
Decoding Oncofusions: Unveiling Mechanisms, Clinical Impact, and Prospects for Personalized Cancer Therapies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
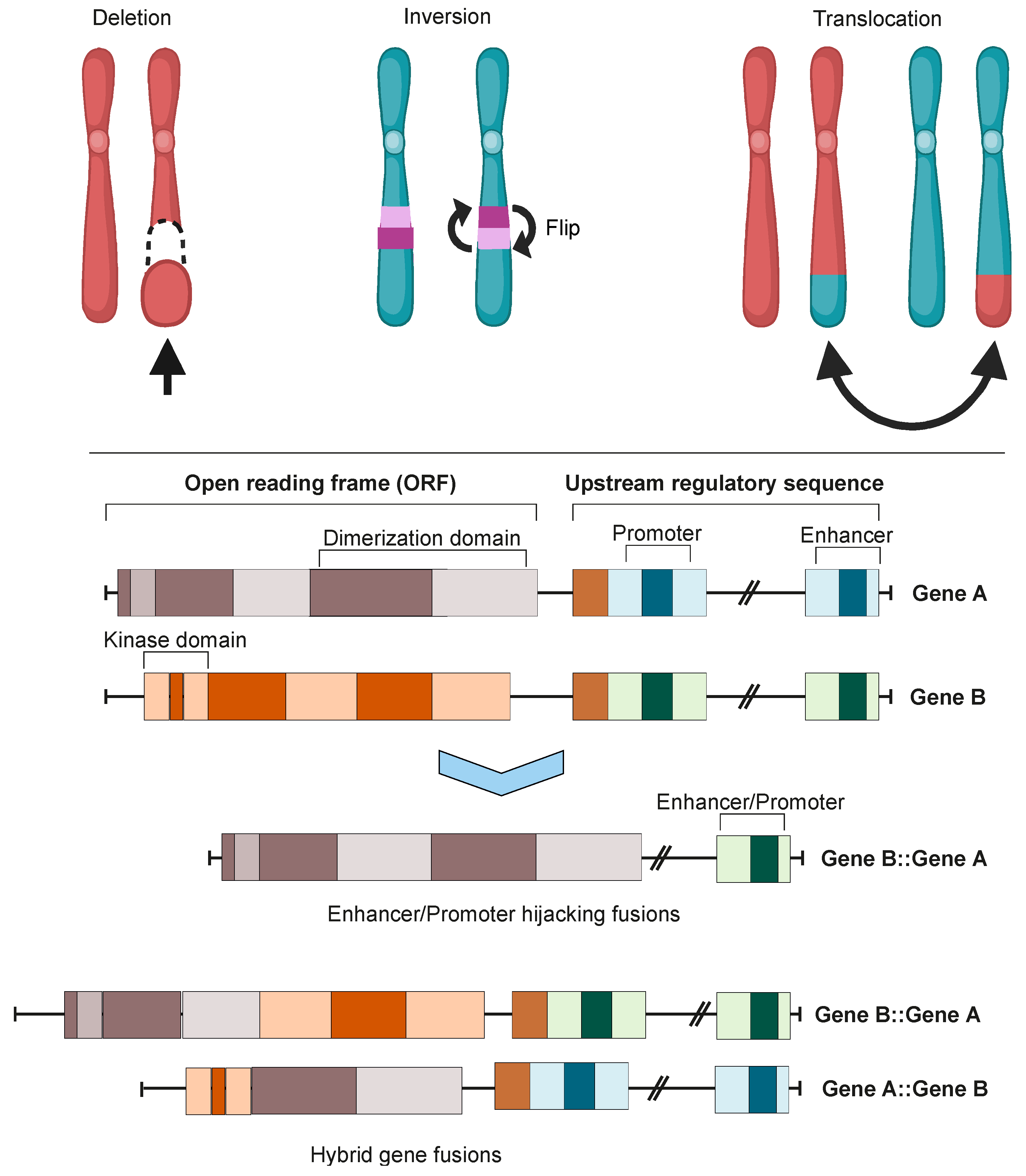
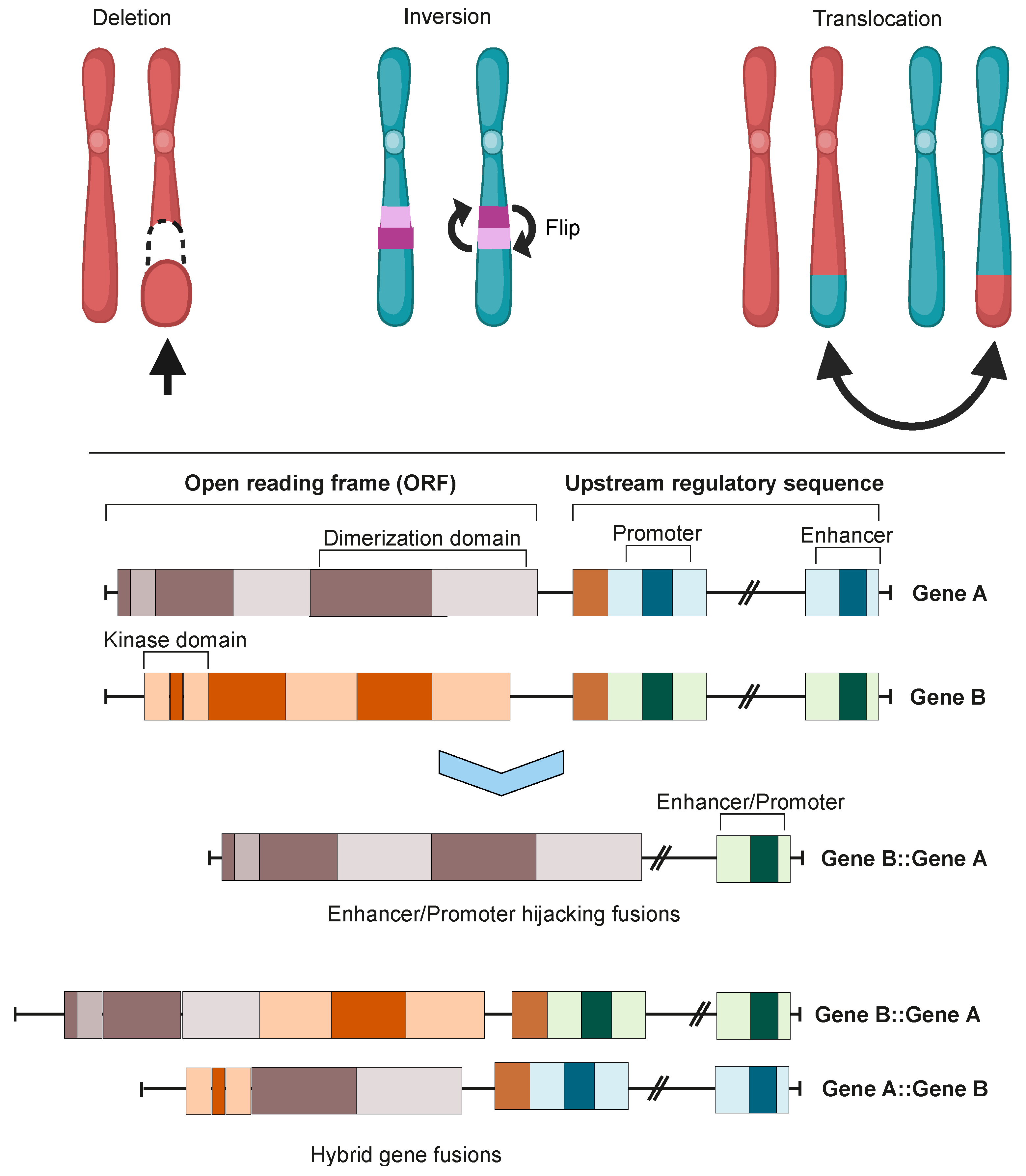
2. Oncofusion Formation
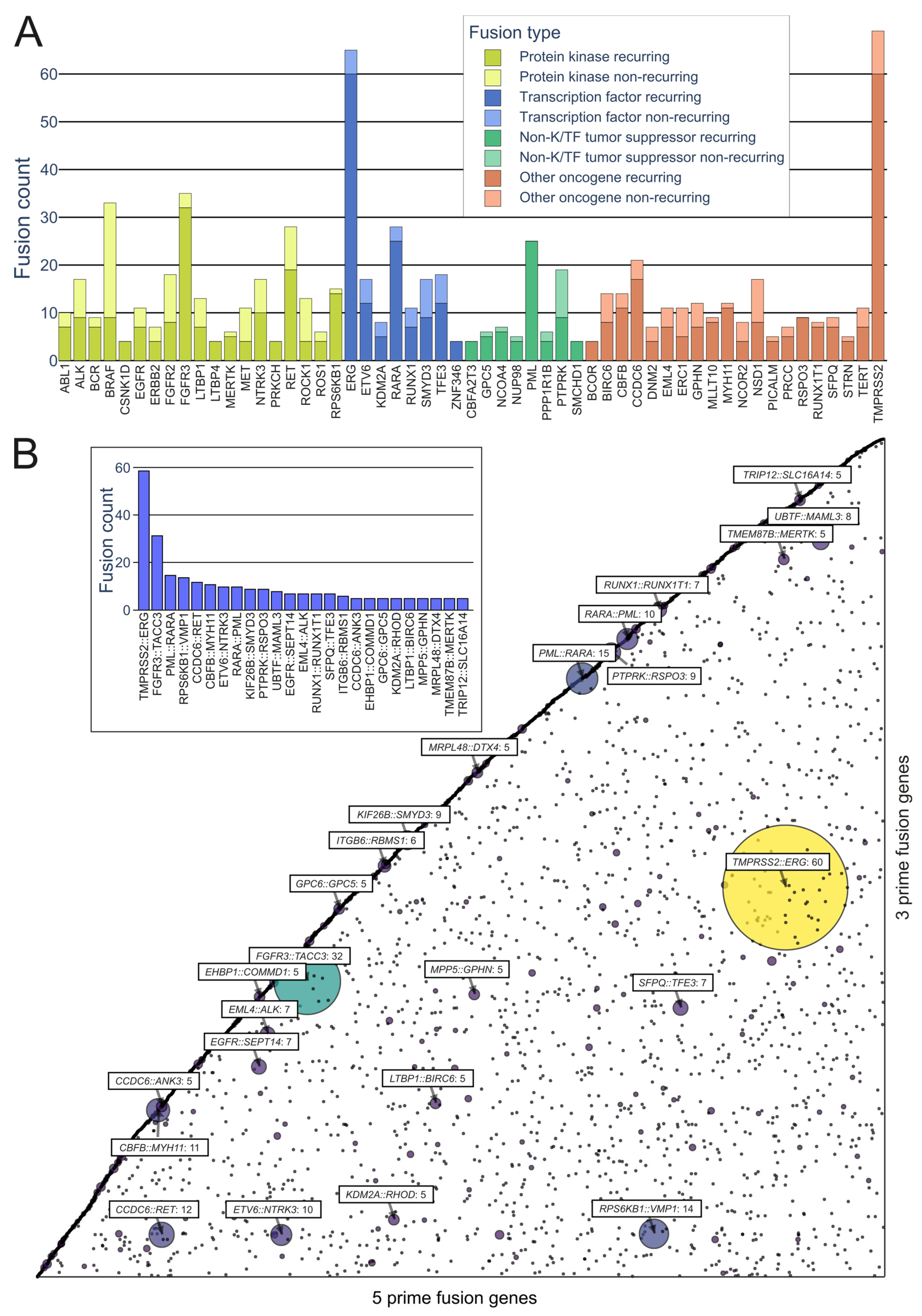
3. Oncofusions’ Role in Cancer Development
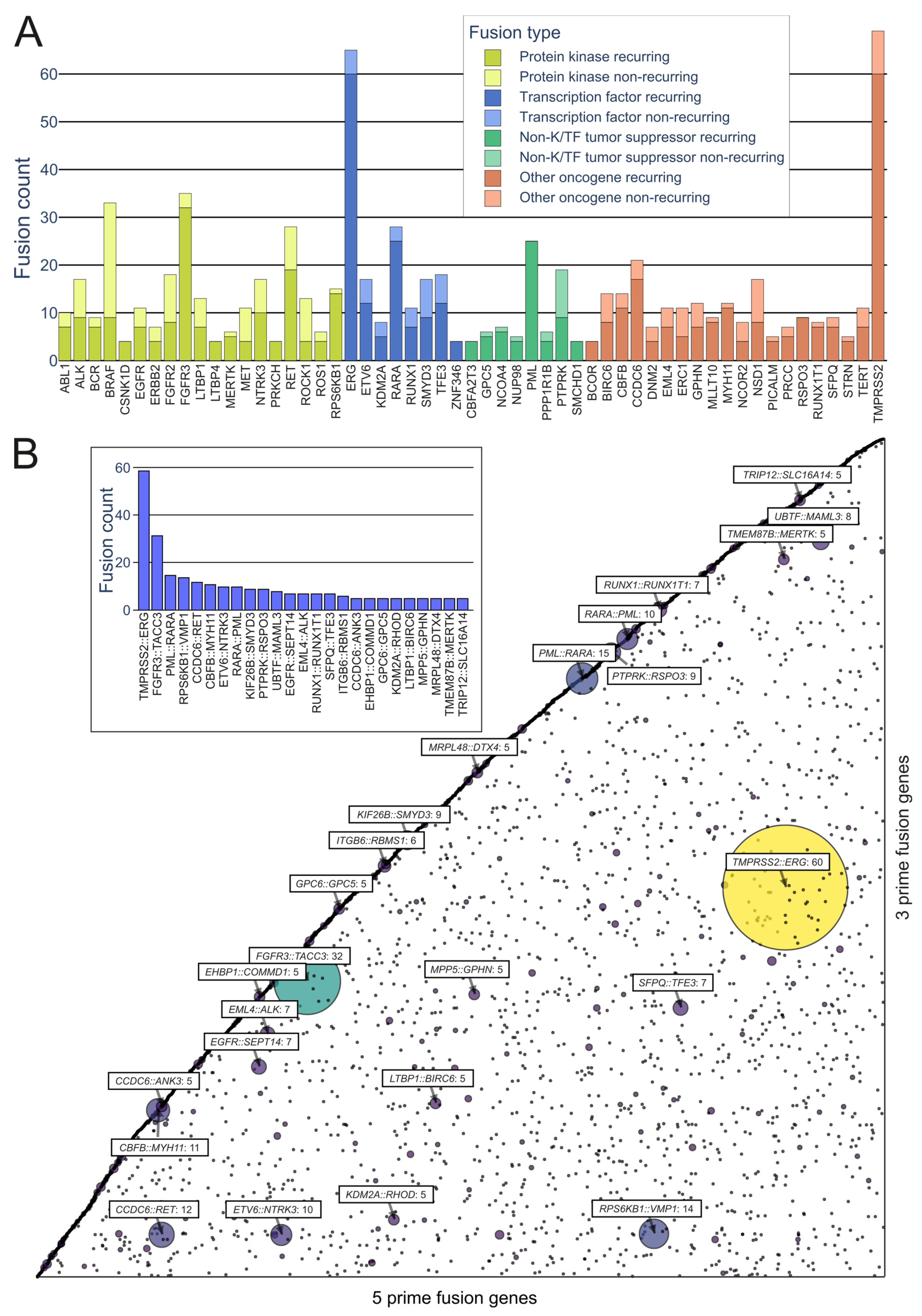
Prominent Oncofusions in Cancer
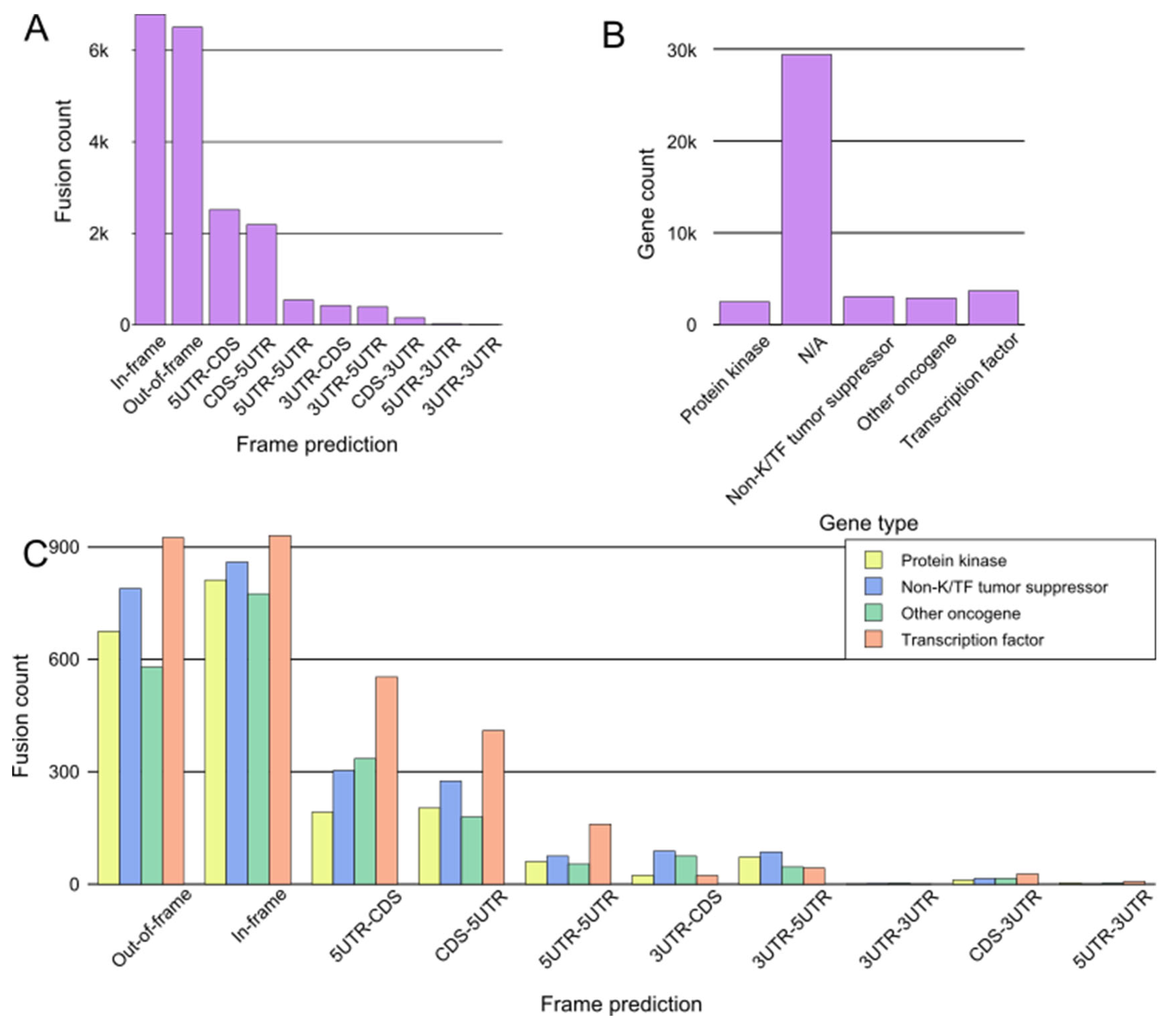
4. Fusion Identification
4.1. Fusion Callers
4.2. Protein-Level Analysis of Oncofusions
5. Functional Validation of Gene Fusions
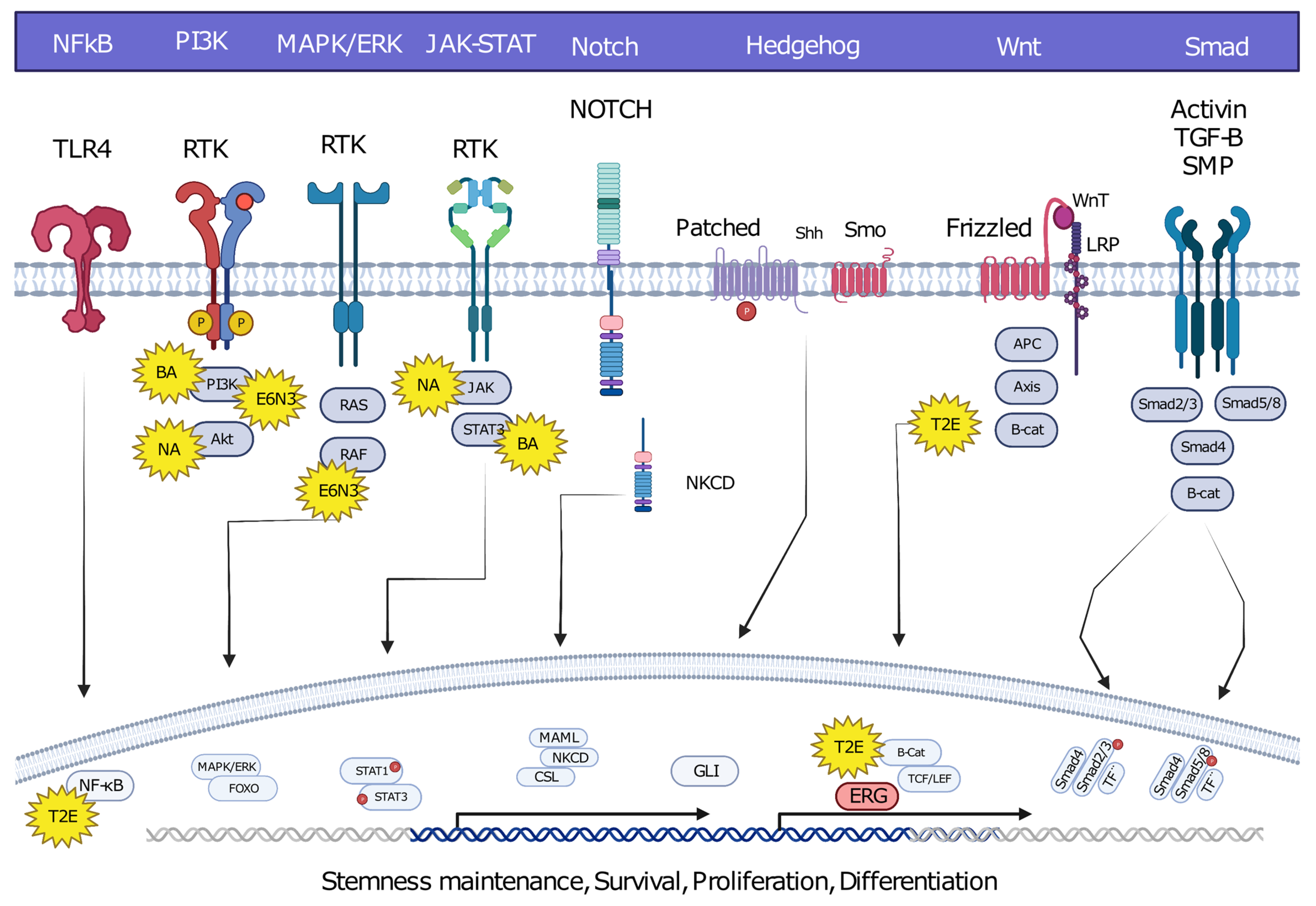
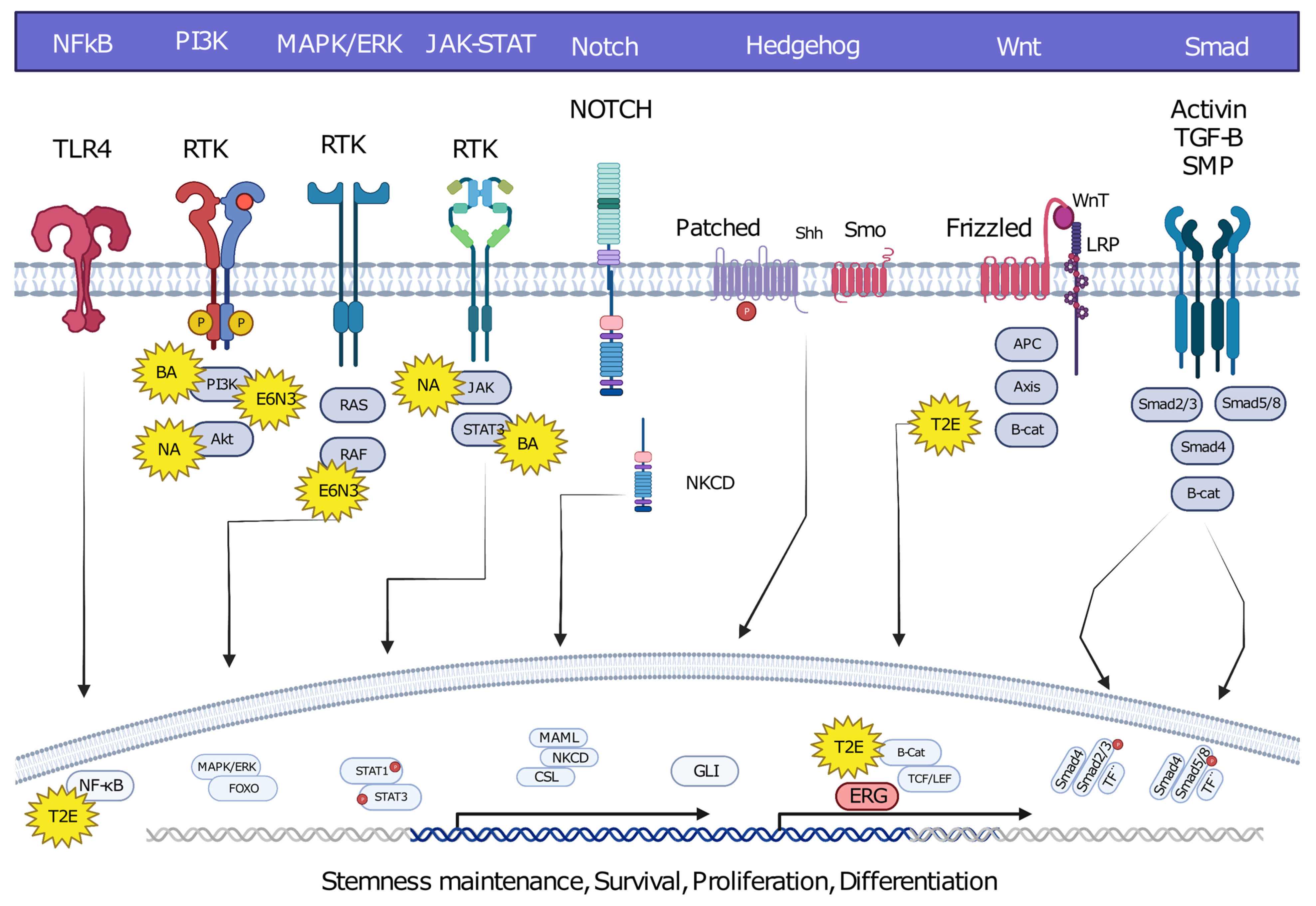
6. Oncofusion-Specific Molecular Mechanisms
7. The Importance of Oncofusion-Specific Molecular Mechanisms
7.1. BCR::ABL
7.2. ETV6::NTRK3
7.3. NPM::ALK
7.4. TMPRSS2::ERG
8. Gene Fusions as Neoantigens
8.1. Neoantigen Immunogenicity
8.2. Treatment Potential for T-Cell Therapies
9. Clinical Implications
10. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Latysheva, N.S.; Babu, M.M. Discovering and Understanding Oncogenic Gene Fusions through Data Intensive Computational Approaches. Nucleic Acids Res. 2016, 44, 4487–4503. [Google Scholar] [CrossRef] [Green Version]
- Nowell, P.; Hungerford, D. Chromosome Studies on Normal and Leukemic Human Leukocytes. J. Natl. Cancer Inst. 1960, 25, 85–109. [Google Scholar]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, T.; Yamaguchi, K.; Urakami, K.; Shimoda, Y.; Ohnami, S.; Ohshima, K.; Tanabe, T.; Naruoka, A.; Kamada, F.; Serizawa, M.; et al. Japanese Version of The Cancer Genome Atlas, JCGA, Established Using Fresh Frozen Tumors Obtained from 5143 Cancer Patients. Cancer Sci. 2020, 111, 687–699. [Google Scholar] [CrossRef]
- Salokas, K.; Weldatsadik, R.G.; Varjosalo, M. Human Transcription Factor and Protein Kinase Gene Fusions in Human Cancer. Sci. Rep. 2020, 10, 14169. [Google Scholar] [CrossRef] [PubMed]
- Erikson, J.; Nishikura, K.; ar-Rushdi, A.; Finan, J.; Emanuel, B.; Lenoir, G.; Nowell, P.C.; Croce, C.M. Translocation of an Immunoglobulin Kappa Locus to a Region 3′ of an Unrearranged c-Myc Oncogene Enhances c-Myc Transcription. Proc. Natl. Acad. Sci. USA 1983, 80, 7581–7585. [Google Scholar] [CrossRef]
- Dalla-Favera, R.; Bregni, M.; Erikson, J.; Patterson, D.; Gallo, R.C.; Croce, C.M. Human C-Myc Onc Gene Is Located on the Region of Chromosome 8 That Is Translocated in Burkitt Lymphoma Cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7824–7827. [Google Scholar] [CrossRef]
- Taub, R.; Kirsch, I.; Morton, C.; Lenoir, G.; Swan, D.; Tronick, S.; Aaronson, S.; Leder, P. Translocation of the C-Myc Gene into the Immunoglobulin Heavy Chain Locus in Human Burkitt Lymphoma and Murine Plasmacytoma Cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7837–7841. [Google Scholar] [CrossRef]
- Heisterkamp, N.; Stam, K.; Groffen, J.; de Klein, A.; Grosveld, G. Structural Organization of the Bcr Gene and Its Role in the Ph’ Translocation. Nature 1985, 315, 758–761. [Google Scholar] [CrossRef]
- Shtivelman, E.; Lifshitz, B.; Gale, R.P.; Canaani, E. Fused Transcript of Abl and Bcr Genes in Chronic Myelogenous Leukaemia. Nature 1985, 315, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Daley, G.Q.; Mes-Masson, A.M.; Witte, O.N.; Baltimore, D. The Chronic Myelogenous Leukemia-Specific P210 Protein Is the Product of the Bcr/Abl Hybrid Gene. Science 1986, 233, 212–214. [Google Scholar] [CrossRef] [PubMed]
- Fainstein, E.; Marcelle, C.; Rosner, A.; Canaani, E.; Gale, R.P.; Dreazen, O.; Smith, S.D.; Croce, C.M. A New Fused Transcript in Philadelphia Chromosome Positive Acute Lymphocytic Leukaemia. Nature 1987, 330, 386–388. [Google Scholar] [CrossRef]
- Reckel, S.; Hamelin, R.; Georgeon, S.; Armand, F.; Jolliet, Q.; Chiappe, D.; Moniatte, M.; Hantschel, O. Differential Signaling Networks of Bcr-Abl P210 and P190 Kinases in Leukemia Cells Defined by Functional Proteomics. Leukemia 2017, 31, 1502–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutler, J.A.; Tahir, R.; Sreenivasamurthy, S.K.; Mitchell, C.; Renuse, S.; Nirujogi, R.S.; Patil, A.H.; Heydarian, M.; Wong, X.; Wu, X.; et al. Differential Signaling through P190 and P210 BCR-ABL Fusion Proteins Revealed by Interactome and Phosphoproteome Analysis. Leukemia 2017, 31, 1513–1524. [Google Scholar] [CrossRef]
- Efremov, G.D. Hemoglobins Lepore and Anti-Lepore. Hemoglobin 1978, 2, 197–233. [Google Scholar] [CrossRef] [PubMed]
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The Emerging Complexity of Gene Fusions in Cancer. Nat. Rev. Cancer 2015, 15, 371–381. [Google Scholar] [CrossRef]
- Kumar-Sinha, C.; Kalyana-Sundaram, S.; Chinnaiyan, A.M. Landscape of Gene Fusions in Epithelial Cancers: Seq and Ye Shall Find. Genome Med. 2015, 7, 129. [Google Scholar] [CrossRef] [Green Version]
- Johansson, B.; Mertens, F.; Schyman, T.; Björk, J.; Mandahl, N.; Mitelman, F. Most Gene Fusions in Cancer Are Stochastic Events. Genes Chromosomes Cancer 2019, 58, 607–611. [Google Scholar] [CrossRef]
- Zhang, J.; Mardis, E.R.; Maher, C.A. INTEGRATE-Neo: A Pipeline for Personalized Gene Fusion Neoantigen Discovery. Bioinformatics 2017, 33, 555–557. [Google Scholar] [CrossRef] [Green Version]
- Yi-Mi, W.; Cieslik, M.; Lonigro, R.J.; Pankaj, V.; Reimers, M.A.; Xuhong, C.; Yu, N.; Lisha, W.; Kunju, L.P.; de Sarkar, N.; et al. Inactivation of CDK12 Delineates a Distinct Immunogenic Class of Advanced Prostate Cancer. Cell 2018, 173, 1770–1782.e14. [Google Scholar] [CrossRef]
- So, A.; Le Guen, T.; Lopez, B.S.; Guirouilh-Barbat, J. Genomic Rearrangements Induced by Unscheduled DNA Double Strand Breaks in Somatic Mammalian Cells. FEBS J. 2017, 284, 2324–2344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, J.W.; Yang, L.; Park, H.-Y.; Lee, C.-W.; Cha, H.; Shin, H.-T.; Noh, K.-W.; Choi, Y.-L.; Park, W.-Y.; Park, P.J. Dysregulation of Cancer Genes by Recurrent Intergenic Fusions. Genome Biol. 2020, 21, 166. [Google Scholar] [CrossRef] [PubMed]
- Namba, S.; Ueno, T.; Kojima, S.; Kobayashi, K.; Kawase, K.; Tanaka, Y.; Inoue, S.; Kishigami, F.; Kawashima, S.; Maeda, N.; et al. Transcript-Targeted Analysis Reveals Isoform Alterations and Double-Hop Fusions in Breast Cancer. Commun. Biol. 2021, 4, 1320. [Google Scholar] [CrossRef]
- Ferrad, M.; Ghazzaui, N.; Issaoui, H.; Cook-Moreau, J.; Denizot, Y. Mouse Models of C-Myc Deregulation Driven by IgH Locus Enhancers as Models of B-Cell Lymphomagenesis. Front. Immunol. 2020, 11, 1564. [Google Scholar] [CrossRef]
- Panagopoulos, I.; Heim, S. Neoplasia-Associated Chromosome Translocations Resulting in Gene Truncation. Cancer Genom. Proteom. 2022, 19, 647–672. [Google Scholar] [CrossRef]
- Wang, T.; Wei, L.; Lu, Q.; Shao, Y.; You, S.; Yin, J.C.; Wang, S.; Shao, Y.; Chen, Z.; Wang, Z. Landscape of Potentially Targetable Receptor Tyrosine Kinase Fusions in Diverse Cancers by DNA-Based Profiling. NPJ Precis. Oncol. 2022, 6, 84. [Google Scholar] [CrossRef]
- Gagos, S.; Irminger-Finger, I. Chromosome Instability in Neoplasia: Chaotic Roots to Continuous Growth. Int. J. Biochem. Cell Biol. 2005, 37, 1014–1033. [Google Scholar] [CrossRef]
- Takeuchi, K. Discovery Stories of RET Fusions in Lung Cancer: A Mini-Review. Front. Physiol. 2019, 10, 216. [Google Scholar] [CrossRef]
- Long, M.; Langley, C.H. Natural Selection and the Origin of Jingwei, a Chimeric Processed Functional Gene in Drosophila. Science 1993, 260, 91–95. [Google Scholar] [CrossRef]
- Mitelman, F.; Johansson, B.; Mertens, F. The Impact of Translocations and Gene Fusions on Cancer Causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-K.; Choi, Y.-L.; Kwon, M.; Park, P.J. Mechanisms and Consequences of Cancer Genome Instability: Lessons from Genome Sequencing Studies. Annu. Rev. Pathol. 2016, 11, 283–312. [Google Scholar] [CrossRef] [PubMed]
- Lobato, M.N.; Metzler, M.; Drynan, L.; Forster, A.; Pannell, R.; Rabbitts, T.H. Modeling Chromosomal Translocations Using Conditional Alleles to Recapitulate Initiating Events in Human Leukemias. J. Natl. Cancer Inst. Monogr. 2008, 2008, 58–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertens, F.; Antonescu, C.R.; Mitelman, F. Gene Fusions in Soft Tissue Tumors: Recurrent and Overlapping Pathogenetic Themes. Genes Chromosomes Cancer 2016, 55, 291–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Q.; Liang, W.-W.; Foltz, S.M.; Mutharasu, G.; Jayasinghe, R.G.; Cao, S.; Liao, W.-W.; Reynolds, S.M.; Wyczalkowski, M.A.; Yao, L.; et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 2018, 23, 227–238.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, Z.; Jeselsohn, R. ESR1 Fusions and Therapeutic Resistance in Metastatic Breast Cancer. Front. Oncol. 2022, 12, 1037531. [Google Scholar] [CrossRef]
- Yu, L.; Davis, I.J.; Liu, P. Regulation of EWSR1-FLI1 Function by Post-Transcriptional and Post-Translational Modifications. Cancers 2023, 15, 382. [Google Scholar] [CrossRef]
- Apfelbaum, A.A.; Wrenn, E.D.; Lawlor, E.R. The Importance of Fusion Protein Activity in Ewing Sarcoma and the Cell Intrinsic and Extrinsic Factors That Regulate It: A Review. Front. Oncol. 2022, 12, 1044707. [Google Scholar] [CrossRef]
- Bowling, G.C.; Rands, M.G.; Dobi, A.; Eldhose, B. Emerging Developments in ETS-Positive Prostate Cancer Therapy. Mol. Cancer Ther. 2023, 22, 168–178. [Google Scholar] [CrossRef]
- Shen, Z.; Qiu, B.; Li, L.; Yang, B.; Li, G. Targeted Therapy of RET Fusion-Positive Non-Small Cell Lung Cancer. Front. Oncol. 2022, 12, 1033484. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, M.; Rabushko, E.; Rozenberg, J.M.; Mohammad, T.; Seryakov, A.; Sekacheva, M.; Buzdin, A. Clinically Relevant Fusion Oncogenes: Detection and Practical Implications. Ther. Adv. Med. Oncol. 2022, 14, 17588359221144108. [Google Scholar] [CrossRef]
- Hagstrom, M.; Fumero-Velázquez, M.; Dhillon, S.; Olivares, S.; Gerami, P. An Update on Genomic Aberrations in Spitz Naevi and Tumours. Pathology 2023, 55, 196–205. [Google Scholar] [CrossRef]
- Chu, Y.-H. This Is Your Thyroid on Drugs: Targetable Mutations and Fusions in Thyroid Carcinoma. Surg. Pathol. Clin. 2023, 16, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.G.; Peiris, M.N.; Meyer, A.N.; Nelson, K.N.; Donoghue, D.J. Oncogenic Driver FGFR3-TACC3 Requires Five Coiled-Coil Heptads for Activation and Disulfide Bond Formation for Stability. Oncotarget 2023, 14, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lalazar, G.; Houlihan, S.L.; Tschaharganeh, D.F.; Baslan, T.; Chen, C.-C.; Requena, D.; Tian, S.; Bosbach, B.; Wilkinson, J.E.; et al. DNAJB1-PRKACA Fusion Kinase Interacts with β-Catenin and the Liver Regenerative Response to Drive Fibrolamellar Hepatocellular Carcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, 13076–13084. [Google Scholar] [CrossRef] [Green Version]
- Matissek, K.J.; Onozato, M.L.; Sun, S.; Zheng, Z.; Schultz, A.; Lee, J.; Patel, K.; Jerevall, P.-L.; Saladi, S.V.; Macleay, A.; et al. Expressed Gene Fusions as Frequent Drivers of Poor Outcomes in Hormone Receptor-Positive Breast Cancer. Cancer Discov. 2018, 8, 336–353. [Google Scholar] [CrossRef] [Green Version]
- Lei, J.T.; Shao, J.; Zhang, J.; Iglesia, M.; Chan, D.W.; Cao, J.; Anurag, M.; Singh, P.; He, X.; Kosaka, Y.; et al. Functional Annotation of ESR1 Gene Fusions in Estrogen Receptor-Positive Breast Cancer. Cell Rep. 2018, 24, 1434–1444.e7. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kim, S.; Ko, S.; In, Y.; Moon, H.-G.; Ahn, S.K.; Kim, M.K.; Lee, M.; Hwang, J.-H.; Ju, Y.S.; et al. Recurrent Fusion Transcripts Detected by Whole-Transcriptome Sequencing of 120 Primary Breast Cancer Samples. Genes Chromosomes Cancer 2015, 54, 681–691. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Finger, L.R.; Yunis, J.; Nowell, P.C.; Croce, C.M. Cloning of the Chromosome Breakpoint of Neoplastic B Cells with the t(14;18) Chromosome Translocation. Science 1984, 226, 1097–1099. [Google Scholar] [CrossRef]
- Vaux, D.L.; Cory, S.; Adams, J.M. Bcl-2 Gene Promotes Haemopoietic Cell Survival and Cooperates with c-Myc to Immortalize Pre-B Cells. Nature 1988, 335, 440–442. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.-W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent Fusion of TMPRSS2 and ETS Transcription Factor Genes in Prostate Cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.-P.; Liu, P.; Nelson, J.; Hamilton, R.L.; Bhargava, R.; Michalopoulos, G.; Chen, Q.; Zhang, J.; Ma, D.; Pennathur, A.; et al. Identification of Recurrent Fusion Genes across Multiple Cancer Types. Sci. Rep. 2019, 9, 1074. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.P.; Ding, Y.; Chen, Z.; Liu, S.; Michalopoulos, A.; Chen, R.; Gulzar, Z.G.; Yang, B.; Cieply, K.M.; Luvison, A.; et al. Novel Fusion Transcripts Associate with Progressive Prostate Cancer. Am. J. Pathol. 2014, 184, 2840–2849. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Yu, J.; Mani, R.-S.; Cao, Q.; Brenner, C.J.; Cao, X.; Wang, G.X.; Wu, L.; Li, J.; Hu, M.; et al. An Integrated Network of Androgen Receptor, Polycomb, and TMPRSS2-ERG Gene Fusions in Prostate Cancer Progression. Cancer Cell 2010, 17, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Gedman, A.L.; Dang, J.; Nakitandwe, J.; Holmfeldt, L.; Parker, M.; Easton, J.; et al. The Landscape of Somatic Mutations in Infant MLL-Rearranged Acute Lymphoblastic Leukemias. Nat. Genet. 2015, 47, 330–337. [Google Scholar] [CrossRef]
- Milne, T.A. Mouse Models of MLL Leukemia: Recapitulating the Human Disease. Blood 2017, 129, 2217–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babiceanu, M.; Qin, F.; Xie, Z.; Jia, Y.; Lopez, K.; Janus, N.; Facemire, L.; Kumar, S.; Pang, Y.; Qi, Y.; et al. Recurrent Chimeric Fusion RNAs in Non-Cancer Tissues and Cells. Nucleic Acids Res. 2016, 44, 2859–2872. [Google Scholar] [CrossRef] [Green Version]
- Forsberg, L.A.; Rasi, C.; Razzaghian, H.R.; Pakalapati, G.; Waite, L.; Thilbeault, K.S.; Ronowicz, A.; Wineinger, N.E.; Tiwari, H.K.; Boomsma, D.; et al. Age-Related Somatic Structural Changes in the Nuclear Genome of Human Blood Cells. Am. J. Hum. Genet. 2012, 90, 217–228. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, Y.; Tsukamoto, T.; Chinen, Y.; Shimura, Y.; Sasaki, N.; Nagoshi, H.; Sato, R.; Adachi, H.; Nakano, M.; Horiike, S.; et al. Detection of Novel and Recurrent Conjoined Genes in Non-Hodgkin B-Cell Lymphoma. J. Clin. Exp. Hematop. JCEH 2021, 61, 71–77. [Google Scholar] [CrossRef]
- Mukherjee, S.; Frenkel-Morgenstern, M. Evolutionary Impact of Chimeric RNAs on Generating Phenotypic Plasticity in Human Cells. Trends Genet. TIG 2022, 38, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dean, D.C.; Hornicek, F.J.; Shi, H.; Duan, Z. RNA Sequencing (RNA-Seq) and Its Application in Ovarian Cancer. Gynecol. Oncol. 2019, 152, 194–201. [Google Scholar] [CrossRef]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. Cancer Genome Atlas Research Network The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, P.; Zhou, X. FusionGDB: Fusion Gene Annotation DataBase. Nucleic Acids Res. 2019, 47, D994–D1004. [Google Scholar] [CrossRef] [PubMed]
- Argani, P.; Palsgrove, D.N.; Anders, R.A.; Smith, S.C.; Saoud, C.; Kwon, R.; Voltaggio, L.; Assarzadegan, N.; Oshima, K.; Rooper, L.; et al. A Novel NIPBL-NACC1 Gene Fusion Is Characteristic of the Cholangioblastic Variant of Intrahepatic Cholangiocarcinoma. Am. J. Surg. Pathol. 2021, 45, 1550–1560. [Google Scholar] [CrossRef]
- Haas, B.J.; Dobin, A.; Li, B.; Stransky, N.; Pochet, N.; Regev, A. Accuracy Assessment of Fusion Transcript Detection via Read-Mapping and de Novo Fusion Transcript Assembly-Based Methods. Genome Biol. 2019, 20, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Tsai, W.-H.; Ding, Y.; Chen, R.; Fang, Z.; Huo, Z.; Kim, S.; Ma, T.; Chang, T.-Y.; Priedigkeit, N.M.; et al. Comprehensive Evaluation of Fusion Transcript Detection Algorithms and a Meta-Caller to Combine Top Performing Methods in Paired-End RNA-Seq Data. Nucleic Acids Res. 2016, 44, e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorney, R.; Dhungel, B.P.; Rasko, J.E.J.; Hebbard, L.; Schmitz, U. Recent Advances in Cancer Fusion Transcript Detection. Brief. Bioinform. 2022, 24, bbac519. [Google Scholar] [CrossRef]
- Nattestad, M.; Goodwin, S.; Ng, K.; Baslan, T.; Sedlazeck, F.J.; Rescheneder, P.; Garvin, T.; Fang, H.; Gurtowski, J.; Hutton, E.; et al. Complex Rearrangements and Oncogene Amplifications Revealed by Long-Read DNA and RNA Sequencing of a Breast Cancer Cell Line. Genome Res. 2018, 28, 1126–1135. [Google Scholar] [CrossRef] [Green Version]
- Apostolides, M.; Jiang, Y.; Husić, M.; Siddaway, R.; Hawkins, C.; Turinsky, A.L.; Brudno, M.; Ramani, A.K. MetaFusion: A High-Confidence Metacaller for Filtering and Prioritizing RNA-Seq Gene Fusion Candidates. Bioinformatics 2021, 37, 3144–3151. [Google Scholar] [CrossRef]
- Thomas, B.B.; Mou, Y.; Keeler, L.; Magnan, C.; Funari, V.; Weiss, L.; Brown, S.; Agersborg, S. A Highly Sensitive and Specific Gene Fusion Algorithm Based on Multiple Fusion Callers and an Ensemble Machine Learning Approach. Blood 2020, 136, 12–13. [Google Scholar] [CrossRef]
- Hedges, D.J. RNA-Seq Fusion Detection in Clinical Oncology. Adv. Exp. Med. Biol. 2022, 1361, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Kuksin, M.; Morel, D.; Aglave, M.; Danlos, F.-X.; Marabelle, A.; Zinovyev, A.; Gautheret, D.; Verlingue, L. Applications of Single-Cell and Bulk RNA Sequencing in Onco-Immunology. Eur. J. Cancer 2021, 149, 193–210. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Bahleda, R.; Hierro, C.; Sanson, M.; Bridgewater, J.; Arkenau, H.-T.; Tran, B.; Kelley, R.K.; Park, J.O.; Javle, M.; et al. Futibatinib, an Irreversible FGFR1-4 Inhibitor, in Patients with Advanced Solid Tumors Harboring FGF/FGFR Aberrations: A Phase I Dose-Expansion Study. Cancer Discov. 2022, 12, 402–415. [Google Scholar] [CrossRef] [PubMed]
- Loo, S.K.; Yates, M.E.; Yang, S.; Oesterreich, S.; Lee, A.V.; Wang, X.-S. Fusion-Associated Carcinomas of the Breast: Diagnostic, Prognostic, and Therapeutic Significance. Genes Chromosomes Cancer 2022, 61, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.; Guo, L.; Zhao, R.; Teng, H.; Wang, Y.; Xiong, L.; Han, Y. Identification and Validation of Noncanonical RET Fusions in Non-Small-Cell Lung Cancer through DNA and RNA Sequencing. J. Mol. Diagn. JMD 2022, 24, 374–385. [Google Scholar] [CrossRef]
- Li, W.; Liu, Y.; Li, W.; Chen, L.; Ying, J. Intergenic Breakpoints Identified by DNA Sequencing Confound Targetable Kinase Fusion Detection in NSCLC. J. Thorac. Oncol. 2020, 15, 1223–1231. [Google Scholar] [CrossRef]
- Rathe, S.K.; Popescu, F.E.; Johnson, J.E.; Watson, A.L.; Marko, T.A.; Moriarity, B.S.; Ohlfest, J.R.; Largaespada, D.A. Identification of Candidate Neoantigens Produced by Fusion Transcripts in Human Osteosarcomas. Sci. Rep. 2019, 9, 358. [Google Scholar] [CrossRef] [Green Version]
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; de Jong, P.; Rouleau, G. Gene Fusion with an ETS DNA-Binding Domain Caused by Chromosome Translocation in Human Tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef]
- Sand, L.G.L.; Szuhai, K.; Hogendoorn, P.C.W. Sequencing Overview of Ewing Sarcoma: A Journey across Genomic, Epigenomic and Transcriptomic Landscapes. Int. J. Mol. Sci. 2015, 16, 16176–16215. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.R.; Kalyana-Sundaram, S.; Wu, Y.-M.; Shankar, S.; Cao, X.; Ateeq, B.; Asangani, I.A.; Iyer, M.; Maher, C.A.; Grasso, C.S.; et al. Functionally Recurrent Rearrangements of the MAST Kinase and Notch Gene Families in Breast Cancer. Nat. Med. 2011, 17, 1646–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.Y.; McCusker, M.G.; Russo, A.; Scilla, K.A.; Gittens, A.; Arensmeyer, K.; Mehra, R.; Adamo, V.; Rolfo, C. RET Fusions in Solid Tumors. Cancer Treat. Rev. 2019, 81, 101911. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.; Moccia, M.; Federico, G.; Carlomagno, F. RET Gene Fusions in Malignancies of the Thyroid and Other Tissues. Genes 2020, 11, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Zhang, Z.; Sun, Y.; Wang, S.; Wu, M.; Ou, Q.; Xu, Y.; Chen, Z.; Shao, Y.; Liu, H.; et al. RET Fusions as Primary Oncogenic Drivers and Secondary Acquired Resistance to EGFR Tyrosine Kinase Inhibitors in Patients with Non-Small-Cell Lung Cancer. J. Transl. Med. 2022, 20, 390. [Google Scholar] [CrossRef]
- Yau, D.T.-W.; Lacambra, M.D.; Chow, C.; To, K.-F. The Novel Finding of an FGFR1::TACC1 Fusion in an Undifferentiated Spindle Cell Sarcoma of Soft Tissue with Aggressive Clinical Course. Genes Chromosomes Cancer 2022, 61, 206–211. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, Q.; Wang, Y.; Dai, X.; Chen, P.; Chen, A.; Zhou, S.; Dai, C.; Zhao, S.; Xiao, S.; et al. Case Report: A Novel LHFPL3::NTRK2 Fusion in Dysembryoplastic Neuroepithelial Tumor. Front. Oncol. 2022, 12, 1064817. [Google Scholar] [CrossRef]
- Deland, L.; Keane, S.; Bontell, T.O.; Fagman, H.; Sjögren, H.; Lind, A.E.; Carén, H.; Tisell, M.; Nilsson, J.A.; Ejeskär, K.; et al. Novel TPR::ROS1 Fusion Gene Activates MAPK, PI3K and JAK/STAT Signaling in an Infant-Type Pediatric Glioma. Cancer Genom. Proteom. 2022, 19, 711–726. [Google Scholar] [CrossRef]
- Hiwatari, M.; Seki, M.; Matsuno, R.; Yoshida, K.; Nagasawa, T.; Sato-Otsubo, A.; Yamamoto, S.; Kato, M.; Watanabe, K.; Sekiguchi, M.; et al. Novel TENM3-ALK Fusion Is an Alternate Mechanism for ALK Activation in Neuroblastoma. Oncogene 2022, 41, 2789–2797. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK Fusion-Positive Cancers and TRK Inhibitor Therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Amatu, A.; Sartore-Bianchi, A.; Bencardino, K.; Pizzutilo, E.G.; Tosi, F.; Siena, S. Tropomyosin Receptor Kinase (TRK) Biology and the Role of NTRK Gene Fusions in Cancer. Ann. Oncol. 2019, 30, viii5–viii15. [Google Scholar] [CrossRef]
- Amatu, A.; Sartore-Bianchi, A.; Siena, S. NTRK Gene Fusions as Novel Targets of Cancer Therapy across Multiple Tumour Types. ESMO Open 2016, 1, e000023. [Google Scholar] [CrossRef] [Green Version]
- Ardini, E.; Bosotti, R.; Borgia, A.L.; De Ponti, C.; Somaschini, A.; Cammarota, R.; Amboldi, N.; Raddrizzani, L.; Milani, A.; Magnaghi, P.; et al. The TPM3-NTRK1 Rearrangement Is a Recurring Event in Colorectal Carcinoma and Is Associated with Tumor Sensitivity to TRKA Kinase Inhibition. Mol. Oncol. 2014, 8, 1495–1507. [Google Scholar] [CrossRef] [PubMed]
- Vaishnavi, A.; Capelletti, M.; Le, A.T.; Kako, S.; Butaney, M.; Ercan, D.; Mahale, S.; Davies, K.D.; Aisner, D.L.; Pilling, A.B.; et al. Oncogenic and Drug-Sensitive NTRK1 Rearrangements in Lung Cancer. Nat. Med. 2013, 19, 1469–1472. [Google Scholar] [CrossRef] [Green Version]
- Dermawan, J.K.; Vanderbilt, C.M.; Chang, J.C.; Untch, B.R.; Singer, S.; Chi, P.; Tap, W.D.; Antonescu, C.R. FGFR2::TACC2 Fusion as a Novel KIT-Independent Mechanism of Targeted Therapy Failure in a Multidrug-Resistant Gastrointestinal Stromal Tumor. Genes Chromosomes Cancer 2022, 61, 412–419. [Google Scholar] [CrossRef]
- Wu, Y.-M.; Su, F.; Kalyana-Sundaram, S.; Khazanov, N.; Ateeq, B.; Cao, X.; Lonigro, R.J.; Vats, P.; Wang, R.; Lin, S.-F.; et al. Identification of Targetable FGFR Gene Fusions in Diverse Cancers. Cancer Discov. 2013, 3, 636–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, T.-M.; Arnau, G.M.; Ryland, G.L.; Huang, S.; Lira, M.E.; Emmanuel, Y.; Perez, O.D.; Irwin, D.; Fellowes, A.P.; Wong, S.Q.; et al. Multiplexed Transcriptome Analysis to Detect ALK, ROS1 and RET Rearrangements in Lung Cancer. Sci. Rep. 2017, 7, 42259. [Google Scholar] [CrossRef] [Green Version]
- Chuang, T.-P.; Lai, W.-Y.; Gabre, J.L.; Lind, D.E.; Umapathy, G.; Bokhari, A.A.; Bergman, B.; Kristenson, L.; Thorén, F.B.; Le, A.; et al. ALK Fusion NSCLC Oncogenes Promote Survival and Inhibit NK Cell Responses via SERPINB4 Expression. Proc. Natl. Acad. Sci. USA 2023, 120, e2216479120. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.-P.; Tsung, A.; Liu, S.; Nalesnick, M.; Geller, D.; Michalopoulos, G.; Luo, J.-H. Detection of Fusion Transcripts in the Serum Samples of Patients with Hepatocellular Carcinoma. Oncotarget 2019, 10, 3352–3360. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.-P.; Liu, S.; Nelson, J.; Luo, J.-H. Detection of Fusion Gene Transcripts in the Blood Samples of Prostate Cancer Patients. Sci. Rep. 2021, 11, 16995. [Google Scholar] [CrossRef]
- Chen, Z.-H.; Yu, Y.P.; Tao, J.; Liu, S.; Tseng, G.; Nalesnik, M.; Hamilton, R.; Bhargava, R.; Nelson, J.B.; Pennathur, A.; et al. MAN2A1–FER Fusion Gene Is Expressed by Human Liver and Other Tumor Types and Has Oncogenic Activity in Mice. Gastroenterology 2017, 153, 1120–1132.e15. [Google Scholar] [CrossRef]
- Glenfield, C.; Innan, H. Gene Duplication and Gene Fusion Are Important Drivers of Tumourigenesis during Cancer Evolution. Genes 2021, 12, 1376. [Google Scholar] [CrossRef] [PubMed]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-Spondin Fusions in Colon Cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slape, C.; Aplan, P.D. The Role of NUP98 Gene Fusions in Hematologic Malignancy. Leuk. Lymphoma 2004, 45, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Gough, S.M.; Slape, C.I.; Aplan, P.D. NUP98 Gene Fusions and Hematopoietic Malignancies: Common Themes and New Biologic Insights. Blood 2011, 118, 6247–6257. [Google Scholar] [CrossRef] [Green Version]
- Lanic, M.-D.; Guérin, R.; Sater, V.; Durdilly, P.; Ruminy, P.; Skálová, A.; Laé, M. A Novel SMARCA2-CREM Fusion Expending the Molecular Spectrum of Salivary Gland Hyalinazing Clear Cell Carcinoma beyond the FET Genes. Genes Chromosomes Cancer 2022, 62, 231–236. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Gao, X.; Zhao, W.; Zhou, F.; Liu, H.; Wang, W. Identification of Novel PIEZO1::CBFA2T3 and INO80C::SETBP1 Fusion Genes in an Acute Myeloid Leukemia Patient by RNA-Seq. Mol. Biol. Rep. 2022, 50, 1961–1966. [Google Scholar] [CrossRef]
- Ryzhova, M.V.; Shaikhaev, E.G.; Snigireva, G.P.; Gorelyshev, S.K.; Zheludkova, O.G.; Golanov, A.V. Novel BRAF::EPB41L2 gene fusion in posterior fossa pilocytic astrocytoma. Brief communication. Arkh. Patol. 2022, 84, 40–42. [Google Scholar] [CrossRef]
- Trubini, S.; Ubiali, A.; Paties, C.T.; Cavanna, L. Novel BRAF Mutation in Melanoma: A Case Report. Mol. Clin. Oncol. 2018, 8, 460–462. [Google Scholar] [CrossRef] [Green Version]
- Gong, L.-H.; Liu, W.-F.; Niu, X.-H.; Ding, Y. Two Cases of Spindle Cell Tumors with S100 and CD34 Co-Expression Showing Novel RAF1 Fusions. Diagn. Pathol. 2022, 17, 80. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Wang, T.; Wang, H.; Chen, X.; Cao, P.; Ma, X.; Liu, M.; Xu, P.; Bi, H.; et al. Competitive Evolved Sub-Clonal BCR::ABL1 and Novel MSI2::PC Fusion Genes in Myelodysplastic Syndrome with Isolated Del(5q). Hematol. Oncol. 2022, 41, 178–181. [Google Scholar] [CrossRef]
- Jain, P.; Iyer, S.; Straka, J.; Surrey, L.F.; Pogoriler, J.; Han, H.; Smith, T.; Busch, C.; Fox, E.; Li, M.; et al. Discovery and Functional Characterization of the Oncogenicity and Targetability of a Novel NOTCH1-ROS1 Gene Fusion in Pediatric Angiosarcoma. Cold Spring Harb. Mol. Case Stud. 2022, 8, a006222. [Google Scholar] [CrossRef]
- Zong, X.; Kang, Z.; Huang, D.; Zhang, X.; Gao, Y.; Wang, H.; Li, W.; Yan, J. One Novel ACOT7-NPHP4 Fusion Gene Identified in One Patient with Acute Lymphoblastic Leukemia: A Case Report. BMC Med. Genom. 2022, 15, 226. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.-G.; Xia, T.-L.; Fu, J.-C.; Li, T.; Zhong, Q.; Han, F. BCL6-SPECC1L: A Novel Fusion Gene in Nasopharyngeal Carcinoma. Technol. Cancer Res. Treat. 2022, 21, 15330338221139980. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.-H.; Zhang, F.-B.; Yan, H.; Yu, W.-Y.; Chen, M.; Guan, Y.-T. A Novel CDKN1A-JAZF1 Gene Fusion in Low-Grade Endometrial Stromal Sarcoma Arising from Endometriosis in Abdominal Wall Cesarean Section Scar: A Case Report and Literature Review. Taiwan. J. Obstet. Gynecol. 2022, 61, 1082–1085. [Google Scholar] [CrossRef] [PubMed]
- Patton, A.; Speeckaert, A.; Zeltman, M.; Cui, X.; Oghumu, S.; Iwenofu, O.H. A Novel IRF2BP2::CDX2 Gene Fusion in Digital Intravascular Myoepithelioma of Soft Tissue: An Enigma! Genes Chromosomes Cancer 2023, 62, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Guo, S.; Liu, D.; Chu, J.; Li, Y.; Wang, X.; Zhang, X.; Song, C.; Huang, Q. Pediatric Meningioma with a Novel MAML2-YAP1 Fusion Variant: A Case Report and Literature Review. BMC Pediatr. 2022, 22, 694. [Google Scholar] [CrossRef]
- Rai, S.; Singh, M.P.; Srivastava, S. Integrated Analysis Identifies Novel Fusion Transcripts in Laterally Spreading Tumors Suggestive of Distinct Etiology Than Colorectal Cancers. J. Gastrointest. Cancer 2022. [Google Scholar] [CrossRef]
- Georgantzoglou, N.; Shen, G.; Jour, G.; Linos, K. A Case of FN1-Fused Calcified Chondroid Mesenchymal Neoplasm of the Hand with Novel FGFR3 Partner Gene. Genes Chromosomes Cancer 2022, 62, 237–241. [Google Scholar] [CrossRef]
- Lipplaa, A.; Meijer, D.; van de Sande, M.A.J.; Gelderblom, H.; Bovée, J.V.M.G.; Mei, H.; Szuhai, K. A Novel CSF1 Translocation Involving Human Endogenous Retroviral Element (ERV) in a Tenosynovial Giant Cell Tumour. Genes Chromosomes Cancer 2023, 62, 223–230. [Google Scholar] [CrossRef]
- Goto, H.; Koga, Y.; Kohashi, K.; Ono, H.; Takemoto, J.; Matsuura, T.; Tajiri, T.; Ihara, K.; Oda, Y.; Ohga, S. Pancreatoblastoma with a Novel Fusion Gene of IQSEC1-RAF1. Pediatr. Blood Cancer 2023, 70, e30155. [Google Scholar] [CrossRef]
- Hu, X.; Wang, Q.; Tang, M.; Barthel, F.; Amin, S.; Yoshihara, K.; Lang, F.M.; Martinez-Ledesma, E.; Lee, S.H.; Zheng, S.; et al. TumorFusions: An Integrative Resource for Cancer-Associated Transcript Fusions. Nucleic Acids Res. 2018, 46, D1144–D1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, Y.E.; Jang, I.; Kim, S.; Cho, S.; Kim, D.; Kim, K.; Kim, J.; Hwang, J.; Kim, S.; Kim, J.; et al. ChimerDB 4.0: An Updated and Expanded Database of Fusion Genes. Nucleic Acids Res. 2020, 48, D817–D824. [Google Scholar] [CrossRef]
- Kerrien, S.; Aranda, B.; Breuza, L.; Bridge, A.; Broackes-Carter, F.; Chen, C.; Duesbury, M.; Dumousseau, M.; Feuermann, M.; Hinz, U.; et al. The IntAct Molecular Interaction Database in 2012. Nucleic Acids Res. 2011, 40, D841–D846. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Aebersold, R.; Mann, M. Mass-Spectrometric Exploration of Proteome Structure and Function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Meissner, F.; Geddes-McAlister, J.; Mann, M.; Bantscheff, M. The Emerging Role of Mass Spectrometry-Based Proteomics in Drug Discovery. Nat. Rev. Drug Discov. 2022, 21, 637–654. [Google Scholar] [CrossRef]
- Sharifi Tabar, M.; Francis, H.; Yeo, D.; Bailey, C.G.; Rasko, J.E.J. Mapping Oncogenic Protein Interactions for Precision Medicine. Int. J. Cancer 2022, 151, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Stangl, C.; Post, J.B.; van Roosmalen, M.J.; Hami, N.; Verlaan-Klink, I.; Vos, H.R.; van Es, R.M.; Koudijs, M.J.; Voest, E.E.; Snippert, H.J.G.; et al. Diverse BRAF Gene Fusions Confer Resistance to EGFR-Targeted Therapy via Differential Modulation of BRAF Activity. Mol. Cancer Res. MCR 2020, 18, 537–548. [Google Scholar] [CrossRef] [Green Version]
- McCoach, C.E.; Le, A.T.; Gowan, K.; Jones, K.; Schubert, L.; Doak, A.; Estrada-Bernal, A.; Davies, K.D.; Merrick, D.T.; Bunn, P.A.; et al. Resistance Mechanisms to Targeted Therapies in ROS1+ and ALK+ Non-Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 3334–3347. [Google Scholar] [CrossRef] [Green Version]
- Szulzewsky, F.; Holland, E.C.; Vasioukhin, V. YAP1 and Its Fusion Proteins in Cancer Initiation, Progression and Therapeutic Resistance. Dev. Biol. 2021, 475, 205–221. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, Y.; Yang, B.; Shao, X.; Ying, M. Driving the Degradation of Oncofusion Proteins for Targeted Cancer Therapy. Drug Discov. Today 2023, 28, 103584. [Google Scholar] [CrossRef] [PubMed]
- Brien, G.L.; Stegmaier, K.; Armstrong, S.A. Targeting Chromatin Complexes in Fusion Protein-Driven Malignancies. Nat. Rev. Cancer 2019, 19, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Gröschel, S.; Sanders, M.A.; Hoogenboezem, R.; de Wit, E.; Bouwman, B.A.M.; Erpelinck, C.; van der Velden, V.H.J.; Havermans, M.; Avellino, R.; van Lom, K.; et al. A Single Oncogenic Enhancer Rearrangement Causes Concomitant EVI1 and GATA2 Deregulation in Leukemia. Cell 2014, 157, 369–381. [Google Scholar] [CrossRef] [Green Version]
- Miller, P.J.; Hollenbach, A.D. The Oncogenic Fusion Protein Pax3-FKHR Has a Greater Post-Translational Stability Relative to Pax3 during Early Myogenesis. Biochim. Biophys. Acta 2007, 1770, 1450–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Tokheim, C.; Lee, J.D.; Gan, W.; North, B.J.; Liu, X.S.; Pandolfi, P.P.; Wei, W. Genetic Fusions Favor Tumorigenesis through Degron Loss in Oncogenes. Nat. Commun. 2021, 12, 6704. [Google Scholar] [CrossRef]
- Tan, Y.; Wang, X.; Song, H.; Zhang, Y.; Zhang, R.; Li, S.; Jin, W.; Chen, S.; Fang, H.; Chen, Z.; et al. A PML/RARα Direct Target Atlas Redefines Transcriptional Deregulation in Acute Promyelocytic Leukemia. Blood 2021, 137, 1503–1516. [Google Scholar] [CrossRef]
- Kim, P.; Jia, P.; Zhao, Z. Kinase Impact Assessment in the Landscape of Fusion Genes That Retain Kinase Domains: A Pan-Cancer Study. Brief. Bioinform. 2016, 19, 450–460. [Google Scholar] [CrossRef] [Green Version]
- An, J.; Ren, S.; Murphy, S.J.; Dalangood, S.; Chang, C.; Pang, X.; Cui, Y.; Wang, L.; Pan, Y.; Zhang, X.; et al. Truncated ERG Oncoproteins from TMPRSS2-ERG Fusions Are Resistant to SPOP-Mediated Proteasome Degradation. Mol. Cell 2015, 59, 904–916. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Wang, F.; Cao, W.-B.; Lv, X.-X.; Hua, F.; Cui, B.; Yu, J.-J.; Zhang, X.-W.; Shang, S.; Liu, S.-S.; et al. TRIB3 Promotes APL Progression through Stabilization of the Oncoprotein PML-RARα and Inhibition of P53-Mediated Senescence. Cancer Cell 2017, 31, 697–710.e7. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.-C.; Shih, H.-M. The Deubiquitinating Enzyme USP37 Regulates the Oncogenic Fusion Protein PLZF/RARA Stability. Oncogene 2013, 32, 5167–5175. [Google Scholar] [CrossRef] [Green Version]
- Hong, Z.; Zhang, W.; Ding, D.; Huang, Z.; Yan, Y.; Cao, W.; Pan, Y.; Hou, X.; Weroha, S.J.; Karnes, R.J.; et al. DNA Damage Promotes TMPRSS2-ERG Oncoprotein Destruction and Prostate Cancer Suppression via Signaling Converged by GSK3β and WEE1. Mol. Cell 2020, 79, 1008–1023.e4. [Google Scholar] [CrossRef] [PubMed]
- Touriol, C.; Greenland, C.; Lamant, L.; Pulford, K.; Bernard, F.; Rousset, T.; Mason, D.Y.; Delsol, G. Further Demonstration of the Diversity of Chromosomal Changes Involving 2p23 in ALK-Positive Lymphoma: 2 Cases Expressing ALK Kinase Fused to CLTCL (Clathrin Chain Polypeptide-Like). Blood 2000, 95, 3204–3207. [Google Scholar] [CrossRef]
- Schneider, J.L.; Lin, J.J.; Shaw, A.T. ALK-Positive Lung Cancer: A Moving Target. Nat. Cancer 2023, 4, 330–343. [Google Scholar] [CrossRef]
- Qin, Z.; Sun, H.; Yue, M.; Pan, X.; Chen, L.; Feng, X.; Yan, X.; Zhu, X.; Ji, H. Phase Separation of EML4–ALK in Firing Downstream Signaling and Promoting Lung Tumorigenesis. Cell Discov. 2021, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Tulpule, A.; Guan, J.; Neel, D.S.; Allegakoen, H.R.; Lin, Y.P.; Brown, D.; Chou, Y.-T.; Heslin, A.; Chatterjee, N.; Perati, S.; et al. Kinase-Mediated RAS Signaling via Membraneless Cytoplasmic Protein Granules. Cell 2021, 184, 2649–2664.e18. [Google Scholar] [CrossRef]
- Sampson, J.; Richards, M.W.; Choi, J.; Fry, A.M.; Bayliss, R. Phase-separated Foci of EML4-ALK Facilitate Signalling and Depend upon an Active Kinase Conformation. EMBO Rep. 2021, 22, e53693. [Google Scholar] [CrossRef]
- Okamura, R.; Boichard, A.; Kato, S.; Sicklick, J.K.; Bazhenova, L.; Kurzrock, R. Analysis of NTRK Alterations in Pan-Cancer Adult and Pediatric Malignancies: Implications for NTRK-Targeted Therapeutics. JCO Precis. Oncol. 2018, 2018, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cremolini, C.; Morano, F.; Moretto, R.; Berenato, R.; Tamborini, E.; Perrone, F.; Rossini, D.; Gloghini, A.; Busico, A.; Zucchelli, G.; et al. Negative Hyper-Selection of Metastatic Colorectal Cancer Patients for Anti-EGFR Monoclonal Antibodies: The PRESSING Case–Control Study. Ann. Oncol. 2017, 28, 3009–3014. [Google Scholar] [CrossRef]
- Rolle, A.-F.L.; Klempner, S.J.; Garrett, C.R.; Seery, T.; Sanford, E.M.; Balasubramanian, S.; Ross, J.S.; Stephens, P.J.; Miller, V.A.; Ali, S.M.; et al. Identification and Characterization of RET Fusions in Advanced Colorectal Cancer. Oncotarget 2015, 6, 28929–28937. [Google Scholar] [CrossRef] [Green Version]
- Declercq, J.; Van Dyck, F.; Van Damme, B.; Van de Ven, W.J.M. Upregulation of Igf and Wnt Signalling Associated Genes in Pleomorphic Adenomas of the Salivary Glands in PLAG1 Transgenic Mice. Int. J. Oncol. 2008, 32, 1041–1047. [Google Scholar] [CrossRef] [Green Version]
- Schram, A.M.; Chang, M.T.; Jonsson, P.; Drilon, A. Fusions in Solid Tumours: Diagnostic Strategies, Targeted Therapy, and Acquired Resistance. Nat. Rev. Clin. Oncol. 2017, 14, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Stransky, N.; Cerami, E.; Schalm, S.; Kim, J.L.; Lengauer, C. The Landscape of Kinase Fusions in Cancer. Nat. Commun. 2014, 5, 4846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amarante-Mendes, G.P.; Rana, A.; Datoguia, T.S.; Hamerschlak, N.; Brumatti, G. BCR-ABL1 Tyrosine Kinase Complex Signaling Transduction: Challenges to Overcome Resistance in Chronic Myeloid Leukemia. Pharmaceutics 2022, 14, 215. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.M.; Hediyeh-zadeh, S.; Sadras, T.; Huckstep, H.; Sandow, J.J.; Bartolo, R.C.; Kosasih, H.J.; Davidson, N.M.; Schmidt, B.; Bjelosevic, S.; et al. SFPQ-ABL1 and BCR-ABL1 Use Different Signaling Networks to Drive B-Cell Acute Lymphoblastic Leukemia. Blood Adv. 2022, 6, 2373–2387. [Google Scholar] [CrossRef]
- Latysheva, N.S.; Oates, M.E.; Maddox, L.; Flock, T.; Gough, J.; Buljan, M.; Weatheritt, R.J.; Babu, M.M. Molecular Principles of Gene Fusion Mediated Rewiring of Protein Interaction Networks in Cancer. Mol. Cell 2016, 63, 579–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baserga, A.; Gastoldi, G.L. The Philadelptia Chromosome. Biomedicine 1973, 18, 89–94. [Google Scholar]
- Stam, K.; Heisterkamp, N.; Grosveld, G.; de Klein, A.; Verma, R.S.; Coleman, M.; Dosik, H.; Groffen, J. Evidence of a New Chimeric Bcr/c-Abl MRNA in Patients with Chronic Myelocytic Leukemia and the Philadelphia Chromosome. N. Engl. J. Med. 1985, 313, 1429–1433. [Google Scholar] [CrossRef]
- Naldini, L.; Stacchini, A.; Cirillo, D.M.; Aglietta, M.; Gavosto, F.; Comoglio, P.M. Phosphotyrosine Antibodies Identify the P210c-Abl Tyrosine Kinase and Proteins Phosphorylated on Tyrosine in Human Chronic Myelogenous Leukemia Cells. Mol. Cell. Biol. 1986, 6, 1803–1811. [Google Scholar]
- Adnan-Awad, S.; Kim, D.; Hohtari, H.; Javarappa, K.K.; Brandstoetter, T.; Mayer, I.; Potdar, S.; Heckman, C.A.; Kytölä, S.; Porkka, K.; et al. Characterization of P190-Bcr-Abl Chronic Myeloid Leukemia Reveals Specific Signaling Pathways and Therapeutic Targets. Leukemia 2021, 35, 1964–1975. [Google Scholar] [CrossRef]
- McWhirter, J.R.; Wang, J.Y. An Actin-Binding Function Contributes to Transformation by the Bcr-Abl Oncoprotein of Philadelphia Chromosome-Positive Human Leukemias. EMBO J. 1993, 12, 1533–1546. [Google Scholar] [CrossRef]
- Aloisi, A.; Di Gregorio, S.; Stagno, F.; Guglielmo, P.; Mannino, F.; Sormani, M.P.; Bruzzi, P.; Gambacorti-Passerini, C.; Saglio, G.; Venuta, S.; et al. BCR-ABL Nuclear Entrapment Kills Human CML Cells: Ex Vivo Study on 35 Patients with the Combination of Imatinib Mesylate and Leptomycin B. Blood 2006, 107, 1591–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrà, G.; Russo, I.; Guerrasio, A.; Morotti, A. Nuclear-Cytoplasmic Shuttling in Chronic Myeloid Leukemia: Implications in Leukemia Maintenance and Therapy. Cells 2019, 8, 1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cilloni, D.; Saglio, G. Molecular Pathways: BCR-ABL. Clin. Cancer Res. 2012, 18, 930–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, K.; Gong, D.; He, C.; Xiao, M.; Zhang, M.; Huang, W. Targeted Therapy Using Larotrectinib and Venetoclax for the Relapsed/Refractory T-Cell Acute Lymphoblastic Leukemia Harboring a Cryptic ETV6-NTRK3 Fusion. Mol. Carcinog. 2023, 62, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Zhang, Z.; He, X.; Chen, M.; Pang, X.; Chen, C.; Du, T.; Zhang, H. Primary Inflammatory Myofibroblastic Tumour of the Liver: A Clinicopathological and Genetic Study Including a Subset with ETV6::NTRK3 Fusion. Histopathology 2023, 82, 925–936. [Google Scholar] [CrossRef]
- Jiang, H. A Novel ETV6-NTRK3 Gene Fusion in Primary Renal Fibrosarcoma. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 4705–4708. [Google Scholar] [CrossRef]
- Knezevich, S.R.; McFadden, D.E.; Tao, W.; Lim, J.F.; Sorensen, P.H. A Novel ETV6-NTRK3 Gene Fusion in Congenital Fibrosarcoma. Nat. Genet. 1998, 18, 184–187. [Google Scholar] [CrossRef]
- Lannon, C.L.; Sorensen, P.H.B. ETV6-NTRK3: A Chimeric Protein Tyrosine Kinase with Transformation Activity in Multiple Cell Lineages. Semin. Cancer Biol. 2005, 15, 215–223. [Google Scholar] [CrossRef]
- Tognon, C.; Knezevich, S.R.; Huntsman, D.; Roskelley, C.D.; Melnyk, N.; Mathers, J.A.; Becker, L.; Carneiro, F.; MacPherson, N.; Horsman, D.; et al. Expression of the ETV6-NTRK3 Gene Fusion as a Primary Event in Human Secretory Breast Carcinoma. Cancer Cell 2002, 2, 367–376. [Google Scholar] [CrossRef] [Green Version]
- ETV6-NTRK3 Transformation Requires Insulin-like Growth Factor 1 Receptor Signaling and Is Associated with Constitutive IRS-1 Tyrosine Phosphorylation—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/12173038/ (accessed on 3 May 2023).
- Tognon, C.E.; Somasiri, A.M.; Evdokimova, V.E.; Trigo, G.; Uy, E.E.; Melnyk, N.; Carboni, J.M.; Gottardis, M.M.; Roskelley, C.D.; Pollak, M.; et al. ETV6-NTRK3-Mediated Breast Epithelial Cell Transformation Is Blocked by Targeting the IGF1R Signaling Pathway. Cancer Res. 2011, 71, 1060–1070. [Google Scholar] [CrossRef] [Green Version]
- Bai, R.Y.; Dieter, P.; Peschel, C.; Morris, S.W.; Duyster, J. Nucleophosmin-Anaplastic Lymphoma Kinase of Large-Cell Anaplastic Lymphoma Is a Constitutively Active Tyrosine Kinase That Utilizes Phospholipase C-Gamma to Mediate Its Mitogenicity. Mol. Cell. Biol. 1998, 18, 6951–6961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duyster, J.; Bai, R.Y.; Morris, S.W. Translocations Involving Anaplastic Lymphoma Kinase (ALK). Oncogene 2001, 20, 5623–5637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buetti-Dinh, A.; O’Hare, T.; Friedman, R. Sensitivity Analysis of the NPM-ALK Signalling Network Reveals Important Pathways for Anaplastic Large Cell Lymphoma Combination Therapy. PLoS ONE 2016, 11, e0163011. [Google Scholar] [CrossRef] [Green Version]
- Chiarle, R.; Simmons, W.J.; Cai, H.; Dhall, G.; Zamo, A.; Raz, R.; Karras, J.G.; Levy, D.E.; Inghirami, G. Stat3 Is Required for ALK-Mediated Lymphomagenesis and Provides a Possible Therapeutic Target. Nat. Med. 2005, 11, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Cussac, D.; Greenland, C.; Roche, S.; Bai, R.-Y.; Duyster, J.; Morris, S.W.; Delsol, G.; Allouche, M.; Payrastre, B. Nucleophosmin-Anaplastic Lymphoma Kinase of Anaplastic Large-Cell Lymphoma Recruits, Activates, and Uses Pp60c-Src to Mediate Its Mitogenicity. Blood 2004, 103, 1464–1471. [Google Scholar] [CrossRef]
- Khosh Kish, E.; Choudhry, M.; Gamallat, Y.; Buharideen, S.M.; Bismar, T.A. The Expression of Proto-Oncogene ETS-Related Gene (ERG) Plays a Central Role in the Oncogenic Mechanism Involved in the Development and Progression of Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 4772. [Google Scholar] [CrossRef]
- Zoma, M.; Curti, L.; Shinde, D.; Albino, D.; Mitra, A.; Sgrignani, J.; Mapelli, S.N.; Sandrini, G.; Civenni, G.; Merulla, J.; et al. EZH2-Induced Lysine K362 Methylation Enhances TMPRSS2-ERG Oncogenic Activity in Prostate Cancer. Nat. Commun. 2021, 12, 4147. [Google Scholar] [CrossRef]
- Wu, L.; Zhao, J.C.; Kim, J.; Jin, H.-J.; Wang, C.-Y.; Yu, J. ERG Is a Critical Regulator of Wnt/LEF1 Signaling in Prostate Cancer. Cancer Res. 2013, 73, 6068–6079. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, Z.; Zielske, S.P.; Ibrahim, K.G.; Cackowski, F.C. Wnt and β-Catenin Signaling in the Bone Metastasis of Prostate Cancer. Life 2021, 11, 1099. [Google Scholar] [CrossRef]
- Abou-Ouf, H.; Assem, H.; Ghosh, S.; Karnes, R.J.; Stoletov, K.; Palanisamy, N.; Lewis, J.D.; Bismar, T.A. High Serine-Arginine Protein Kinase 1 Expression with PTEN Loss Defines Aggressive Phenotype of Prostate Cancer Associated with Lethal Outcome and Decreased Overall Survival. Eur. Urol. Open Sci. 2021, 23, 1–8. [Google Scholar] [CrossRef]
- Ruiz Moreno, J.M.; Medrano López, M.; Rodriguez Prats, J.L. Cavernous angioma of the retina: Therapeutic approach. J. Fr. Ophtalmol. 1987, 10, 731–733. [Google Scholar] [PubMed]
- Meisel Sharon, S.; Pozniak, Y.; Geiger, T.; Werner, H. TMPRSS2-ERG Fusion Protein Regulates Insulin-like Growth Factor-1 Receptor (IGF1R) Gene Expression in Prostate Cancer: Involvement of Transcription Factor Sp1. Oncotarget 2016, 7, 51375–51392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate Cancer. Nat. Rev. Dis. Primer 2021, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lu, M.; Qin, Y.; Gao, W.; Tao, L.; Su, W.; Zhong, J. Neoantigen: A New Breakthrough in Tumor Immunotherapy. Front. Immunol. 2021, 12, 672356. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Shen, G.; Gao, W.; Huang, Z.; Huang, C.; Fu, L. Neoantigens: Promising Targets for Cancer Therapy. Signal Transduct. Target. Ther. 2023, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wu, J.; Chen, S.; Zhou, Z. Shared Neoantigens: Ideal Targets for off-the-Shelf Cancer Immunotherapy. Pharmacogenomics 2020, 21, 637–645. [Google Scholar] [CrossRef]
- Smith, B.; Kennedy, J.W. Thrombolysis in the Treatment of Acute Transmural Myocardial Infarction. Ann. Intern. Med. 1987, 106, 414–420. [Google Scholar] [CrossRef]
- Liu, Y.; Klein, J.; Bajpai, R.; Dong, L.; Tran, Q.; Kolekar, P.; Smith, J.L.; Ries, R.E.; Huang, B.J.; Wang, Y.-C.; et al. Etiology of Oncogenic Fusions in 5,190 Childhood Cancers and Its Clinical and Therapeutic Implication. Nat. Commun. 2023, 14, 1739. [Google Scholar] [CrossRef]
- Wei, Z.; Zhou, C.; Zhang, Z.; Guan, M.; Zhang, C.; Liu, Z.; Liu, Q. The Landscape of Tumor Fusion Neoantigens: A Pan-Cancer Analysis. iScience 2019, 21, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Shi, T.; Song, X.; Liu, B.; Wei, J. Gene Fusion Neoantigens: Emerging Targets for Cancer Immunotherapy. Cancer Lett. 2021, 506, 45–54. [Google Scholar] [CrossRef]
- Lin, Y.-Y.; Gawronski, A.; Hach, F.; Li, S.; Numanagić, I.; Sarrafi, I.; Mishra, S.; McPherson, A.; Collins, C.C.; Radovich, M.; et al. Computational Identification of Micro-Structural Variations and Their Proteogenomic Consequences in Cancer. Bioinformatics 2018, 34, 1672–1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howarth, K.D.; Mirza, T.; Cooke, S.L.; Chin, S.-F.; Pole, J.C.; Turro, E.; Eldridge, M.D.; Garcia, R.M.; Rueda, O.M.; Boursnell, C.; et al. NRG1 Fusions in Breast Cancer. Breast Cancer Res. BCR 2021, 23, 3. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Koh, H.H.; Chang, E.S.; Choi, J.; Song, J.-Y.; Lee, M.-S.; Choi, Y.-L. Unearthing Novel Fusions as Therapeutic Targets in Solid Tumors Using Targeted RNA Sequencing. Front. Oncol. 2022, 12, 892918. [Google Scholar] [CrossRef] [PubMed]
- Mackall, C.L.; Rhee, E.H.; Read, E.J.; Khuu, H.M.; Leitman, S.F.; Bernstein, D.; Tesso, M.; Long, L.M.; Grindler, D.; Merino, M.; et al. A Pilot Study of Consolidative Immunotherapy in Patients with High-Risk Pediatric Sarcomas. Clin. Cancer Res. 2008, 14, 4850–4858. [Google Scholar] [CrossRef] [Green Version]
- Biernacki, M.A.; Foster, K.A.; Woodward, K.B.; Coon, M.E.; Cummings, C.; Cunningham, T.M.; Dossa, R.G.; Brault, M.; Stokke, J.; Olsen, T.M.; et al. CBFB-MYH11 Fusion Neoantigen Enables T Cell Recognition and Killing of Acute Myeloid Leukemia. J. Clin. Investig. 2020, 130, 5127–5141. [Google Scholar] [CrossRef]
- Yang, W.; Lee, K.-W.; Srivastava, R.M.; Kuo, F.; Krishna, C.; Chowell, D.; Makarov, V.; Hoen, D.; Dalin, M.G.; Wexler, L.; et al. Immunogenic Neoantigens Derived from Gene Fusions Stimulate T Cell Responses. Nat. Med. 2019, 25, 767–775. [Google Scholar] [CrossRef]
- Kalina, J.L.; Neilson, D.S.; Lin, Y.-Y.; Hamilton, P.T.; Comber, A.P.; Loy, E.M.H.; Sahinalp, S.C.; Collins, C.C.; Hach, F.; Lum, J.J. Mutational Analysis of Gene Fusions Predicts Novel MHC Class I-Restricted T-Cell Epitopes and Immune Signatures in a Subset of Prostate Cancer. Clin. Cancer Res. 2017, 23, 7596–7607. [Google Scholar] [CrossRef] [Green Version]
- Pavet, V.; Portal, M.M.; Moulin, J.C.; Herbrecht, R.; Gronemeyer, H. Towards Novel Paradigms for Cancer Therapy. Oncogene 2011, 30, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Powers, M.P. The Ever-Changing World of Gene Fusions in Cancer: A Secondary Gene Fusion and Progression. Oncogene 2019, 38, 7197–7199. [Google Scholar] [CrossRef]
- Wong, S.; Witte, O.N. The BCR-ABL Story: Bench to Bedside and Back. Annu. Rev. Immunol. 2004, 22, 247–306. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Aubin, S.M.J.; Siddiqui, J.; Lonigro, R.J.; Sefton-Miller, L.; Miick, S.; Williamsen, S.; Hodge, P.; Meinke, J.; Blase, A.; et al. Urine TMPRSS2:ERG Fusion Transcript Stratifies Prostate Cancer Risk in Men with Elevated Serum PSA. Sci. Transl. Med. 2011, 3, 94ra72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villani, A.; Davidson, S.; Kanwar, N.; Lo, W.W.; Li, Y.; Cohen-Gogo, S.; Fuligni, F.; Edward, L.-M.; Light, N.; Layeghifard, M.; et al. The Clinical Utility of Integrative Genomics in Childhood Cancer Extends beyond Targetable Mutations. Nat. Cancer 2022, 4, 203–221. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.; Ibn-Salem, J.; Sorn, P.; Suchan, M.; Holtsträter, C.; Lahrmann, U.; Vogler, I.; Schmoldt, K.; Lang, F.; Schrörs, B.; et al. Accurate Detection of Tumor-Specific Gene Fusions Reveals Strongly Immunogenic Personal Neo-Antigens. Nat. Biotechnol. 2022, 40, 1276–1284. [Google Scholar] [CrossRef]
- Larkin, R.; Hermsen, M.A.; London, N.R., Jr. Translocations and Gene Fusions in Sinonasal Malignancies. Curr. Oncol. Rep. 2023, 25, 269–278. [Google Scholar] [CrossRef]
- Nikanjam, M.; Okamura, R.; Barkauskas, D.A.; Kurzrock, R. Targeting Fusions for Improved Outcomes in Oncology Treatment. Cancer 2020, 126, 1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudzinski, E.R.; Drilon, A.; Moore, A.; Spinosa, S.; Willi, M.; Laetsch, T.W. Testing Methods to Diagnose TRK Fusion Cancer: A Plain Language Summary and Patient Perspective. Future Oncol. Lond. Engl. 2023, 18, 4141–4151. [Google Scholar] [CrossRef]
- Li, W.; Guo, L.; Liu, Y.; Dong, L.; Yang, L.; Chen, L.; Liu, K.; Shao, Y.; Ying, J. Potential Unreliability of Uncommon ALK, ROS1, and RET Genomic Breakpoints in Predicting the Efficacy of Targeted Therapy in NSCLC. J. Thorac. Oncol. 2021, 16, 404–418. [Google Scholar] [CrossRef]
- Dankner, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF Alterations in Cancer: New Rational Therapeutic Strategies for Actionable Mutations. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef]
- Zhang, L.; Zheng, L.; Yang, Q.; Sun, J. The Evolution of BRAF Activation in Non-Small-Cell Lung Cancer. Front. Oncol. 2022, 12, 882940. [Google Scholar] [CrossRef]
- Vojnic, M.; Kubota, D.; Kurzatkowski, C.; Offin, M.; Suzawa, K.; Benayed, R.; Schoenfeld, A.J.; Plodkowski, A.J.; Poirier, J.T.; Rudin, C.M.; et al. Acquired BRAF Rearrangements Induce Secondary Resistance to EGFR Therapy in EGFR-Mutated Lung Cancers. J. Thorac. Oncol. 2019, 14, 802–815. [Google Scholar] [CrossRef]
- Servetto, A.; Esposito, D.; Ferrara, R.; Signorelli, D.; Belli, S.; Napolitano, F.; Santaniello, A.; Ciciola, P.; Formisano, L.; Bianco, R. RET Rearrangements in Non-Small Cell Lung Cancer: Evolving Treatment Landscape and Future Challenges. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188810. [Google Scholar] [CrossRef] [PubMed]
- Barnes, E.J.; Eide, C.A.; Kaempf, A.; Bottomly, D.; Romine, K.A.; Wilmot, B.; Saunders, D.; McWeeney, S.K.; Tognon, C.E.; Druker, B.J. Secondary Fusion Proteins as a Mechanism of BCR::ABL1 Kinase-Independent Resistance in Chronic Myeloid Leukaemia. Br. J. Haematol. 2023, 200, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Liu, Y.; Liang, Z.; Wang, W.; Qin, T.; Liu, S.V.; Um, S.-W.; Luo, F.; Liu, J. Classical ALK G1202R Resistance Mutation Was Identified in a Lung Adenocarcinoma Patient with Rare LOC388942-ALK Fusion after Sequential Treatment with ALK-TKIs and Anlotinib: A Case Report. Ann. Transl. Med. 2022, 10, 1180. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Oxnard, G.R.; Cohen, E.F.; Mahadevan, N.R.; Alessi, J.V.; Hung, Y.P.; Bertram, A.A.; Heppner, D.E.; Ribeiro, M.F.; Sacardo, K.P.; et al. Genomic and Biological Study of Fusion Genes as Resistance Mechanisms to EGFR Inhibitors. Nat. Commun. 2022, 13, 5614. [Google Scholar] [CrossRef] [PubMed]
- Jonna, S.; Feldman, R.A.; Swensen, J.; Gatalica, Z.; Korn, W.M.; Borghaei, H.; Ma, P.C.; Nieva, J.J.; Spira, A.I.; Vanderwalde, A.M.; et al. Detection of NRG1 Gene Fusions in Solid Tumors. Clin. Cancer Res. 2019, 25, 4966–4972. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.H.; Lee, D.; Hong, D.W.; Hong, S.H.; Hwang, J.-A.; Lee, B.I.; You, H.J.; Lee, G.K.; Kim, I.-H.; Lee, Y.-S.; et al. Oncogenic Function and Clinical Implications of SLC3A2-NRG1 Fusion in Invasive Mucinous Adenocarcinoma of the Lung. Oncotarget 2016, 7, 69450–69465. [Google Scholar] [CrossRef] [Green Version]
- Nam, R.K.; Sugar, L.; Wang, Z.; Yang, W.; Kitching, R.; Klotz, L.H.; Venkateswaran, V.; Narod, S.A.; Seth, A. Expression of TMPRSS2:ERG Gene Fusion in Prostate Cancer Cells Is an Important Prognostic Factor for Cancer Progression. Cancer Biol. Ther. 2007, 6, 40–45. [Google Scholar] [CrossRef] [Green Version]
- Kumar-Sinha, C.; Tomlins, S.A.; Chinnaiyan, A.M. Recurrent Gene Fusions in Prostate Cancer. Nat. Rev. Cancer 2008, 8, 497–511. [Google Scholar] [CrossRef] [Green Version]
- Demichelis, F.; Fall, K.; Perner, S.; Andrén, O.; Schmidt, F.; Setlur, S.R.; Hoshida, Y.; Mosquera, J.-M.; Pawitan, Y.; Lee, C.; et al. TMPRSS2:ERG Gene Fusion Associated with Lethal Prostate Cancer in a Watchful Waiting Cohort. Oncogene 2007, 26, 4596–4599. [Google Scholar] [CrossRef] [Green Version]
- Hägglöf, C.; Hammarsten, P.; Strömvall, K.; Egevad, L.; Josefsson, A.; Stattin, P.; Granfors, T.; Bergh, A. TMPRSS2-ERG Expression Predicts Prostate Cancer Survival and Associates with Stromal Biomarkers. PLoS ONE 2014, 9, e86824. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Chen, H. Predictive Significance of TMRPSS2-ERG Fusion in Prostate Cancer: A Meta-Analysis. Cancer Cell Int. 2018, 18, 177. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wan, R.; Guo, L.; Chang, G.; Jiang, D.; Meng, L.; Ying, J. Reliability Analysis of Exonic-Breakpoint Fusions Identified by DNA Sequencing for Predicting the Efficacy of Targeted Therapy in Non-Small Cell Lung Cancer. BMC Med. 2022, 20, 160. [Google Scholar] [CrossRef]
- Pagani, F.; Randon, G.; Guarini, V.; Raimondi, A.; Prisciandaro, M.; Lobefaro, R.; Di Bartolomeo, M.; Sozzi, G.; de Braud, F.; Gasparini, P.; et al. The Landscape of Actionable Gene Fusions in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 5319. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; El-Bahrawy, M. Gene Fusions in Tumourigenesis with Particular Reference to Ovarian Cancer. J. Med. Genet. 2021, 58, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Heyer, E.E.; Deveson, I.W.; Wooi, D.; Selinger, C.I.; Lyons, R.J.; Hayes, V.M.; O’Toole, S.A.; Ballinger, M.L.; Gill, D.; Thomas, D.M.; et al. Diagnosis of Fusion Genes Using Targeted RNA Sequencing. Nat. Commun. 2019, 10, 1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heydt, C.; Wölwer, C.B.; Velazquez Camacho, O.; Wagener-Ryczek, S.; Pappesch, R.; Siemanowski, J.; Rehker, J.; Haller, F.; Agaimy, A.; Worm, K.; et al. Detection of Gene Fusions Using Targeted Next-Generation Sequencing: A Comparative Evaluation. BMC Med. Genom. 2021, 14, 62. [Google Scholar] [CrossRef] [PubMed]
- Carter, T.C.; He, M.M. Challenges of Identifying Clinically Actionable Genetic Variants for Precision Medicine. J. Healthc. Eng. 2016, 2016, 3617572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budczies, J.; Kirchner, M.; Kluck, K.; Kazdal, D.; Glade, J.; Allgäuer, M.; Kriegsmann, M.; Heußel, C.-P.; Herth, F.J.; Winter, H.; et al. Deciphering the Immunosuppressive Tumor Microenvironment in ALK- and EGFR-Positive Lung Adenocarcinoma. Cancer Immunol. Immunother. 2022, 71, 251–265. [Google Scholar] [CrossRef]
- Galkin, A.V.; Melnick, J.S.; Kim, S.; Hood, T.L.; Li, N.; Li, L.; Xia, G.; Steensma, R.; Chopiuk, G.; Jiang, J.; et al. Identification of NVP-TAE684, a Potent, Selective, and Efficacious Inhibitor of NPM-ALK. Proc. Natl. Acad. Sci. USA 2007, 104, 270–275. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Baek, J.H.; Sun, J.-M.; Min, Y.J.; Cho, E.K.; Cho, B.C.; Kim, J.-H.; Ahn, M.-J.; Park, K. Efficacy of EGFR Tyrosine Kinase Inhibitors in Patients with EGFR-Mutated Non-Small Cell Lung Cancer except Both Exon 19 Deletion and Exon 21 L858R: A Retrospective Analysis in Korea. Lung Cancer 2015, 87, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and Safety of a Specific Inhibitor of the BCR-ABL Tyrosine Kinase in Chronic Myeloid Leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pemovska, T.; Johnson, E.; Kontro, M.; Repasky, G.A.; Chen, J.; Wells, P.; Cronin, C.N.; McTigue, M.; Kallioniemi, O.; Porkka, K.; et al. Axitinib Effectively Inhibits BCR-ABL1(T315I) with a Distinct Binding Conformation. Nature 2015, 519, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal Evolution in Cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase Drug Discovery 20 Years after Imatinib: Progress and Future Directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef]
- Meador, C.B.; Hata, A.N. Acquired Resistance to Targeted Therapies in NSCLC: Updates and Evolving Insights. Pharmacol. Ther. 2020, 210, 107522. [Google Scholar] [CrossRef]
- Viossat, Y.; Noble, R. A Theoretical Analysis of Tumour Containment. Nat. Ecol. Evol. 2021, 5, 826–835. [Google Scholar] [CrossRef]
- Naik, R.R.; Shakya, A.K. Exploring the Chemotherapeutic Potential of Currently Used Kinase Inhibitors: An Update. Front. Pharmacol. 2023, 13, 1064472. [Google Scholar] [CrossRef]
- Qin, S.; Li, A.; Yi, M.; Yu, S.; Zhang, M.; Wu, K. Recent Advances on Anti-Angiogenesis Receptor Tyrosine Kinase Inhibitors in Cancer Therapy. J. Hematol. Oncol. 2019, 12, 27. [Google Scholar] [CrossRef] [Green Version]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jérusalem, G.; Longuespée, R.; Sounni, N.E. Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef] [Green Version]
- Murumägi, A.; Ungureanu, D.; Arjama, M.; Bützow, R.; Lohi, J.; Sariola, H.; Kanerva, J.; Koskenvuo, M.; Kallioniemi, O. STRN-ALK Rearranged Pediatric Malignant Peritoneal Mesothelioma—Functional Testing of 527 Cancer Drugs in Patient-Derived Cancer Cells. Transl. Oncol. 2021, 14, 101027. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 Signalling Axis in Cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- A Phase I Clinical Trial of Ruxolitinib in Combination with Nilotinib in Chronic Myeloid Leukemia Patients with Molecular Evidence of Disease—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/30340199/ (accessed on 3 May 2023).
- Chirnomas, D.; Hornberger, K.R.; Crews, C.M. Protein Degraders Enter the Clinic—A New Approach to Cancer Therapy. Nat. Rev. Clin. Oncol. 2023, 20, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zhou, Y.; Sun, D.; Yang, Y.; Liu, Y.; Li, X.; Li, H.; Chen, L. PROTACs: New Method to Degrade Transcription Regulating Proteins. Eur. J. Med. Chem. 2020, 207, 112698. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhao, J.; Zhong, K.; Tong, A.; Jia, D. Targeted Protein Degradation: Mechanisms, Strategies and Application. Signal Transduct. Target. Ther. 2022, 7, 113. [Google Scholar] [CrossRef]
- Lai, A.C.; Toure, M.; Hellerschmied, D.; Salami, J.; Jaime-Figueroa, S.; Ko, E.; Hines, J.; Crews, C.M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem. Int. Ed. 2016, 55, 807–810. [Google Scholar] [CrossRef] [Green Version]
- Burslem, G.M.; Schultz, A.R.; Bondeson, D.P.; Eide, C.A.; Savage Stevens, S.L.; Druker, B.J.; Crews, C.M. Targeting BCR-ABL1 in Chronic Myeloid Leukemia by PROTAC-Mediated Targeted Protein Degradation. Cancer Res. 2019, 79, 4744–4753. [Google Scholar] [CrossRef]
- Kang, C.H.; Lee, D.H.; Lee, C.O.; Du Ha, J.; Park, C.H.; Hwang, J.Y. Induced Protein Degradation of Anaplastic Lymphoma Kinase (ALK) by Proteolysis Targeting Chimera (PROTAC). Biochem. Biophys. Res. Commun. 2018, 505, 542–547. [Google Scholar] [CrossRef]
- Powell, C.E.; Gao, Y.; Tan, L.; Donovan, K.A.; Nowak, R.P.; Loehr, A.; Bahcall, M.; Fischer, E.S.; Jänne, P.A.; George, R.E.; et al. Chemically Induced Degradation of Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2018, 61, 4249–4255. [Google Scholar] [CrossRef]
- Song, X.; Zhong, H.; Qu, X.; Yang, L.; Jiang, B. Two Novel Strategies to Overcome the Resistance to ALK Tyrosine Kinase Inhibitor Drugs: Macrocyclic Inhibitors and Proteolysis-Targeting Chimeras. MedComm 2021, 2, 341–350. [Google Scholar] [CrossRef]
- Sun, N.; Ren, C.; Kong, Y.; Zhong, H.; Chen, J.; Li, Y.; Zhang, J.; Zhou, Y.; Qiu, X.; Lin, H.; et al. Development of a Brigatinib Degrader (SIAIS117) as a Potential Treatment for ALK Positive Cancer Resistance. Eur. J. Med. Chem. 2020, 193, 112190. [Google Scholar] [CrossRef] [PubMed]
- Mayor-Ruiz, C.; Bauer, S.; Brand, M.; Kozicka, Z.; Siklos, M.; Imrichova, H.; Kaltheuner, I.H.; Hahn, E.; Seiler, K.; Koren, A.; et al. Rational Discovery of Molecular Glue Degraders via Scalable Chemical Profiling. Nat. Chem. Biol. 2020, 16, 1199–1207. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salokas, K.; Dashi, G.; Varjosalo, M. Decoding Oncofusions: Unveiling Mechanisms, Clinical Impact, and Prospects for Personalized Cancer Therapies. Cancers 2023, 15, 3678. https://doi.org/10.3390/cancers15143678
Salokas K, Dashi G, Varjosalo M. Decoding Oncofusions: Unveiling Mechanisms, Clinical Impact, and Prospects for Personalized Cancer Therapies. Cancers. 2023; 15(14):3678. https://doi.org/10.3390/cancers15143678
Chicago/Turabian StyleSalokas, Kari, Giovanna Dashi, and Markku Varjosalo. 2023. "Decoding Oncofusions: Unveiling Mechanisms, Clinical Impact, and Prospects for Personalized Cancer Therapies" Cancers 15, no. 14: 3678. https://doi.org/10.3390/cancers15143678
APA StyleSalokas, K., Dashi, G., & Varjosalo, M. (2023). Decoding Oncofusions: Unveiling Mechanisms, Clinical Impact, and Prospects for Personalized Cancer Therapies. Cancers, 15(14), 3678. https://doi.org/10.3390/cancers15143678