
Regulation of Kinase Signaling Pathways by α6β4-Integrins and Plectin in Prostate Cancer


Abstract
Simple Summary
Abstract

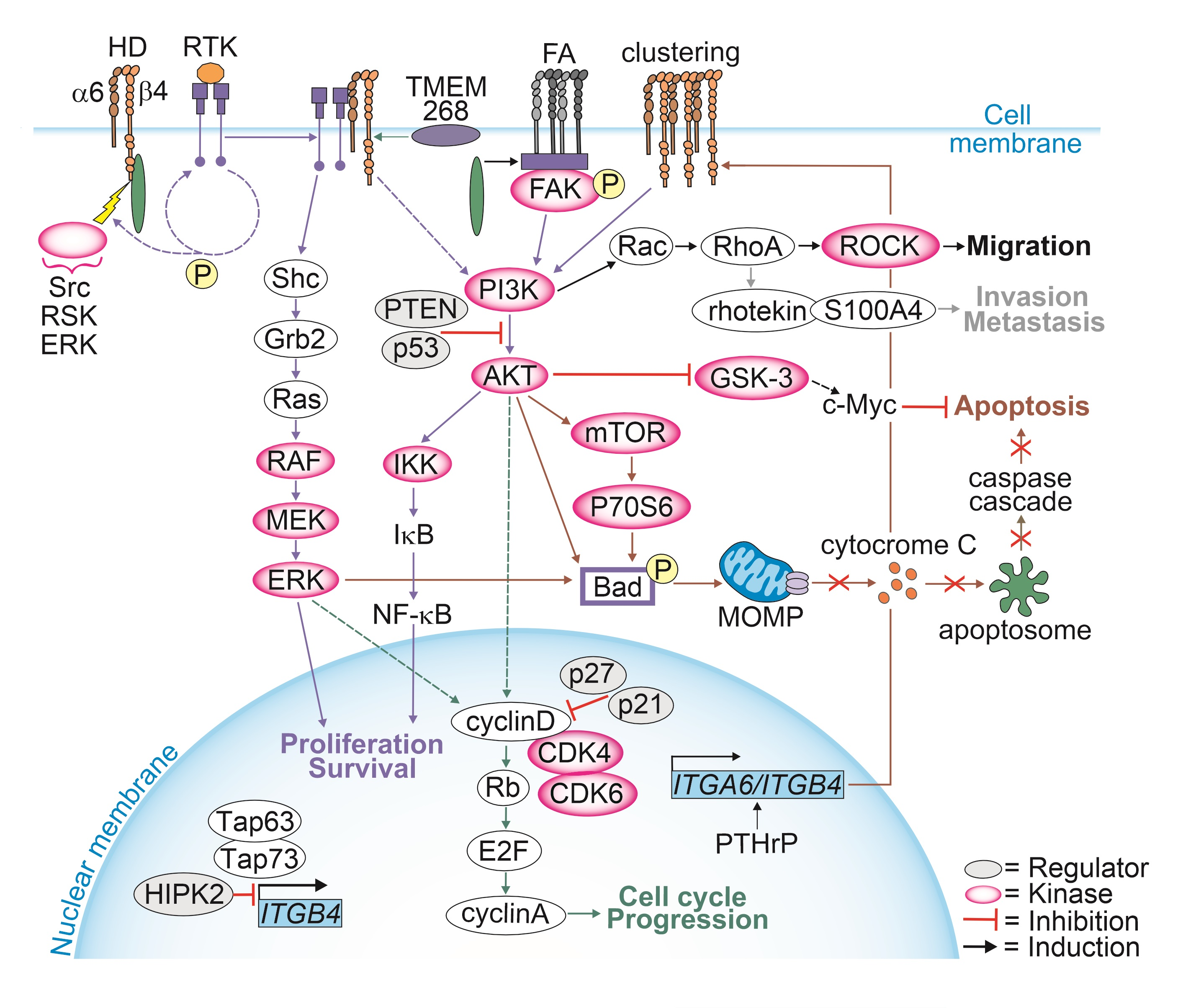{kind=link}
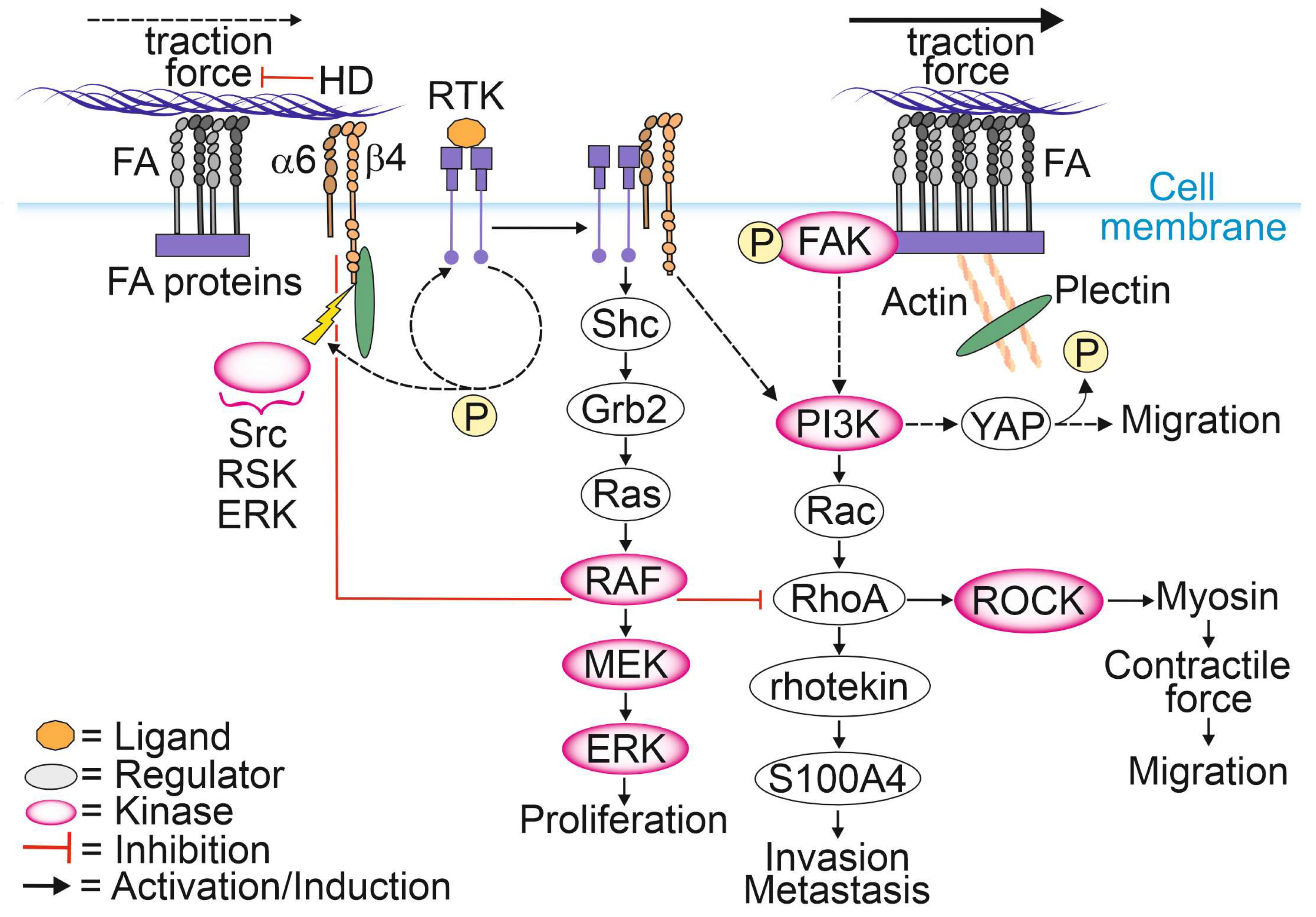{kind=link}
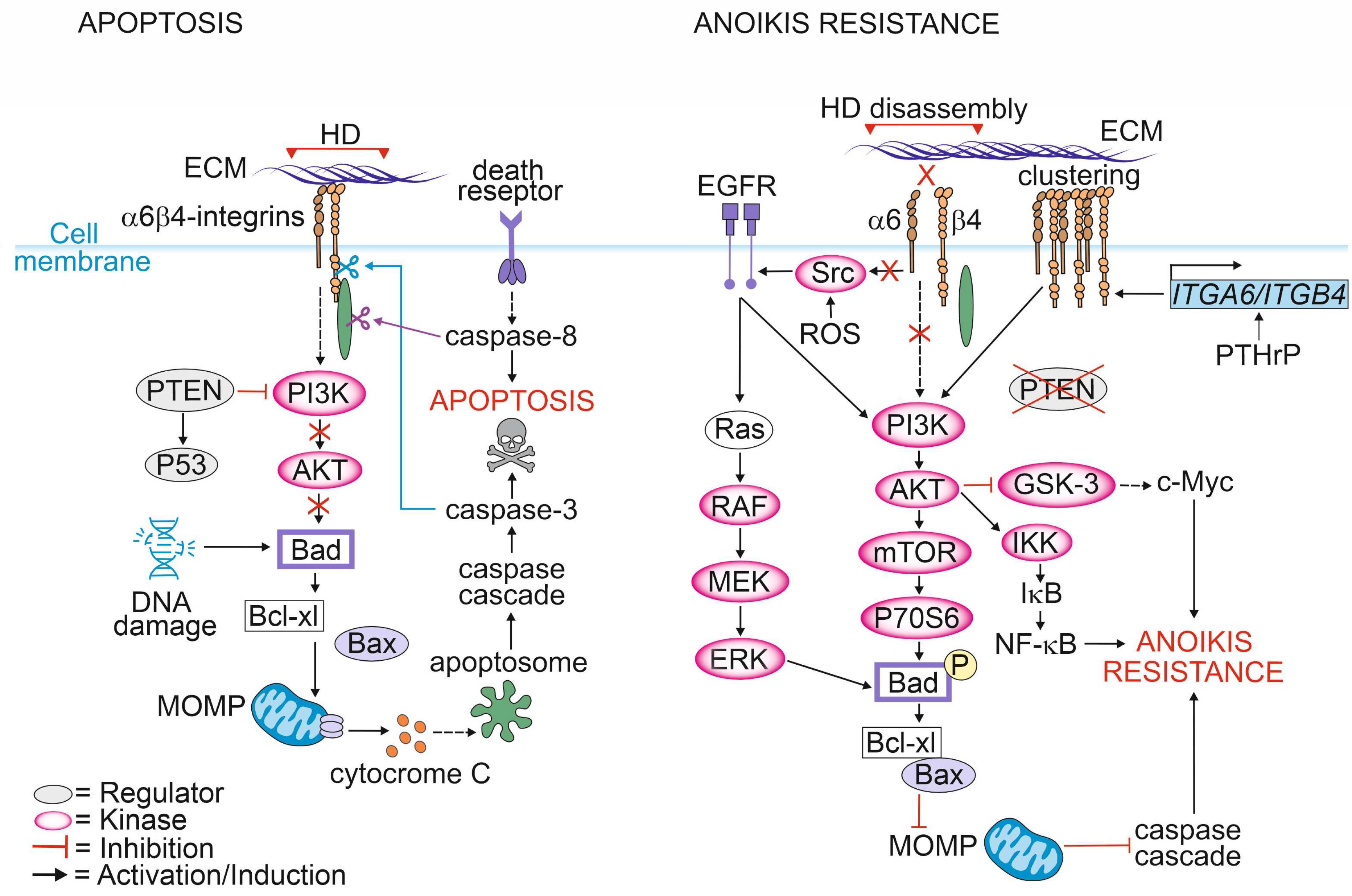{kind=link}
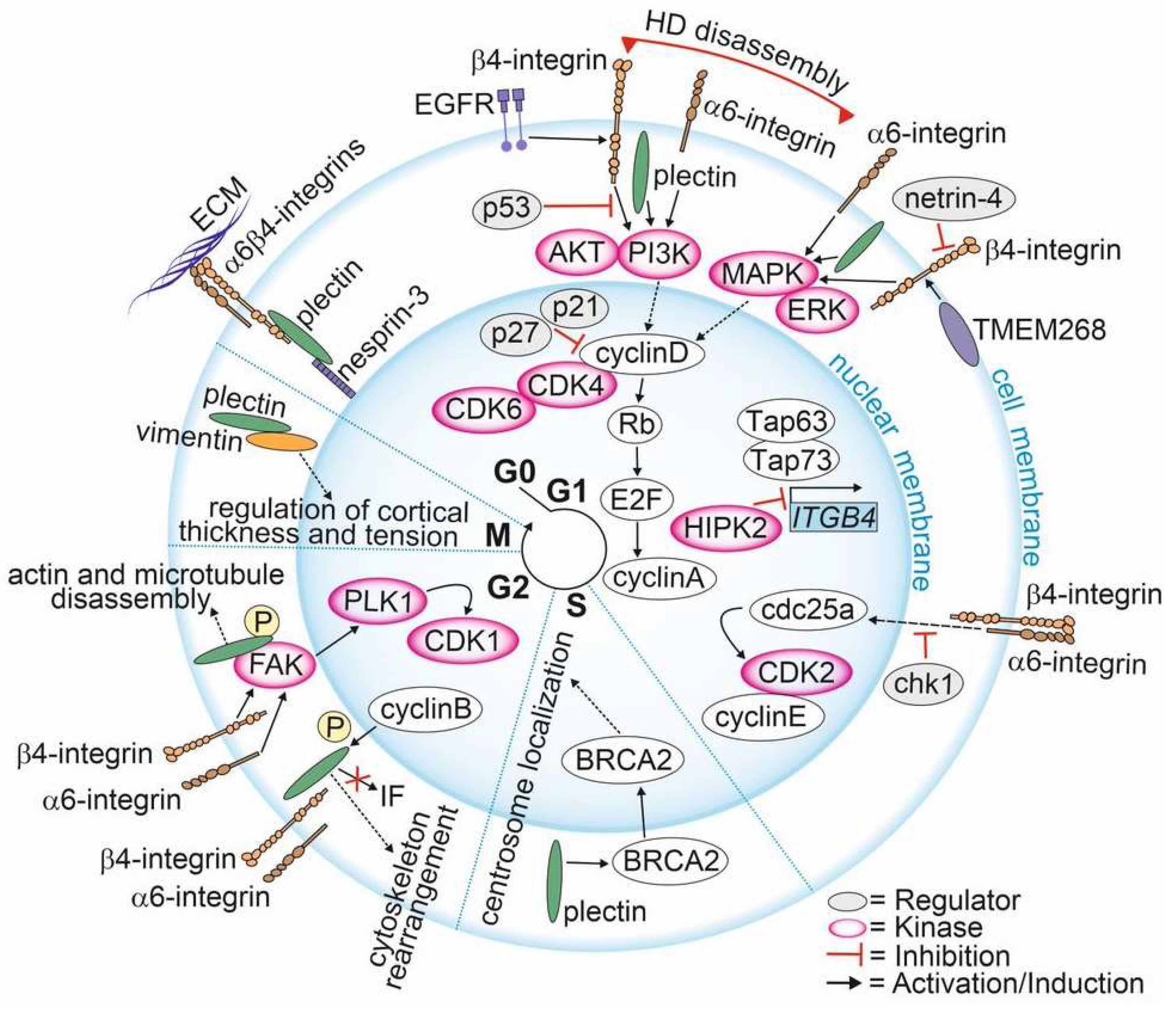{kind=link}
1. Introduction
2. Hemidesmosomal Components Regulate the Tumorigenic Properties of Cancer Cells
2.1. Disassembly of HDs Is a Prerequisite for Initiation of Carcinogenesis Driven by Hemidesmosomal Proteins
2.2. Regulation of Cell Proliferation
2.3. Regulation of Cell Migration
2.4. Regulation of Cell-Death-Associated Pathways
Role of p53 and PTEN in the Regulation of Hemidesmosomal-Components-Mediated Cell Death
2.5. Regulation of Cell Cycle
3. Drugs Targeting Hemidesmosomal Components as Potential Anti-Cancer Therapy
4. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate Cancer. Nat. Rev. Dis. Primer 2021, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, B.S.; Vasioukhin, V. Mechanisms of Prostate Cancer Initiation and Progression. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2010; Volume 109, pp. 1–50. ISBN 978-0-12-380890-5. [Google Scholar]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the Global Cancer Incidence and Mortality in 2018: GLOBOCAN Sources and Methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular Principles of Metastasis: A Hallmark of Cancer Revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Izzi, V.; Heljasvaara, R.; Heikkinen, A.; Karppinen, S.-M.; Koivunen, J.; Pihlajaniemi, T. Exploring the Roles of MACIT and Multiplexin Collagens in Stem Cells and Cancer. Semin. Cancer Biol. 2020, 62, 134–148. [Google Scholar] [CrossRef]
- Hamidi, H.; Ivaska, J. Every Step of the Way: Integrins in Cancer Progression and Metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef]
- Ye, X.; Weinberg, R.A. Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef]
- Cooper, J.; Giancotti, F.G. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef]
- Tsuruta, D.; Hashimoto, T.; Hamill, K.J.; Jones, J.C.R. Hemidesmosomes and Focal Contact Proteins: Functions and Cross-Talk in Keratinocytes, Bullous Diseases and Wound Healing. J. Dermatol. Sci. 2011, 62, 1–7. [Google Scholar] [CrossRef][Green Version]
- Schmidt, A.; Kaakinen, M.; Wenta, T.; Manninen, A. Loss of A6β4 Integrin-Mediated Hemidesmosomes Promotes Prostate Epithelial Cell Migration by Stimulating Focal Adhesion Dynamics. Front. Cell Dev. Biol. 2022, 10, 886569. [Google Scholar] [CrossRef]
- Myllymäki, S.-M.; Kämäräinen, U.-R.; Liu, X.; Cruz, S.P.; Miettinen, S.; Vuorela, M.; Varjosalo, M.; Manninen, A. Assembly of the B4-Integrin Interactome Based on Proximal Biotinylation in the Presence and Absence of Heterodimerization. Mol. Cell. Proteom. MCP 2019, 18, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Pora, A.; Yoon, S.; Windoffer, R.; Leube, R.E. Hemidesmosomes and Focal Adhesions Treadmill as Separate but Linked Entities during Keratinocyte Migration. J. Investig. Dermatol. 2019, 139, 1876–1888.e4. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.C.R.; Hopkinson, S.B.; Goldfinger, L.E. Structure and Assembly of Hemidesmosomes. BioEssays 1998, 20, 488–494. [Google Scholar] [CrossRef]
- Knox, J.D.; Cress, A.E.; Clark, V.; Manriquez, L.; Affinito, K.S.; Dalkin, B.L.; Nagle, R.B. Differential Expression of Extracellular Matrix Molecules and the Alpha 6-Integrins in the Normal and Neoplastic Prostate. Am. J. Pathol. 1994, 145, 167–174. [Google Scholar]
- Allen, M.V.; Smith, G.J.; Juliano, R.; Maygarden, S.J.; Mohler, J.L. Downregulation of the B4 Integrin Subunit in Prostatic Carcinoma and Prostatic Intraepithelial Neoplasia. Hum. Pathol. 1998, 29, 311–318. [Google Scholar] [CrossRef]
- Nievers, M.G.; Schaapveld, R.Q.; Sonnenberg, A. Biology and Function of Hemidesmosomes. Matrix Biol. J. Int. Soc. Matrix Biol. 1999, 18, 5–17. [Google Scholar] [CrossRef]
- Walko, G.; Castañón, M.J.; Wiche, G. Molecular Architecture and Function of the Hemidesmosome. Cell Tissue Res. 2015, 360, 529–544. [Google Scholar] [CrossRef]
- Wenta, T.; Schmidt, A.; Zhang, Q.; Devarajan, R.; Singh, P.; Yang, X.; Ahtikoski, A.; Vaarala, M.; Wei, G.-H.; Manninen, A. Disassembly of A6β4-Mediated Hemidesmosomal Adhesions Promotes Tumorigenesis in PTEN-Negative Prostate Cancer by Targeting Plectin to Focal Adhesions. Oncogene 2022, 41, 3804–3820. [Google Scholar] [CrossRef]
- Davis, T.L.; Cress, A.E.; Dalkin, B.L.; Nagle, R.B. Unique Expression Pattern of the Alpha6beta4 Integrin and Laminin-5 in Human Prostate Carcinoma. Prostate 2001, 46, 240–248. [Google Scholar] [CrossRef]
- Huang, W.; Fan, L.; Tang, Y.; Chi, Y.; Li, J. A Pan-Cancer Analysis of the Oncogenic Role of Integrin Beta4 (ITGB4) in Human Tumors. Int. J. Gen. Med. 2021, 14, 9629–9645. [Google Scholar] [CrossRef]
- Bigoni-Ordóñez, G.D.; Czarnowski, D.; Parsons, T.; Madlambayan, G.J.; Villa-Diaz, L.G. Integrin A6 (CD49f), The Microenvironment and Cancer Stem Cells. Curr. Stem Cell Res. Ther. 2019, 14, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Perez, S.M.; Brinton, L.T.; Kelly, K.A. Plectin in Cancer: From Biomarker to Therapeutic Target. Cells 2021, 10, 2246. [Google Scholar] [CrossRef] [PubMed]
- Buckup, M.; Rice, M.A.; Hsu, E.-C.; Garcia-Marques, F.; Liu, S.; Aslan, M.; Bermudez, A.; Huang, J.; Pitteri, S.J.; Stoyanova, T. Plectin Is a Regulator of Prostate Cancer Growth and Metastasis. Oncogene 2021, 40, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Theivendren, P.; Kunjiappan, S.; Hegde, Y.M.; Vellaichamy, S.; Gopal, M.; Dhramalingam, S.R.; Kumar, S. Importance of Protein Kinase and Its Inhibitor: A Review. In Biochemistry; Kumar Singh, R., Ed.; IntechOpen: London, UK, 2021; Volume 24, ISBN 978-1-83880-906-5. [Google Scholar]
- Bagheri, S.; Rahban, M.; Bostanian, F.; Esmaeilzadeh, F.; Bagherabadi, A.; Zolghadri, S.; Stanek, A. Targeting Protein Kinases and Epigenetic Control as Combinatorial Therapy Options for Advanced Prostate Cancer Treatment. Pharmaceutics 2022, 14, 515. [Google Scholar] [CrossRef]
- Miller, K.J.; Asim, M. Unravelling the Role of Kinases That Underpin Androgen Signalling in Prostate Cancer. Cells 2022, 11, 952. [Google Scholar] [CrossRef]
- Whitworth, H.; Bhadel, S.; Ivey, M.; Conaway, M.; Spencer, A.; Hernan, R.; Holemon, H.; Gioeli, D. Identification of Kinases Regulating Prostate Cancer Cell Growth Using an RNAi Phenotypic Screen. PLoS ONE 2012, 7, e38950. [Google Scholar] [CrossRef]
- Manning, G.; Plowman, G.D.; Hunter, T.; Sudarsanam, S. Evolution of Protein Kinase Signaling from Yeast to Man. Trends Biochem. Sci. 2002, 27, 514–520. [Google Scholar] [CrossRef]
- Stewart, R.L.; O’Connor, K.L. Clinical Significance of the Integrin A6β4 in Human Malignancies. Lab. Investig. 2015, 95, 976–986. [Google Scholar] [CrossRef]
- Yang, H.; Xu, Z.; Peng, Y.; Wang, J.; Xiang, Y. Integrin B4 as a Potential Diagnostic and Therapeutic Tumor Marker. Biomolecules 2021, 11, 1197. [Google Scholar] [CrossRef]
- Masugi, Y.; Yamazaki, K.; Emoto, K.; Effendi, K.; Tsujikawa, H.; Kitago, M.; Itano, O.; Kitagawa, Y.; Sakamoto, M. Upregulation of Integrin B4 Promotes Epithelial–Mesenchymal Transition and Is a Novel Prognostic Marker in Pancreatic Ductal Adenocarcinoma. Lab. Investig. 2015, 95, 308–319. [Google Scholar] [CrossRef]
- Dalton, G.N.; Massillo, C.; Scalise, G.D.; Duca, R.; Porretti, J.; Farré, P.L.; Gardner, K.; Paez, A.; Gueron, G.; De Luca, P.; et al. CTBP1 Depletion on Prostate Tumors Deregulates MiRNA/MRNA Expression and Impairs Cancer Progression in Metabolic Syndrome Mice. Cell Death Dis. 2019, 10, 299. [Google Scholar] [CrossRef] [PubMed]
- Cheng, I.; Plummer, S.J.; Neslund-Dudas, C.; Klein, E.A.; Casey, G.; Rybicki, B.A.; Witte, J.S. Prostate Cancer Susceptibility Variants Confer Increased Risk of Disease Progression. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2124–2132. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aumailley, M.; Bruckner-Tuderman, L.; Carter, W.G.; Deutzmann, R.; Edgar, D.; Ekblom, P.; Engel, J.; Engvall, E.; Hohenester, E.; Jones, J.C.R.; et al. A Simplified Laminin Nomenclature. Matrix Biol. J. Int. Soc. Matrix Biol. 2005, 24, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Litjens, S.H.M.; de Pereda, J.M.; Sonnenberg, A. Current Insights into the Formation and Breakdown of Hemidesmosomes. Trends Cell Biol. 2006, 16, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Décline, F.; Okamoto, O.; Mallein-Gerin, F.; Helbert, B.; Bernaud, J.; Rigal, D.; Rousselle, P. Keratinocyte Motility Induced by TGF-Beta1 Is Accompanied by Dramatic Changes in Cellular Interactions with Laminin 5. Cell Motil. Cytoskelet. 2003, 54, 64–80. [Google Scholar] [CrossRef]
- Henning, K.; Berndt, A.; Katenkamp, D.; Kosmehl, H. Loss of Laminin-5 in the Epithelium-Stroma Interface: An Immunohistochemical Marker of Malignancy in Epithelial Lesions of the Breast: Laminin-5 in Epithelial Tumours of the Breast. Histopathology 1999, 34, 305–309. [Google Scholar] [CrossRef]
- Shinto, E.; Tsuda, H.; Ueno, H.; Hashiguchi, Y.; Hase, K.; Tamai, S.; Mochizuki, H.; Inazawa, J.; Matsubara, O. Prognostic Implication of Laminin-5 Gamma 2 Chain Expression in the Invasive Front of Colorectal Cancers, Disclosed by Area-Specific Four-Point Tissue Microarrays. Lab. Investig. 2005, 85, 257–266. [Google Scholar] [CrossRef]
- Hao, J.; Jackson, L.; Calaluce, R.; McDaniel, K.; Dalkin, B.L.; Nagle, R.B. Investigation into the Mechanism of the Loss of Laminin 5 (A3β3γ2) Expression in Prostate Cancer. Am. J. Pathol. 2001, 158, 1129–1135. [Google Scholar] [CrossRef]
- Mainiero, F.; Pepe, A.; Yeon, M.; Ren, Y.; Giancotti, F.G. The Intracellular Functions of Alpha6beta4 Integrin Are Regulated by EGF. J. Cell Biol. 1996, 134, 241–253. [Google Scholar] [CrossRef]
- Wilkinson, E.J.; Woodworth, A.M.; Parker, M.; Phillips, J.L.; Malley, R.C.; Dickinson, J.L.; Holloway, A.F. Epigenetic Regulation of the ITGB4 Gene in Prostate Cancer. Exp. Cell Res. 2020, 392, 112055. [Google Scholar] [CrossRef]
- Tuxhorn, J.A.; Ayala, G.E.; Rowley, D.R. Reactive Stroma in Prostate Cancer Progression. J. Urol. 2001, 166, 2472–2483. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Franco, O.E.; Hayward, S.W. Interaction of Prostate Carcinoma-Associated Fibroblasts with Human Epithelial Cell Lines In Vivo. Differentiation 2017, 96, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Dans, M.; Gagnoux-Palacios, L.; Blaikie, P.; Klein, S.; Mariotti, A.; Giancotti, F.G. Tyrosine Phosphorylation of the B4 Integrin Cytoplasmic Domain Mediates Shc Signaling to Extracellular Signal-Regulated Kinase and Antagonizes Formation of Hemidesmosomes. J. Biol. Chem. 2001, 276, 1494–1502. [Google Scholar] [CrossRef] [PubMed]
- Giancotti, F.G. Signal Transduction by the A6β4 Integrin: Charting the Path between Laminin Binding and Nuclear Events. J. Cell Sci. 1996, 109, 1165–1172. [Google Scholar] [CrossRef]
- Luo, B.-H.; Carman, C.V.; Springer, T.A. Structural Basis of Integrin Regulation and Signaling. Annu. Rev. Immunol. 2007, 25, 619–647. [Google Scholar] [CrossRef]
- Askari, J.A.; Buckley, P.A.; Mould, A.P.; Humphries, M.J. Linking Integrin Conformation to Function. J. Cell Sci. 2009, 122, 165–170. [Google Scholar] [CrossRef]
- Mainiero, F.; Pepe, A.; Wary, K.K.; Spinardi, L.; Mohammadi, M.; Schlessinger, J.; Giancotti, F.G. Signal Transduction by the Alpha 6 Beta 4 Integrin: Distinct Beta 4 Subunit Sites Mediate Recruitment of Shc/Grb2 and Association with the Cytoskeleton of Hemidesmosomes. EMBO J. 1995, 14, 4470–4481. [Google Scholar] [CrossRef]
- Guo, Y.; Pan, W.; Liu, S.; Shen, Z.; Xu, Y.; Hu, L. ERK/MAPK Signalling Pathway and Tumorigenesis (Review). Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Tran, T.H.; Chan, A.H.; Young, L.C.; Bindu, L.; Neale, C.; Messing, S.; Dharmaiah, S.; Taylor, T.; Denson, J.-P.; Esposito, D.; et al. KRAS Interaction with RAF1 RAS-Binding Domain and Cysteine-Rich Domain Provides Insights into RAS-Mediated RAF Activation. Nat. Commun. 2021, 12, 1176. [Google Scholar] [CrossRef]
- Weinstein-Oppenheimer, C.R.; Blalock, W.L.; Steelman, L.S.; Chang, F.; McCubrey, J.A. The Raf Signal Transduction Cascade as a Target for Chemotherapeutic Intervention in Growth Factor-Responsive Tumors. Pharmacol. Ther. 2000, 88, 229–279. [Google Scholar] [CrossRef]
- Fu, Z.; Smith, P.C.; Zhang, L.; Rubin, M.A.; Dunn, R.L.; Yao, Z.; Keller, E.T. Effects of Raf Kinase Inhibitor Protein Expression on Suppression of Prostate Cancer Metastasis. JNCI J. Natl. Cancer Inst. 2003, 95, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.-Y.; Choi, M.; Kim, B.-H.; Cho, Y.-M.; Moon, K.C.; Kang, G.H. BRAF AndKRAS Mutations in Prostatic Adenocarcinoma. Int. J. Cancer 2006, 119, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Shaw, L.M. Identification of Insulin Receptor Substrate 1 (IRS-1) and IRS-2 as Signaling Intermediates in the A6β4 Integrin-Dependent Activation of Phosphoinositide 3-OH Kinase and Promotion of Invasion. Mol. Cell. Biol. 2001, 21, 5082–5093. [Google Scholar] [CrossRef]
- Gu, H.; Maeda, H.; Moon, J.J.; Lord, J.D.; Yoakim, M.; Nelson, B.H.; Neel, B.G. New Role for Shc in Activation of the Phosphatidylinositol 3-Kinase/Akt Pathway. Mol. Cell. Biol. 2000, 20, 7109–7120. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the Phosphoinositide 3-Kinase Pathway in Cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, I.; Chen, Z.C.; Tanos, B.; Oldrini, B.; Hsieh, W.-Y.; Yannuzzi, N.; Campos, C.; Mellinghoff, I.K. A Kinase-Independent Function of AKT Promotes Cancer Cell Survival. eLife 2014, 3, e03751. [Google Scholar] [CrossRef]
- Danilkovitch-Miagkova, A. Oncogenic Signaling Pathways Activated by RON Receptor Tyrosine Kinase. Curr. Cancer Drug Targets 2003, 3, 31–40. [Google Scholar] [CrossRef]
- Benight, N.M.; Waltz, S.E. Ron Receptor Tyrosine Kinase Signaling as a Therapeutic Target. Expert Opin. Ther. Targets 2012, 16, 921–931. [Google Scholar] [CrossRef][Green Version]
- Yin, B.; Liu, Z.; Wang, Y.; Wang, X.; Liu, W.; Yu, P.; Duan, X.; Liu, C.; Chen, Y.; Zhang, Y.; et al. RON and C-Met Facilitate Metastasis through the ERK Signaling Pathway in Prostate Cancer Cells. Oncol. Rep. 2017, 37, 3209–3218. [Google Scholar] [CrossRef]
- O’Toole, J.M.; Rabenau, K.E.; Burns, K.; Lu, D.; Mangalampalli, V.; Balderes, P.; Covino, N.; Bassi, R.; Prewett, M.; Gottfredsen, K.J.; et al. Therapeutic Implications of a Human Neutralizing Antibody to the Macrophage-Stimulating Protein Receptor Tyrosine Kinase (RON), a c-MET Family Member. Cancer Res. 2006, 66, 9162–9170. [Google Scholar] [CrossRef]
- Thobe, M.N.; Gurusamy, D.; Pathrose, P.; Waltz, S.E. The Ron Receptor Tyrosine Kinase Positively Regulates Angiogenic Chemokine Production in Prostate Cancer Cells. Oncogene 2010, 29, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Ye, H.; Gerrin, S.; Wang, H.; Sharma, A.; Chen, S.; Patnaik, A.; Sowalsky, A.G.; Voznesensky, O.; Han, W.; et al. ErbB2 Signaling Increases Androgen Receptor Expression in Abiraterone-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 3672–3682. [Google Scholar] [CrossRef] [PubMed]
- Frijns, E.; Sachs, N.; Kreft, M.; Wilhelmsen, K.; Sonnenberg, A. EGF-Induced MAPK Signaling Inhibits Hemidesmosome Formation through Phosphorylation of the Integrin B4. J. Biol. Chem. 2010, 285, 37650–37662. [Google Scholar] [CrossRef] [PubMed]
- Osmanagic-Myers, S.; Wiche, G. Plectin-RACK1 (Receptor for Activated C Kinase 1) Scaffolding. J. Biol. Chem. 2004, 279, 18701–18710. [Google Scholar] [CrossRef]
- Burch, T.C.; Watson, M.T.; Nyalwidhe, J.O. Variable Metastatic Potentials Correlate with Differential Plectin and Vimentin Expression in Syngeneic Androgen Independent Prostate Cancer Cells. PLoS ONE 2013, 8, e65005. [Google Scholar] [CrossRef]
- Solt, L.A.; May, M.J. The IκB Kinase Complex: Master Regulator of NF-ΚB Signaling. Immunol. Res. 2008, 42, 3–18. [Google Scholar] [CrossRef]
- Albensi, B.C. What Is Nuclear Factor Kappa B (NF-ΚB) Doing in and to the Mitochondrion? Front. Cell Dev. Biol. 2019, 7, 154. [Google Scholar] [CrossRef]
- Zahir, N.; Lakins, J.N.; Russell, A.; Ming, W.; Chatterjee, C.; Rozenberg, G.I.; Marinkovich, M.P.; Weaver, V.M. Autocrine Laminin-5 Ligates A6β4 Integrin and Activates RAC and NFκB to Mediate Anchorage-Independent Survival of Mammary Tumors. J. Cell Biol. 2003, 163, 1397–1407. [Google Scholar] [CrossRef]
- Elaimy, A.L.; Sheel, A.; Brown, C.W.; Walker, M.R.; Wang, M.; Amante, J.J.; Xue, W.; Chan, A.; Baer, C.E.; Goel, H.L.; et al. Real-Time Imaging of Integrin B4 Dynamics Using a Reporter Cell Line Generated by Crispr/Cas9 Genome Editing. J. Cell Sci. 2019, 132, jcs231241. [Google Scholar] [CrossRef]
- Rabinovitz, I.; Mercurio, A.M. The Integrin A6β4 Functions in Carcinoma Cell Migration on Laminin-1 by Mediating the Formation and Stabilization of Actin-Containing Motility Structures. J. Cell Biol. 1997, 139, 1873–1884. [Google Scholar] [CrossRef]
- Geuijen, C.A.W.; Sonnenberg, A. Dynamics of the A6β4 Integrin in Keratinocytes. Mol. Biol. Cell 2002, 13, 3845–3858. [Google Scholar] [CrossRef] [PubMed]
- te Molder, L.; de Pereda, J.M.; Sonnenberg, A. Regulation of Hemidesmosome Dynamics and Cell Signaling by Integrin A6β4. J. Cell Sci. 2021, 134, jcs259004. [Google Scholar] [CrossRef] [PubMed]
- Lipscomb, E.A.; Mercurio, A.M. Mobilization and Activation of a Signaling Competent A6β4integrin Underlies Its Contribution to Carcinoma Progression. Cancer Metastasis Rev. 2005, 24, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Frijns, E.; Kuikman, I.; Litjens, S.; Raspe, M.; Jalink, K.; Ports, M.; Wilhelmsen, K.; Sonnenberg, A. Phosphorylation of Threonine 1736 in the C-Terminal Tail of Integrin B4 Contributes to Hemidesmosome Disassembly. Mol. Biol. Cell 2012, 23, 1475–1485. [Google Scholar] [CrossRef]
- te Molder, L.; Sonnenberg, A. PKD2 and RSK1 Regulate Integrin B4 Phosphorylation at Threonine 1736. PLoS ONE 2015, 10, e0143357. [Google Scholar] [CrossRef]
- Wang, W.; Zuidema, A.; te Molder, L.; Nahidiazar, L.; Hoekman, L.; Schmidt, T.; Coppola, S.; Sonnenberg, A. Hemidesmosomes Modulate Force Generation via Focal Adhesions. J. Cell Biol. 2020, 219, e201904137. [Google Scholar] [CrossRef]
- Ozawa, T.; Tsuruta, D.; Jones, J.C.R.; Ishii, M.; Ikeda, K.; Harada, T.; Aoyama, Y.; Kawada, A.; Kobayashi, H. Dynamic Relationship of Focal Contacts and Hemidesmosome Protein Complexes in Live Cells. J. Investig. Dermatol. 2010, 130, 1624–1635. [Google Scholar] [CrossRef]
- Lachowski, D.; Cortes, E.; Robinson, B.; Rice, A.; Rombouts, K.; Del Río Hernández, A.E. FAK Controls the Mechanical Activation of YAP, a Transcriptional Regulator Required for Durotaxis. FASEB J. 2018, 32, 1099–1107. [Google Scholar] [CrossRef]
- Kim, N.-G.; Gumbiner, B.M. Adhesion to Fibronectin Regulates Hippo Signaling via the FAK–Src–PI3K Pathway. J. Cell Biol. 2015, 210, 503–515. [Google Scholar] [CrossRef]
- Lessey, E.C.; Guilluy, C.; Burridge, K. From Mechanical Force to RhoA Activation. Biochemistry 2012, 51, 7420–7432. [Google Scholar] [CrossRef]
- Amano, M.; Ito, M.; Kimura, K.; Fukata, Y.; Chihara, K.; Nakano, T.; Matsuura, Y.; Kaibuchi, K. Phosphorylation and Activation of Myosin by Rho-Associated Kinase (Rho-Kinase). J. Biol. Chem. 1996, 271, 20246–20249. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, S.; Matsui, T.S.; Ohashi, K.; Deguchi, S.; Mizuno, K. Solo, a RhoA-Targeting Guanine Nucleotide Exchange Factor, Is Critical for Hemidesmosome Formation and Acinar Development in Epithelial Cells. PLoS ONE 2018, 13, e0195124. [Google Scholar] [CrossRef] [PubMed]
- Nardone, G.; Oliver-De La Cruz, J.; Vrbsky, J.; Martini, C.; Pribyl, J.; Skládal, P.; Pešl, M.; Caluori, G.; Pagliari, S.; Martino, F.; et al. YAP Regulates Cell Mechanics by Controlling Focal Adhesion Assembly. Nat. Commun. 2017, 8, 15321. [Google Scholar] [CrossRef] [PubMed]
- Halder, G.; Dupont, S.; Piccolo, S. Transduction of Mechanical and Cytoskeletal Cues by YAP and TAZ. Nat. Rev. Mol. Cell Biol. 2012, 13, 591–600. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in Mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Chen, M.; Bresnick, A.R.; O’Connor, K.L. Coupling S100A4 to Rhotekin Alters Rho Signaling Output in Breast Cancer Cells. Oncogene 2013, 32, 3754–3764. [Google Scholar] [CrossRef]
- Chen, M.; Sinha, M.; Luxon, B.A.; Bresnick, A.R.; O’Connor, K.L. Integrin A6β4 Controls the Expression of Genes Associated with Cell Motility, Invasion, and Metastasis, Including S100A4/Metastasin. J. Biol. Chem. 2009, 284, 1484–1494. [Google Scholar] [CrossRef]
- O’Connor, K.L.; Nguyen, B.-K.; Mercurio, A.M. Rhoa Function in Lamellae Formation and Migration Is Regulated by the A6β4 Integrin and Camp Metabolism. J. Cell Biol. 2000, 148, 253–258. [Google Scholar] [CrossRef]
- Gilmore, A.P.; Owens, T.W.; Foster, F.M.; Lindsay, J. How Adhesion Signals Reach a Mitochondrial Conclusion—ECM Regulation of Apoptosis. Curr. Opin. Cell Biol. 2009, 21, 654–661. [Google Scholar] [CrossRef]
- Popgeorgiev, N.; Jabbour, L.; Gillet, G. Subcellular Localization and Dynamics of the Bcl-2 Family of Proteins. Front. Cell Dev. Biol. 2018, 6, 13. [Google Scholar] [CrossRef]
- Driak, D.; Dvorska, M.; Bolehovska, P.; Svandova, I.; Novotny, J.; Halaska, M. Bad and Bid—Potential Background Players in Preneoplastic to Neoplastic Shift in Human Endometrium. Neoplasma 2014, 61, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.C.; Muñoz-Pinedo, C.; Ricci, J.-E.; Adams, S.R.; Kelekar, A.; Schuler, M.; Tsien, R.Y.; Green, D.R. Cytochrome c Is Released in a Single Step during Apoptosis. Cell Death Differ. 2005, 12, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.E.; Chen, F.; Moyano, J.V.; Yehiely, F.; Jones, J.C.R.; Cryns, V.L. Caspase Proteolysis of the Integrin B4 Subunit Disrupts Hemidesmosome Assembly, Promotes Apoptosis, and Inhibits Cell Migration. J. Biol. Chem. 2007, 282, 5560–5569. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, E.; Eaves, C.J. Paradoxical Roles of Caspase-3 in Regulating Cell Survival, Proliferation, and Tumorigenesis. J. Cell Biol. 2022, 221, e202201159. [Google Scholar] [CrossRef] [PubMed]
- Stegh, A.H.; Herrmann, H.; Lampel, S.; Weisenberger, D.; Andrä, K.; Seper, M.; Wiche, G.; Krammer, P.H.; Peter, M.E. Identification of the Cytolinker Plectin as a Major Early In Vivo Substrate for Caspase 8 during CD95- and Tumor Necrosis Factor Receptor-Mediated Apoptosis. Mol. Cell. Biol. 2000, 20, 5665–5679. [Google Scholar] [CrossRef]
- Aho, S. Plakin Proteins Are Coordinately Cleaved during Apoptosis but Preferentially through the Action of Different Caspases. Exp. Dermatol. 2004, 13, 700–707. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Cheng, C.-C.; Ho, C.-C.; Chao, W.-T.; Pei, R.-J.; Hsu, Y.-H.; Yeh, K.-T.; Ho, L.-C.; Tsai, M.-C.; Lai, Y.-S. Degradation of Plectin with Modulation of Cytokeratin 18 in Human Liver Cells during Staurosporine-Induced Apoptosis. Vivo Athens Greece 2008, 22, 543–548. [Google Scholar]
- Ni, Y.; Wang, X.; Yin, X.; Li, Y.; Liu, X.; Wang, H.; Liu, X.; Zhang, J.; Gao, H.; Shi, B.; et al. Plectin Protects Podocytes from Adriamycin-Induced Apoptosis and F-Actin Cytoskeletal Disruption through the Integrin A6β4/FAK/P38 MAPK Pathway. J. Cell. Mol. Med. 2018, 22, 5450–5467. [Google Scholar] [CrossRef]
- Giannoni, E.; Fiaschi, T.; Ramponi, G.; Chiarugi, P. Redox Regulation of Anoikis Resistance of Metastatic Prostate Cancer Cells: Key Role for Src and EGFR-Mediated pro-Survival Signals. Oncogene 2009, 28, 2074–2086. [Google Scholar] [CrossRef]
- Heppner, D.E.; Dustin, C.M.; Liao, C.; Hristova, M.; Deng, B.; Lam, Y.-W.; Li, J.; van der Vliet, A. Molecular Basis for the Redox Regulation of the Src Kinase. Free Radic. Biol. Med. 2016, 100, S40. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt Phosphorylation of BAD Couples Survival Signals to the Cell-Intrinsic Death Machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Goc, A.; Kochuparambil, S.T.; Al-Husein, B.; Al-Azayzih, A.; Mohammad, S.; Somanath, P.R. Simultaneous Modulation of the Intrinsic and Extrinsic Pathways by Simvastatin in Mediating Prostate Cancer Cell Apoptosis. BMC Cancer 2012, 12, 409. [Google Scholar] [CrossRef] [PubMed]
- Chao, O.S.P.; Clément, M.-V. Epidermal Growth Factor and Serum Activate Distinct Pathways to Inhibit the BH3 Only Protein BAD in Prostate Carcinoma LNCaP Cells. Oncogene 2006, 25, 4458–4469. [Google Scholar] [CrossRef] [PubMed]
- Sastry, K.S.R.; Karpova, Y.; Kulik, G. Epidermal Growth Factor Protects Prostate Cancer Cells from Apoptosis by Inducing BAD Phosphorylation via Redundant Signaling Pathways. J. Biol. Chem. 2006, 281, 27367–27377. [Google Scholar] [CrossRef]
- Balta, E.; Kramer, J.; Samstag, Y. Redox Regulation of the Actin Cytoskeleton in Cell Migration and Adhesion: On the Way to a Spatiotemporal View. Front. Cell Dev. Biol. 2021, 8, 618261. [Google Scholar] [CrossRef] [PubMed]
- Ketema, M.; Wilhelmsen, K.; Kuikman, I.; Janssen, H.; Hodzic, D.; Sonnenberg, A. Requirements for the Localization of Nesprin-3 at the Nuclear Envelope and Its Interaction with Plectin. J. Cell Sci. 2007, 120, 3384–3394. [Google Scholar] [CrossRef]
- Allison, S.J. Benefits of Actin Cytoskeleton Stabilization. Nat. Rev. Nephrol. 2022, 18, 413. [Google Scholar] [CrossRef]
- Spurny, R.; Abdoulrahman, K.; Janda, L.; Ruönzler, D.; Koöhler, G.; Castañón, M.J.; Wiche, G. Oxidation and Nitrosylation of Cysteines Proximal to the Intermediate Filament (IF)-Binding Site of Plectin. J. Biol. Chem. 2007, 282, 8175–8187. [Google Scholar] [CrossRef]
- Giannoni, E.; Buricchi, F.; Raugei, G.; Ramponi, G.; Chiarugi, P. Intracellular Reactive Oxygen Species Activate Src Tyrosine Kinase during Cell Adhesion and Anchorage-Dependent Cell Growth. Mol. Cell. Biol. 2005, 25, 6391–6403. [Google Scholar] [CrossRef]
- Basuroy, S.; Dunagan, M.; Sheth, P.; Seth, A.; Rao, R.K. Hydrogen Peroxide Activates Focal Adhesion Kinase and C-Src by a Phosphatidylinositol 3 Kinase-Dependent Mechanism and Promotes Cell Migration in Caco-2 Cell Monolayers. Am. J. Physiol.-Gastrointest. Liver Physiol. 2010, 299, G186–G195. [Google Scholar] [CrossRef]
- Heppner, D.E.; Dustin, C.M.; Liao, C.; Hristova, M.; Veith, C.; Little, A.C.; Ahlers, B.A.; White, S.L.; Deng, B.; Lam, Y.-W.; et al. Direct Cysteine Sulfenylation Drives Activation of the Src Kinase. Nat. Commun. 2018, 9, 4522. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, V.; Mula, R.V.; Weigel, N.L.; Falzon, M. Parathyroid Hormone-Related Protein Regulates Cell Survival Pathways via Integrin A6β4-Mediated Activation of Phosphatidylinositol 3-Kinase/Akt Signaling. Mol. Cancer Res. 2009, 7, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Thrasher, J.B.; Terranova, P. Glycogen Synthase Kinase-3: A Potential Preventive Target for Prostate Cancer Management. Urol. Oncol. Semin. Orig. Investig. 2015, 33, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Hahn-Windgassen, A.; Nogueira, V.; Chen, C.-C.; Skeen, J.E.; Sonenberg, N.; Hay, N. Akt Activates the Mammalian Target of Rapamycin by Regulating Cellular ATP Level and AMPK Activity. J. Biol. Chem. 2005, 280, 32081–32089. [Google Scholar] [CrossRef]
- Harada, H.; Andersen, J.S.; Mann, M.; Terada, N.; Korsmeyer, S.J. P70S6 Kinase Signals Cell Survival as Well as Growth, Inactivating the pro-Apoptotic Molecule BAD. Proc. Natl. Acad. Sci. USA 2001, 98, 9666–9670. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Beltran, H.; Yelensky, R.; Frampton, G.M.; Park, K.; Downing, S.R.; MacDonald, T.Y.; Jarosz, M.; Lipson, D.; Tagawa, S.T.; Nanus, D.M.; et al. Targeted Next-Generation Sequencing of Advanced Prostate Cancer Identifies Potential Therapeutic Targets and Disease Heterogeneity. Eur. Urol. 2013, 63, 920–926. [Google Scholar] [CrossRef]
- van Dessel, L.F.; van Riet, J.; Smits, M.; Zhu, Y.; Hamberg, P.; van der Heijden, M.S.; Bergman, A.M.; van Oort, I.M.; de Wit, R.; Voest, E.E.; et al. The Genomic Landscape of Metastatic Castration-Resistant Prostate Cancers Reveals Multiple Distinct Genotypes with Potential Clinical Impact. Nat. Commun. 2019, 10, 5251. [Google Scholar] [CrossRef]
- Hamid, A.A.; Gray, K.P.; Shaw, G.; MacConaill, L.E.; Evan, C.; Bernard, B.; Loda, M.; Corcoran, N.M.; Van Allen, E.M.; Choudhury, A.D.; et al. Compound Genomic Alterations of TP53, PTEN, and RB1 Tumor Suppressors in Localized and Metastatic Prostate Cancer. Eur. Urol. 2019, 76, 89–97. [Google Scholar] [CrossRef]
- Vinall, R.L.; Chen, J.Q.; Hubbard, N.E.; Sulaimon, S.S.; DeVere White, R.W.; Borowsky, A.D. Evidence for an Alternate Molecular Progression in Prostate Cancer. Dis. Model. Mech. 2012, dmm-008995. [Google Scholar] [CrossRef]
- Bachelder, R.E.; Ribick, M.J.; Marchetti, A.; Falcioni, R.; Soddu, S.; Davis, K.R.; Mercurio, A.M. P53 Inhibits A6β4 Integrin Survival Signaling by Promoting the Caspase 3–Dependent Cleavage of Akt/PKB. J. Cell Biol. 1999, 147, 1063–1072. [Google Scholar] [CrossRef]
- Persad, S.; Attwell, S.; Gray, V.; Delcommenne, M.; Troussard, A.; Sanghera, J.; Dedhar, S. Inhibition of Integrin-Linked Kinase (ILK) Suppresses Activation of Protein Kinase B/Akt and Induces Cell Cycle Arrest and Apoptosis of PTEN-Mutant Prostate Cancer Cells. Proc. Natl. Acad. Sci. USA 2000, 97, 3207–3212. [Google Scholar] [CrossRef] [PubMed]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical Implications of PTEN Loss in Prostate Cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, A.; Kitagishi, Y.; Ogura, Y.; Matsuda, S. The Tumor Suppressor PTEN Interacts with P53 in Hereditary Cancer. Int. J. Oncol. 2014, 44, 1813–1819. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Zhang, X.; Li, R.; Yu, J.; Lou, Y.; He, Q.; Li, X.; Xu, D.; Lv, P.; Lin, J.; et al. Deletion of TMEM268 Inhibits Growth of Gastric Cancer Cells by Downregulating the ITGB4 Signaling Pathway. Cell Death Differ. 2019, 26, 1453–1466. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Ohtsubo, M.; Böhmer, R.M.; Roberts, J.M.; Assoian, R.K. Adhesion-Dependent Cell Cycle Progression Linked to the Expression of Cyclin D1, Activation of Cyclin E-Cdk2, and Phosphorylation of the Retinoblastoma Protein. J. Cell Biol. 1996, 133, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Shanmugasundaram, K.; Block, K.; Nayak, B.K.; Livi, C.B.; Venkatachalam, M.A.; Sudarshan, S. PI3K Regulation of the SKP-2/P27 Axis through MTORC2. Oncogene 2013, 32, 2027–2036. [Google Scholar] [CrossRef]
- Guo, Z.-Y.; Hao, X.-H.; Tan, F.-F.; Pei, X.; Shang, L.-M.; Jiang, X.-L.; Yang, F. The Elements of Human Cyclin D1 Promoter and Regulation Involved. Clin. Epigenet. 2011, 2, 63–76. [Google Scholar] [CrossRef]
- Kamranvar, S.A.; Rani, B.; Johansson, S. Cell Cycle Regulation by Integrin-Mediated Adhesion. Cells 2022, 11, 2521. [Google Scholar] [CrossRef]
- Zhao, J.; Bian, Z.C.; Yee, K.; Chen, B.P.C.; Chien, S.; Guan, J.-L. Identification of Transcription Factor KLF8 as a Downstream Target of Focal Adhesion Kinase in Its Regulation of Cyclin D1 and Cell Cycle Progression. Mol. Cell 2003, 11, 1503–1515. [Google Scholar] [CrossRef]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen Synthase Kinase-3β Regulates Cyclin D1 Proteolysis and Subcellular Localization. Genes Dev. 1998, 12, 3499–3511. [Google Scholar] [CrossRef] [PubMed]
- Kamranvar, S.A.; Gupta, D.K.; Wasberg, A.; Liu, L.; Roig, J.; Johansson, S. Integrin-Mediated Adhesion Promotes Centrosome Separation in Early Mitosis. Cells 2022, 11, 1360. [Google Scholar] [CrossRef] [PubMed]
- Gheghiani, L.; Loew, D.; Lombard, B.; Mansfeld, J.; Gavet, O. PLK1 Activation in Late G2 Sets Up Commitment to Mitosis. Cell Rep. 2017, 19, 2060–2073. [Google Scholar] [CrossRef] [PubMed]
- Steinböck, F.A.; Wiche, G. Plectin: A Cytolinker by Design. Biol. Chem. 1999, 380, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Dong, Z.; Zhang, Y.; Miao, J. The Roles of Integrin B4 in Vascular Endothelial Cells. J. Cell. Physiol. 2012, 227, 474–478. [Google Scholar] [CrossRef]
- Lewis, J.M.; Truong, T.N.; Schwartz, M.A. Integrins Regulate the Apoptotic Response to DNA Damage through Modulation of P53. Proc. Natl. Acad. Sci. USA 2002, 99, 3627–3632. [Google Scholar] [CrossRef]
- Giancotti, F.G. Targeting Integrin Beta4 for Cancer and Anti-Angiogenic Therapy. Trends Pharmacol. Sci. 2007, 28, 506–511. [Google Scholar] [CrossRef]
- Yuan, L.; Du, X.; Tang, S.; Wu, S.; Wang, L.; Xiang, Y.; Qu, X.; Liu, H.; Qin, X.; Liu, C. ITGB 4 Deficiency Induces Senescence of Airway Epithelial Cells through P53 Activation. FEBS J. 2019, 286, 1191–1203. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. P21 in Cancer: Intricate Networks and Multiple Activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Sun, X.; Liu, B.; Wang, J.; Li, J.; Ji, W.-Y. Inhibition of P21-Activated Kinase 4 Expression Suppresses the Proliferation of Hep-2 Laryngeal Carcinoma Cells via Activation of the ATM/Chk1/2/P53 Pathway. Int. J. Oncol. 2013, 42, 683–689. [Google Scholar] [CrossRef]
- Bon, G.; Di Carlo, S.E.; Folgiero, V.; Avetrani, P.; Lazzari, C.; D’Orazi, G.; Brizzi, M.F.; Sacchi, A.; Soddu, S.; Blandino, G.; et al. Negative Regulation of B4 Integrin Transcription by Homeodomain-Interacting Protein Kinase 2 and P53 Impairs Tumor Progression. Cancer Res. 2009, 69, 5978–5986. [Google Scholar] [CrossRef] [PubMed]
- Sombroek, D.; Hofmann, T.G. How Cells Switch HIPK2 on and Off. Cell Death Differ. 2009, 16, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Vikhreva, P.; Annicchiarico-Petruzzelli, M.; Amelio, I.; Barlev, N.; Knight, R.A.; Melino, G. Integrin-B4 Is a Novel Transcriptional Target of TAp73. Cell Cycle 2018, 17, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Maas, A.-M.; Bretz, A.C.; Mack, E.; Stiewe, T. Targeting P73 in Cancer. Cancer Lett. 2013, 332, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Ylivinkka, I.; Chen, P.; Li, L.; Hautaniemi, S.; Nyman, T.A.; Keski-Oja, J.; Hyytiäinen, M. Netrin-4 Promotes Glioblastoma Cell Proliferation through Integrin B4 Signaling. Neoplasia 2012, 14, 219IN23. [Google Scholar] [CrossRef]
- Zhang, X.; Rozengurt, E.; Reed, E.F. HLA Class I Molecules Partner with Integrin β 4 to Stimulate Endothelial Cell Proliferation and Migration. Sci. Signal. 2010, 3, ra85. [Google Scholar] [CrossRef]
- Wang, Y.; Shenouda, S.; Baranwal, S.; Rathinam, R.; Jain, P.; Bao, L.; Hazari, S.; Dash, S.; Alahari, S.K. Integrin Subunits Alpha5 and Alpha6 Regulate Cell Cycle by Modulating the Chk1 and Rb/E2F Pathways to Affect Breast Cancer Metastasis. Mol. Cancer 2011, 10, 84. [Google Scholar] [CrossRef]
- Inoue, Y.; Kitagawa, M.; Taya, Y. Phosphorylation of PRB at Ser612 by Chk1/2 Leads to a Complex between PRB and E2F-1 after DNA Damage. EMBO J. 2007, 26, 2083–2093. [Google Scholar] [CrossRef]
- Kreutzer, J.; Guerra, B. The Regulatory Beta-Subunit of Protein Kinase CK2 Accelerates the Degradation of CDC25A Phosphatase through the Checkpoint Kinase Chk1. Int. J. Oncol. 2007, 31, 1251–1259. [Google Scholar]
- Chen, M.-S.; Ryan, C.E.; Piwnica-Worms, H. Chk1 Kinase Negatively Regulates Mitotic Function of Cdc25A Phosphatase through 14-3-3 Binding. Mol. Cell. Biol. 2003, 23, 7488–7497. [Google Scholar] [CrossRef]
- Hu, T.; Zhou, R.; Zhao, Y.; Wu, G. Integrin A6/Akt/Erk Signaling Is Essential for Human Breast Cancer Resistance to Radiotherapy. Sci. Rep. 2016, 6, 33376. [Google Scholar] [CrossRef] [PubMed]
- Mizuta, K.; Matsubara, T.; Goto, A.; Addison, W.N.; Nakatomi, M.; Matsuo, K.; Tada-Shigeyama, Y.; Yaginuma, T.; Honda, H.; Yoshioka, I.; et al. Plectin Promotes Tumor Formation by B16 Mouse Melanoma Cells via Regulation of Rous Sarcoma Oncogene Activity. BMC Cancer 2022, 22, 936. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Saito, H.; Imajoh-ohmi, S.; Kaminishi, M.; Seto, Y.; Miki, Y.; Nakanishi, A. BRCA2 Interacts with the Cytoskeletal Linker Protein Plectin to Form a Complex Controlling Centrosome Localization. Cancer Sci. 2009, 100, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Foisner, R.; Malecz, N.; Dressel, N.; Stadler, C.; Wiche, G. M-Phase-Specific Phosphorylation and Structural Rearrangement of the Cytoplasmic Cross-Linking Protein Plectin Involve P34cdc2 Kinase. Mol. Biol. Cell 1996, 7, 273–288. [Google Scholar] [CrossRef]
- Valencia, R.G.; Walko, G.; Janda, L.; Novacek, J.; Mihailovska, E.; Reipert, S.; Andrä-Marobela, K.; Wiche, G. Intermediate Filament-Associated Cytolinker Plectin 1c Destabilizes Microtubules in Keratinocytes. Mol. Biol. Cell 2013, 24, 768–784. [Google Scholar] [CrossRef]
- Burgstaller, G.; Gregor, M.; Winter, L.; Wiche, G. Keeping the Vimentin Network under Control: Cell-Matrix Adhesion-Associated Plectin 1f Affects Cell Shape and Polarity of Fibroblasts. Mol. Biol. Cell 2010, 21, 3362–3375. [Google Scholar] [CrossRef]
- Perez, S.M.; Dimastromatteo, J.; Landen, C.N.; Kelly, K.A. A Novel Monoclonal Antibody Targeting Cancer-Specific Plectin Has Potent Antitumor Activity in Ovarian Cancer. Cells 2021, 10, 2218. [Google Scholar] [CrossRef]
- Wilhelmsen, K.; Litjens, S.H.M.; Kuikman, I.; Tshimbalanga, N.; Janssen, H.; van den Bout, I.; Raymond, K.; Sonnenberg, A. Nesprin-3, a Novel Outer Nuclear Membrane Protein, Associates with the Cytoskeletal Linker Protein Plectin. J. Cell Biol. 2005, 171, 799–810. [Google Scholar] [CrossRef]
- Foisner, R.; Traub, P.; Wiche, G. Protein Kinase A- and Protein Kinase C-Regulated Interaction of Plectin with Lamin B and Vimentin. Proc. Natl. Acad. Sci. USA 1991, 88, 3812–3816. [Google Scholar] [CrossRef]
- Serres, M.P.; Samwer, M.; Quang, B.A.T.; Lavoie, G.; Perera, U.; Görlich, D.; Charras, G.; Petronczki, M.; Roux, P.P.; Paluch, E.K. F-Actin Interactome Reveals Vimentin as a Key Regulator of Actin Organization and Cell Mechanics in Mitosis. Dev. Cell 2020, 52, 210–222. [Google Scholar] [CrossRef]
- Wei, L.; Yin, F.; Chen, C.; Li, L. Expression of Integrin A-6 Is Associated with Multi Drug Resistance and Prognosis in Ovarian Cancer. Oncol. Lett. 2019, 17, 3974–3980. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, N.; Kaneda, K.; Saito, Y.; Ichihara, E.; Morishita, K. The Increased Expression of Integrin A6 (ITGA6) Enhances Drug Resistance in EVI1high Leukemia. PLoS ONE 2012, 7, e30706. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Calin, G.A.; Lopez-Berestein, G.; Sood, A.K. MiRNA Deregulation in Cancer Cells and the Tumor Microenvironment. Cancer Discov. 2016, 6, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Zoni, E.; van der Horst, G.; van de Merbel, A.F.; Chen, L.; Rane, J.K.; Pelger, R.C.M.; Collins, A.T.; Visakorpi, T.; Snaar-Jagalska, B.E.; Maitland, N.J.; et al. MiR-25 Modulates Invasiveness and Dissemination of Human Prostate Cancer Cells via Regulation of Av- and A6-Integrin Expression. Cancer Res. 2015, 75, 2326–2336. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.-P.; Hu, Y.-P.; Wu, X.-S.; Wu, Y.-S.; Ye, Y.-Y.; Li, H.-F.; Liu, Y.-C.; Jiang, L.; Liu, F.-T.; Zhang, Y.-J.; et al. MiR-143-3p Targeting of ITGA6 Suppresses Tumour Growth and Angiogenesis by Downregulating PLGF Expression via the PI3K/AKT Pathway in Gallbladder Carcinoma. Cell Death Dis. 2018, 9, 182. [Google Scholar] [CrossRef]
- Laudato, S.; Patil, N.; Abba, M.L.; Leupold, J.H.; Benner, A.; Gaiser, T.; Marx, A.; Allgayer, H. P53-Induced MiR-30e-5p Inhibits Colorectal Cancer Invasion and Metastasis by Targeting ITGA6 and ITGB1: MiR-30e-5p Inhibits CRC Metastasis. Int. J. Cancer 2017, 141, 1879–1890. [Google Scholar] [CrossRef]
- Wang, Y.; Li, L.; Zhang, X.; Zhao, X. Inhibited Long Non-Coding RNA OIP5-AS1 Elevated MicroRNA-92a to Suppress Proliferation and Metastasis of Ovarian Cancer Cells by Silencing ITGA6. 2020; in review. [Google Scholar]
- Hozaka, Y.; Seki, N.; Tanaka, T.; Asai, S.; Moriya, S.; Idichi, T.; Wada, M.; Tanoue, K.; Kawasaki, Y.; Mataki, Y.; et al. Molecular Pathogenesis and Regulation of the MiR-29-3p-Family: Involvement of ITGA6 and ITGB1 in Intra-Hepatic Cholangiocarcinoma. Cancers 2021, 13, 2804. [Google Scholar] [CrossRef]
- Landowski, T.H.; Gard, J.; Pond, E.; Pond, G.D.; Nagle, R.B.; Geffre, C.P.; Cress, A.E. Targeting Integrin A6 Stimulates Curative-Type Bone Metastasis Lesions in a Xenograft Model. Mol. Cancer Ther. 2014, 13, 1558–1566. [Google Scholar] [CrossRef]
- Hogervorst, F.; Kuikman, I.; Noteboom, E.; Sonnenberg, A. The Role of Phosphorylation in Activation of the Alpha 6A Beta 1 Laminin Receptor. J. Biol. Chem. 1993, 268, 18427–18430. [Google Scholar] [CrossRef]
- Damsky, C.H.; Librach, C.; Lim, K.H.; Fitzgerald, M.L.; McMaster, M.T.; Janatpour, M.; Zhou, Y.; Logan, S.K.; Fisher, S.J. Integrin Switching Regulates Normal Trophoblast Invasion. Development 1994, 120, 3657–3666. [Google Scholar] [CrossRef]
- Chen, X.; Zhou, H.; Li, X.; Duan, N.; Hu, S.; Liu, Y.; Yue, Y.; Song, L.; Zhang, Y.; Li, D.; et al. Plectin-1 Targeted Dual-Modality Nanoparticles for Pancreatic Cancer Imaging. EBioMedicine 2018, 30, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Bausch, D.; Thomas, S.; Mino-Kenudson, M.; Fernández-del, C.C.; Bauer, T.W.; Williams, M.; Warshaw, A.L.; Thayer, S.P.; Kelly, K.A. Plectin-1 as a Novel Biomarker for Pancreatic Cancer. Clin. Cancer Res. 2011, 17, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Wernitznig, D.; Meier-Menches, S.M.; Cseh, K.; Theiner, S.; Wenisch, D.; Schweikert, A.; Jakupec, M.A.; Koellensperger, G.; Wernitznig, A.; Sommergruber, W.; et al. Plecstatin-1 Induces an Immunogenic Cell Death Signature in Colorectal Tumour Spheroids. Metallomics 2020, 12, 2121–2133. [Google Scholar] [CrossRef] [PubMed]
- Meier, S.M.; Kreutz, D.; Winter, L.; Klose, M.H.M.; Cseh, K.; Weiss, T.; Bileck, A.; Alte, B.; Mader, J.C.; Jana, S.; et al. An Organoruthenium Anticancer Agent Shows Unexpected Target Selectivity For Plectin. Angew. Chem. Int. Ed. 2017, 56, 8267–8271. [Google Scholar] [CrossRef] [PubMed]
- Meier-Menches, S.M.; Zappe, K.; Bileck, A.; Kreutz, D.; Tahir, A.; Cichna-Markl, M.; Gerner, C. Time-Dependent Shotgun Proteomics Revealed Distinct Effects of an Organoruthenium Prodrug and Its Activation Product on Colon Carcinoma Cells. Metallomics 2019, 11, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Dowling, J.; Yu, Q.C.; Fuchs, E. Beta4 Integrin Is Required for Hemidesmosome Formation, Cell Adhesion and Cell Survival. J. Cell Biol. 1996, 134, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Murgia, C.; Blaikie, P.; Kim, N.; Dans, M.; Petrie, H.T.; Giancotti, F.G. Cell Cycle and Adhesion Defects in Mice Carrying a Targeted Deletion of the Integrin B4 Cytoplasmic Domain. EMBO J. 1998, 17, 3940–3951. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Price, T.T.; Cantelli, G.; Ngo, B.; Warner, M.J.; Olivere, L.; Ridge, S.M.; Jablonski, E.M.; Therrien, J.; Tannheimer, S.; et al. Leukaemia Hijacks a Neural Mechanism to Invade the Central Nervous System. Nature 2018, 560, 55–60. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koivusalo, S.; Schmidt, A.; Manninen, A.; Wenta, T. Regulation of Kinase Signaling Pathways by α6β4-Integrins and Plectin in Prostate Cancer. Cancers 2023, 15, 149. https://doi.org/10.3390/cancers15010149
Koivusalo S, Schmidt A, Manninen A, Wenta T. Regulation of Kinase Signaling Pathways by α6β4-Integrins and Plectin in Prostate Cancer. Cancers. 2023; 15(1):149. https://doi.org/10.3390/cancers15010149
Chicago/Turabian StyleKoivusalo, Saara, Anette Schmidt, Aki Manninen, and Tomasz Wenta. 2023. "Regulation of Kinase Signaling Pathways by α6β4-Integrins and Plectin in Prostate Cancer" Cancers 15, no. 1: 149. https://doi.org/10.3390/cancers15010149
APA StyleKoivusalo, S., Schmidt, A., Manninen, A., & Wenta, T. (2023). Regulation of Kinase Signaling Pathways by α6β4-Integrins and Plectin in Prostate Cancer. Cancers, 15(1), 149. https://doi.org/10.3390/cancers15010149