murata, R.; Ito, S.; Miyamoto, T.; Yokoyama, K.; et al. Prognostic Value of Sequencing-Based Minimal Residual Disease Detection in Patients with Multiple Myeloma Who Underwent Autologous Stem-Cell Transplantation. Ann. Oncol. 2017, 28, 2503–2510. [Google Scholar] [CrossRef] [PubMed]
- González-Calle, V.; Cerdá, S.; Labrador, J.; Sobejano, E.; González-Mena, B.; Aguilera, C.; Ocio, E.M.; Vidriales, M.B.; Puig, N.; Gutiérrez, N.C.; et al. Recovery of Polyclonal Immunoglobulins One Year after Autologous Stem Cell Transplantation as a Long-Term Predictor Marker of Progression and Survival in Multiple Myeloma. Haematologica 2017, 102, 922–931. [Google Scholar] [CrossRef]
- Frerichs, K.A.; Bosman, P.W.C.; Van Velzen, J.F.; Fraaij, P.L.A.; Koopmans, M.P.G.; Rimmelzwaan, G.F.; Nijhof, I.S.; Bloem, A.C.; Mutis, T.; Zweegman, S.; et al. Effect of Daratumumab on Normal Plasma Cells, Polyclonal Immunoglobulin Levels, and Vaccination Responses in Extensively Pre-Treated Multiple Myeloma Patients. Haematologica 2020, 105, e302–e306. [Google Scholar] [CrossRef]
- Tschautscher, M.A.; Jevremovic, D.; Rajkumar, V.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Dingli, D.; Hwa, Y.L.; Fonder, A.L.; et al. Prognostic Value of Minimal Residual Disease and Polyclonal Plasma Cells in Myeloma Patients Achieving a Complete Response to Therapy. Am. J. Hematol. 2019, 94, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Dávila, J.; González-Calle, V.; Escalante, F.; Cerdá, S.; Puig, N.; García-Sanz, R.; Bárez, A.; Montes, C.; López, R.; Alonso, J.M.; et al. Recovery of Polyclonal Immunoglobulins during Treatment in Patients Ineligible for Autologous Stem-cell Transplantation Is a Prognostic Marker of Longer Progression-free Survival and Overall Survival. Br. J. Haematol. 2022, 198, 278–287. [Google Scholar] [CrossRef]
- Alonso, R.; Cedena, M.T.; Gómez-Grande, A.; Ríos, R.; Moraleda, J.M.; Cabañas, V.; Moreno, M.J.; López-Jiménez, J.; Martín, F.; Sanz, A.; et al. Imaging and Bone Marrow Assessments Improve Minimal Residual Disease Prediction in Multiple Myeloma. Am. J. Hematol. 2019, 94, 853–861. [Google Scholar] [CrossRef]
- Rasche, L.; Alapat, D.; Kumar, M.; Gershner, G.; McDonald, J.; Wardell, C.P.; Samant, R.; Van Hemert, R.; Epstein, J.; Williams, A.F.; et al. Combination of Flow Cytometry and Functional Imaging for Monitoring of Residual Disease in Myeloma. Leukemia 2019, 33, 1713–1722. [Google Scholar] [CrossRef]
- Sanoja-Flores, L.; Flores-Montero, J.; Garcés, J.J.; Paiva, B.; Puig, N.; García-Mateo, A.; García-Sánchez, O.; Corral-Mateos, A.; Burgos, L.; Blanco, E.; et al. Next Generation Flow for Minimally-Invasive Blood Characterization of MGUS and Multiple Myeloma at Diagnosis Based on Circulating Tumor Plasma Cells (CTPC). Blood Cancer J. 2018, 8, 117. [Google Scholar] [CrossRef] [PubMed]
- Sanoja-Flores, L.; Flores-Montero, J.; Puig, N.; Contreras-Sanfeliciano, T.; Pontes, R.; Corral-Mateos, A.; García-Sánchez, O.; Díez-Campelo, M.; Pessoa De Magalhães, R.J.; García-Martín, L.; et al. Blood Monitoring of Circulating Tumor Plasma Cells by next Generation Flow in Multiple Myeloma after Therapy. Blood 2019, 134, 2218–2222. [Google Scholar] [CrossRef] [PubMed]
- Garcés, J.-J.; Cedena, M.-T.; Puig, N.; Burgos, L.; Perez, J.J.; Cordon, L.; Flores-Montero, J.; Sanoja-Flores, L.; Calasanz, M.-J.; Ortiol, A.; et al. Circulating Tumor Cells for the Staging of Patients With Newly Diagnosed Transplant-Eligible Multiple Myeloma. J. Clin. Oncol. 2022, 40, 3151–3161. [Google Scholar] [CrossRef]
- Bertamini, L.; Oliva, S.; Rota-Scalabrini, D.; Paris, L.; Morè, S.; Corradini, P.; Ledda, A.; Gentile, M.; De Sabbata, G.; Pietrantuono, G.; et al. High Levels of Circulating Tumor Plasma Cells as a Key Hallmark of Aggressive Disease in Transplant-Eligible Patients With Newly Diagnosed Multiple Myeloma. J. Clin. Oncol. 2022, 40, 3120–3131. [Google Scholar] [CrossRef]
- EuroClonality-NGS Working Group; Knecht, H.; Reigl, T.; Kotrová, M.; Appelt, F.; Stewart, P.; Bystry, V.; Krejci, A.; Grioni, A.; Pal, K.; et al. Quality Control and Quantification in IG/TR next-Generation Sequencing Marker Identification: Protocols and Bioinformatic Functionalities by EuroClonality-NGS. Leukemia 2019, 33, 2254–2265. [Google Scholar] [CrossRef]
- Scherer, F.; Kurtz, D.M.; Diehn, M.; Alizadeh, A.A. High-Throughput Sequencing for Noninvasive Disease Detection in Hematologic Malignancies. Blood 2017, 130, 440–452. [Google Scholar] [CrossRef]
- Roschewski, M.; Dunleavy, K.; Pittaluga, S.; Moorhead, M.; Pepin, F.; Kong, K.; Shovlin, M.; Jaffe, E.S.; Staudt, L.M.; Lai, C.; et al. Circulating Tumour DNA and CT Monitoring in Patients with Untreated Diffuse Large B-Cell Lymphoma: A Correlative Biomarker Study. Lancet Oncol. 2015, 16, 541–549. [Google Scholar] [CrossRef]
- Kurtz, D.M.; Green, M.R.; Bratman, S.V.; Scherer, F.; Liu, C.L.; Kunder, C.A.; Takahashi, K.; Glover, C.; Keane, C.; Kihira, S.; et al. Noninvasive Monitoring of Diffuse Large B-Cell Lymphoma by Immunoglobulin High-Throughput Sequencing. Blood 2015, 125, 3679–3687. [Google Scholar] [CrossRef]
- Mithraprabhu, S.; Chen, M.; Savvidou, I.; Reale, A.; Spencer, A. Liquid Biopsy: An Evolving Paradigm for the Biological Characterisation of Plasma Cell Disorders. Leukemia 2021, 35, 2771–2783. [Google Scholar] [CrossRef] [PubMed]
- Oberle, A.; Brandt, A.; Voigtlaender, M.; Thiele, B.; Radloff, J.; Schulenkorf, A.; Alawi, M.; Akyüz, N.; März, M.; Ford, C.T.; et al. Monitoring Multiple Myeloma by Next-Generation Sequencing of V(D)J Rearrangements from Circulating Myeloma Cells and Cell-Free Myeloma DNA. Haematologica 2017, 102, 1105–1111. [Google Scholar] [CrossRef]
- Mithraprabhu, S.; Khong, T.; Ramachandran, M.; Chow, A.; Klarica, D.; Mai, L.; Walsh, S.; Broemeling, D.; Marziali, A.; Wiggin, M.; et al. Circulating Tumour DNA Analysis Demonstrates Spatial Mutational Heterogeneity That Coincides with Disease Relapse in Myeloma. Leukemia 2017, 31, 1695–1705. [Google Scholar] [CrossRef]
- Kis, O.; Kaedbey, R.; Chow, S.; Danesh, A.; Dowar, M.; Li, T.; Li, Z.; Liu, J.; Mansour, M.; Masih-Khan, E.; et al. Circulating Tumour DNA Sequence Analysis as an Alternative to Multiple Myeloma Bone Marrow Aspirates. Nat. Commun. 2017, 8, 15086. [Google Scholar] [CrossRef] [PubMed]
- Biancon, G.; Gimondi, S.; Vendramin, A.; Carniti, C.; Corradini, P. Noninvasive Molecular Monitoring in Multiple Myeloma Patients Using Cell-Free Tumor DNA. J. Mol. Diagn. 2018, 20, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Waldschmidt, J.M.; Yee, A.J.; Vijaykumar, T.; Pinto, R.A.; Frede, J.; Anand, P.; Bianchi, G.; Guo, G.; Potdar, S.; Seifer, C.; et al. Cell-Free DNA for the Detection of Emerging Treatment Failure in Relapsed/ Refractory Multiple Myeloma. Leukemia 2022, 36, 1078–1087. [Google Scholar] [CrossRef]
- Mazzotti, C.; Buisson, L.; Maheo, S.; Perrot, A.; Chretien, M.-L.; Leleu, X.; Hulin, C.; Manier, S.; Hébraud, B.; Roussel, M.; et al. Myeloma MRD by Deep Sequencing from Circulating Tumor DNA Does Not Correlate with Results Obtained in the Bone Marrow. Blood Adv. 2018, 2, 2811–2813. [Google Scholar] [CrossRef]
- Mithraprabhu, S.; Sirdesai, S.; Chen, M.; Khong, T.; Spencer, A. Circulating Tumour DNA Analysis for Tumour Genome Characterisation and Monitoring Disease Burden in Extramedullary Multiple Myeloma. Int. J. Mol. Sci. 2018, 19, 1858. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Xu, Q.; Lou, Y.; Li, C.; Gu, J.; Cai, H.; Wang, D.; Xu, J.; Li, T.; Zhou, X.; et al. The Utility of Non-invasive Liquid Biopsy for Mutational Analysis and Minimal Residual Disease Assessment in Extramedullary Multiple Myeloma. Br. J. Haematol. 2020, 189, e45–e48. [Google Scholar] [CrossRef]
- Jelinek, T.; Bezdekova, R.; Zihala, D.; Sevcikova, T.; Anilkumar Sithara, A.; Pospisilova, L.; Sevcikova, S.; Polackova, P.; Stork, M.; Knechtova, Z.; et al. More Than 2% of Circulating Tumor Plasma Cells Defines Plasma Cell Leukemia–Like Multiple Myeloma. J. Clin. Oncol. 2023, 41, 1383–1392. [Google Scholar] [CrossRef]
- Termini, R.; Žihala, D.; Terpos, E.; Perez-Montaña, A.; Jelínek, T.; Raab, M.; Weinhold, N.; Mai, E.K.; Grab, A.L.; Corre, J.; et al. Circulating Tumor and Immune Cells for Minimally Invasive Risk Stratification of Smoldering Multiple Myeloma. Clin. Cancer Res. 2022, 28, 4771–4781. [Google Scholar] [CrossRef]
- Lohr, J.G.; Kim, S.; Gould, J.; Knoechel, B.; Drier, Y.; Cotton, M.J.; Gray, D.; Birrer, N.; Wong, B.; Ha, G.; et al. Genetic Interrogation of Circulating Multiple Myeloma Cells at Single-Cell Resolution. Sci. Transl. Med. 2016, 8, 363ra147. [Google Scholar] [CrossRef]
- GEM/PETHEMA (Grupo Español de Mieloma/Programa para el Estudio de la Terapéutica en Hemopatías Malignas) Cooperative Study Group; Garcés, J.-J.; Bretones, G.; Burgos, L.; Valdes-Mas, R.; Puig, N.; Cedena, M.-T.; Alignani, D.; Rodriguez, I.; Puente, D.Á.; et al. Circulating Tumor Cells for Comprehensive and Multiregional Non-Invasive Genetic Characterization of Multiple Myeloma. Leukemia 2020, 34, 3007–3018. [Google Scholar] [CrossRef]
- Dutta, A.K.; Alberge, J.-B.; Lightbody, E.D.; Boehner, C.J.; Dunford, A.; Sklavenitis-Pistofidis, R.; Mouhieddine, T.H.; Cowan, A.N.; Su, N.K.; Horowitz, E.M.; et al. MinimuMM-Seq: Genome Sequencing of Circulating Tumor Cells for Minimally Invasive Molecular Characterization of Multiple Myeloma Pathology. Cancer Discov. 2023, 13, 348–363. [Google Scholar] [CrossRef]
- Allegra, A.; Cancemi, G.; Mirabile, G.; Tonacci, A.; Musolino, C.; Gangemi, S. Circulating Tumour Cells, Cell Free DNA and Tumour-Educated Platelets as Reliable Prognostic and Management Biomarkers for the Liquid Biopsy in Multiple Myeloma. Cancers 2022, 14, 4136. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, E.; Cai, Z. Liquid Biopsy by Analysis of Circulating Myeloma Cells and Cell-Free Nucleic Acids: A Novel Noninvasive Approach of Disease Evaluation in Multiple Myeloma. Biomark. Res. 2023, 11, 27. [Google Scholar] [CrossRef]
- Hillengass, J.; Moulopoulos, L.A.; Delorme, S.; Koutoulidis, V.; Mosebach, J.; Hielscher, T.; Drake, M.; Rajkumar, S.V.; Oestergaard, B.; Abildgaard, N.; et al. Whole-Body Computed Tomography versus Conventional Skeletal Survey in Patients with Multiple Myeloma: A Study of the International Myeloma Working Group. Blood Cancer J. 2017, 7, e599. [Google Scholar] [CrossRef] [PubMed]
- Regelink, J.C.; Minnema, M.C.; Terpos, E.; Kamphuis, M.H.; Raijmakers, P.G.; Pieters-Van Den Bos, I.C.; Heggelman, B.G.F.; Nievelstein, R.-J.; Otten, R.H.J.; Van Lammeren-Venema, D.; et al. Comparison of Modern and Conventional Imaging Techniques in Establishing Multiple Myeloma-Related Bone Disease: A Systematic Review. Br. J. Haematol. 2013, 162, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Messiou, C.; Hillengass, J.; Delorme, S.; Lecouvet, F.E.; Moulopoulos, L.A.; Collins, D.J.; Blackledge, M.D.; Abildgaard, N.; Østergaard, B.; Schlemmer, H.-P.; et al. Guidelines for Acquisition, Interpretation, and Reporting of Whole-Body MRI in Myeloma: Myeloma Response Assessment and Diagnosis System (MY-RADS). Radiology 2019, 291, 5–13. [Google Scholar] [CrossRef]
- Belotti, A.; Ribolla, R.; Cancelli, V.; Villanacci, A.; Angelini, V.; Chiarini, M.; Giustini, V.; Facchetti, G.V.; Roccaro, A.M.; Ferrari, S.; et al. Predictive Role of Diffusion-weighted Whole-body MRI (DW-MRI) Imaging Response According to MY-RADS Criteria after Autologous Stem Cell Transplantation in Patients with Multiple Myeloma and Combined Evaluation with MRD Assessment by Flow Cytometry. Cancer Med. 2021, 10, 5859–5865. [Google Scholar] [CrossRef]
- Zamagni, E.; Nanni, C.; Dozza, L.; Carlier, T.; Bailly, C.; Tacchetti, P.; Versari, A.; Chauvie, S.; Gallamini, A.; Gamberi, B.; et al. Standardization of 18F-FDG–PET/CT According to Deauville Criteria for Metabolic Complete Response Definition in Newly Diagnosed Multiple Myeloma. J. Clin. Oncol. 2021, 39, 116–125. [Google Scholar] [CrossRef]
- Zamagni, E.; Nanni, C.; Mancuso, K.; Tacchetti, P.; Pezzi, A.; Pantani, L.; Zannetti, B.; Rambaldi, I.; Brioli, A.; Rocchi, S.; et al. PET/CT Improves the Definition of Complete Response and Allows to Detect Otherwise Unidentifiable Skeletal Progression in Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4384–4390. [Google Scholar] [CrossRef]
- Moreau, P.; Attal, M.; Caillot, D.; Macro, M.; Karlin, L.; Garderet, L.; Facon, T.; Benboubker, L.; Escoffre-Barbe, M.; Stoppa, A.-M.; et al. Prospective Evaluation of Magnetic Resonance Imaging and [18F]Fluorodeoxyglucose Positron Emission Tomography-Computed Tomography at Diagnosis and Before Maintenance Therapy in Symptomatic Patients With Multiple Myeloma Included in the IFM/DFCI 2009 Trial: Results of the IMAJEM Study. J. Clin. Oncol. 2017, 35, 2911–2918. [Google Scholar] [CrossRef] [PubMed]
- Charalampous, C.; Goel, U.; Broski, S.M.; Dingli, D.; Kapoor, P.; Gertz, M.A.; Lacy, M.Q.; Dispenzieri, A.; Hayman, S.R.; Buadi, F.; et al. Utility of PET/CT in Assessing Early Treatment Response in Patients with Newly Diagnosed Multiple Myeloma. Blood Adv. 2022, 6, 2763–2772. [Google Scholar] [CrossRef]
- Hillengass, J.; Usmani, S.; Rajkumar, S.V.; Durie, B.G.M.; Mateos, M.-V.; Lonial, S.; Joao, C.; Anderson, K.C.; García-Sanz, R.; Riva, E.; et al. International Myeloma Working Group Consensus Recommendations on Imaging in Monoclonal Plasma Cell Disorders. Lancet Oncol. 2019, 20, e302–e312. [Google Scholar] [CrossRef]
- Nanni, C.; Kobe, C.; Baeßler, B.; Baues, C.; Boellaard, R.; Borchmann, P.; Buck, A.; Buvat, I.; Chapuy, B.; Cheson, B.D.; et al. European Association of Nuclear Medicine (EANM) Focus 4 Consensus Recommendations: Molecular Imaging and Therapy in Haematological Tumours. Lancet Haematol. 2023, 10, e367–e381. [Google Scholar] [CrossRef] [PubMed]
- Kraeber-Bodéré, F.; Zweegman, S.; Perrot, A.; Hulin, C.; Caillot, D.; Facon, T.; Leleu, X.; Belhadj, K.; Itti, E.; Karlin, L.; et al. Prognostic Value of Positron Emission Tomography/Computed Tomography in Transplant-Eligible Newly Diagnosed Multiple Myeloma Patients from CASSIOPEIA: The CASSIOPET Study. Haematologica 2022, 108, 621–626. [Google Scholar] [CrossRef]
- Lapa, C.; Kircher, M.; Da Via, M.; Schreder, M.; Rasche, L.; Kortüm, K.M.; Einsele, H.; Buck, A.K.; Hänscheid, H.; Samnick, S. Comparison of 11C-Choline and 11C-Methionine PET/CT in Multiple Myeloma. Clin. Nucl. Med. 2019, 44, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Stokke, C.; Nørgaard, J.N.; Feiring Phillips, H.; Sherwani, A.; Nuruddin, S.; Connelly, J.; Schjesvold, F.; Revheim, M.-E. Comparison of [18F]Fluciclovine and [18F]FDG PET/CT in Newly Diagnosed Multiple Myeloma Patients. Mol. Imaging Biol. 2022, 24, 842–851. [Google Scholar] [CrossRef]
- Okasaki, M.; Kubota, K.; Minamimoto, R.; Miyata, Y.; Morooka, M.; Ito, K.; Ishiwata, K.; Toyohara, J.; Inoue, T.; Hirai, R.; et al. Comparison of 11C-4′-Thiothymidine, 11C-Methionine, and 18F-FDG PET/CT for the Detection of Active Lesions of Multiple Myeloma. Ann. Nucl. Med. 2015, 29, 224–232. [Google Scholar] [CrossRef]
- Kuyumcu, S.; Isik, E.G.; Tiryaki, T.O.; Has-Simsek, D.; Sanli, Y.; Buyukkaya, F.; Özkan, Z.G.; Kalayoglu-Besisik, S.; Unal, S.N. Prognostic Significance of 68Ga-Pentixafor PET/CT in Multiple Myeloma Recurrence: A Comparison to 18F-FDG PET/CT and Laboratory Results. Ann. Nucl. Med. 2021, 35, 1147–1156. [Google Scholar] [CrossRef]
- Zamagni, E.; Nanni, C.; Patriarca, F.; Englaro, E.; Castellucci, P.; Geatti, O.; Tosi, P.; Tacchetti, P.; Cangini, D.; Perrone, G.; et al. A Prospective Comparison of 18F-Fluorodeoxyglucose Positron Emission Tomography-Computed Tomography, Magnetic Resonance Imaging and Whole-Body Planar Radiographs in the Assessment of Bone Disease in Newly Diagnosed Multiple Myeloma. Haematologica 2007, 92, 50–55. [Google Scholar] [CrossRef]
- Rasche, L.; Angtuaco, E.; McDonald, J.E.; Buros, A.; Stein, C.; Pawlyn, C.; Thanendrarajan, S.; Schinke, C.; Samant, R.; Yaccoby, S.; et al. Low Expression of Hexokinase-2 Is Associated with False-Negative FDG–Positron Emission Tomography in Multiple Myeloma. Blood 2017, 130, 30–34. [Google Scholar] [CrossRef]
- Pawlyn, C.; Fowkes, L.; Otero, S.; Jones, J.R.; Boyd, K.D.; Davies, F.E.; Morgan, G.J.; Collins, D.J.; Sharma, B.; Riddell, A.; et al. Whole-Body Diffusion-Weighted MRI: A New Gold Standard for Assessing Disease Burden in Patients with Multiple Myeloma? Leukemia 2016, 30, 1446–1448. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.L.; Puig, N.; Kristinsson, S.; Usmani, S.Z.; Dispenzieri, A.; Bianchi, G.; Kumar, S.; Chng, W.J.; Hajek, R.; Paiva, B.; et al. Mass Spectrometry for the Evaluation of Monoclonal Proteins in Multiple Myeloma and Related Disorders: An International Myeloma Working Group Mass Spectrometry Committee Report. Blood Cancer J. 2021, 11, 24. [Google Scholar] [CrossRef]
- Bergen, H.R.; Dasari, S.; Dispenzieri, A.; Mills, J.R.; Ramirez-Alvarado, M.; Tschumper, R.C.; Jelinek, D.F.; Barnidge, D.R.; Murray, D.L. Clonotypic Light Chain Peptides Identified for Monitoring Minimal Residual Disease in Multiple Myeloma without Bone Marrow Aspiration. Clin. Chem. 2016, 62, 243–251. [Google Scholar] [CrossRef]
- Zajec, M.; Jacobs, J.F.M.; De Kat Angelino, C.M.; Dekker, L.J.M.; Stingl, C.; Luider, T.M.; De Rijke, Y.B.; VanDuijn, M.M. Integrating Serum Protein Electrophoresis with Mass Spectrometry, A New Workflow for M-Protein Detection and Quantification. J. Proteome Res. 2020, 19, 2845–2853. [Google Scholar] [CrossRef] [PubMed]
- Langerhorst, P.; Noori, S.; Zajec, M.; De Rijke, Y.B.; Gloerich, J.; Van Gool, A.J.; Caillon, H.; Joosten, I.; Luider, T.M.; Corre, J.; et al. Multiple Myeloma Minimal Residual Disease Detection: Targeted Mass Spectrometry in Blood vs Next-Generation Sequencing in Bone Marrow. Clin. Chem. 2021, 67, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Noori, S.; Verkleij, C.P.M.; Zajec, M.; Langerhorst, P.; Bosman, P.W.C.; De Rijke, Y.B.; Zweegman, S.; VanDuijn, M.; Luider, T.; Van De Donk, N.W.C.J.; et al. Monitoring the M-Protein of Multiple Myeloma Patients Treated with a Combination of Monoclonal Antibodies: The Laboratory Solution to Eliminate Interference. Clin. Chem. Lab. Med. CCLM 2021, 59, 1963–1971. [Google Scholar] [CrossRef]
- Mills, J.R.; Barnidge, D.R.; Murray, D.L. Detecting Monoclonal Immunoglobulins in Human Serum Using Mass Spectrometry. Methods 2015, 81, 56–65. [Google Scholar] [CrossRef]
- Mills, J.R.; Kohlhagen, M.C.; Dasari, S.; Vanderboom, P.M.; Kyle, R.A.; Katzmann, J.A.; Willrich, M.A.V.; Barnidge, D.R.; Dispenzieri, A.; Murray, D.L. Comprehensive Assessment of M-Proteins Using Nanobody Enrichment Coupled to MALDI-TOF Mass Spectrometry. Clin. Chem. 2016, 62, 1334–1344. [Google Scholar] [CrossRef]
- Abeykoon, J.P.; Murray, D.L.; Murray, I.; Jevremovic, D.; Otteson, G.E.; Dispenzieri, A.; Arendt, B.K.; Dasari, S.; Gertz, M.; Gonsalves, W.I.; et al. Implications of Detecting Serum Monoclonal Protein by MASS-fix Following Stem Cell Transplantation in Multiple Myeloma. Br. J. Haematol. 2021, 193, 380–385. [Google Scholar] [CrossRef]
- Fatica, E.M.; Martinez, M.; Ladwig, P.M.; Murray, J.D.; Kohlhagen, M.C.; Kyle, R.A.; Kourelis, T.; Lust, J.A.; Snyder, M.R.; Dispenzieri, A.; et al. MALDI-TOF Mass Spectrometry Can Distinguish Immunofixation Bands of the Same Isotype as Monoclonal or Biclonal Proteins. Clin. Biochem. 2021, 97, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Eveillard, M.; Rustad, E.; Roshal, M.; Zhang, Y.; Ciardiello, A.; Korde, N.; Hultcrantz, M.; Lu, S.; Shah, U.; Hassoun, H.; et al. Comparison of MALDI-TOF Mass Spectrometry Analysis of Peripheral Blood and Bone Marrow-based Flow Cytometry for Tracking Measurable Residual Disease in Patients with Multiple Myeloma. Br. J. Haematol. 2020, 189, 904–907. [Google Scholar] [CrossRef] [PubMed]
- Derman, B.A.; Stefka, A.T.; Jiang, K.; McIver, A.; Kubicki, T.; Jasielec, J.K.; Jakubowiak, A.J. Measurable Residual Disease Assessed by Mass Spectrometry in Peripheral Blood in Multiple Myeloma in a Phase II Trial of Carfilzomib, Lenalidomide, Dexamethasone and Autologous Stem Cell Transplantation. Blood Cancer J. 2021, 11, 19. [Google Scholar] [CrossRef]
- Dispenzieri, A.; Krishnan, A.; Arendt, B.; Blackwell, B.; Wallace, P.K.; Dasari, S.; Vogl, D.T.; Efebera, Y.; Fei, M.; Geller, N.; et al. Mass-Fix Better Predicts for PFS and OS than Standard Methods among Multiple Myeloma Patients Participating on the STAMINA Trial (BMT CTN 0702/07LT). Blood Cancer J. 2022, 12, 27. [Google Scholar] [CrossRef]
- Puig, N.; Contreras, M.-T.; Agulló, C.; Martínez-López, J.; Oriol, A.; Blanchard, M.-J.; Ríos, R.; Martín, J.; Iñigo, M.-B.; Sureda, A.; et al. Mass Spectrometry vs Immunofixation for Treatment Monitoring in Multiple Myeloma. Blood Adv. 2022, 6, 3234–3239. [Google Scholar] [CrossRef]
- Campbell, L.; Simpson, D.; Ramasamy, K.; Sadler, R. Using Quantitative Immunoprecipitation Mass Spectrometry (QIP-MS) to Identify Low Level Monoclonal Proteins. Clin. Biochem. 2021, 95, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Puig, N.; Agulló, C.; Contreras Sanfeliciano, T.; Paiva, B.; Cedena, M.T.; Rosinol Dachs, L.; García-Sanz, R.; Martínez-López, J.; Oriol, A.; Blanchard, M.J.; et al. Clinical Impact of Next Generation Flow in Bone Marrow Vs Qip-Mass Spectrometry in Peripheral Blood to Assess Minimal Residual Disease in Newly Diagnosed Multiple Myeloma Patients Receiving Maintenance as Part of the GEM2014MAIN Trial. Blood 2022, 140, 2098–2100. [Google Scholar] [CrossRef]
- Gu, J.; Liu, J.; Chen, M.; Huang, B.; Li, J. Longitudinal Flow Cytometry Identified “Minimal Residual Disease” (MRD) Evolution Patterns for Predicting the Prognosis of Patients with Transplant-Eligible Multiple Myeloma. Biol. Blood Marrow Transpl. 2018, 24, 2568–2574. [Google Scholar] [CrossRef]
- Paiva, B.; Manrique, I.; Dimopoulos, M.A.; Gay, F.; Min, C.-K.; Zweegman, S.; Špička, I.; Teipel, R.; Mateos, M.-V.; Giuliani, N.; et al. MRD Dynamics during Maintenance for Improved Prognostication of 1280 Patients with Myeloma in the TOURMALINE-MM3 and -MM4 Trials. Blood 2023, 141, 579–591. [Google Scholar] [CrossRef]
- Martinez-Lopez, J.; Alonso, R.; Wong, S.W.; Rios, R.; Shah, N.; Ruiz-Heredia, Y.; Sanchez-Pina, J.M.; Sanchez, R.; Bahri, N.; Zamanillo, I.; et al. Making Clinical Decisions Based on Measurable Residual Disease Improves the Outcome in Multiple Myeloma. J. Hematol. Oncol. 2021, 14, 126. [Google Scholar] [CrossRef]
- Costa, L.J.; Chhabra, S.; Medvedova, E.; Dholaria, B.R.; Schmidt, T.M.; Godby, K.N.; Silbermann, R.; Dhakal, B.; Bal, S.; Giri, S.; et al. Daratumumab, Carfilzomib, Lenalidomide, and Dexamethasone with Minimal Residual Disease Response-Adapted Therapy in Newly Diagnosed Multiple Myeloma. J. Clin. Oncol. 2022, 40, 2901–2912. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Risk Marker | Prognostic Impact | References |
|---|---|---|
| Classical | ||
| Age | Survival decrease with each decade of life | [12] |
| Baseline HR cytogenetics | OS < 3 years | [13] |
| ISS, OS (months) | [14] | |
| I | 62 | |
| II | 44 | |
| III | 29 | |
| R-ISS, 5-year OS rate | [15] | |
| I | 82% | |
| II | 62% | |
| III | 40% | |
| Novel | ||
| R2-ISS, OS (months) | [16] | |
| I | NR | |
| II | 109.2 | |
| III | 68.5 | |
| IV | 37.9 | |
| Double-hit, OS (months) | 20.7 | [17] |
| Study | Sensitivity (Median) | Treatment Algorithm | MRDneg Rates |
|---|---|---|---|
| ASCT-eligible | |||
| Myeloma XI | 4 × 10−5 | ASCT + R vs. | 65.6% |
| (NCT01554852) [55] | no maintenance | 34.4% | |
| GEM2012MENOS65 | 3 × 10−6 | VRd + ASCT + VRd | 50.2% |
| (NCT01916252) [52,53] | |||
| CASSIOPEIA | 10−5 | Part 1: Dara − VTd + ASCT + Dara − VTd vs. | 64% |
| (NCT02541383) [57] | VTd + ASCT + VTd | 44% | |
| 10−5 | Part 2: Maintenance with Dara vs. | 66% | |
| observation | 55.2% | ||
| EMN02/HO95 | 10−5 | Consolidation with VRd vs. | 9.8% |
| (NCT01208766) [54] | No consolidation | 8.2% |
| Study | Sensitivity (Median) | Treatment Algorithm | Mrdneg Rates |
|---|---|---|---|
| ASCT-eligible | |||
| IFM2009 | 10−6 | VRd, 8 cycles vs. | 20% |
| (NCT01191060) [89] | VRd + ASCT | 30% | |
| CASSIOPEIA | 10−5 | Part 1: Dara − VTd + ASCT + Dara − VTd vs. | 57% |
| (NCT02541383) [57] | VTd + ASCT + VTd | 37% | |
| 10−6 | Part 2: Maintenance with Dara vs. | 49.5% | |
| observation | 36.7% | ||
| GRIFFIN | 10−5 | Dara − VRd + ASCT + Dara − VRd vs. | 51% |
| (NCT02874742) [96] | VRd + ASCT + VRd | 20.4% | |
| ASCT-non-eligible | |||
| ALCYONE | 10−5 | Dara − VMP vs. | 22% |
| (NCT02195479) [97,98] | VMP | 6% | |
| MAIA | 10−5 | Dara − Rd vs. | 24.2% |
| (NCT02252172) [98,99] | Rd | 7.3% | |
| Relapsed/refractory | |||
| CASTOR | 10−5 | Dara − Vd vs. | 15% |
| (NCT02136134) [100] | Vd | 1.6% | |
| POLLUX | 10−5 | Dara − Rd vs. | 33.2% |
| (NCT02076009) [101] | Rd | 6.7% | |
| IKEMA | 10−5 | Isa–Kd vs. | 29.6% |
| (NCT03275285) [102] | Kd | 13% |
| Standard MFC | NGF | ASOqPCR | NGS | ddPCR | |
|---|---|---|---|---|---|
| Applicability | 90–100% | 90–100% | 40–75% | ~90% | Comparable to qPCR |
| Sensitivity | 10−4–10−5 | 10−5–10−6 | 10−4–10−5 | 10−5–10−6 | At least 10−5 |
| Standardization | No | EuroFlow | EuroMRD | ClonoSEQ * | Ongoing |
| Turnaround time | 1 day | 1 day | ≥1 week | 4 days–1 week | ≥1 week |
| Specific primers/probes | Not applicable | Not applicable | Yes | No | Yes |
| Standard curve | Not applicable | Not applicable | Yes | No | No |
| Influenced by SHM | No | No | Yes | Yes | Yes |
| Baseline BM | No | No | Yes | Yes | Yes |
| Fresh sample (processing time) | Yes (24–48 h) | Yes (24 h) | No | No | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medina-Herrera, A.; Sarasquete, M.E.; Jiménez, C.; Puig, N.; García-Sanz, R. Minimal Residual Disease in Multiple Myeloma: Past, Present, and Future. Cancers 2023, 15, 3687. https://doi.org/10.3390/cancers15143687
Medina-Herrera A, Sarasquete ME, Jiménez C, Puig N, García-Sanz R. Minimal Residual Disease in Multiple Myeloma: Past, Present, and Future. Cancers. 2023; 15(14):3687. https://doi.org/10.3390/cancers15143687
Chicago/Turabian StyleMedina-Herrera, Alejandro, María Eugenia Sarasquete, Cristina Jiménez, Noemí Puig, and Ramón García-Sanz. 2023. "Minimal Residual Disease in Multiple Myeloma: Past, Present, and Future" Cancers 15, no. 14: 3687. https://doi.org/10.3390/cancers15143687
APA StyleMedina-Herrera, A., Sarasquete, M. E., Jiménez, C., Puig, N., & García-Sanz, R. (2023). Minimal Residual Disease in Multiple Myeloma: Past, Present, and Future. Cancers, 15(14), 3687. https://doi.org/10.3390/cancers15143687