

A Novel Bispecific Antibody for EpCAM-Directed Inhibition of the CD73/Adenosine Immune Checkpoint in Ovarian Cancer
 , ,
, ,
Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
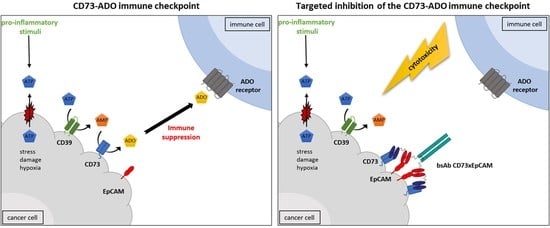
1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Cell Lines, Primary Patient-Derived OC Cells, and Transfectants
2.3. Construction of bsAb CD73xEpCAM-IgG2silent
2.4. Production of Recombinant bsAbs
2.5. Assessment Dual Binding Activity of bsAb CD73xEpCAM
2.6. Assessment of Internalization of bsAb CD73xEpCAM/Antigen Complexes
2.7. Assessment Capacity of bsAb CD73xEpCAM to Inhibit the Enzyme Activity of CD73
2.8. Assessment Capacity of bsAb CD73xEpCAM to Restore Proliferation Capacity of ADO-Suppressed T Cells
2.9. Assessment Capacity of bsAb CD73xEpCAM to Restore Anticancer Activity of ADO-Suppressed T Cells
2.10. Assessment Inhibitory Effect of bsAb CD73xEpCAM on Proliferation of Cancer Cells
2.11. Assessment Sensitization of Cancer Cells by bsAb CD73xEpCAM for Chemotherapeutic Agents and Ionizing Radiation
2.12. Statistical Analysis
3. Results
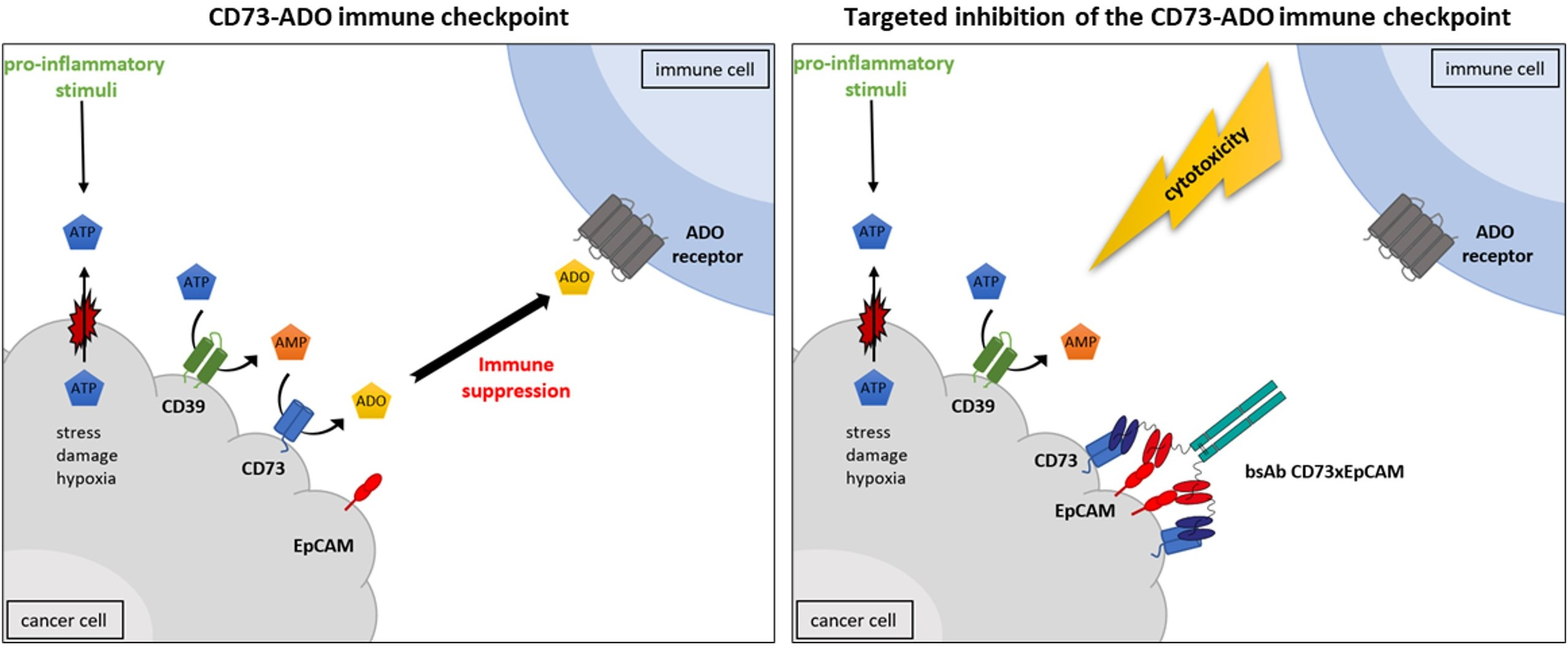
3.1. BsAb CD73xEpCAM Has Dual Binding Specificity for OC-Exposed CD73 and EpCAM
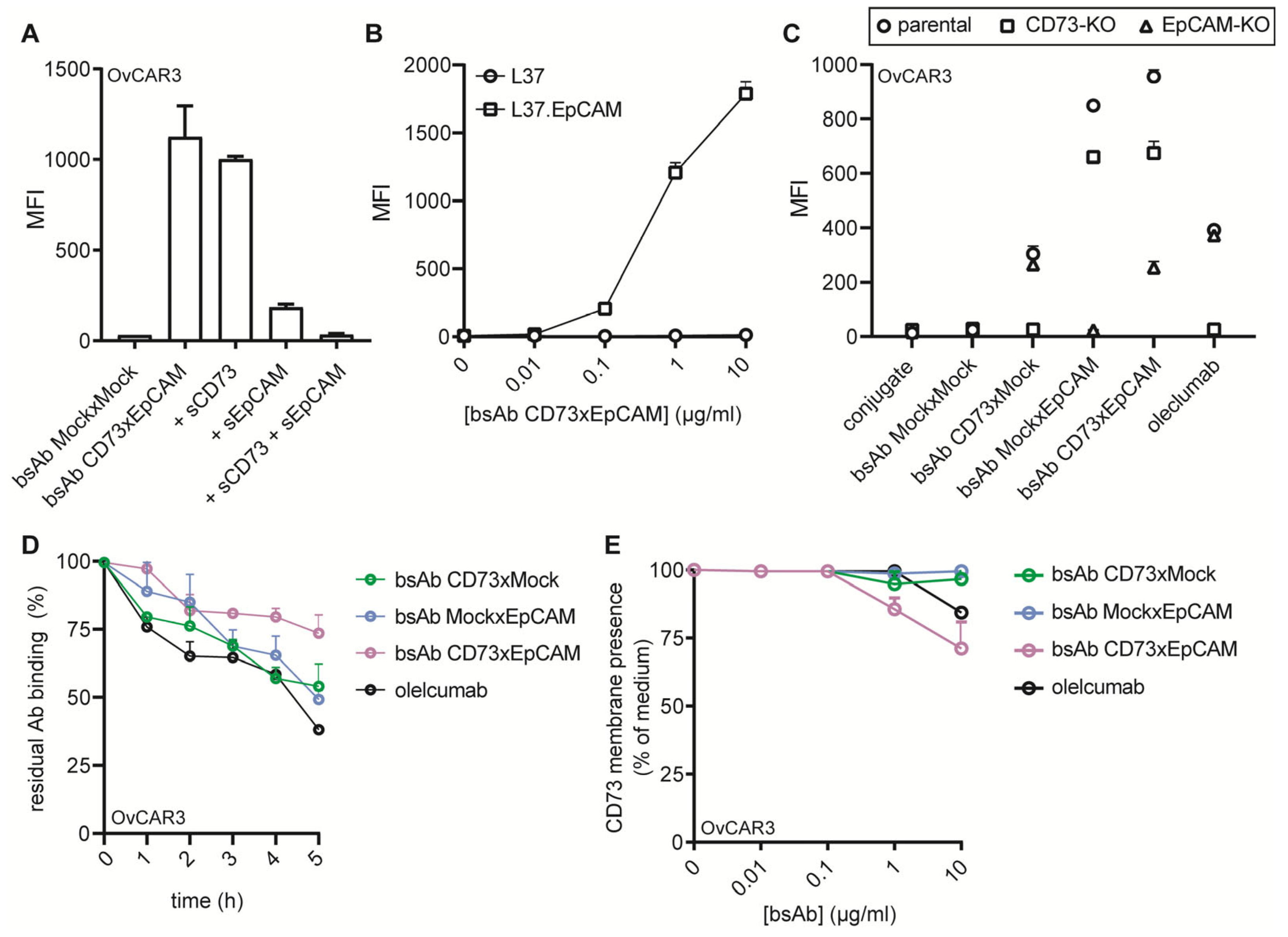
3.2. BsAb CD73xEpCAM Treatment Potently Inhibits CD73 Enzyme Activity in an EpCAM-Directed Manner
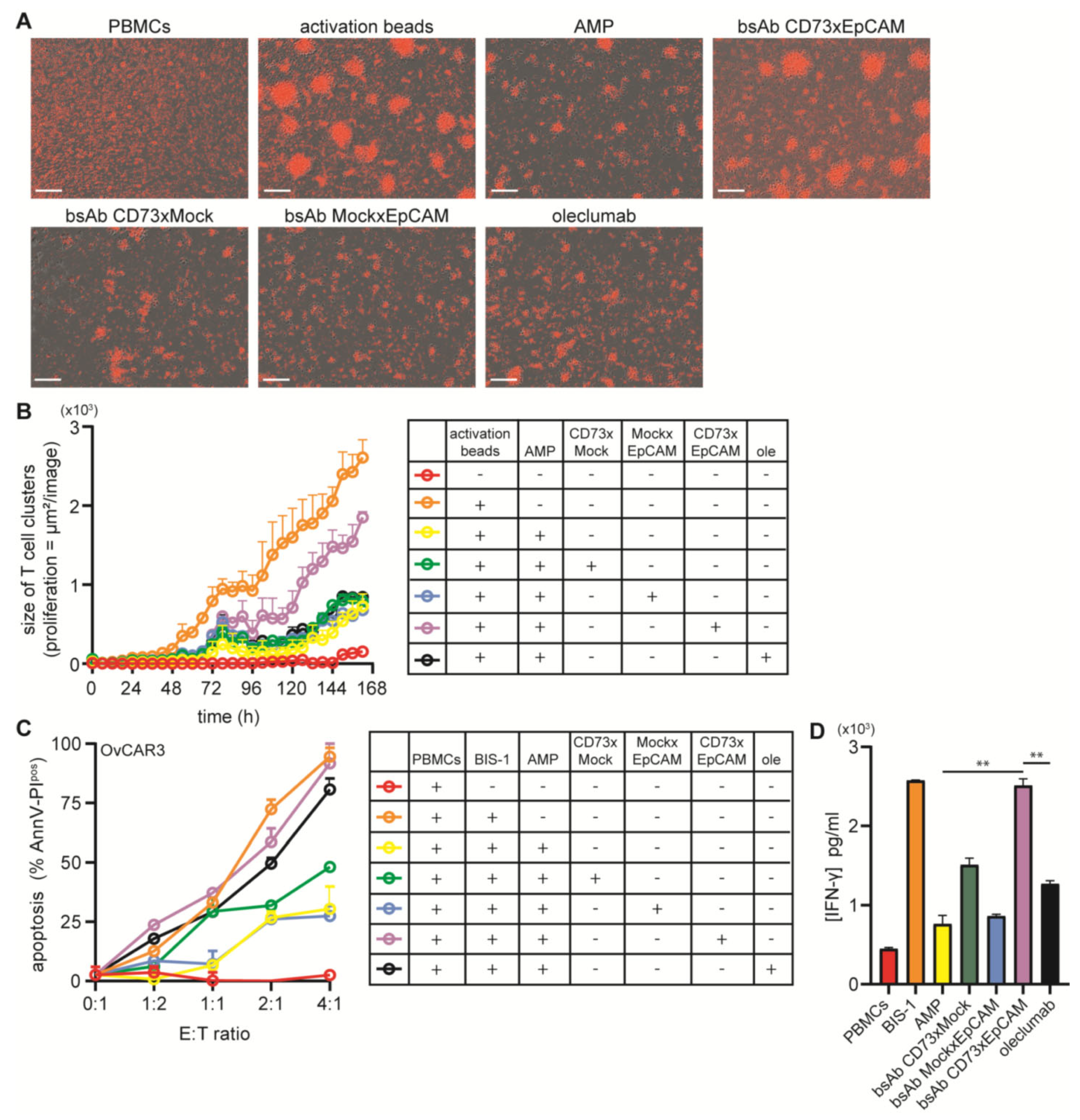
3.3. BsAb CD73xEpCAM Treatment Overcomes ADO-Mediated Suppression of T Cell Proliferation
3.4. BsAb CD73xEpCAM Treatment Restores Anticancer Activity of ADO-Suppressed Cytotoxic T Cells
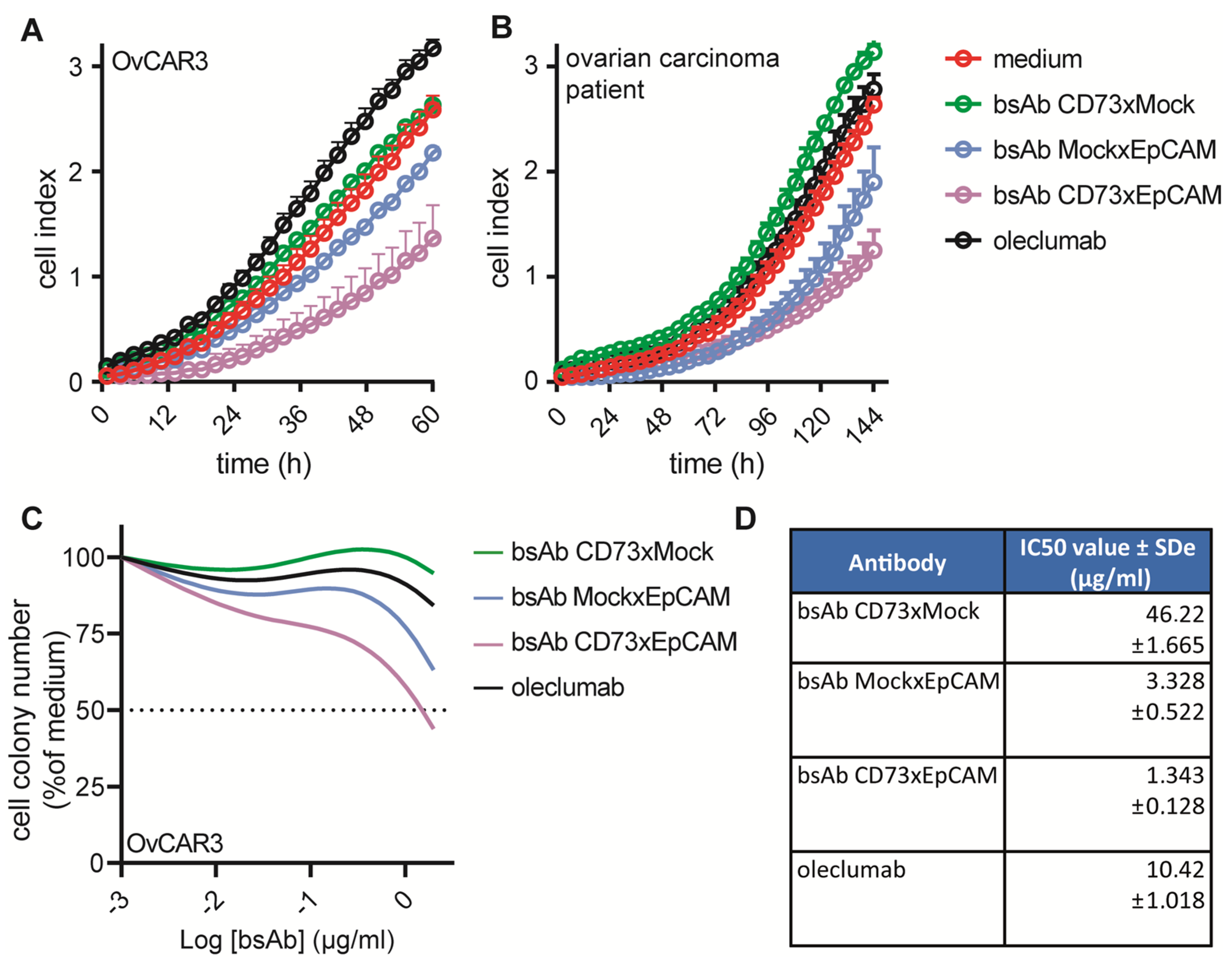
3.5. BsAb CD73xEpCAM Treatment Inhibits the Proliferative and Colony-Forming Capacity of OC Cells
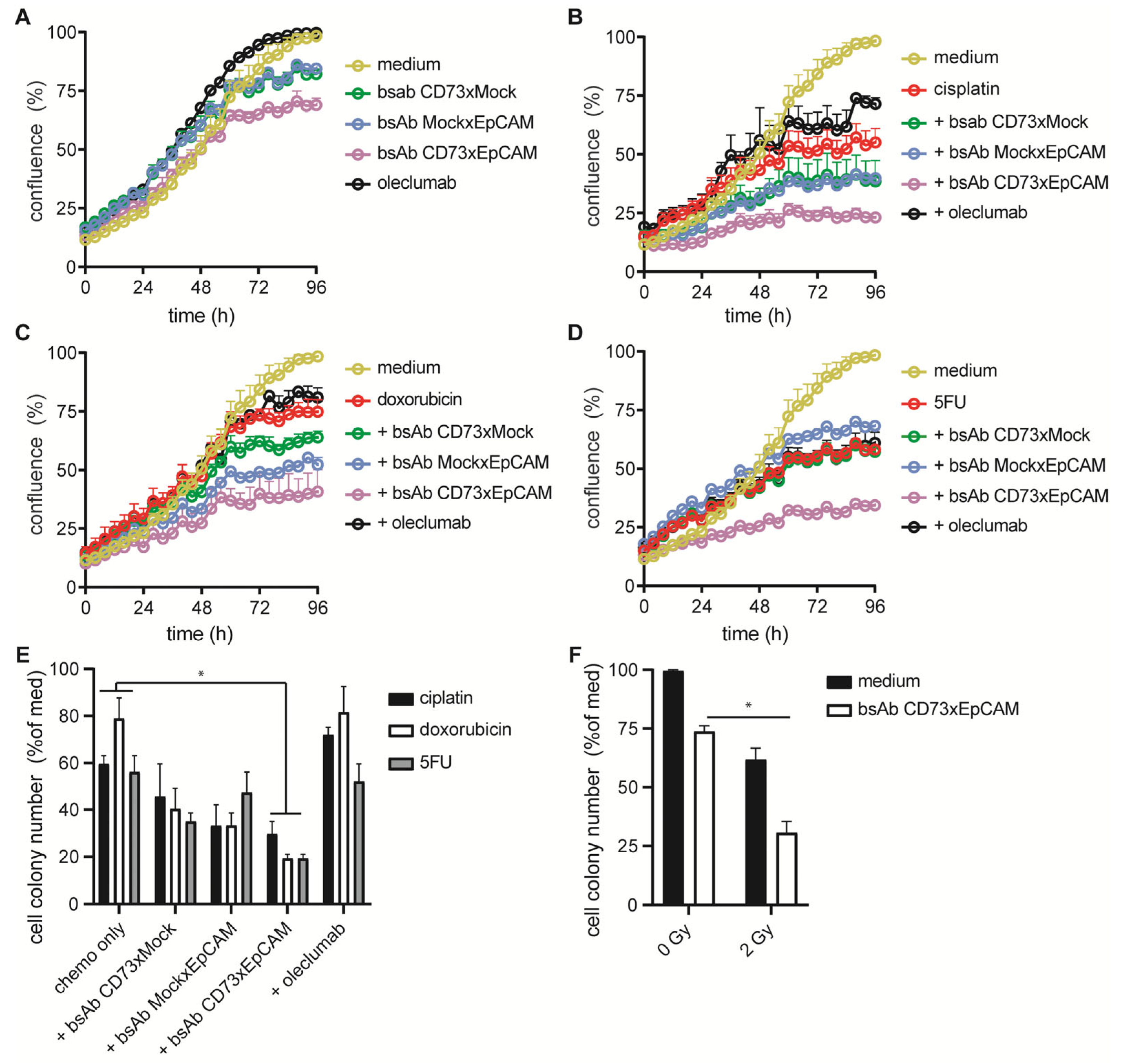
3.6. BsAb CD73xEpCAM Treatment Sensitizes OC Cells towards Chemotherapeutic Agents and Ionizing Radiation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Allard, D.; Allard, B.; Gaudreau, P.-O.; Chrobak, P.; Stagg, J. CD73–adenosine: A next-generation target in immuno-oncology. Immunotherapy 2016, 8, 145–163. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Allard, D.; Buisseret, L.; Stagg, J. The adenosine pathway in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Blandizzi, C.; Malavasi, F.; Ferrari, D.; Haskó, G. Anti-CD73 immunotherapy: A viable way to reprogram the tumor microenvironment. Oncoimmunology 2016, 5, e1216292. [Google Scholar] [CrossRef] [PubMed]
- LaFleur, M.W.; Muroyama, Y.; Drake, C.G.; Sharpe, A.H. Inhibitors of the PD-1 Pathway in Tumor Therapy. J. Immunol. 2018, 200, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Shaw, A.T.; Sequist, L.V.; Fu, X.; Azzoli, C.G.; Piotrowska, Z.; Huynh, T.G.; Zhao, L.; Fulton, L.; Schultz, K.R.; et al. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non-Small Cell Lung Cancer: A Retrospective Analysis. Clin. Cancer Res. 2016, 22, 4585–4593. [Google Scholar] [CrossRef]
- Antonios, J.P.; Soto, H.; Everson, R.G.; Moughon, D.; Orpilla, J.R.; Shin, N.P.; Sedighim, S.; Treger, J.; Odesa, S.; Tucker, A.; et al. Immunosuppressive tumor-infiltrating myeloid cells mediate adaptive immune resistance via a PD-1/PD-L1 mechanism in glioblastoma. Neuro Oncol. 2017, 19, 796–807. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Hay, C.M.; Sult, E.; Huang, Q.; Mulgrew, K.; Fuhrmann, S.R.; McGlinchey, K.A.; Hammond, S.A.; Rothstein, R.; Rios-Doria, J.; Poon, E.; et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology 2016, 5, e1208875. [Google Scholar] [CrossRef]
- Keizer, R.J.; Huitema, A.D.R.; Schellens, J.H.M.; Beijnen, J.H. Clinical Pharmacokinetics of Therapeutic Monoclonal Antibodies. Clin. Pharmacokinet. 2010, 49, 493–507. [Google Scholar] [CrossRef]
- Liu, B.; Song, S.; Setroikromo, R.; Chen, S.; Hu, W.; Chen, D.; van der Wekken, A.J.; Melgert, B.N.; Timens, W.; Berg, A.V.D.; et al. CX Chemokine Receptor 7 Contributes to Survival of KRAS-Mutant Non-Small Cell Lung Cancer upon Loss of Epidermal Growth Factor Receptor. Cancers 2019, 11, 455. [Google Scholar] [CrossRef]
- Zhang, J.-P.; Li, X.-L.; Neises, A.; Chen, W.; Hu, L.-P.; Ji, G.-Z.; Yu, J.-Y.; Xu, J.; Yuan, W.-P.; Cheng, T.; et al. Different Effects of sgRNA Length on CRISPR-mediated Gene Knockout Efficiency. Sci. Rep. 2016, 6, 28566. [Google Scholar] [CrossRef] [PubMed]
- Rossotti, M.A.; Henry, K.A.; van Faassen, H.; Tanha, J.; Callaghan, D.; Hussack, G.; Arbabi-Ghahroudi, M.; MacKenzie, C.R. Camelid single-domain antibodies raised by DNA immunization are potent inhibitors of EGFR signaling. Biochem. J. 2019, 476, 39–50. [Google Scholar]
- Ploeg, E.M.; Ke, X.; Britsch, I.; Hendriks, M.A.J.M.; Van der Zant, F.A.; Kruijff, S.; Samplonius, D.F.; Zhang, H.; Helfrich, W. Bispecific antibody CD73xEpCAM selectively inhibits the adenosine-mediated immunosuppressive activity of carcinoma-derived extracellular vesicles. Cancer Lett. 2021, 521, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Ke, X.; Wang, L.; Lin, Y.; Wang, S.; Yao, Z.; Li, K.; Luo, Y.; Liu, F.; Pan, Y.; et al. Neoantigen-based cancer vaccination using chimeric RNA-loaded dendritic cell-derived extracellular vesicles. J. Extracell. Vesicles 2022, 11, e12243. [Google Scholar] [CrossRef]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. mAbs 2017, 9, 182–212. [Google Scholar] [CrossRef]
- Gao, Z.w.; Dong, K.; Zhang, H.z. The roles of CD73 in cancer. Biomed. Res. Int. 2014, 2014, 460654. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Iwasa, S.; Koyama, T.; Fujita, T.; Sugibayashi, K.; Murayama, K.; Yamamoto, N. Safety, tolerability, pharmacokinetics, and antitumour activity of oleclumab in Japanese patients with advanced solid malignancies: A phase I, open-label study. Int. J. Clin. Oncol. 2022, 27, 1795–1804. [Google Scholar] [CrossRef]
- Mirza, M.; Henriksen, J.R.; Maenpaa, J.; Depont Christensen, R.; Waldstroem, M.; Tandaric, L.; Lindemann, K.; Roed, H.; Auranen, A.; Akslen, L.; et al. 1195 Results of NSGO-OV-UMB1/ENGOT-OV30 study: A phase II study of durvalumab and oleclumab in patients with relapsed ovarian cancer (OC). Int. J. Gynecol. Cancer 2021, 31 (Suppl. 3), A376. Available online: http://ijgc.bmj.com/content/31/Suppl_3/A376.2.abstract (accessed on 25 October 2021).
- Koopmans, I.; Hendriks, D.; Samplonius, D.F.; van Ginkel, R.J.; Heskamp, S.; Wierstra, P.J.; Bremer, E.; Helfrich, W. A novel bispecific antibody for EGFR-directed blockade of the PD-1/PD-L1 immune checkpoint. Oncoimmunology 2018, 7, e1466016. [Google Scholar] [CrossRef]
- Koopmans, I.; Hendriks, M.A.J.M.; van Ginkel, R.J.; Samplonius, D.F.; Bremer, E.; Helfrich, W. Bispecific Antibody Approach for Improved Melanoma-Selective PD-L1 Immune Checkpoint Blockade. J. Invest. Dermatol. 2019, 139, 2343–2351. [Google Scholar] [CrossRef]
- Van Bommel, P.E.; He, Y.; Schepel, I.; Hendriks, M.A.J.M.; Wiersma, V.R.; van Ginkel, R.J.; van Meerten, T.; Ammatuna, E.; Huls, G.; Samplonius, D.F.; et al. CD20-selective inhibition of CD47-SIRPα “don’t eat me” signaling with a bispecific antibody-derivative enhances the anticancer activity of daratumumab, alemtuzumab and obinutuzumab. Oncoimmunology 2018, 7, e1386361. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, M.A.J.M.; Ploeg, E.M.; Koopmans, I.; Britsch, I.; Ke, X.; Samplonius, D.F.; Helfrich, W. Bispecific antibody approach for EGFR-directed blockade of the CD47-SIRPα “don’t eat me” immune checkpoint promotes neutrophil-mediated trogoptosis and enhances antigen cross-presentation. Oncoimmunology 2020, 9, 1824323. [Google Scholar] [CrossRef] [PubMed]
- Somaiah, N.; Livingston, J.A.A.; Ravi, V.; Lin, H.Y.; Amini, B.; Solis, L.M.; Conley, A.P.; Zarzour, M.A.; Ludwig, J.A.; Ratan, R.; et al. A phase II multi-arm study to test the efficacy of oleclumab and durvalumab in specific sarcoma subtypes. J. Clin. Oncol. 2022, 40, TPS11594. [Google Scholar] [CrossRef]
- Overman, M.J.; Lorusso, P.; Strickler, J.H.; Patel, S.P.; Clarke, S.J.; Noonan, A.M.; Prasanna, T.; Amin, M.A.; Nemunaitis, J.J.; Desai, J.; et al. Safety, efficacy and pharmacodynamics (PD) of MEDI9447 (oleclumab) alone or in combination with durvalumab in advanced colorectal cancer (CRC) or pancreatic cancer (panc). J. Clin. Oncol. 2018, 36, 4123. [Google Scholar] [CrossRef]
- Selvaggi, G.; Novello, S.; Torri, V.; Leonardo, E.; De Giuli, P.; Borasio, P.; Mossetti, C.; Ardissone, F.; Lausi, P.; Scagliotti, G.V. Epidermal growth factor receptor overexpression correlates with a poor prognosis in completely resected non-small-cell lung cancer. Ann. Oncol. 2004, 15, 28–32. [Google Scholar] [CrossRef]
- Yoshida, T.; Okamoto, I.; Okabe, T.; Iwasa, T.; Satoh, T.; Nishio, K.; Fukuoka, M.; Nakagawa, K. Matuzumab and cetuximab activate the epidermal growth factor receptor but fail to trigger downstream signaling by Akt or Erk. Int. J. Cancer 2008, 122, 1530–1538. [Google Scholar] [CrossRef]
- Zhi, X.; Wang, Y.; Yu, J.; Yu, J.; Zhang, L.; Yin, L.; Zhou, P. Potential prognostic biomarker CD73 regulates epidermal growth factor receptor expression in human breast cancer. IUBMB Life 2012, 64, 911–920. [Google Scholar] [CrossRef]
- Griesing, S.; Liao, B.-C.; Yang, J.C.-H. CD73 Is Regulated by the EGFR-ERK Signaling Pathway in Non-small Cell Lung Cancer. Anticancer Res. 2021, 41, 1231–1242. [Google Scholar] [CrossRef]
- Tu, E.; McGlinchey, K.; Wang, J.; Martin, P.; Ching, S.L.; Floc’h, N.; Kurasawa, J.; Starrett, J.H.; Lazdun, Y.; Wetzel, L.; et al. Anti–PD-L1 and anti-CD73 combination therapy promotes T cell response to EGFR-mutated NSCLC. J. Clin. Investig. 2022, 7, e142843. [Google Scholar] [CrossRef] [PubMed]
- Vafa, O.; Gilliland, G.L.; Brezski, R.J.; Strake, B.; Wilkinson, T.; Lacy, E.R.; Scallon, B.; Teplyakov, A.; Malia, T.J.; Strohl, W.R. An engineered Fc variant of an IgG eliminates all immune effector functions via structural perturbations. Methods 2014, 65, 114–126. [Google Scholar] [CrossRef]
- Gao, Z.-W.; Wang, H.-P.; Lin, F.; Wang, X.; Long, M.; Zhang, H.-Z.; Dong, K. CD73 promotes proliferation and migration of human cervical cancer cells independent of its enzyme activity. BMC Cancer 2017, 17, 135. [Google Scholar] [CrossRef] [PubMed]
- Biscardi, J.S.; Maa, M.-C.; Tice, D.A.; Cox, M.E.; Leu, T.-H.; Parsons, S.J. c-Src-mediated Phosphorylation of the Epidermal Growth Factor Receptor on Tyr845 and Tyr1101 Is Associated with Modulation of Receptor Function. J. Biol. Chem. 1999, 274, 8335–8343. [Google Scholar] [CrossRef] [PubMed]
- Dianzani, U.; Redoglia, V.; Bragardo, M.; Attisano, C.; Bianchi, A.; Di Franco, D.; Ramenghi, U.; Wolff, H.; Thompson, L.F.; Pileri, A.; et al. Co-stimulatory signal delivered by CD73 molecule to human CD45RAhiCD45ROlo (naive) CD8+ T lymphocytes. J. Immunol. 1993, 151, 3961–3970. [Google Scholar] [CrossRef]
- Airas, L.; Niemelä, J.; Salmi, M.; Puurunen, T.; Smith, D.J.; Jalkanen, S. Differential regulation and function of CD73, a glycosyl-phosphatidylinositol-linked 70-kD adhesion molecule, on lymphocytes and endothelial cells. J. Cell Biol. 1997, 136, 421–431. [Google Scholar] [CrossRef]
- Samanta, D.; Park, Y.; Ni, X.; Li, H.; Zahnow, C.A.; Gabrielson, E.; Pan, F.; Semenza, G.L. Chemotherapy induces enrichment of CD47+/CD73+/PDL1+ immune evasive triple-negative breast cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, e1239–e1248. [Google Scholar] [CrossRef] [PubMed]
- Ujházy, P.; Klobusická, M.; Babusíková, O.; Strausbauch, P.; Mihich, E.; Ehrke, M.J. Ecto-5′-nucleotidase (CD73) in multidrug-resistant cell lines generated by doxorubicin. Int. J. Cancer 1994, 59, 83–93. [Google Scholar] [CrossRef]
- Loi, S.; Pommey, S.; Haibe-Kains, B.; Beavis, P.A.; Darcy, P.K.; Smyth, M.J.; Stagg, J. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 11091–11096. [Google Scholar] [CrossRef]
- Dietrich, F.; Figueiró, F.; Filippi-Chiela, E.C.; Cappellari, A.R.; Rockenbach, L.; Tremblay, A.; de Paula, P.B.; Roesler, R.; Filho, A.B.; Sévigny, J.; et al. Ecto-5’-nucleotidase/CD73 contributes to the radiosensitivity of T24 human bladder cancer cell line. J. Cancer Res. Clin. Oncol. 2018, 144, 469–482. [Google Scholar] [CrossRef]
- Mazor, Y.; Hansen, A.; Yang, C.; Chowdhury, P.S.; Wang, J.; Stephens, G.; Wu, H.; Dall’Acqua, W.F. Insights into the molecular basis of a bispecific antibody’s target selectivity. MAbs 2015, 7, 461–469. [Google Scholar] [CrossRef]
- Dong, J.; Sereno, A.; Aivazian, D.; Langley, E.; Miller, B.R.; Snyder, W.B.; Chan, E.; Cantele, M.; Morena, R.; Joseph, I.B.; et al. A stable IgG-like bispecific antibody targeting the epidermal growth factor receptor and the type I insulin-like growth factor receptor demonstrates superior anti-tumor activity. MAbs 2011, 3, 273–288. [Google Scholar] [CrossRef]
- Winter, M.J.; Nagelkerken, B.; Mertens, A.E.; Rees-Bakker, H.A.; Bruijn, I.H.B.-D.; Litvinov, S.V. Expression of Ep-CAM shifts the state of cadherin-mediated adhesions from strong to weak. Exp. Cell Res. 2003, 285, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Tayama, S.; Motohara, T.; Narantuya, D.; Li, C.; Fujimoto, K.; Sakaguchi, I.; Tashiro, H.; Saya, H.; Nagano, O.; Katabuchi, H. The impact of EpCAM expression on response to chemotherapy and clinical outcomes in patients with epithelial ovarian cancer. Oncotarget 2017, 8, 44312–44325. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Cozzi, P.; Hao, J.; Beretov, J.; Chang, L.; Duan, W.; Shigdar, S.; Delprado, W.; Graham, P.; Bucci, J.; et al. Epithelial cell adhesion molecule (EpCAM) is associated with prostate cancer metastasis and chemo/radioresistance via the PI3K/Akt/mTOR signaling pathway. Int. J. Biochem. Cell Biol. 2013, 45, 2736–2748. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.M.; Zhou, J.; Sicairos, B.; Sonney, S.; Du, Y. Upregulation of CD73 Confers Acquired Radioresistance and is Required for Maintaining Irradiation-selected Pancreatic Cancer Cells in a Mesenchymal State. Mol. Cell. Proteom. 2020, 19, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Yan, Q.; Liu, S.; Yang, X. Knockdown of EpCAM Enhances the Chemosensitivity of Breast Cancer Cells to 5-fluorouracil by Downregulating the Antiapoptotic Factor Bcl-2. PLoS ONE 2014, 9, e102590. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ploeg, E.M.; Britsch, I.; van Wijngaarden, A.P.; Ke, X.; Hendriks, M.A.J.M.; Samplonius, D.F.; Helfrich, W. A Novel Bispecific Antibody for EpCAM-Directed Inhibition of the CD73/Adenosine Immune Checkpoint in Ovarian Cancer. Cancers 2023, 15, 3651. https://doi.org/10.3390/cancers15143651
Ploeg EM, Britsch I, van Wijngaarden AP, Ke X, Hendriks MAJM, Samplonius DF, Helfrich W. A Novel Bispecific Antibody for EpCAM-Directed Inhibition of the CD73/Adenosine Immune Checkpoint in Ovarian Cancer. Cancers. 2023; 15(14):3651. https://doi.org/10.3390/cancers15143651
Chicago/Turabian StylePloeg, Emily Maria, Isabel Britsch, Anne Paulien van Wijngaarden, Xiurong Ke, Mark Alexander Johannes Martinus Hendriks, Douwe Freerk Samplonius, and Wijnand Helfrich. 2023. "A Novel Bispecific Antibody for EpCAM-Directed Inhibition of the CD73/Adenosine Immune Checkpoint in Ovarian Cancer" Cancers 15, no. 14: 3651. https://doi.org/10.3390/cancers15143651
APA StylePloeg, E. M., Britsch, I., van Wijngaarden, A. P., Ke, X., Hendriks, M. A. J. M., Samplonius, D. F., & Helfrich, W. (2023). A Novel Bispecific Antibody for EpCAM-Directed Inhibition of the CD73/Adenosine Immune Checkpoint in Ovarian Cancer. Cancers, 15(14), 3651. https://doi.org/10.3390/cancers15143651