1. Introduction
Natural killer (NK) cells are crucial in anticancer immunotherapy. The current trends of research on NK cell-mediated immunotherapy have focused on NK cell expansion, cytotoxicity, improved targeting, and lifespan extension [
1]. To be applied clinically, NK cells need to be expanded via a mass culture for 2 weeks or more. However, in patients with terminal cancer, this activation induction period is insufficient for NK cell expansion. According to recent studies, the culture period can be shortened by culturing cells in a medium containing cytokines, such as IL-2 and IL-15 [
2,
3], or by using a formulation containing feeder cells and cytokines [
4,
5]. However, despite the induction of proliferation and activation, large-scale proliferation is limited, owing to the cost and cellular phenotype/functional differences [
6]. Therefore, new strategies that can activate NK cell expansion are needed.
NK cell proliferation depends on the mitochondrial metabolic pathway [
7,
8]. Inactive NK cells mainly produce ATP through mitochondrial oxidative phosphorylation (OXPHOS) and have a relatively low basal metabolic rate [
7,
8]. However, when activated, glycolysis and OXPHOS increase ATP production [
7,
8]. Recent studies have shown that NK cell proliferation is inhibited when mitochondria-related signaling mechanisms are suppressed [
9]. In addition, the metabolic performance of normal and cytokine-stimulated NK cells is different, with both of these showing distinct mitochondrial polarization patterns [
10]. Therefore, mitochondrial remodeling of OXPHOS has been proposed to be an essential gatekeeper of NK cell function.
The decreased antitumor capacity of NK cells is associated with damaged mitochondria [
10]. Tumor-infiltrating NK cells exhibit highly fragmented mitochondria and reduced cytotoxicity and proliferation [
9]. Furthermore, NK cells from the tumor site of patients with liver cancer have small and fragmented mitochondria in their cytoplasm, whereas those from healthy individuals and the peripheral NK cells outside the tumor have normal, large, and tubular mitochondria [
11]. Mitochondrial fragmentation is correlated with reduced cytotoxicity and NK cell loss that leads to tumor evasion and a decreased NK cell-mediated tumor surveillance, reducing the survival rates of patients with liver cancer [
10,
11]. By regulating mitochondrial activity and effector functions, recent studies have demonstrated a reduction in cell cytotoxicity and cytokine production in PGC-1α-deficient-NK cells compared to those in naïve NK cells [
12]. In addition, the transcriptional regulation of mitochondria mediated by PGC-1α plays an important role in anticancer immunity [
12]. This evidence suggests that the mitochondrial function in NK cells plays a key role in innate immunity.
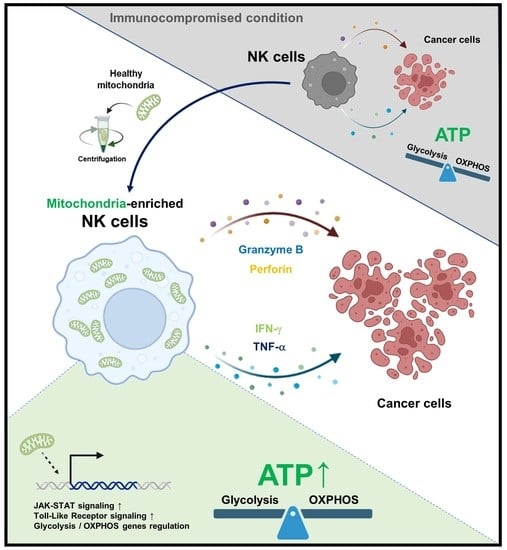
Recently, the transfer of healthy exogenous mitochondria to target cells has emerged as an attractive therapeutic strategy to treat damaged mitochondria. Studies have shown that mitochondria isolated from various sources can be transferred to damaged cells or tissues to restore the loss of function [
13,
14,
15,
16,
17]. A recent study on immune cells demonstrated the activation of T cells and the promotion of regulatory T cell differentiation through the mesenchymal stem cell-mediated transfer of mitochondria [
18]. However, to the best of our knowledge, no study to date has verified the efficacy of the direct transfer of mitochondria into NK cells. Therefore, in this study, we aimed to investigate whether the transfer of healthy exogenous mitochondria into NK cells affected their activity, in particular, NK cell expansion and anticancer regulatory ability. To enhance the mitochondrial capacity, mitochondria were isolated from normal hepatic cells (WRL-68) derived from liver tissue with high energy metabolism activity. The isolated mitochondria were transferred into NK cells using a simple centrifugation method described in a previous study [
19]. After the transfer, we evaluated the proliferative and cytotoxic capacities of NK cells and the changes in the NK cell surface receptors. We demonstrated that mitochondria-enriched NK cells, which do not require in vitro cytokine-induced culture, have the potential to be used as a novel solid cancer treatment agent.
2. Materials and Methods
2.1. Cell Culture
This in vitro study was approved by the Institutional Review Board (IRB) of CHA University (Seongnam, Republic of Korea; IRB No.202204-BR-018-02). For the cell culture experiments, NK-92MI and WRL-68 cells were purchased from the American Type Culture Collection (Manassas, VA, USA). K562, MCF-7, HCT-116, OVCAR-3, and A549 cells were purchased from the Korean Cell Line Bank (Seoul, Republic of Korea). Primary NK cells were obtained from donors using an NK cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany) and the Ficoll method. The NK-92MI cells were cultured in Minimum Essential Medium Eagle Alpha Modification (HyClone Laboratories Inc., Logan, UT, USA) supplemented with 12.5% fetal bovine serum (FBS; Gibco, Waltham, MA, USA), 12.5% horse serum (Gibco), 0.1 mM 2-mercaptoethanol (Gibco), 0.2 mM myo-inositol (Sigma-Aldrich, St. Louis, MO, USA), and 0.02 mM folic acid (Sigma-Aldrich). The WRL-68 cells were cultured in Dulbecco’s Eagle’s minimal essential medium (HyClone) supplemented with 10% FBS. The K562, MCF-7, HCT-116, OVCAR-3, and A549 cells were cultured in the RPMI-1640 medium (HyClone) supplemented with 10% FBS. Primary NK cells were cultured in ALys505NK (CSTI, Sendai, Japan) supplemented with 10% FBS, 200 IU/mL IL-2, 5 ng/mL IL-12, and 50 ng/mL IL-18. All cells were cultured at 37 °C under 5% CO2.
2.2. Isolation of the Mitochondria
Mitochondria were isolated using a method described in a previous study, with slight modifications [
19]. Briefly, the WRL-68 cells (1 × 10
7) were homogenized in the SHE buffer (0.25 M sucrose, 20 mM HEPES, 2 mM EGTA, 10 mM KCl, 1.5 mM MgCl
2, and 0.1% defatted bovine serum albumin; pH 7.4) using a 1 mL disposable syringe and centrifuged at 1500×
g for 5 min. To obtain the mitochondria, the supernatant was centrifuged at 20,000×
g for 10 min. The mitochondrial pellet was resuspended in 1.8 mL of the SHE buffer and centrifuged at 20,000×
g for 5 min. Then, the pellet was resuspended in 1.8 mL of the respiration buffer and centrifuged at 20,000×
g for 5 min to obtain pure mitochondria. All procedures were carried out on ice, and the freshly isolated mitochondria were stored at 4 °C.
2.3. Quality Control of the Mitochondria
The mitochondrial content was measured using the Pierce bicinchoninic acid protein assay kit (Thermo Scientific, Waltham, MA, USA). The ATP content of the isolated mitochondria was measured using the CellTiter-Glo 2.0 (Promega, Madison, WI, USA) reagent, following the manufacturer’s protocol. To assess its purity, the isolated mitochondria were labeled with MitoTracker Green (Invitrogen, Waltham, MA, USA) and APC-conjugated TOMM20 (Abcam, Cambridge, UK; ab225341), and were analyzed using the CytoFLEX flow cytometry (Beckman Coulter, CA, USA). Isolated mitochondria were characterized using Western blot analysis.
2.4. Transfer of Mitochondria to the Target Cells
The isolated mitochondria were transferred to the target cells, according to a method described in a previous study [
19]. Briefly, recipient cells were added to a tube containing 100 µL of phosphate-buffered saline (PBS). The number of mitochondria refers to the mass of mitochondria (µg of protein) per 1 × 10
5 recipient cells. The mitochondrial suspension in 10 µL of PBS was added slowly to each tube containing the recipient cells, and the tubes were then centrifuged at 1500×
g for 15 min. All the tubes were stored at 4 °C.
After mitochondrial transfer, the NK-92MI cells were seeded at a density of 1 × 105 cells/mL and cultured at 37 °C under 5% CO2. The NK-92MI cells were sub-cultured every 48 h. To track the intracellular retention period after the mitochondrial transfer, the NK-92MI cell mitochondrial DNA (mtDNA) expression, proliferative capacity, and cytotoxicity against K562 cells were analyzed at each passage point.
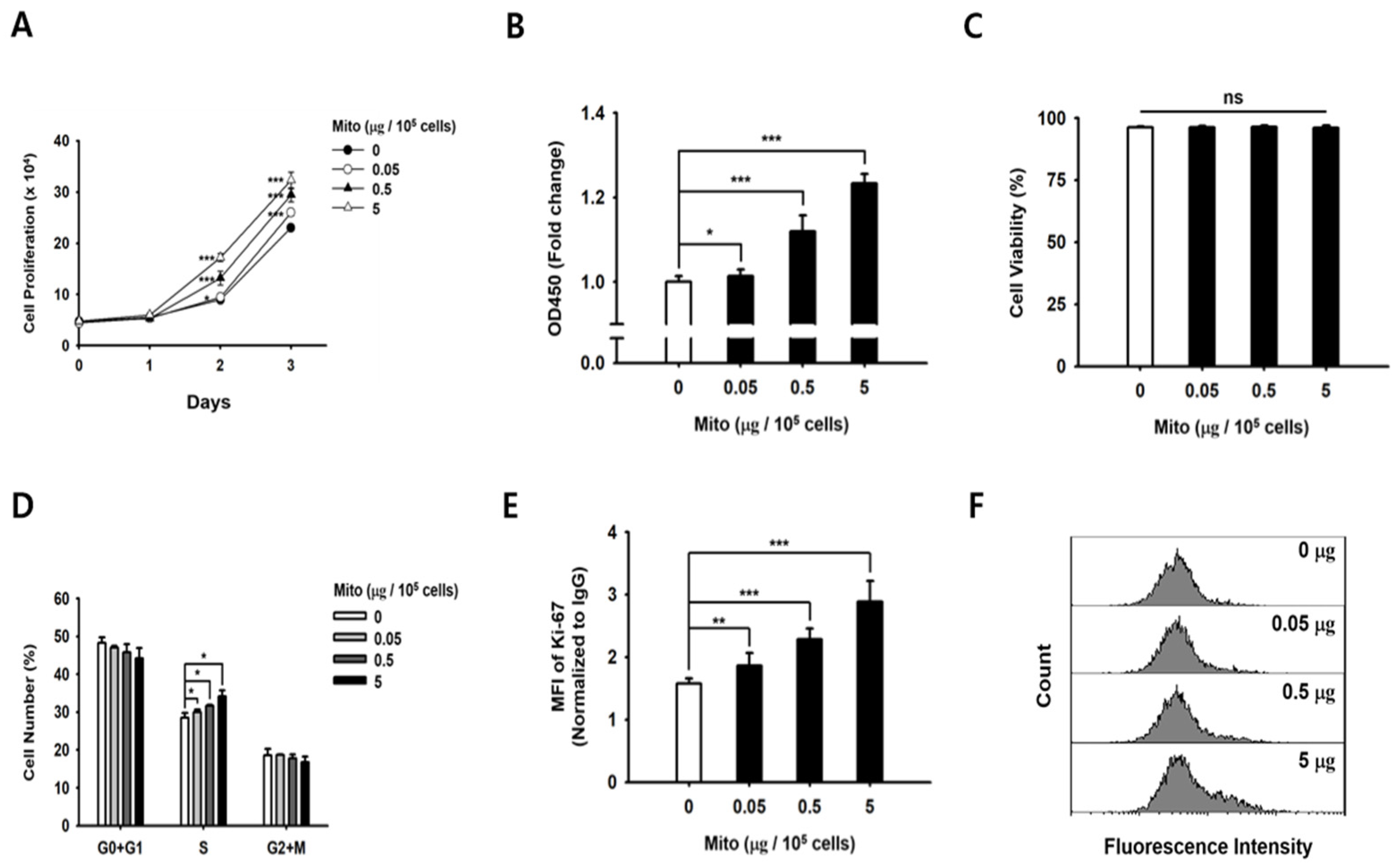
2.5. Cell Proliferation Assay
To determine the changes in cell proliferation after the mitochondrial transfer, the NK-92MI cells were seeded in 48- and 96-well plates at a density of 1 × 105 cells/mL and incubated at 37 °C. Cell proliferation in each group was assessed using the WST-based EZ Cyto-X assay (DoGenbio, Seoul, Republic of Korea) every 24 h. At each time point, the WST solution was added to each well (final dilution: 1:10), followed by incubation for 2 h. The optical density at 450 nm (OD 450 nm) was measured using a Synergy HTX microplate reader (BioTek, Winooski, VT, USA). The WST-based EZ Cyto-X assay is a colorimetric method widely used to measure cell proliferation and viability. This assay was employed in our study to evaluate the impact of the mitochondrial transfer on cell proliferation.
2.6. Flow Cytometry Analysis
To validate the quantification results, the mitochondria isolated using MitoTracker Red CMXRos were transferred into the NK-92MI cells. The quantification of mitochondria was performed using the CytoFLEX flow cytometry. The mean fluorescence intensity (MFI) values were recorded in the PE channel, and regression analysis was performed using SigmaPlot 10.0 (Systat Software, San Jose, CA, USA).
After mitochondrial transfer, we analyzed cell viability, Ki-67, and cell cycle assays using flow cytometry. For the cell viability assay, the NK-92MI cells were cultured at 37 °C for 48 h after mitochondrial transfer. Then, they were resuspended in PBS containing 10 µg/mL propidium iodide (PI; Sigma-Aldrich, St. Louis, MO, USA) and analyzed using the CytExpert software, version 2.4. For the Ki-67 intracellular staining, the NK-92MI cells were fixed/permeated using a fixation/permeabilization kit (BD Biosciences, San Jose, CA, USA), following the manufacturer’s protocol. The NK-92MI cells were suspended in perm/wash buffer (BD Biosciences, Franklin Lakes, NJ, USA) with PE-conjugated Ki-67 (BioLegend; 350504, San Diego, CA, USA), and analyzed using the CytExpert software. For the cell cycle assay, the NK-92MI cells were analyzed 48 h after mitochondrial transfer. They were permeabilized in 70% ethanol at 4 °C for 30 min. The DNA was stained with 50 µg/mL PI and 200 µg/mL RNase A (iNtRON Biotechnology, Seoul, Republic of Korea) at 37 °C for 45 min. The cells were analyzed using the CytExpert software, and data analysis was performed using FlowJo 10.7.1 (BD Biosciences).
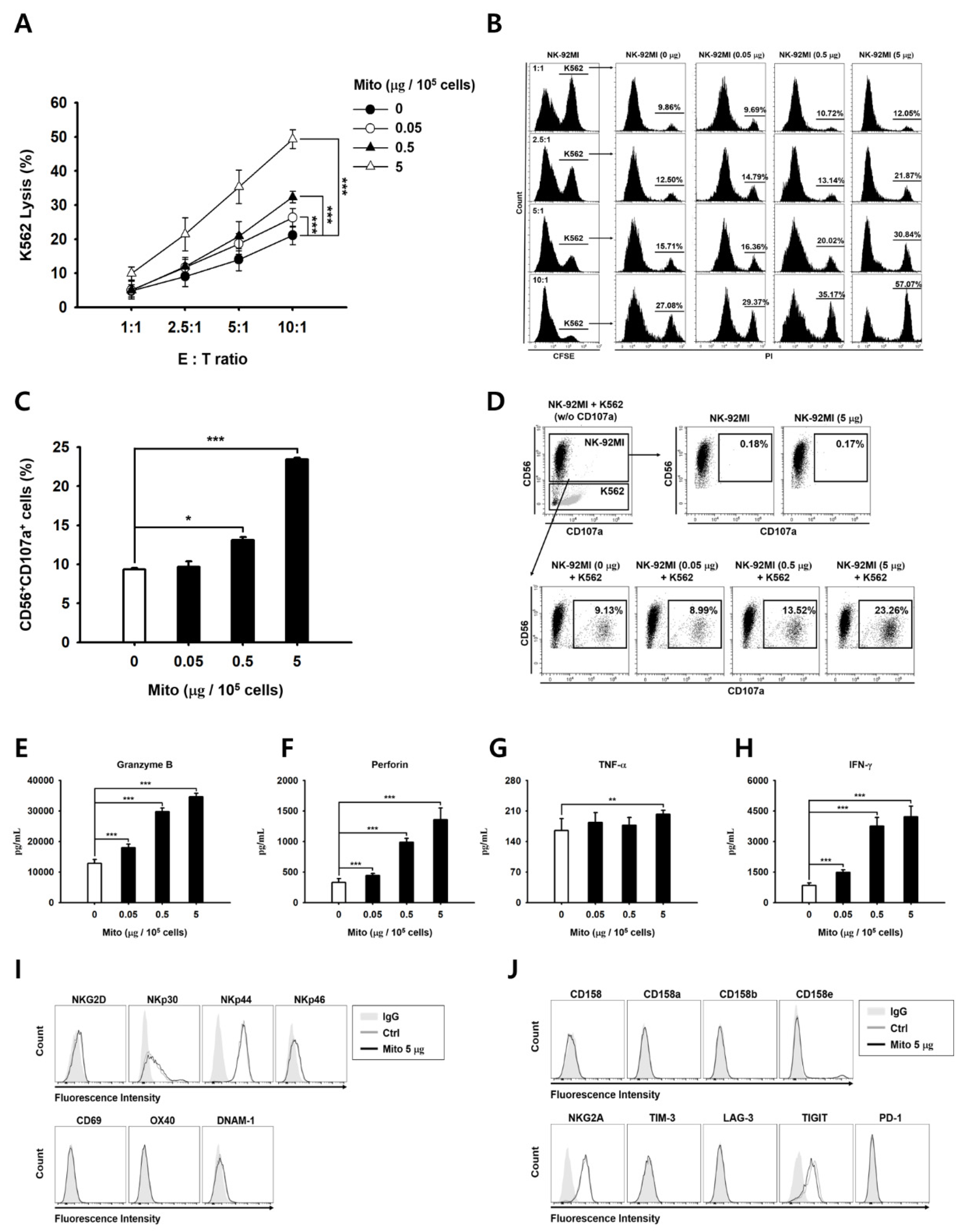
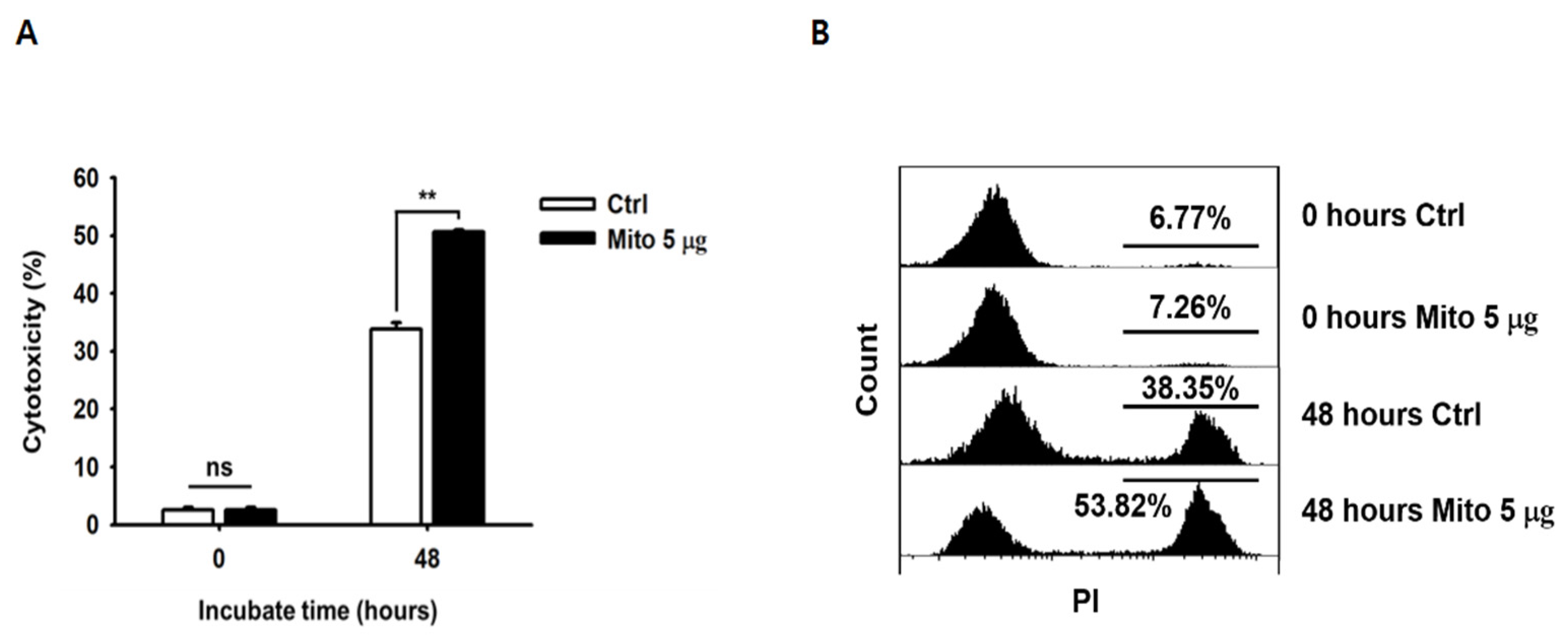
To analyze their cytotoxicity against cancer cells, the specific lysis ability of NK cells was analyzed using flow cytometry. Target tumor cells (K562 cells) were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE; Invitrogen). Then, 48 h after mitochondrial transfer, NK cells and CFSE-labeled target cells were co-cultured for 4 h. Then, 10 µg/mL PI was added to the co-cultured samples to identify the dead cells, which were analyzed using the CytExpert software. The specific cell lysis percentage was calculated using Equation (1):
For the CD107a degranulation assay, 48 h after mitochondria transfer, NK-92MI cells were co-cultured with the same number of K562 target cells for 2 h. All the cells were harvested, stained with APC-conjugated CD56 (Invitrogen; 17-0567-42) and PE-conjugated CD107a (Invitrogen; 12-1079-42) for 30 min, and analyzed using the CytExpert software.
For cell phenotype analysis, the NK cells were stained using the monoclonal antibodies against the following proteins: NKG2D (Invitrogen; 11-5878-42), NKp30 (BioLegend, San Diego, CA, USA; 325210), NKp44 (BioLegend; 325108), NKp46 (BD Biosciences; 557991), CD69 (Invitrogen; 12-0699-42), OX40 (BioLegend; 350004), DNAM-1 (BioLegend; 338306), CD158 (BioLegend; 339504), CD158a (Invitrogen; 12-1589-42), CD158b (BioLegend; 312614), CD158e (Miltenyi Biotec; 130-116-822), NKG2A (Beckman Coulter; IM3291U), TIM-3 (Invitrogen; 11-3109-42), LAG-3 (BioLegend; 369306), TIGIT (BD, Ashland, OR, USA; 747846), and PD-1 (BioLegend; 367406). Data analysis was performed using FlowJo 10.7.1 (BD Biosciences).
2.7. Determining Granzyme B, Perforin, and Cytokine Levels
The amounts of proteins released were determined using ELISA kits (for granzyme B, IFN-γ, and TNF-α; R&D Systems, Minneapolis, MN, USA, and for perforin; Invitrogen) following the manufacturer’s protocol. Briefly, 48 h after mitochondrial transfer, NK-92MI cells were seeded in 48-well plates (1 × 105 cells/well) and co-cultured with 1 × 104 K562 cells for 4 h. Then, the culture medium was collected, and the protein levels were determined.
2.8. PCR Analysis
To validate the quantification results, the total RNA from NK-92MI cells was isolated using an easy-spin total RNA extraction kit (iNtRON Biotechnology), and cDNA synthesis was performed using the Maxime RT premix kit (iNtRON Biotechnology) according to the manufacturer’s protocol. cDNA was added to a PCR reaction mix containing the PCR master mix solution (iNtRON Biotechnology) and primers (NK-92MI mtDNA: forward, 5′-TTAACTCCACCATTAGCACC-3′ and reverse, 5′-GAGGATGGTGGTCAAGGGA-3′; WRL-68 mtDNA: forward, 5′-TGCCAGCCACCATGAATATC-3′ and reverse, 5′-GGTGGGTAGGTTTGTTGA-3′; and GAPDH: forward, 5′-GGAAGGTGAAGGTCGGAG-3′ and reverse, 5′-GGCAACAATATCCACTTTACC-3′). Products were loaded onto a 1.5 % agarose gel, and after agarose gel electrophoresis, they were observed under ultraviolet light.
2.9. Fluorescence Analysis
To confirm the mitochondrial transfer, NK-92MI cells were labeled with MitoTracker Green, and the mitochondria of WRL-68 cells were labeled with MitoTracker Red CMXRos (Invitrogen). After transferring the mitochondria into NK-92MI cells by centrifugation, the cell nuclei were counterstained with 4′,6-diamidino-2-phenylindole (Sigma-Aldrich). Image analysis was performed using the Axiovert 200M fluorescence microscope (Carl Zeiss, Jena, Germany). Digital images were generated using the Axiovision SE64 Rel 4.9.1 software (Carl Zeiss).
2.10. Western Blot Analysis
The isolated mitochondria were heat treated at 95 °C for 7 min using the SDS-PAGE loading buffer (LPS solution, Daejeon, Republic of Korea). After separating the proteins by size using a 12% SDS-PAGE gel, they were transferred to PVDF membranes. The membranes were treated with 3% BSA for 1 h, in order to block non-specific interactions, and then incubated overnight at 4 °C with each of the following primary antibodies: anti-AIF (sc-13116), anti-cytochrome C (sc-13156), anti-PCNA (sc-56) (all from Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), anti-COX IV (#4844s), and anti-GAPDH (#2118s) (all from Cell Signaling Technology, Beverly, MA, USA). After washing with TBST, the membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies, goat anti-mouse IgG (1:1000; Santa Cruz Biotechnology; sc-516102-CM), and goat anti-rabbit IgG (Santa Cruz; sc-2357). The expression of the target proteins was visualized using an enhanced chemiluminescence system (ECL component from Pierce Clarity and Western ECL Substrate; Bio-Rad Laboratories, Hercules, CA, USA) and LAS-4000 camera (Fuji Photo Film, Tokyo, Japan).
2.11. Statistical Analyses
All statistical analyses were performed using SigmaPlot 12.0 (Systat Software, San Jose, CA, USA). Significant differences between groups were evaluated using Student’s t-test. Data are expressed as the mean ± standard deviation, and statistical significance was defined as p < 0.05.
4. Discussion
The immune surveillance capacity of NK cells is closely related to the number and activity of the endogenous mitochondria [
20]. We demonstrated that transferring exogenous allogeneic mitochondria to NK cells can increase their immune activity. The transfer of exogenous mitochondria to NK cells promotes their proliferation and anticancer activity. Therefore, NK cells with an increased anticancer ability can be produced in a short period of time by mitochondrial transfer, without the need for a 2-week culture.
Recently, we demonstrated that impaired cellular energy metabolism can be repaired by the transfer of foreign mitochondria into cells [
19]. After the transfer of healthy exogenous mitochondria, the proliferation of mtDNA-damaged ρ
0 cells was restored [
19]. Similarly, the proliferative capacity of NK cells treated with exogenous mitochondria was improved in a dose-dependent manner (
Figure 1). In addition, as the proliferation rate of NK cells increased, the glucose consumption and lactic acid production in the medium increased rapidly. Therefore, we demonstrated that NK cell proliferation and ATP synthesis could be improved by exogenous mitochondrial transfer without the need for culture with cytokines.
The anticancer effect of NK-92MI cells increased in a dose-dependent manner after mitochondrial transfer (
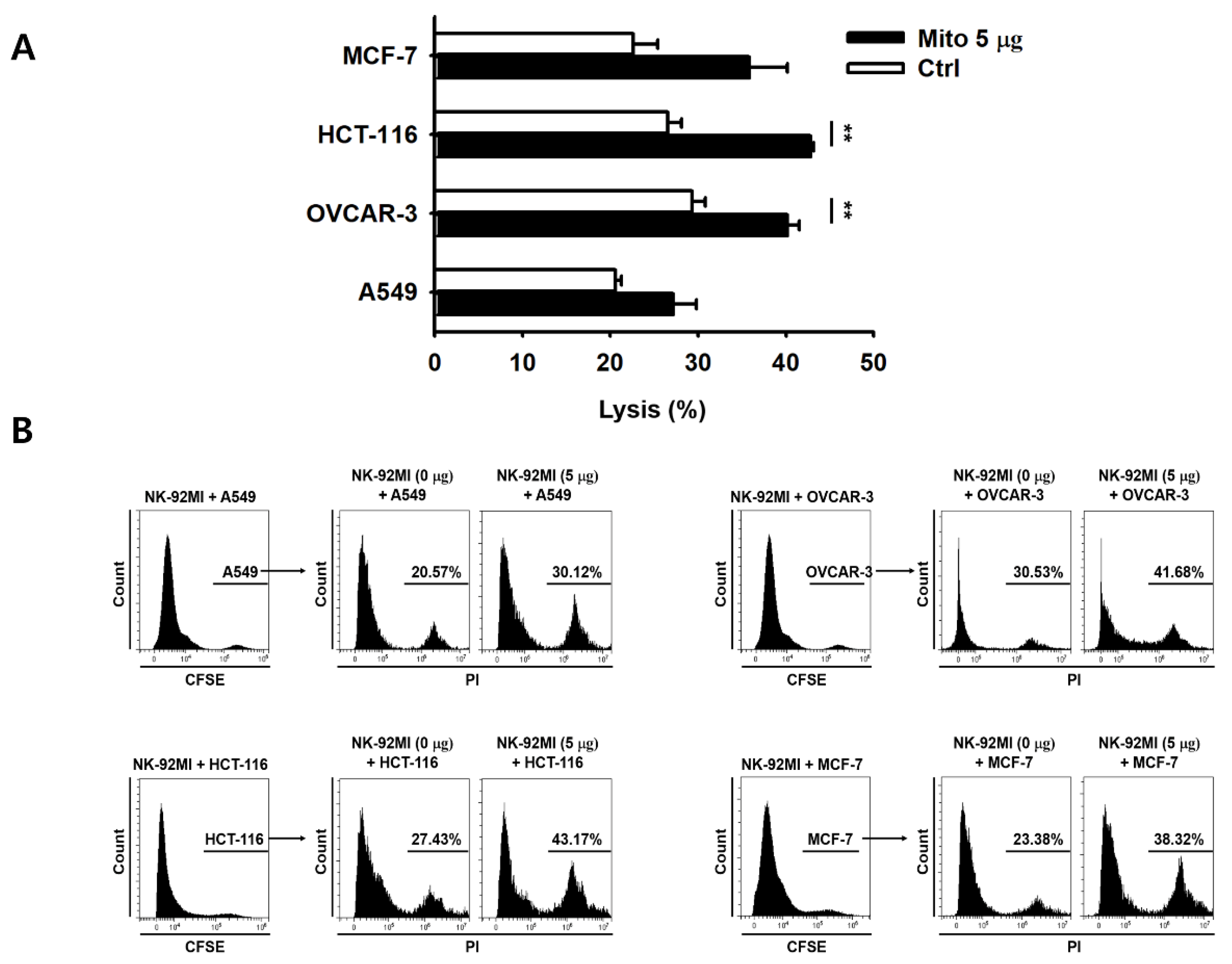
Figure 2). The cell anticancer effect did not increase immediately after mitochondrial transfer, but gradually increased over time. In addition, this effect differed to some extent depending on the type of target cell. However, the killing ability of NK-92MI cells treated with mitochondria proved to be effective not only against hematological cancer cells (K562) but also against solid tumor cells (
Figure 3).
The problem with chimeric antigen receptor T-cell therapy, an anticancer immune cell therapy, is that side effects, such as an excessive cytokine storm, can persist for a long time in the body [
20]. In addition, culturing or activating NK cells using cytokines is problematic because NK cells undergo apoptosis due to excessive stimulation. In the case of mitochondrial-enriched NK-92MI cells, no change in cell viability was observed, depending on the amount of mitochondria transferred (
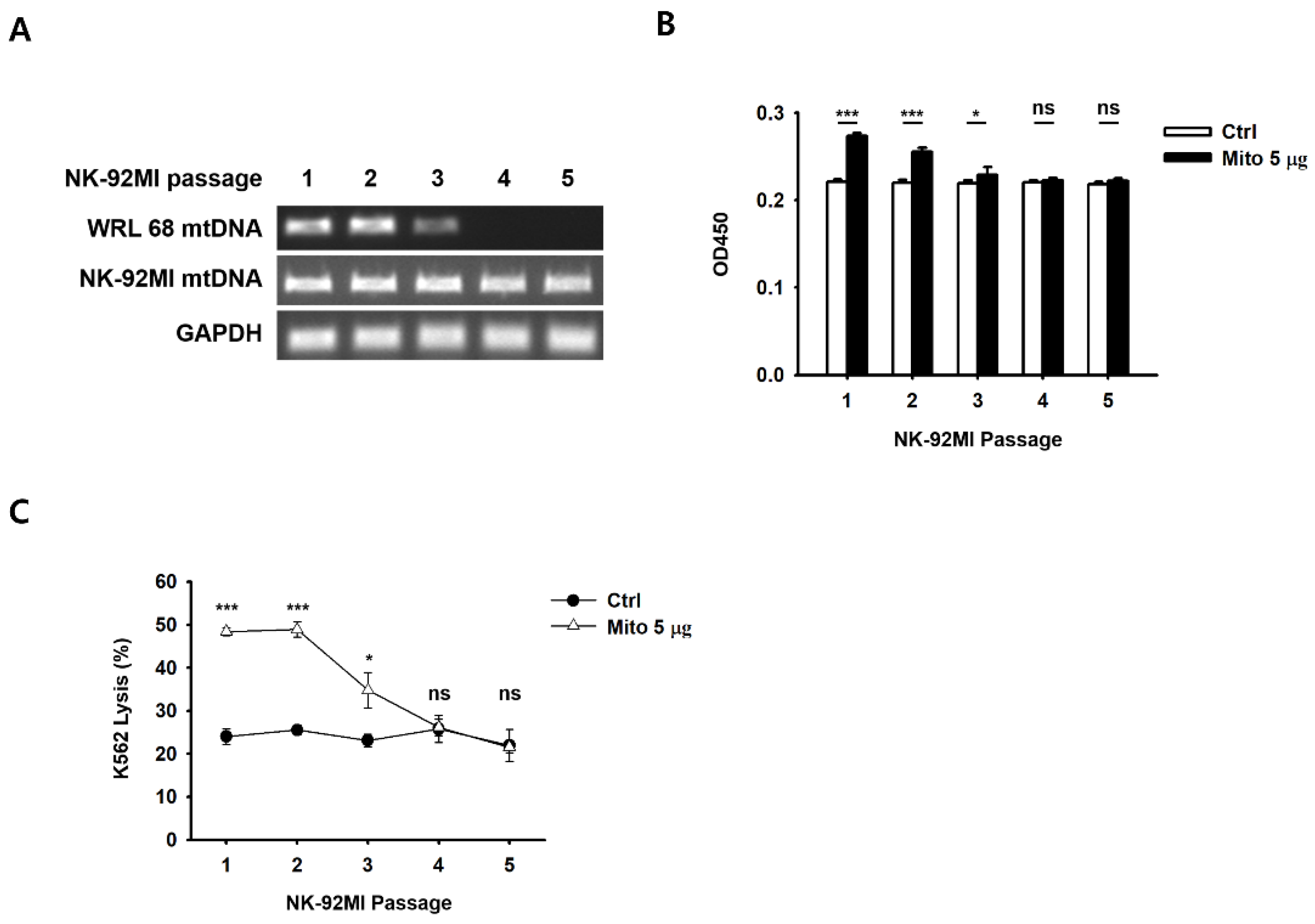
Figure S3), and the exogenous mitochondria were eliminated approximately 8 days after the transfer (
Figure S4). Simultaneously, cell proliferation and the cell anticancer effects were restored to pre-mitochondrial transfer levels. As a result, mitochondria-enriched NK cells naturally returned to their original state after a week.
NK cell receptors play an important role in regulating NK cell activity [
21]. When NK cells are cultured using cytokines, such as IL-2, IL-12, and IL-15, the expression of the activation receptors increases. The proliferation and activity of NK cells are enhanced by external signals. However, when exogenous mitochondria were transferred into cells, no changes in the activation receptors of the NK cells were observed (
Figure 2). Therefore, the anticancer effect of mitochondria-enriched NK cells is due to an increase in the metabolic activity inside the cell, rather than an external signal. Conversely, if the number of mitochondria or their function decreases, the anticancer effect is also reduced.
As mentioned in previous studies, cell function enhancement is associated with mitochondrial states [
22,
23]. Although it is unclear how, the mitochondrial states may be improved via induction of mitochondrial biogenesis or fusion after mitochondrial transfer [
24,
25]. The artificial and natural transfer of mitochondria improves the function of normal and damaged cells [
18,
26,
27,
28]. These phenomena may explain the changes in cell metabolism associated with the mitochondria. First, studies have shown that mitochondrial transfer changes OXPHOS activity and ATP production [
29,
30]. The cytotoxicity and proliferation of NK cells involve the production of cytolytic granules and proliferative factors, respectively, which require a lot of ATP [
8,
31,
32,
33]. Our results suggest that an increased rate of ATP synthesis due to the transfer of mitochondria enhances NK cell functions, such as their killing activity, the release of cytokines and granules, and the cell cycle. Second, studies have reported that the changes in the mTOR signaling pathway mechanism induced by mitochondria are closely related to cell metabolism and the immune response [
34,
35,
36]. In an mTOR inhibition model, the function and proliferation of NK cells decreased remarkably [
35]. In addition, the hyperactivation of mTOR signaling leads to a decrease in NK cell function due to the mitochondrial fragmentation caused by autophagy [
28]. Hence, we suggest that mitochondrial transfer might trigger an optimum regulation of the mTOR signaling pathway.
Gene set enrichment analysis (GSEA) using RNA-seq data also supports this observation at the transcriptome level (
Figure S5). Based on these results, it can be concluded that the cytotoxicity and immune sensitivity of NK-92MI cells increased by upregulating gene expression in the Toll-like receptor and JAK-STAT signaling pathways (
Figure S5C,D). Interestingly, the observation that exogenous mitochondria upregulated glycolysis genes but downregulated OXPHOS genes suggests that there may be a balance between energy efficiency and productivity that is being optimized by the cell (
Figure S5E,F). One possibility is that the proportion of active versus inactive NK cells may play a role in this regulation. It is known that Glycolysis is more critical than OXPHOS for NK-receptor-activated cytotoxicity [
8]. We observed that both pathways were upregulated at the protein level, but the transcriptomic level of OXPHOS was relatively downregulated. It is possible that the activation of NK cell function was acquired independently of the gene transcription involved in the OXPHOS pathway. This activation may be due to either sufficient protein abundance in OXPHOS, or other unknown mechanisms that altered the overall signal strength between protein and RNA levels in response to the proportion of active and inactive NK cell populations that already existed.
Further studies are needed to investigate whether the primary NK cells isolated from peripheral blood also exhibit anticancer activity against various solid cancer cells. Furthermore, whether mitochondria-enriched primary NK cells derived from peripheral blood naturally return to their original state needs to be confirmed in the future. Additionally, the mechanism by which the transferred mitochondria enhance the proliferation and killing ability of NK-92MI cells needs to be elucidated. Additionally, an important point to be considered is that the difference in cell anticancer effect according to the type of transferred mitochondria and the presentation of information on the donor of mitochondria should be supplemented.
5. Conclusions
In this study, we provide a concept at the in vitro level for the necessity of in vivo studies to accurately evaluate the toxicity of mitochondria-transferred NK cells to anticancer cells, and a general discussion of the results. Here, we demonstrated that the artificial transfer of allogenic mitochondria can improve the anticancer effect and proliferation of NK cells. When applying this strategy as an autologous NK cell therapy, we suggest transferring the mitochondria without additional cell culture. Moreover, we can combine this strategy with previous methods to expand and activate cells. Since this strategy leads to an increased cell proliferative capacity, the period of cell culture required to ensure a sufficient number of cells is produced can be shortened, and its associated cost can be lower. In addition, the results demonstrated that not only blood cancer cells, but also solid cancers, are affected by NK-92MI cells treated with mitochondria, suggesting that clinical trials are possible.
Furthermore, we demonstrated the functional changes in NK cells induced by exogenous mitochondrial transfer. The transfer of mitochondria with a relatively high ATP-producing ability into NK cells led to a mitochondrial function improvement. In addition, the mitochondrial transfer increased the proliferation of NK cells and enhanced their cytotoxicity. Collectively, mitochondrial transfer can be used to enhance NK cell activity in a short period and can be used as a novel therapeutic strategy for clinical application.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




