
New Approaches to Targeted Therapy in Melanoma
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
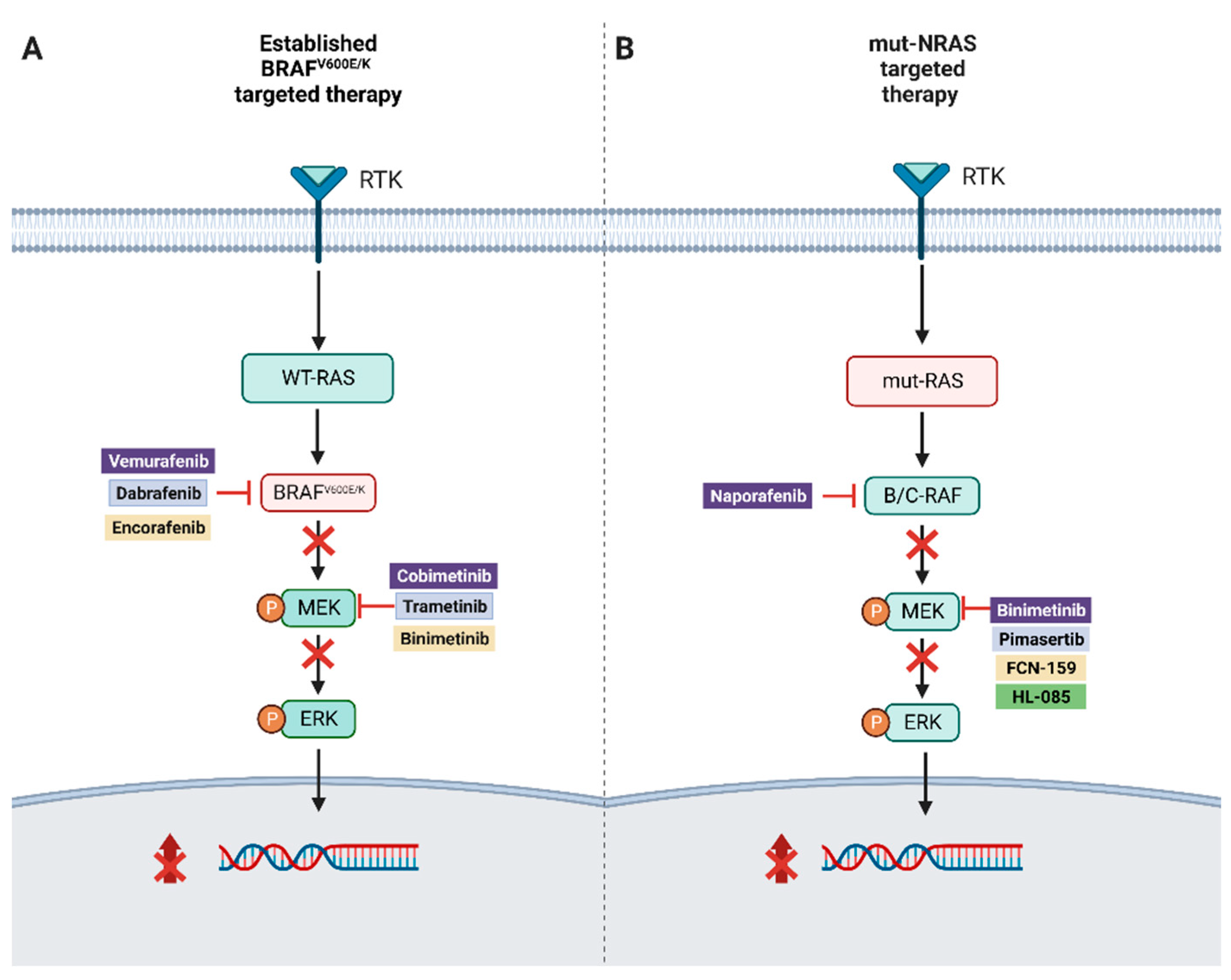
2. RAF Mutations, MAPK Pathway
3. BRAF Mutant Resistance
4. Pan-RAF Inhibitors
5. NRAS
6. ERK
7. Atypical BRAF Mutations
8. CDK4/6 + MEK Inhibition
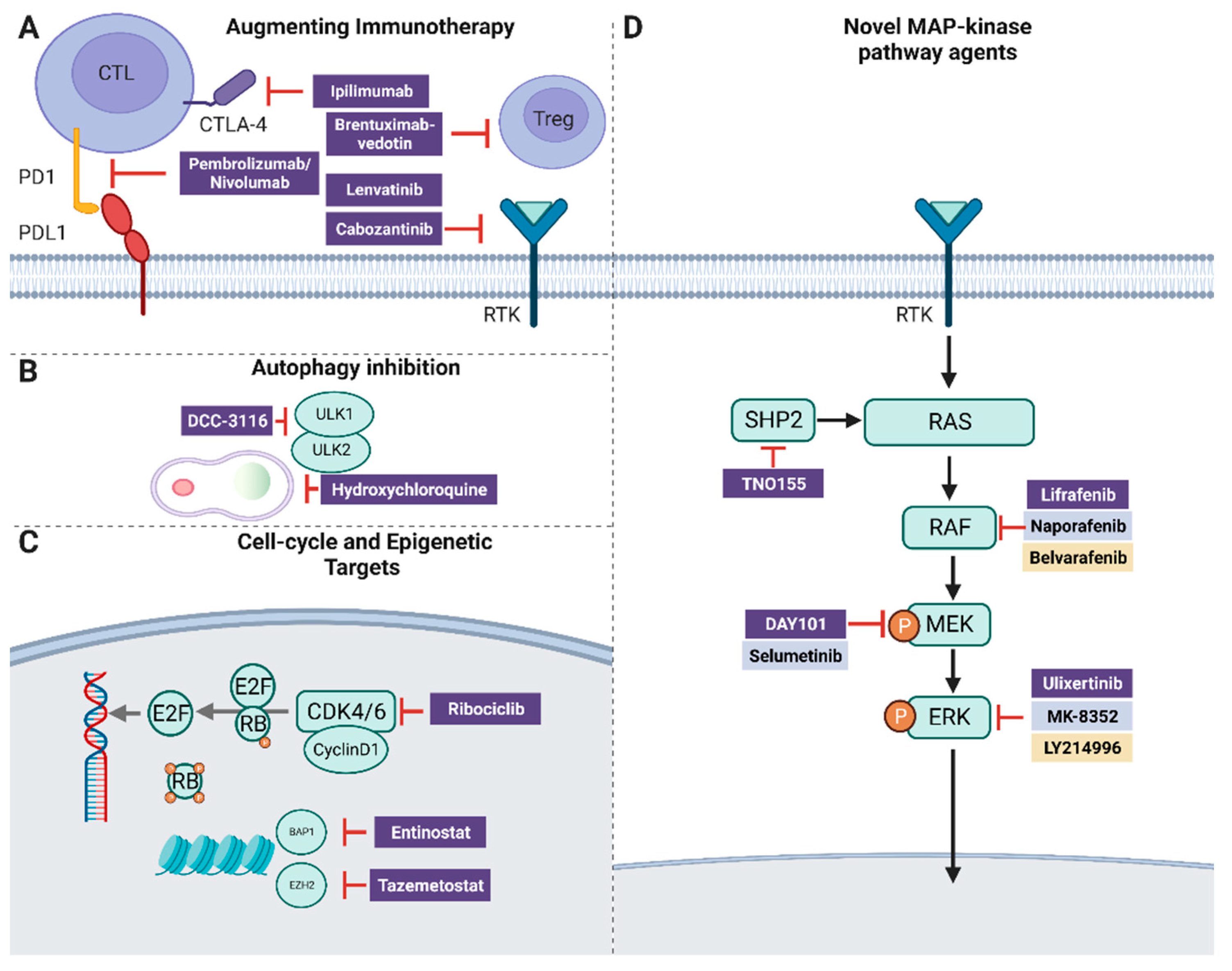
9. SHP2
10. Autophagy
11. Epigenetic Targets
12. BAP1 and HDAC
13. Augmenting Immunotherapy
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Surveillance Research Program, National Cancer Institute. Data Source(s): SEER Incidence Data, November 2022 Submission (1975–2020), SEER 22 Registries. 19 April 2023. Available online: https://seer.cancer.gov/statistics-network/explorer/ (accessed on 19 April 2023).
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF -mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-Term Outcomes With Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab in Patients With Advanced Melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Larkin, J.; Sosman, J.A.; Lebbé, C.; Brady, B.; Neyns, B.; Schmidt, H.; Hassel, J.C.; Hodi, F.S.; Lorigan, P.; et al. Efficacy and Safety of Nivolumab Alone or in Combination With Ipilimumab in Patients With Mucosal Melanoma: A Pooled Analysis. J. Clin. Oncol. 2017, 35, 226–235. [Google Scholar] [CrossRef]
- Van Zeijl, M.C.T.; Boer, F.L.; Van Poelgeest, M.I.E.; Van Den Eertwegh, A.J.M.; Wouters, M.W.J.M.; De Wreede, L.C.; Aarts, M.J.B.; Van Den Berkmortel, F.W.P.J.; De Groot, J.W.B.; Hospers, G.A.P.; et al. Survival outcomes of patients with advanced mucosal melanoma diagnosed from 2013 to 2017 in the Netherlands—A nationwide population-based study. Eur. J. Cancer 2020, 137, 127–135. [Google Scholar] [CrossRef]
- Shoushtari, A.N.; Wagstaff, J.; Ascierto, P.A.; Butler, M.O.; Lao, C.D.; Marquez-Rodas, I.; Chiarion-Sileni, V.; Dummer, R.; Ferrucci, P.F.; Lorigan, P.; et al. CheckMate 067: Long-term outcomes in patients with mucosal melanoma. J. Clin. Oncol. 2020, 38, 10019. [Google Scholar] [CrossRef]
- Pelster, M.S.; Gruschkus, S.K.; Bassett, R.; Gombos, D.S.; Shephard, M.; Posada, L.; Glover, M.S.; Simien, R.; Diab, A.; Hwu, P.; et al. Nivolumab and Ipilimumab in Metastatic Uveal Melanoma: Results From a Single-Arm Phase II Study. J. Clin. Oncol. 2021, 39, 599–607. [Google Scholar] [CrossRef]
- Lavoie, H.; Therrien, M. Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol. 2015, 16, 281–298. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Lee, J.H.; Choi, J.W.; Kim, Y.S. Frequencies of BRAF and NRAS mutations are different in histological types and sites of origin of cutaneous melanoma: A meta-analysis. Br. J. Dermatol. 2011, 164, 776–784. [Google Scholar] [CrossRef]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.-H.; Aiba, S.; Bröcker, E.-B.; Leboit, P.E.; et al. Distinct Sets of Genetic Alterations in Melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 2017, 28, 1631–1639. [Google Scholar] [CrossRef]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined Vemurafenib and Cobimetinib in BRAF-Mutated Melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Dummer, R.; Flaherty, K.; Robert, C.; Arance, A.M.; Groot, J.W.d.; Garbe, C.; Gogas, H.; Gutzmer, R.; Krajsová, I.; Liszkay, G.; et al. Five-year overall survival (OS) in COLUMBUS: A randomized phase 3 trial of encorafenib plus binimetinib versus vemurafenib or encorafenib in patients (pts) with BRAF V600-mutant melanoma. J. Clin. Oncol. 2021, 39, 9507. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef]
- Tian, Y.; Guo, W. A Review of the Molecular Pathways Involved in Resistance to BRAF Inhibitors in Patients with Advanced-Stage Melanoma. Med. Sci. Monit. 2020, 26, e920957-13. [Google Scholar] [CrossRef]
- Proietti, I.; Skroza, N.; Bernardini, N.; Tolino, E.; Balduzzi, V.; Marchesiello, A.; Michelini, S.; Volpe, S.; Mambrin, A.; Mangino, G.; et al. Mechanisms of Acquired BRAF Inhibitor Resistance in Melanoma: A Systematic Review. Cancers 2020, 12, 2801. [Google Scholar] [CrossRef]
- Liu, L.; Lee, M.R.; Kim, J.L.; Whittington, D.A.; Bregman, H.; Hua, Z.; Lewis, R.T.; Martin, M.W.; Nishimura, N.; Potashman, M.; et al. Purinylpyridinylamino-based DFG-in/αC-helix-out B-Raf inhibitors: Applying mutant versus wild-type B-Raf selectivity indices for compound profiling. Bioorg. Med. Chem. 2016, 24, 2215–2234. [Google Scholar] [CrossRef]
- Okaniwa, M.; Hirose, M.; Arita, T.; Yabuki, M.; Nakamura, A.; Takagi, T.; Kawamoto, T.; Uchiyama, N.; Sumita, A.; Tsutsumi, S.; et al. Discovery of a selective kinase inhibitor (TAK-632) targeting pan-RAF inhibition: Design, synthesis, and biological evaluation of C-7-substituted 1,3-benzothiazole derivatives. J. Med. Chem. 2013, 56, 6478–6494. [Google Scholar] [CrossRef] [PubMed]
- PPeng, S.-B.; Henry, J.R.; Kaufman, M.D.; Lu, W.-P.; Smith, B.D.; Vogeti, S.; Rutkoski, T.J.; Wise, S.; Chun, L.; Zhang, Y.; et al. Inhibition of RAF Isoforms and Active Dimers by LY3009120 Leads to Anti-tumor Activities in RAS or BRAF Mutant Cancers. Cancer Cell 2015, 28, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Izar, B.; Sharfman, W.; Hodi, F.S.; Lawrence, D.; Flaherty, K.T.; Amaravadi, R.; Kim, K.B.; Puzanov, I.; Sosman, J.; Dummer, R.; et al. A first-in-human phase I, multicenter, open-label, dose-escalation study of the oral RAF/VEGFR-2 inhibitor (RAF265) in locally advanced or metastatic melanoma independent from BRAF mutation status. Cancer Med. 2017, 6, 1904–1914. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Hollebecque, A.; Flaherty, K.T.; Shapiro, G.I.; Rodon Ahnert, J.; Millward, M.J.; Zhang, W.; Gao, L.; Sykes, A.; Willard, M.D.; et al. A Phase I Study of LY3009120, a Pan-RAF Inhibitor, in Patients with Advanced or Metastatic Cancer. Mol. Cancer Ther. 2020, 19, 460–467. [Google Scholar] [CrossRef]
- Desai, J.; Gan, H.; Barrow, C.; Jameson, M.; Atkinson, V.; Haydon, A.; Millward, M.; Begbie, S.; Brown, M.; Markman, B.; et al. Phase I, Open-Label, Dose-Escalation/Dose-Expansion Study of Lifirafenib (BGB-283), an RAF Family Kinase Inhibitor, in Patients With Solid Tumors. J. Clin. Oncol. 2020, 38, 2140–2150. [Google Scholar] [CrossRef] [PubMed]
- Monaco, K.-A.; Delach, S.; Yuan, J.; Mishina, Y.; Fordjour, P.; Labrot, E.; McKay, D.; Guo, R.; Higgins, S.; Wang, H.Q.; et al. LXH254, a Potent and Selective ARAF-Sparing Inhibitor of BRAF and CRAF for the Treatment of MAPK-Driven Tumors. Clin. Cancer Res. 2021, 27, 2061–2073. [Google Scholar] [CrossRef] [PubMed]
- Yen, I.; Shanahan, F.; Lee, J.; Hong, Y.S.; Shin, S.J.; Moore, A.R.; Sudhamsu, J.; Chang, M.T.; Bae, I.; Dela Cruz, D.; et al. ARAF mutations confer resistance to the RAF inhibitor belvarafenib in melanoma. Nature 2021, 594, 418–423. [Google Scholar] [CrossRef]
- Hong, A.; Piva, M.; Liu, S.; Hugo, W.; Lomeli, S.H.; Zoete, V.; Randolph, C.E.; Yang, Z.; Wang, Y.; Lee, J.J.; et al. Durable Suppression of Acquired MEK Inhibitor Resistance in Cancer by Sequestering MEK from ERK and Promoting Antitumor T-cell Immunity. Cancer Discov. 2021, 11, 714–735. [Google Scholar] [CrossRef]
- Chen, S.H.; Gong, X.; Zhang, Y.; Van Horn, R.D.; Yin, T.; Huber, L.; Burke, T.F.; Manro, J.; Iversen, P.W.; Wu, W.; et al. RAF inhibitor LY3009120 sensitizes RAS or BRAF mutant cancer to CDK4/6 inhibition by abemaciclib via superior inhibition of phospho-RB and suppression of cyclin D1. Oncogene 2018, 37, 821–832. [Google Scholar] [CrossRef]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Adari, H.; Lowy, D.R.; Willumsen, B.M.; Der, C.J.; McCormick, F. Guanosine triphosphatase activating protein (GAP) interacts with the p21 ras effector binding domain. Science 1988, 240, 518–521. [Google Scholar] [CrossRef] [PubMed]
- Jakob, J.A.; Bassett, R.L.; Ng, C.S.; Curry, J.L.; Joseph, R.W.; Alvarado, G.C.; Rohlfs, M.L.; Richard, J.; Gershenwald, J.E.; Kim, K.B.; et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer 2012, 118, 4014–4023. [Google Scholar] [CrossRef] [PubMed]
- Mandalà, M.; Merelli, B.; Massi, D. Nras in melanoma: Targeting the undruggable target. Crit. Rev. Oncol./Hematol. 2014, 92, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.H.; Lee, M. The siRNA-mediated downregulation of N-Ras sensitizes human melanoma cells to apoptosis induced by selective BRAF inhibitors. Mol. Cell Biochem. 2014, 392, 239–247. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Al Mahi, A.; Ablain, J. RAS pathway regulation in melanoma. Dis. Model. Mech. 2022, 15, dmm049229. [Google Scholar] [CrossRef]
- van Herpen, C.M.L.; Agarwala, S.S.; Hauschild, A.; Berking, C.; Beck, J.T.; Schadendorf, D.; Jansen, R.; Queirolo, P.; Ascierto, P.A.; Blank, C.U.; et al. Biomarker results from a phase II study of MEK1/2 inhibitor binimetinib (MEK162) in patients with advanced NRAS- or BRAF-mutated melanoma. Oncotarget 2019, 10, 1850–1859. [Google Scholar] [CrossRef]
- Lebbé, C.; Dutriaux, C.; Lesimple, T.; Kruit, W.; Kerger, J.; Thomas, L.; Guillot, B.; De Braud, F.; Garbe, C.; Grob, J.-J.; et al. Pimasertib Versus Dacarbazine in Patients With Unresectable NRAS-Mutated Cutaneous Melanoma: Phase II, Randomized, Controlled Trial with Crossover. Cancers 2020, 12, 1727. [Google Scholar] [CrossRef]
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del Vecchio, M.; Gutzmer, R.; Mandala, M.; et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef]
- Wang, X.; Luo, Z.; Chen, J.; Chen, Y.; Ji, D.; Fan, L.; Chen, L.; Zhao, Q.; Hu, P.; Sun, P.; et al. First-in-human phase I dose-escalation and dose-expansion trial of the selective MEK inhibitor HL-085 in patients with advanced melanoma harboring NRAS mutations. BMC Med. 2023, 21, 2. [Google Scholar] [CrossRef]
- Mao, L.; Guo, J.; Zhu, L.; Jiang, Y.; Yan, W.; Zhang, J.; Hui, A.-M.; Yang, Y.; Diao, L.; Tan, Y.; et al. A first-in-human, phase 1a dose-escalation study of the selective MEK1/2 inhibitor FCN-159 in patients with advanced NRAS-mutant melanoma. Eur. J. Cancer 2022, 175, 125–135. [Google Scholar] [CrossRef] [PubMed]
- de Braud, F.; Dooms, C.; Heist, R.S.; Lebbe, C.; Wermke, M.; Gazzah, A.; Schadendorf, D.; Rutkowski, P.; Wolf, J.; Ascierto, P.A.; et al. Initial Evidence for the Efficacy of Naporafenib in Combination With Trametinib in NRAS-Mutant Melanoma: Results From the Expansion Arm of a Phase Ib, Open-Label Study. J. Clin. Oncol. 2023, 41, 2651–2660. [Google Scholar] [CrossRef] [PubMed]
- Lake, D.; Corrêa, S.A.L.; Müller, J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell. Mol. Life Sci. 2016, 73, 4397–4413. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Moriceau, G.; Kong, X.; Lee, M.-K.; Lee, H.; Koya, R.C.; Ng, C.; Chodon, T.; Scolyer, R.A.; Dahlman, K.B.; et al. Melanoma whole-exome sequencing identifies V600EB-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat. Commun. 2012, 3, 724. [Google Scholar] [CrossRef] [PubMed]
- Kidger, A.M.; Munck, J.M.; Saini, H.K.; Balmanno, K.; Minihane, E.; Courtin, A.; Graham, B.; O’Reilly, M.; Odle, R.; Cook, S.J. Dual-Mechanism ERK1/2 Inhibitors Exploit a Distinct Binding Mode to Block Phosphorylation and Nuclear Accumulation of ERK1/2. Mol. Cancer Ther. 2020, 19, 525–539. [Google Scholar] [CrossRef]
- Sammons, R.M.; Ghose, R.; Tsai, K.Y.; Dalby, K.N. Targeting ERK beyond the boundaries of the kinase active site in melanoma. Mol. Carcinog. 2019, 58, 1551–1570. [Google Scholar] [CrossRef]
- Bhagwat, S.V.; McMillen, W.T.; Cai, S.; Zhao, B.; Whitesell, M.; Shen, W.; Kindler, L.; Flack, R.S.; Wu, W.; Anderson, B.; et al. ERK Inhibitor LY3214996 Targets ERK Pathway-Driven Cancers: A Therapeutic Approach Toward Precision Medicine. Mol. Cancer Ther. 2020, 19, 325–336. [Google Scholar] [CrossRef]
- Germann, U.A.; Furey, B.F.; Markland, W.; Hoover, R.R.; Aronov, A.M.; Roix, J.J.; Hale, M.; Boucher, D.M.; Sorrell, D.A.; Martinez-Botella, G.; et al. Targeting the MAPK Signaling Pathway in Cancer: Promising Preclinical Activity with the Novel Selective ERK1/2 Inhibitor BVD-523 (Ulixertinib). Mol. Cancer Ther. 2017, 16, 2351–2363. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Infante, J.R.; Janku, F.; Wong, D.J.L.; Sosman, J.A.; Keedy, V.; Patel, M.R.; Shapiro, G.I.; Mier, J.W.; Tolcher, A.W.; et al. First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer Discov. 2018, 8, 184–195. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Cohen, J.V.; Haq, R.; Hodi, F.S.; Lawrence, D.P.; Giobbie-Hurder, A.; Knoerzer, D.; Sullivan, R.J. A phase II study of ERK inhibition by ulixertinib (BVD-523) in metastatic uveal melanoma. J. Clin. Oncol. 2020, 38, 10036. [Google Scholar] [CrossRef]
- Wu, J.; Liu, D.; Offin, M.; Lezcano, C.; Torrisi, J.M.; Brownstein, S.; Hyman, D.M.; Gounder, M.M.; Abida, W.; Drilon, A.; et al. Characterization and management of ERK inhibitor associated dermatologic adverse events: Analysis from a nonrandomized trial of ulixertinib for advanced cancers. Investig. New Drugs 2021, 39, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, B.S.; Durinck, S.; Stawiski, E.W.; Yin, J.; Wang, W.; Lin, E.; Moffat, J.; Martin, S.E.; Modrusan, Z.; Seshagiri, S. ERK Mutations and Amplification Confer Resistance to ERK-Inhibitor Therapy. Clin. Cancer Res. 2018, 24, 4044–4055. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; Morris, E.J.; Hruza, A.; Mansueto, M.S.; Schroeder, G.K.; Arbanas, J.; McMasters, D.; Restaino, C.R.; Dayananth, P.; Black, S.; et al. Dissecting Therapeutic Resistance to ERK Inhibition. Mol. Cancer Ther. 2016, 15, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Moschos, S.J.; Sullivan, R.J.; Hwu, W.-J.; Ramanathan, R.K.; Adjei, A.A.; Fong, P.C.; Shapira-Frommer, R.; Tawbi, H.A.; Rubino, J.; Rush, T.S.; et al. Development of MK-8353, an orally administered ERK1/2 inhibitor, in patients with advanced solid tumors. JCI Insight 2018, 3, e92352. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Martinez, P.; Yeap, B.Y.; Ambrogio, C.; Ferris, L.A.; Lydon, C.; Nguyen, T.; Jessop, N.A.; Iafrate, A.J.; Johnson, B.E.; et al. Impact of BRAF Mutation Class on Disease Characteristics and Clinical Outcomes in BRAF-mutant Lung Cancer. Clin. Cancer Res. 2019, 25, 158–165. [Google Scholar] [CrossRef]
- Dumaz, N.; Jouenne, F.; Delyon, J.; Mourah, S.; Bensussan, A.; Lebbé, C. Atypical BRAF and NRAS Mutations in Mucosal Melanoma. Cancers 2019, 11, 1133. [Google Scholar] [CrossRef]
- Dahlman, K.B.; Xia, J.; Hutchinson, K.; Ng, C.; Hucks, D.; Jia, P.; Atefi, M.; Su, Z.; Branch, S.; Lyle, P.L.; et al. BRAF L597 Mutations in Melanoma Are Associated with Sensitivity to MEK Inhibitors. Cancer Discov. 2012, 2, 791–797. [Google Scholar] [CrossRef]
- Nebhan, C.A.; Johnson, D.B.; Sullivan, R.J.; Amaria, R.N.; Flaherty, K.T.; Sosman, J.A.; Davies, M.A. Efficacy and Safety of Trametinib in Non-V600 BRAF Mutant Melanoma: A Phase II Study. Oncologist 2021, 26, 731-e1498. [Google Scholar] [CrossRef]
- Menzer, C.; Menzies, A.M.; Carlino, M.S.; Reijers, I.; Groen, E.J.; Eigentler, T.; de Groot, J.W.B.; van der Veldt, A.A.M.; Johnson, D.B.; Meiss, F.; et al. Targeted Therapy in Advanced Melanoma With Rare BRAF Mutations. J. Clin. Oncol. 2019, 37, 3142–3151. [Google Scholar] [CrossRef]
- Botton, T.; Yeh, I.; Nelson, T.; Vemula, S.S.; Sparatta, A.; Garrido, M.C.; Allegra, M.; Rocchi, S.; Bahadoran, P.; McCalmont, T.H.; et al. Recurrent BRAF kinase fusions in melanocytic tumors offer an opportunity for targeted therapy. Pigment. Cell Melanoma Res. 2013, 26, 845–851. [Google Scholar] [CrossRef]
- Botton, T.; Talevich, E.; Mishra, V.K.; Zhang, T.; Shain, A.H.; Berquet, C.; Gagnon, A.; Judson, R.L.; Ballotti, R.; Ribas, A.; et al. Genetic Heterogeneity of BRAF Fusion Kinases in Melanoma Affects Drug Responses. Cell Rep. 2019, 29, 573–588.e577. [Google Scholar] [CrossRef] [PubMed]
- Menzies, A.M.; Yeh, I.; Botton, T.; Bastian, B.C.; Scolyer, R.A.; Long, G.V. Clinical activity of the MEK inhibitor trametinib in metastatic melanoma containing BRAF kinase fusion. Pigment. Cell Melanoma Res. 2015, 28, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Chew, S.M.; Lucas, M.; Brady, M.; Kelly, C.M. SKAP2-BRAF fusion and response to an MEK inhibitor in a patient with metastatic melanoma resistant to immunotherapy. BMJ Case Rep. 2021, 14, e238494. [Google Scholar] [CrossRef]
- Posch, C.; Sanlorenzo, M.; Ma, J.; Kim, S.T.; Zekhtser, M.; Ortiz-Urda, S. MEK/CDK4,6 co-targeting is effective in a subset of NRAS, BRAF and ‘wild type’ melanomas. Oncotarget 2018, 9, 34990–34995. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.; Zimmer, L.; Kim, K.B.; Sosman, J.A.; Ascierto, P.A.; Postow, M.A.; De Vos, F.Y.F.L.; van Herpen, C.M.L.; Carlino, M.S.; Johnson, D.B.; et al. Phase Ib/II Trial of Ribociclib in Combination with Binimetinib in Patients with NRAS-mutant Melanoma. Clin. Cancer Res. 2022, 28, 3002–3010. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, F.; Niu, R. Functions of Shp2 in cancer. J. Cell. Mol. Med. 2015, 19, 2075–2083. [Google Scholar] [CrossRef]
- Wei, B.; Xu, L.; Guo, W.; Wang, Y.; Wu, J.; Li, X.; Cai, X.; Hu, J.; Wang, M.; Xu, Q.; et al. SHP2-Mediated Inhibition of DNA Repair Contributes to cGAS-STING Activation and Chemotherapeutic Sensitivity in Colon Cancer. Cancer Res. 2021, 81, 3215–3228. [Google Scholar] [CrossRef]
- Wu, C.J.; O’Rourke, D.M.; Feng, G.S.; Johnson, G.R.; Wang, Q.; Greene, M.I. The tyrosine phosphatase SHP-2 is required for mediating phosphatidylinositol 3-kinase/Akt activation by growth factors. Oncogene 2001, 20, 6018–6025. [Google Scholar] [CrossRef]
- Yang, W.; Klaman, L.D.; Chen, B.; Araki, T.; Harada, H.; Thomas, S.M.; George, E.L.; Neel, B.G. An Shp2/SFK/Ras/Erk Signaling Pathway Controls Trophoblast Stem Cell Survival. Dev. Cell 2006, 10, 317–327. [Google Scholar] [CrossRef]
- Li, J.; Jie, H.B.; Lei, Y.; Gildener-Leapman, N.; Trivedi, S.; Green, T.; Kane, L.P.; Ferris, R.L. PD-1/SHP-2 inhibits Tc1/Th1 phenotypic responses and the activation of T cells in the tumor microenvironment. Cancer Res. 2015, 75, 508–518. [Google Scholar] [CrossRef]
- Brana, I.; Shapiro, G.; Johnson, M.L.; Yu, H.A.; Robbrecht, D.; Tan, D.S.-W.; Siu, L.L.; Minami, H.; Steeghs, N.; Hengelage, T.; et al. Initial results from a dose finding study of TNO155, a SHP2 inhibitor, in adults with advanced solid tumors. J. Clin. Oncol. 2021, 39, 3005. [Google Scholar] [CrossRef]
- Sanduja, S.; Feng, Y.; Mathis, R.A.; Sokol, E.S.; Reinhardt, F.; Halaban, R.; Gupta, P.B. AMPK promotes tolerance to Ras pathway inhibition by activating autophagy. Oncogene 2016, 35, 5295–5303. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Mehnert, J.M.; Mitchell, T.C.; Huang, A.C.; Aleman, T.S.; Kim, B.J.; Schuchter, L.M.; Linette, G.P.; Karakousis, G.C.; Mitnick, S.; Giles, L.; et al. BAMM (BRAF Autophagy and MEK Inhibition in Melanoma): A Phase I/II Trial of Dabrafenib, Trametinib, and Hydroxychloroquine in Advanced BRAFV600-mutant Melanoma. Clin. Cancer Res. 2022, 28, 1098–1106. [Google Scholar] [CrossRef]
- Lee, S.H.; Li, Y.; Kim, H.; Eum, S.; Park, K.; Lee, C.-H. The role of EZH1 and EZH2 in development and cancer. BMB Rep. 2022, 55, 595–601. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef]
- Zingg, D.; Debbache, J.; Schaefer, S.M.; Tuncer, E.; Frommel, S.C.; Cheng, P.; Arenas-Ramirez, N.; Haeusel, J.; Zhang, Y.; Bonalli, M.; et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat. Commun. 2015, 6, 6051. [Google Scholar] [CrossRef]
- Yu, H.; Ma, M.; Yan, J.; Xu, L.; Yu, J.; Dai, J.; Xu, T.; Tang, H.; Wu, X.; Li, S.; et al. Identification of coexistence of BRAF V600E mutation and EZH2 gain specifically in melanoma as a promising target for combination therapy. J. Transl. Med. 2017, 15, 243. [Google Scholar] [CrossRef]
- Souroullas, G.P.; Jeck, W.R.; Parker, J.S.; Simon, J.M.; Liu, J.-Y.; Paulk, J.; Xiong, J.; Clark, K.S.; Fedoriw, Y.; Qi, J.; et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat. Med. 2016, 22, 632–640. [Google Scholar] [CrossRef]
- Matatall, K.A.; Agapova, O.A.; Onken, M.D.; Worley, L.A.; Bowcock, A.M.; Harbour, J.W. BAP1 deficiency causes loss of melanocytic cell identity in uveal melanoma. BMC Cancer 2013, 13, 371. [Google Scholar] [CrossRef]
- Ny, L.; Jespersen, H.; Karlsson, J.; Alsén, S.; Filges, S.; All-Eriksson, C.; Andersson, B.; Carneiro, A.; Helgadottir, H.; Levin, M.; et al. The PEMDAC phase 2 study of pembrolizumab and entinostat in patients with metastatic uveal melanoma. Nat. Commun. 2021, 12, 5155. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Zappasodi, R. A decade of checkpoint blockade immunotherapy in melanoma: Understanding the molecular basis for immune sensitivity and resistance. Nat. Immunol. 2022, 23, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Rahma, O.E.; Hodi, F.S. The Intersection between Tumor Angiogenesis and Immune Suppression. Clin. Cancer Res. 2019, 25, 5449–5457. [Google Scholar] [CrossRef]
- Khan, K.A.; Kerbel, R.S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol. 2018, 15, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Yang, H.; Chon, H.J.; Kim, C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp. Mol. Med. 2020, 52, 1475–1485. [Google Scholar] [CrossRef]
- Arance, A.; de la Cruz-Merino, L.; Petrella, T.M.; Jamal, R.; Ny, L.; Carneiro, A.; Berrocal, A.; Márquez-Rodas, I.; Spreafico, A.; Atkinson, V.; et al. Phase II LEAP-004 Study of Lenvatinib Plus Pembrolizumab for Melanoma With Confirmed Progression on a Programmed Cell Death Protein-1 or Programmed Death Ligand 1 Inhibitor Given as Monotherapy or in Combination. J. Clin. Oncol. 2023, 41, 75–85. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandez, M.F.; Choi, J.; Sosman, J. New Approaches to Targeted Therapy in Melanoma. Cancers 2023, 15, 3224. https://doi.org/10.3390/cancers15123224
Fernandez MF, Choi J, Sosman J. New Approaches to Targeted Therapy in Melanoma. Cancers. 2023; 15(12):3224. https://doi.org/10.3390/cancers15123224
Chicago/Turabian StyleFernandez, Manuel Felipe, Jacob Choi, and Jeffrey Sosman. 2023. "New Approaches to Targeted Therapy in Melanoma" Cancers 15, no. 12: 3224. https://doi.org/10.3390/cancers15123224
APA StyleFernandez, M. F., Choi, J., & Sosman, J. (2023). New Approaches to Targeted Therapy in Melanoma. Cancers, 15(12), 3224. https://doi.org/10.3390/cancers15123224