
Repurposing of Chronically Used Drugs in Cancer Therapy: A Chance to Grasp


{kind=link}
{kind=link}
{kind=link}
{kind=link}
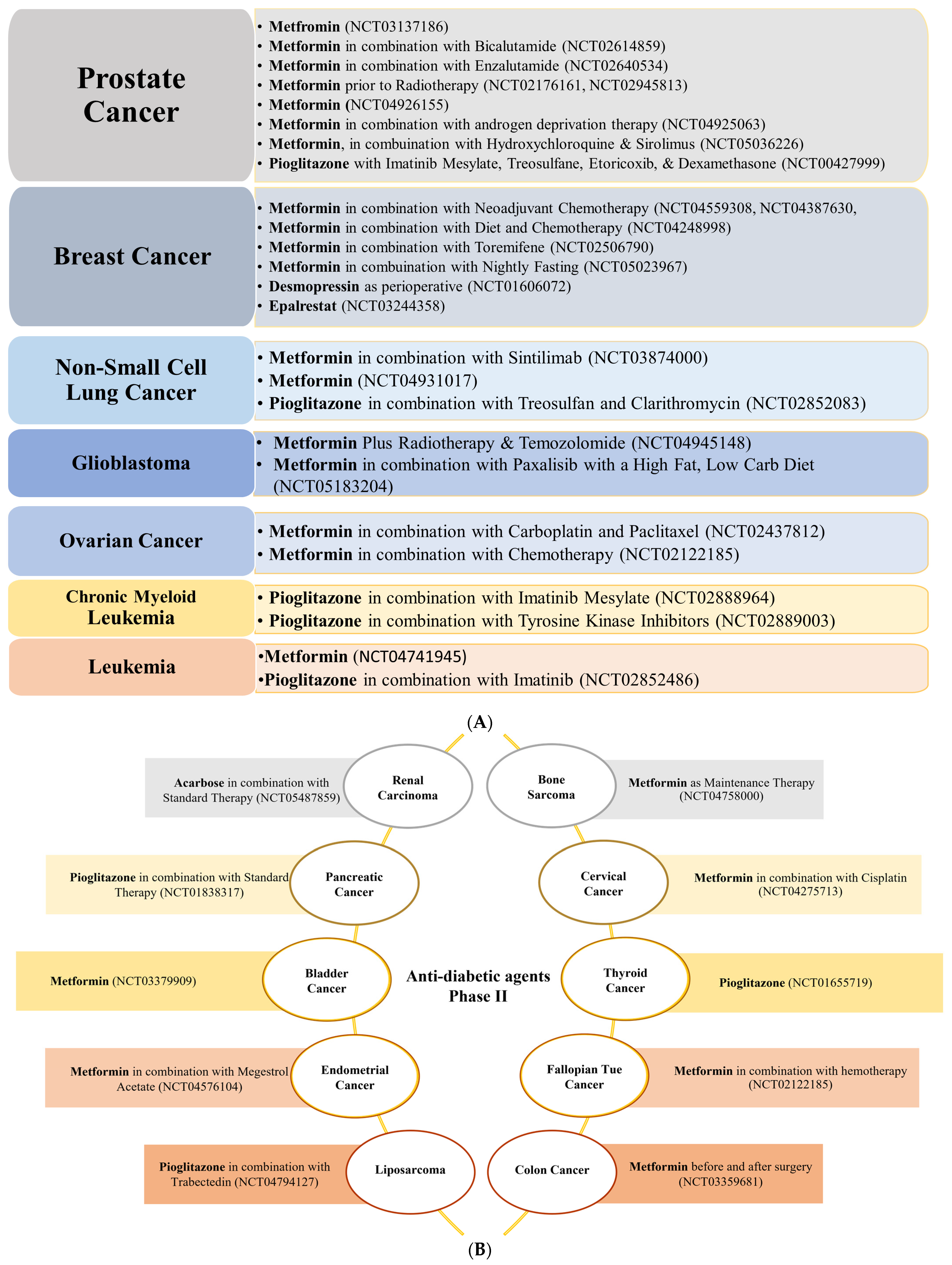{kind=link}
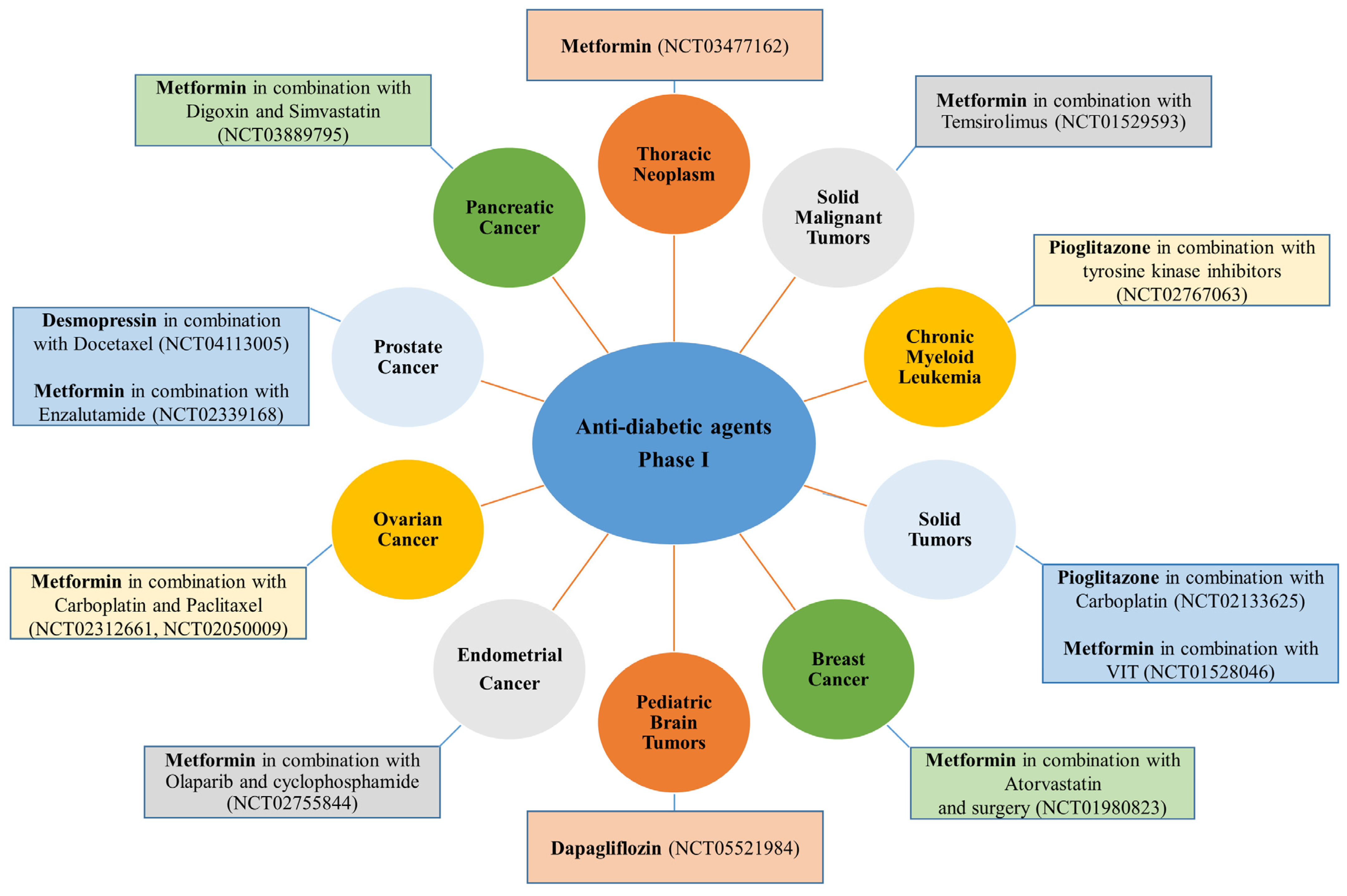{kind=link}
Simple Summary
Abstract
1. Introduction
2. So, Why Is the Repurposing of Anti-Diabetic and Anti-Hypertensive Drugs Exceptional?
2.1. Relation between Diabetes and Cancer
2.2. Relation between Cardiovascular Diseases and Cancer
3. Repurposing of Anti-Hypertensive and Anti-Diabetic Drugs: Current Update
3.1. Colorectal Cancer
3.1.1. Anti-Hypertensive Agents
3.1.2. Anti-Diabetic Agents
3.2. Breast Cancer
3.2.1. Anti-Hypertensive Agents
3.2.2. Anti-Diabetic Agents
3.3. Prostate Cancer
3.3.1. Anti-Hypertensive Agents
3.3.2. Anti-Diabetic Agents
3.4. Pancreatic Cancer
3.4.1. Anti-Hypertensive Agents
3.4.2. Anti-Diabetic Agents
3.5. Lung Cancer
3.5.1. Anti-Hypertensive Agents
3.5.2. Anti-Diabetic Agents
3.6. Ovarian, Cervical, and Endometrial Cancers
3.6.1. Anti-Hypertensive Agents
3.6.2. Anti-Diabetic Agents
3.7. Cancers of the Brain and Spinal Cord, Neuroblastoma, Osteosarcoma, and Head and Neck Squamous Cell Carcinoma
3.7.1. Anti-Hypertensive Agents
3.7.2. Anti-Diabetic Agents
3.8. Liver and Kidney Cancer
3.8.1. Anti-Hypertensive Agents
3.8.2. Anti-Diabetic Agents
3.9. Gastric and Esophageal Cancer
3.9.1. Anti-Hypertensive Agents
3.9.2. Anti-Diabetic Agents
3.10. Skin Cancer
3.10.1. Anti-Hypertensive Agents
3.10.2. Anti-Diabetic Agents
3.11. Blood Cancer
3.11.1. Anti-Hypertensive Agents
3.11.2. Anti-Diabetic Agents
4. Conclusions and Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ma, Y.; He, B.; Jiang, M.; Yang, Y.; Wang, C.; Huang, C.; Han, L. Prevalence and risk factors of cancer-related fatigue: A systematic review and meta-analysis. Int. J. Nurs. Stud. 2020, 111, 103707. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, A.; Rudzki, M.; Rudzki, S.; Lewandowski, T.; Laskowska, B. Environmental risk factors for cancer–review paper. Ann. Agric. Environ. Med. 2019, 26, 1–7. [Google Scholar] [CrossRef]
- Lentz, R.; Benson, A.B., 3rd; Kircher, S. Financial toxicity in cancer care: Prevalence, causes, consequences, and reduction strategies. J. Surg. Oncol. 2019, 120, 85–92. [Google Scholar] [CrossRef]
- Gonzalez-Fierro, A.; Dueñas-González, A. Drug repurposing for cancer therapy, easier said than done. Semin. Cancer Biol. 2019, 68, 123–131. [Google Scholar] [CrossRef]
- Ekinci, E.; Rohondia, S.; Khan, R.; Dou, Q.P. Repurposing Disulfiram as An Anti-Cancer Agent: Updated Review on Literature and Patents. Recent Patents Anti-Cancer Drug Discov. 2019, 14, 113–132. [Google Scholar] [CrossRef]
- Li, Y.-J.; Lei, Y.-H.; Yao, N.; Wang, C.-R.; Hu, N.; Ye, W.-C.; Zhang, D.-M.; Chen, Z.-S. Autophagy and multidrug resistance in cancer. Chin. J. Cancer 2017, 36, 52. [Google Scholar] [CrossRef]
- Navya, P.N.; Kaphle, A.; Srinivas, S.P.; Bhargava, S.K.; Rotello, V.M.; Daima, H.K. Current trends and challenges in cancer management and therapy using designer nanomaterials. Nano Converg. 2019, 6, 23. [Google Scholar] [CrossRef]
- Zugazagoitia, J.; Guedes, C.; Ponce, S.; Ferrer, I.; Molina-Pinelo, S.; Paz-Ares, L. Current Challenges in Cancer Treatment. Clin. Ther. 2016, 38, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Miao, K.; Sun, H.; Deng, C.-X. Tumor heterogeneity reshapes the tumor microenvironment to influence drug resistance. Int. J. Biol. Sci. 2022, 18, 3019–3033. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Gao, W.-Q.; Liu, Y. Metabolic heterogeneity in cancer: An overview and therapeutic implications. Biochim. Et Biophys. Acta (BBA)-Rev. Cancer 2020, 1874, 188421. [Google Scholar] [CrossRef] [PubMed]
- Hirata, A.; Hatano, Y.; Niwa, M.; Hara, A.; Tomita, H. Heterogeneity in Colorectal Cancer Stem CellsColorectal Cancer Stem Cells. Cancer Prev. Res. 2019, 12, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Pyun, W.Y.; Park, H.W. Cancer metabolism: Phenotype, signaling and therapeutic targets. Cells 2020, 9, 2308. [Google Scholar] [CrossRef]
- Oshimori, N.; Oristian, D.; Fuchs, E. TGF-β Promotes Heterogeneity and Drug Resistance in Squamous Cell Carcinoma. Cell 2015, 160, 963–976. [Google Scholar] [CrossRef] [PubMed]
- Poonpanichakul, T.; Shiao, M.-S.; Jiravejchakul, N.; Matangkasombut, P.; Sirachainan, E.; Charoensawan, V.; Jinawath, N. Capturing tumour heterogeneity in pre- and post-chemotherapy colorectal cancer ascites-derived cells using single-cell RNA-sequencing. Biosci. Rep. 2021, 41, BSR20212093. [Google Scholar] [CrossRef] [PubMed]
- Letai, A.; Bhola, P.; Welm, A.L. Functional precision oncology: Testing tumors with drugs to identify vulnerabilities and novel combinations. Cancer Cell 2021, 40, 26–35. [Google Scholar] [CrossRef]
- Maeda, H.; Khatami, M. Analyses of repeated failures in cancer therapy for solid tumors: Poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin. Transl. Med. 2018, 7, 11. [Google Scholar] [CrossRef]
- Lu, C.; Li, X.; Ren, Y.; Zhang, X. Disulfiram: A novel repurposed drug for cancer therapy. Cancer Chemother. Pharmacol. 2021, 87, 159–172. [Google Scholar] [CrossRef]
- Talevi, A. 2.34-Drug Repurposing. In Comprehensive Pharmacology; Kenakin, T., Ed.; Elsevier: Oxford, UK, 2022; pp. 813–824. [Google Scholar]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Jourdan, J.-P.; Bureau, R.; Rochais, C.; Dallemagne, P. Drug repositioning: A brief overview. J. Pharm. Pharmacol. 2020, 72, 1145–1151. [Google Scholar] [CrossRef]
- Parvathaneni, V.; Kulkarni, N.S.; Muth, A.; Gupta, V. Drug repurposing: A promising tool to accelerate the drug discovery process. Drug Discov. Today 2019, 24, 2076–2085. [Google Scholar] [CrossRef]
- Prager, G.W.; Braga, S.; Bystricky, B.; Qvortrup, C.; Criscitiello, C.; Esin, E.; Sonke, G.S.; Martínez, G.A.; Frenel, J.-S.; Karamouzis, M.; et al. Global cancer control: Responding to the growing burden, rising costs and inequalities in access. ESMO Open 2018, 3, e000285. [Google Scholar] [CrossRef] [PubMed]
- Sleire, L.; Førde, H.E.; Netland, I.A.; Leiss, L.; Skeie, B.S.; Enger, P.O. Drug repurposing in cancer. Pharmacol. Res. 2017, 124, 74–91. [Google Scholar] [CrossRef] [PubMed]
- Schein, C.H. Repurposing approved drugs for cancer therapy. Br. Med. Bull. 2021, 137, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Vargesson, N. The teratogenic effects of thalidomide on limbs. J. Hand Surg. 2018, 44, 88–95. [Google Scholar] [CrossRef]
- Mohty, M.; Terpos, E.; Mateos, M.-V.; Cavo, M.; Lejniece, S.; Beksac, M.; Bekadja, M.A.; Legiec, W.; Dimopoulos, M.; Stankovic, S.; et al. Multiple Myeloma Treatment in Real-world Clinical Practice: Results of a Prospective, Multinational, Noninterventional Study. Clin. Lymphoma Myeloma Leuk. 2018, 18, e401–e419. [Google Scholar] [CrossRef]
- Rao, Y.; Li, R.; Zhang, D. A drug from poison: How the therapeutic effect of arsenic trioxide on acute promyelocytic leukemia was discovered. Sci. China Life Sci. 2013, 56, 495–502. [Google Scholar] [CrossRef]
- Sanz, M.A.; Fenaux, P.; Tallman, M.S.; Estey, E.H.; Löwenberg, B.; Naoe, T.; Lengfelder, E.; Döhner, H.; Burnett, A.K.; Chen, S.-J.; et al. Management of acute promyelocytic leukemia: Updated recommendations from an expert panel of the European LeukemiaNet. Blood 2019, 133, 1630–1643. [Google Scholar] [CrossRef]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Adams, D.H.; Sanchez-Fueyo, A.; Samuel, D. From immunosuppression to tolerance. J. Hepatol. 2015, 62, S170–S185. [Google Scholar] [CrossRef]
- Chou, R.; Wagner, J.; Ahmed, A.Y.; Blazina, I.; Brodt, E.; Buckley, D.I.; Cheney, T.P.; Choo, E.; Dana, T.; Gordon, D.; et al. Treatments for Acute Pain: A Systematic Review; Agency for Healthcare Research and Quality (US): Rockville, MD, USA, 2020. [CrossRef]
- Christaki, E.; Marcou, M.; Tofarides, A. Antimicrobial Resistance in Bacteria: Mechanisms, Evolution, and Persistence. J. Mol. Evol. 2019, 88, 26–40. [Google Scholar] [CrossRef]
- Ambrosio, G.; De Ferrari, G.M.; Federici, M.; Filardi, P.P. Safety and tolerability of oral hypoglycemic therapies in type 2 diabetes mellitus patients at high cardiovascular risk. G. Ital. Cardiol. 2017, 18, 485–495. [Google Scholar]
- Schcolnik-Cabrera, A.; Juárez-López, D.; Duenas-Gonzalez, A. Perspectives on Drug Repurposing. Curr. Med. Chem. 2021, 28, 2085–2099. [Google Scholar] [CrossRef]
- Zhu, B.; Qu, S. The Relationship between Diabetes Mellitus and Cancers and Its Underlying Mechanisms. Front. Endocrinol. 2022, 13, 75. [Google Scholar] [CrossRef]
- Shafiei-Irannejad, V.; Samadi, N.; Salehi, R.; Yousefi, B.; Zarghami, N. New insights into antidiabetic drugs: Possible applications in cancer treatment. Chem. Biol. Drug Des. 2017, 90, 1056–1066. [Google Scholar] [CrossRef]
- Olatunde, A.; Nigam, M.; Singh, R.K.; Panwar, A.S.; Lasisi, A.; Alhumaydhi, F.A.; Kumar, V.J.; Mishra, A.P.; Sharifi-Rad, J. Cancer and diabetes: The interlinking metabolic pathways and repurposing actions of antidiabetic drugs. Cancer Cell Int. 2021, 21, 499. [Google Scholar] [CrossRef] [PubMed]
- Tokajuk, A.; Krzyżanowska-Grycel, E.; Tokajuk, A.; Grycel, S.; Sadowska, A.; Car, H. Antidiabetic drugs and risk of cancer. Pharmacol. Rep. 2015, 67, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Kirtonia, A.; Gala, K.; Fernandes, S.G.; Pandya, G.; Pandey, A.K.; Sethi, G.; Khattar, E.; Garg, M. Repurposing of drugs: An attractive pharmacological strategy for cancer therapeutics. Semin. Cancer Biol. 2020, 68, 258–278. [Google Scholar] [CrossRef] [PubMed]
- Dąbrowski, M. Diabetes, Antidiabetic Medications and Cancer Risk in Type 2 Diabetes: Focus on SGLT-2 Inhibitors. Int. J. Mol. Sci. 2021, 22, 1680. [Google Scholar] [CrossRef] [PubMed]
- Heckman-Stoddard, B.M.; DeCensi, A.; Sahasrabuddhe, V.V.; Ford, L.G. Repurposing metformin for the prevention of cancer and cancer recurrence. Diabetologia 2017, 60, 1639–1647. [Google Scholar] [CrossRef]
- Kim, H.M.; Kang, M.J.; Song, S.O. Metformin and Cervical Cancer Risk in Patients with Newly Diagnosed Type 2 Diabetes: A Population-Based Study in Korea. Endocrinol. Metab. 2022, 37, 929–937. [Google Scholar] [CrossRef]
- Søndergaard, C.S.; Esquivel, P.N.; Dalamaga, M.; Magkos, F. Use of Antihyperglycemic Drugs and Risk of Cancer in Patients with Diabetes. Curr. Oncol. Rep. 2022, 25, 29–40. [Google Scholar] [CrossRef]
- Kostapanos, M.S.; Elisaf, M.S.; Mikhailidis, D.P. Pioglitazone and cancer: Angel or demon? Curr. Pharm. Des. 2013, 19, 4913–4929. [Google Scholar] [CrossRef]
- Keith, R.L.; Blatchford, P.J.; Merrick, D.T.; Bunn, P.A.; Bagwell, B.; Dwyer-Nield, L.D.; Jackson, M.K.; Geraci, M.W.; Miller, Y.E. A Randomized Phase II Trial of Pioglitazone for Lung Cancer Chemoprevention in High-Risk Current and Former Smokers. Cancer Prev. Res. 2019, 12, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Hendryx, M.; Dong, Y.; Ndeke, J.M.; Luo, J. Sodium-glucose cotransporter 2 (SGLT2) inhibitor initiation and hepatocellular carcinoma prognosis. PLoS ONE 2022, 17, e0274519. [Google Scholar] [CrossRef]
- Barrea, L.; Annunziata, G.; Bordoni, L.; Muscogiuri, G.; Colao, A.; Savastano, S. Nutrigenetics—Personalized nutrition in obesity and cardiovascular diseases. Int. J. Obes. Suppl. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.; Lee, D.; Shim, H. Metabolic Positron Emission Tomography Imaging in Cancer Detection and Therapy Response. Semin. Oncol. 2011, 38, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Fowler, A.M.; Strigel, R.M. Clinical advances in PET–MRI for breast cancer. Lancet Oncol. 2022, 23, e32–e43. [Google Scholar] [CrossRef]
- Koene, R.J.; Prizment, A.E.; Blaes, A.; Konety, S.H. Shared Risk Factors in Cardiovascular Disease and Cancer. Circulation 2016, 133, 1104–1114. [Google Scholar] [CrossRef]
- Prabhakar, N.R.; Peng, Y.-J.; Nanduri, J. Hypoxia-inducible factors and obstructive sleep apnea. J. Clin. Investig. 2020, 130, 5042–5051. [Google Scholar] [CrossRef]
- Yu, B.; Wang, X.; Song, Y.; Xie, G.; Jiao, S.; Shi, L.; Cao, X.; Han, X.; Qu, A. The role of hypoxia-inducible factors in cardiovascular diseases. Pharmacol. Ther. 2022, 238, 108186. [Google Scholar] [CrossRef]
- Korbecki, J.; Simińska, D.; Gąssowska-Dobrowolska, M.; Listos, J.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. Chronic and cycling hypoxia: Drivers of cancer chronic inflammation through HIF-1 and NF-κB activation: A review of the molecular mechanisms. Int. J. Mol. Sci. 2021, 22, 10701. [Google Scholar] [CrossRef] [PubMed]
- Malekan, M.; Ebrahimzadeh, M.A.; Sheida, F. The role of Hypoxia-Inducible Factor-1alpha and its signaling in melanoma. Biomed. Pharmacother. 2021, 141, 111873. [Google Scholar] [CrossRef]
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Curr. Oncol. Rep. 2019, 21, 6. [Google Scholar] [CrossRef]
- Wicks, E.E.; Semenza, G.L. Hypoxia-inducible factors: Cancer progression and clinical translation. J. Clin. Investig. 2022, 132, e159839. [Google Scholar] [CrossRef]
- Mills, K.T.; Stefanescu, A.; He, J. The global epidemiology of hypertension. Nat. Rev. Nephrol. 2020, 16, 223–237. [Google Scholar] [CrossRef]
- Cho, I.J.; Shin, J.H.; Jung, M.H.; Kang, C.Y.; Hwang, J.; Kwon, C.H.; Kim, W.; Kim, D.H.; Lee, C.J.; Kang, S.-H.; et al. Antihypertensive drugs and the risk of cancer: A nationwide cohort study. J. Clin. Med. 2021, 10, 771. [Google Scholar] [CrossRef]
- Boudreau, D.M.; Yu, O.; Chubak, J.; Wirtz, H.S.; Bowles, E.J.A.; Fujii, M.; Buist, D.S.M. Comparative safety of cardiovascular medication use and breast cancer outcomes among women with early stage breast cancer. Breast Cancer Res. Treat. 2014, 144, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Cronin-Fenton, D.; Lash, T.L.; Ahern, T.P.; Damkier, P.; Christiansen, P.; Ejlertsen, B.; Sørensen, H.T. Concurrent new drug prescriptions and prognosis of early breast cancer: Studies using the Danish Breast Cancer Group clinical database. Acta Oncol. 2017, 57, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Carlos-Escalante, J.A.; de Jesús-Sánchez, M.; Rivas-Castro, A.; Pichardo-Rojas, P.S.; Arce, C.; Wegman-Ostrosky, T. The Use of Antihypertensive Drugs as Coadjuvant Therapy in Cancer. Front. Oncol. 2021, 11, 660943. [Google Scholar] [CrossRef] [PubMed]
- Gales, L.; Forsea, L.; Mitrea, D.; Stefanica, I.; Stanculescu, I.; Mitrica, R.; Georgescu, M.; Trifanescu, O.; Anghel, R.; Serbanescu, L. Antidiabetics, Anthelmintics, Statins, and Beta-Blockers as Co-Adjuvant Drugs in Cancer Therapy. Medicina 2022, 58, 1239. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, T.; Vale, N. Pharmacological Efficacy of Repurposing Drugs in the Treatment of Prostate Cancer. Int. J. Mol. Sci. 2023, 24, 4154. [Google Scholar] [CrossRef] [PubMed]
- Loosen, S.H.; Schöler, D.; Luedde, M.; Eschrich, J.; Luedde, T.; Gremke, N.; Kalder, M.; Kostev, K.; Roderburg, C. Antihypertensive Therapy and Incidence of Cancer. J. Clin. Med. 2022, 11, 6624. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wu, S. Risks and management of hypertension in cancer patients undergoing targeted therapy: A review. Clin. Hypertens. 2022, 28, 14. [Google Scholar] [CrossRef]
- Chang, A.; Yeung, S.; Thakkar, A.; Huang, K.M.; Liu, M.M.; Kanassatega, R.S.; Parsa, C.; Orlando, R.; Jackson, E.K.; Andresen, B.T.; et al. Prevention of Skin Carcinogenesis by the β-Blocker CarvedilolCarvedilol for Skin Cancer Prevention. Cancer Prev. Res. 2015, 8, 27–36. [Google Scholar] [CrossRef]
- Chae, Y.K.; Valsecchi, M.E.; Kim, J.; Bianchi, A.L.; Khemasuwan, D.; Desai, A.; Tester, W. Reduced Risk of Breast Cancer Recurrence in Patients Using ACE Inhibitors, ARBs, and/or Statins. Cancer Investig. 2011, 29, 585–593. [Google Scholar] [CrossRef]
- Keizman, D.; Huang, P.; Eisenberger, M.A.; Pili, R.; Kim, J.J.; Antonarakis, E.S.; Hammers, H.; Carducci, M.A. Angiotensin system inhibitors and outcome of sunitinib treatment in patients with metastatic renal cell carcinoma: A retrospective examination. Eur. J. Cancer 2011, 47, 1955–1961. [Google Scholar] [CrossRef]
- Shahrokhi, M.; Gupta, V. Propranolol; StatPearls: Treasure Island, FL, USA, 2023.
- Banavali, S.; Pasquier, E.; Andre, N. Targeted therapy with propranolol and metronomic chemotherapy combination: Sustained complete response of a relapsing metastatic angiosarcoma. Ecancermedicalscience 2015, 9, 499. [Google Scholar] [CrossRef]
- Chang, P.Y.; Huang, W.Y.; Lin, C.L.; Huang, T.C.; Wu, Y.Y.; Chen, J.H.; Kao, C.H. Propranolol reduces cancer risk: A population-based cohort study. Medicine 2015, 94, e1097. [Google Scholar] [CrossRef]
- Zhang, D.; Ma, Q.; Shen, S.; Hu, H. Inhibition of pancreatic cancer cell proliferation by propranolol occurs through apoptosis induction: The study of β-adrenoceptor antagonist’s anti-cancer effect in pancreatic cancer cell. Pancreas 2009, 38, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Song, K.; Zhu, Z.; Liu, Z.; Zhang, W.; Li, W.; Hu, J.; Hu, Q.; Chen, C.; Chen, B.; et al. Propranolol Suppresses the Growth of Colorectal Cancer Through Simultaneously Activating Autologous CD8 + T Cells and Inhibiting Tumor AKT/MAPK Pathway. Clin. Pharmacol. Ther. 2020, 108, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Ishida, J.; Konishi, M.; Ebner, N.; Springer, J. Repurposing of approved cardiovascular drugs. J. Transl. Med. 2016, 14, 269. [Google Scholar] [CrossRef] [PubMed]
- Neo, J.H.; Malcontenti-Wilson, C.; Muralidharan, V.; Christophi, C. Effect of ACE inhibitors and angiotensin II receptor antagonists in a mouse model. J. Gastroenterol. Hepatol. 2007, 22, 577–584. [Google Scholar] [CrossRef]
- Jones, M.; Schrader, K.; Shen, Y.; Pleasance, E.; Ch’Ng, C.; Dar, N.; Yip, S.; Renouf, D.; Schein, J.; Mungall, A.; et al. Response to angiotensin blockade with irbesartan in a patient with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 801–806. [Google Scholar] [CrossRef]
- Romankiewicz, J.A.; Brogden, R.N.; Heel, R.C.; Speight, T.M.; Avery, G.S. Captopril: An update review of its pharmacological properties and therapeutic efficacy in congestive heart failure. Drugs 1983, 25, 6–40. [Google Scholar] [CrossRef]
- Kinuya, S.; Yokoyama, K.; Kawashima, A.; Hiramatsu, T.; Konishi, S.; Shuke, N.; Watanabe, N.; Takayama, T.; Michigishi, T.; Tonami, N. Pharmacologic intervention with angiotensin II and kininase inhibitor enhanced efficacy of radioimmunotherapy in human colon cancer xenografts. J. Nucl. Med. 2000, 41, 1244–1249. [Google Scholar]
- Yang, Y.; Ma, L.; Xu, Y.; Liu, Y.; Li, W.; Cai, J.; Zhang, Y. Enalapril overcomes chemoresistance and potentiates antitumor efficacy of 5-FU in colorectal cancer by suppressing proliferation, angiogenesis, and NF-κB/STAT3-regulated proteins. Cell Death Dis. 2020, 11, 477. [Google Scholar] [CrossRef]
- Yasumaru, M.; Tsuji, S.; Tsujii, M.; Irie, T.; Komori, M.; Kimura, A.; Nishida, T.; Kakiuchi, Y.; Kawai, N.; Murata, H.; et al. Inhibition of angiotensin II activity enhanced the antitumor effect of cyclooxygenase-2 inhibitors via insulin-like growth factor I receptor pathway. Cancer Res. 2003, 63, 6726–6734. [Google Scholar]
- Wu, L.; Lin, W.; Liao, Q.; Wang, H.; Lin, C.; Tang, L.; Lian, W.; Chen, Z.; Li, K.; Xu, L.; et al. Calcium Channel Blocker Nifedipine Suppresses Colorectal Cancer Progression and Immune Escape by Preventing NFAT2 Nuclear Translocation. Cell Rep. 2020, 33, 108327. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef]
- Ben Sahra, I.; Laurent, K.; Loubat, A.; Giorgetti-Peraldi, S.; Colosetti, P.; Auberger, P.; Tanti, J.F.; Le Marchand-Brustel, Y.; Bost, F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 2008, 27, 3576–3586. [Google Scholar] [CrossRef]
- Ripoll, G.V.; Pifano, M.; Garona, J.; Alonso, D.F. Commentary: Arginine vasopressin receptor 1a is a therapeutic target for castration-resistant prostate cancer. Front. Oncol. 2020, 9, 1490. [Google Scholar] [CrossRef]
- Ripoll, G.V.; Garona, J.; Hermo, G.A.; Gomez, D.E.; Alonso, D.F. Effects of the synthetic vasopressin analog desmopressin in a mouse model of colon cancer. Anticancer Res. 2010, 30, 5049–5054. [Google Scholar]
- Orasanu, G.; Ziouzenkova, O.; Devchand, P.R.; Nehra, V.; Hamdy, O.; Horton, E.S.; Plutzky, J. The peroxisome proliferator-activated receptor-gamma agonist pioglitazone represses inflammation in a peroxisome proliferator-activated receptor-alpha-dependent manner in vitro and in vivo in mice. J. Am. Coll. Cardiol. 2008, 52, 869–881. [Google Scholar] [CrossRef]
- Takano, S.; Kubota, T.; Nishibori, H.; Hasegawa, H.; Ishii, Y.; Nitori, N.; Ochiai, H.; Okabayashi, K.; Kitagawa, Y.; Watanabe, M.; et al. Pioglitazone, a ligand for peroxisome proliferator-activated receptor-γ acts as an inhibitor of colon cancer liver metastasis. Anticancer Res. 2008, 28, 3593–3599. [Google Scholar] [PubMed]
- Kohlmann, O.; Bresnahan, M.; Gavras, H. Central and peripheral indices of sympathetic activity after blood pressure lowering with enalapril (MK-421) or hydralazine in normotensive rats. Hypertension 1984, 6, I1-6. [Google Scholar] [CrossRef] [PubMed]
- Segura-Pacheco, B.; Perez-Cardenas, E.; Taja-Chayeb, L.; Chavez-Blanco, A.; Revilla-Vazquez, A.; Benitez-Bribiesca, L.; Duenas-González, A. Global DNA hypermethylation-associated cancer chemotherapy resistance and its reversion with the demethylating agent hydralazine. J. Transl. Med. 2006, 4, 32. [Google Scholar] [CrossRef] [PubMed]
- Segura-Pacheco, B.; Trejo-Becerril, C.; Perez-Cardenas, E.; Taja-Chayeb, L.; Mariscal, I.; Chavez, A.; Acuña, C.; Salazar, A.M.; Lizano, M.; Dueñas-Gonzalez, A. Reactivation of tumor suppressor genes by the cardiovascular drugs hydralazine and procainamide and their potential use in cancer therapy. Clin. Cancer Res. 2003, 9, 1596–1603. [Google Scholar]
- Pasquier, E.; Ciccolini, J.; Carre, M.; Giacometti, S.; Fanciullino, R.; Pouchy, C.; Montero, M.-P.; Serdjebi, C.; Kavallaris, M.; André, N. Propranolol potentiates the anti-angiogenic effects and anti-tumor efficacy of chemotherapy agents: Implication in breast cancer treatment. Oncotarget 2011, 2, 797–809. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef] [PubMed]
- Coulson, R.; Liew, S.H.; Connelly, A.A.; Yee, N.S.; Deb, S.; Kumar, B.; Vargas, A.C.; O’toole, S.A.; Parslow, A.C.; Poh, A.; et al. The angiotensin receptor blocker, Losartan, inhibits mammary tumor development and progression to invasive carcinoma. Oncotarget 2017, 8, 18640–18656. [Google Scholar] [CrossRef] [PubMed]
- Ni, S.; Chen, X.; Yu, Q.; Xu, Y.; Hu, Z.; Zhang, J.; Zhang, W.; Li, B.; Yang, X.; Mao, F.; et al. Discovery of candesartan cilexetic as a novel neddylation inhibitor for suppressing tumor growth. Eur. J. Med. Chem. 2019, 185, 111848. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, W.A. Multiplicity of effectors of the cardioprotective agent, diazoxide. Pharmacol. Ther. 2013, 140, 167–175. [Google Scholar] [CrossRef]
- Wang, A.; Lim, H.; Cheng, S.-Y.; Xie, L. ANTENNA, a Multi-Rank, Multi-Layered Recommender System for Inferring Reliable Drug-Gene-Disease Associations: Repurposing Diazoxide as a Targeted Anti-Cancer Therapy. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 15, 1960–1967. [Google Scholar] [CrossRef]
- Aljofan, M.; Riethmacher, D. Anticancer activity of metformin: A systematic review of the literature. Futur. Sci. OA 2019, 5, FSO410. [Google Scholar] [CrossRef]
- Wang, Y.-W.; He, S.-J.; Feng, X.; Cheng, J.; Luo, Y.-T.; Tian, L.; Huang, Q. Metformin: A review of its potential indications. Drug Des. Dev. Ther. 2017, 11, 2421–2429. [Google Scholar] [CrossRef]
- Zordoky, B.N.; Bark, D.; Soltys, C.L.; Sung, M.M.; Dyck, J.R. The anti-proliferative effect of metformin in triple-negative MDA-MB-231 breast cancer cells is highly dependent on glucose concentration: Implications for cancer therapy and prevention. Biochim. Et Biophys. Acta (BBA)-Gen. Subj. 2014, 1840, 1943–1957. [Google Scholar] [CrossRef]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin Selectively Targets Cancer Stem Cells, and Acts Together with Chemotherapy to Block Tumor Growth and Prolong Remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef]
- Deng, X.-S.; Wang, S.; Deng, A.; Liu, B.; Edgerton, S.M.; Lind, S.E.; Wahdan-Alaswad, R.; Thor, A.D. Metformin targets Stat3 to inhibit cell growth and induce apoptosis in triple-negative breast cancers. Cell Cycle 2012, 11, 367–376. [Google Scholar] [CrossRef]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin Is an AMP Kinase–Dependent Growth Inhibitor for Breast Cancer Cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef] [PubMed]
- Alonso, D.F.; Skilton, G.; Farías, E.F.; Joffé, E.B.d.K.; Gomez, D.E. Antimetastatic effect of desmopressin in a mouse mammary tumor model. Breast Cancer Res. Treat. 1999, 57, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, M.T.; Garona, J.; Sobol, N.T.; Farina, H.G.; Alonso, D.F.; Álvarez, V.A. In vitro and in vivo evaluation of desmopressin-loaded poly(D,L-lactic-co-glycolic acid) nanoparticles for its potential use in cancer treatment. Nanomedicine 2018, 13, 2835–2849. [Google Scholar] [CrossRef] [PubMed]
- Garona, J.; Pifano, M.; Orlando, U.D.; Pastrian, M.B.; Iannucci, N.B.; Ortega, H.H.; Podesta, E.J.; Gomez, D.E.; Ripoll, G.V.; Alonso, D.F. The novel desmopressin analogue [V4Q5]dDAVP inhibits angiogenesis, tumour growth and metastases in vasopressin type 2 receptor-expressing breast cancer models. Int. J. Oncol. 2015, 46, 2335–2345. [Google Scholar] [CrossRef]
- Ripoll, G.V.; Garona, J.; Pifano, M.; Farina, H.G.; Gomez, D.E.; Alonso, D.F. Reduction of tumor angiogenesis induced by desmopressin in a breast cancer model. Breast Cancer Res. Treat. 2013, 142, 9–18. [Google Scholar] [CrossRef]
- Ripoll, G.V.; Giron, S.; Krzymuski, M.J.; Hermo, G.A.; Gomez, D.E.; Alonso, D.F. Antitumor effects of desmopressin in combination with chemotherapeutic agents in a mouse model of breast cancer. Anticancer Res. 2008, 28, 2607–2611. [Google Scholar]
- Ramirez, M.A.; Borja, N.L. Epalrestat: An Aldose Reductase Inhibitor for the Treatment of Diabetic Neuropathy. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2008, 28, 646–655. [Google Scholar] [CrossRef]
- Wu, X.; Li, X.; Fu, Q.; Cao, Q.; Chen, X.; Wang, M.; Yu, J.; Long, J.; Yao, J.; Liu, H.; et al. AKR1B1 promotes basal-like breast cancer progression by a positive feedback loop that activates the EMT program. J. Exp. Med. 2017, 214, 1065–1079. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, N.; Li, Q.; Zhou, Y.; Luan, Y. A two-pronged photodynamic nanodrug to prevent metastasis of basal-like breast cancer. Chem. Commun. 2021, 57, 2305–2308. [Google Scholar] [CrossRef]
- Kole, L.; Sarkar, M.; Deb, A.; Giri, B. Pioglitazone, an anti-diabetic drug requires sustained MAPK activation for its anti-tumor activity in MCF7 breast cancer cells, independent of PPAR-γ pathway. Pharmacol. Rep. 2016, 68, 144–154. [Google Scholar] [CrossRef]
- Singh, S.; Preuss, C.V. Carvedilol; StatPearls: Treasure Island, FL, USA, 2022.
- Zahalka, A.H.; Fram, E.; Lin, W.; Mohn, L.; Frenette, P.S.; Agalliu, I.; Watts, K.L. Use of beta-blocker types and risk of incident prostate cancer in a multiethnic population. Urol. Oncol. Semin. Orig. Investig. 2020, 38, 794.e11–794.e16. [Google Scholar] [CrossRef] [PubMed]
- Alhusban, A.; Al-Azayzih, A.; Goc, A.; Gao, F.; Fagan, S.C.; Somanath, P.R. Clinically Relevant Doses of Candesartan Inhibit Growth of Prostate Tumor Xenografts In Vivo through Modulation of Tumor Angiogenesis. Experiment 2014, 350, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Shebl, R.I. Anti-cancer Potential of Captopril and Botulinum Toxin Type-A and Associated p53 Gene Apototic Stimulating Activity. Iran. J. Pharm. Res. 2019, 18, 1967–1977. [Google Scholar] [CrossRef]
- Graça, I.; Sousa, E.J.; Costa-Pinheiro, P.; Vieira, F.Q.; Torres-Ferreira, J.; Martins, M.G.; Henrique, R.; Jerónimo, C. Anti-neoplastic properties of hydralazine in prostate cancer. Oncotarget 2014, 5, 5950–5964. [Google Scholar] [CrossRef] [PubMed]
- Coyle, C.; Cafferty, F.; Vale, C.; Langley, R. Metformin as an adjuvant treatment for cancer: A systematic review and meta-analysis. Ann. Oncol. 2016, 27, 2184–2195. [Google Scholar] [CrossRef]
- Ahn, H.K.; Lee, Y.H.; Koo, K.C. Current Status and Application of Metformin for Prostate Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2020, 21, 8540. [Google Scholar] [CrossRef] [PubMed]
- Kao, L.-T.; Xirasagar, S.; Lin, H.-C.; Huang, C.-Y. Association Between Pioglitazone Use and Prostate Cancer: A Population-Based Case-Control Study in the Han Population. J. Clin. Pharmacol. 2018, 59, 344–349. [Google Scholar] [CrossRef]
- Miyazawa, M.; Subbaramaiah, K.; Bhardwaj, P.; Zhou, X.K.; Wang, H.; Falcone, D.J. Pioglitazone Inhibits Periprostatic White Adipose Tissue Inflammation in Obese MicePioglitazone Reverses Periprostatic Fat Inflammation. Cancer Prev. Res. 2018, 11, 215–226. [Google Scholar] [CrossRef]
- Bass, R.; Roberto, D.; Wang, D.Z.; Cantu, F.P.; Mohamadi, R.M.; Kelley, S.O.; Klotz, L.; Venkateswaran, V. Combining Desmopressin and Docetaxel for the Treatment of Castration-Resistant Prostate Cancer in an Orthotopic Model. Anticancer Res. 2019, 39, 113–118. [Google Scholar] [CrossRef]
- Hoffman, A.; Sasaki, H.; Roberto, D.; Mayer, M.J.; Klotz, L.H.; Venkateswaran, V. Effect of Combination therapy of Desmopressin and Docetaxel on prostate cancer cell (DU145) proliferation, migration and tumor growth. J. Cancer Biol. Therap. 2016, 1, 129–136. [Google Scholar]
- Sasaki, H.; Klotz, L.H.; Sugar, L.M.; Kiss, A.; Venkateswaran, V. A combination of desmopressin and docetaxel inhibit cell proliferation and invasion mediated by urokinase-type plasminogen activator (uPA) in human prostate cancer cells. Biochem. Biophys. Res. Commun. 2015, 464, 848–854. [Google Scholar] [CrossRef]
- Gautam, S.K.; Dalal, V.; Sajja, B.R.; Gupta, S.; Gulati, M.; Dwivedi, N.V.; Aithal, A.; Cox, J.L.; Rachagani, S.; Liu, Y.; et al. Endothelin-axis antagonism enhances tumor perfusion in pancreatic cancer. Cancer Lett. 2022, 544, 215801. [Google Scholar] [CrossRef]
- Fitzner, B.; Brock, P.; Holzhüter, S.-A.; Nizze, H.; Sparmann, G.; Emmrich, J.; Liebe, S.; Jaster, R. Synergistic Growth Inhibitory Effects of the Dual Endothelin-1 Receptor Antagonist Bosentan on Pancreatic Stellate and Cancer Cells. Dig. Dis. Sci. 2008, 54, 309–320. [Google Scholar] [CrossRef]
- Ahn, H.-M.; Kim, D.-G.; Kim, Y.-J. Blockade of endothelin receptor A enhances the therapeutic efficacy of gemcitabine in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2020, 527, 568–573. [Google Scholar] [CrossRef]
- Fendrich, V.; Chen, N.-M.; Neef, M.; Waldmann, J.; Buchholz, M.; Feldmann, G.; Slater, E.P.; Maitra, A.; Bartsch, D.K. The angiotensin-I-converting enzyme inhibitor enalapril and aspirin delay progression of pancreatic intraepithelial neoplasia and cancer formation in a genetically engineered mouse model of pancreatic cancer. Gut 2009, 59, 630–637. [Google Scholar] [CrossRef]
- Song, Y.; Zhang, C. Hydralazine inhibits human cervical cancer cell growth in vitro in association with APC demethylation and re-expression. Cancer Chemother. Pharmacol. 2008, 63, 605–613. [Google Scholar] [CrossRef]
- Bhattacharyya, G.S.; Babu, K.G.; Bondarde, S.A.; Biswas, G.; Ranade, A.; Parikh, P.M.; Bascomb, N.F.; Malhotra, H. Effect of coadministered beta blocker and COX-2 inhibitor to patients with pancreatic cancer prior to receiving albumin-bound (Nab) paclitaxel. J. Clin. Oncol. 2015, 33, 302. [Google Scholar] [CrossRef]
- Chow, W.; Amaya, C.N.; Rains, S.; Chow, M.; Dickerson, E.B.; Bryan, B.A. Growth Attenuation of Cutaneous Angiosarcoma With Propranolol-Mediated β-Blockade. JAMA Dermatol. 2015, 151, 1226–1229. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Bouche, G.; Sukhatme, V.; Meheus, L.; Rooman, I.; Sukhatme, V.P. Repurposing Drugs in Oncology (ReDO)—Propranolol as an anti-cancer agent. Ecancermedicalscience 2016, 10, 680. [Google Scholar] [CrossRef] [PubMed]
- Jäger, H.; Dreker, T.; Buck, A.; Giehl, K.; Gress, T.; Grissmer, S. Blockage of Intermediate-Conductance Ca2+-Activated K+Channels Inhibit Human Pancreatic Cancer Cell Growth in Vitro. Mol. Pharmacol. 2004, 65, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Woods, N.; Trevino, J.; Coppola, D.; Chellappan, S.; Yang, S.; Padmanabhan, J. Fendiline inhibits proliferation and invasion of pancreatic cancer cells by interfering with ADAM10 activation and β-catenin signaling. Oncotarget 2015, 6, 35931–35948. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, I.; Yamazaki, K.; Oyama, K.; Hayashi, H.; Tajima, H.; Kitagawa, H.; Fushida, S.; Fujimura, T.; Ohta, T. Pioglitazone inhibits the proliferation and metastasis of human pancreatic cancer cells. Oncol. Lett. 2014, 8, 2709–2714. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Wang, Z.; Ali, S.; Ahmad, A.; Azmi, A.S.; Sarkar, S.H.; Banerjee, S.; Kong, D.; Li, Y.; Thakur, S.; et al. Metformin Inhibits Cell Proliferation, Migration and Invasion by Attenuating CSC Function Mediated by Deregulating miRNAs in Pancreatic Cancer Cells. Cancer Prev. Res. 2012, 5, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Qian, W.; Jiang, Z.; Cheng, L.; Liang, C.; Sun, L.; Zhou, C.; Gao, L.; Lei, M.; Yan, B.; et al. Metformin suppresses cancer initiation and progression in genetic mouse models of pancreatic cancer. Mol. Cancer 2017, 16, 131. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Cárdenas, E.; Taja-Chayeb, L.; Trejo-Becerril, C.; Chanona-Vilchis, J.; Chávez-Blanco, A.; Domínguez-Gómez, G.; Langley, E.; García-Carrancá, A.; Dueñas-González, A. Antimetastatic effect of epigenetic drugs, hydralazine and valproic acid, in Ras-transformed NIH 3T3 cells. OncoTargets Ther. 2018, 11, 8823–8833. [Google Scholar] [CrossRef]
- Carter, C.A.; Zeman, K.; Day, R.M.; Richard, P.; Oronsky, A.; Oronsky, N.; Lybeck, M.; Scicinski, J.; Oronsky, B. Addressing the elephant in the room, therapeutic resistance in non-small cell lung cancer, with epigenetic therapies. Oncotarget 2016, 7, 40781–40791. [Google Scholar] [CrossRef]
- Regan, D.P.; Coy, J.W.; Chahal, K.K.; Chow, L.; Kurihara, J.N.; Guth, A.M.; Kufareva, I.; Dow, S.W. The Angiotensin Receptor Blocker Losartan Suppresses Growth of Pulmonary Metastases via AT1R-Independent Inhibition of CCR2 Signaling and Monocyte Recruitment. J. Immunol. 2019, 202, 3087–3102. [Google Scholar] [CrossRef]
- Li, J.; Chen, L.; Yu, P.; Liu, B.; Zhu, J.; Yang, Y. Telmisartan Exerts Anti-Tumor Effects by Activating Peroxisome Proliferator-Activated Receptor-γ in Human Lung Adenocarcinoma A549 Cells. Molecules 2014, 19, 2862–2876. [Google Scholar] [CrossRef]
- Zhang, W.-M.; Zhou, J.; Ye, Q.-J. Endothelin-1 enhances proliferation of lung cancer cells by increasing intracellular free Ca2+. Life Sci. 2008, 82, 764–771. [Google Scholar] [CrossRef]
- Wang, Y.; He, X.; Li, C.; Ma, Y.; Xue, W.; Hu, B.; Wang, J.; Zhang, T.; Zhang, F. Carvedilol serves as a novel CYP1B1 inhibitor, a systematic drug repurposing approach through structure-based virtual screening and experimental verification. Eur. J. Med. Chem. 2020, 193, 112235. [Google Scholar] [CrossRef]
- Attoub, S.; Gaben, A.M.; Al-Salam, S.; Al Sultan, M.; John, A.; Nicholls, M.G.; Mester, J.; Petroianu, G. Captopril as a Potential Inhibitor of Lung Tumor Growth and Metastasis. Ann. N. Y. Acad. Sci. 2008, 1138, 65–72. [Google Scholar] [CrossRef]
- Han, P.; Zhou, J.; Xiang, J.; Liu, Q.; Sun, K. Research progress on the therapeutic effect and mechanism of metformin for lung cancer (Review). Oncol. Rep. 2022, 49, 1–21. [Google Scholar] [CrossRef]
- Barrios-Bernal, P.; Zatarain-Barrón, Z.L.; Hernández-Pedro, N.; Orozco-Morales, M.; Olivera-Ramírez, A.; Ávila-Moreno, F.; Colín-González, A.L.; Cardona, A.F.; Rosell, R.; Arrieta, O. Will We Unlock the Benefit of Metformin for Patients with Lung Cancer? Lessons from Current Evidence and New Hypotheses. Pharmaceuticals 2022, 15, 786. [Google Scholar] [CrossRef]
- Sun, X.; Dong, M.; Gao, Y.; Wang, Y.; Du, L.; Liu, Y.; Wang, Q.; Ji, K.; He, N.; Wang, J.; et al. Metformin increases the radiosensitivity of non-small cell lung cancer cells by destabilizing NRF2. Biochem. Pharmacol. 2022, 199, 114981. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, J.; Sun, Y.; Song, B.; Zhang, Y.; Lu, Y.; Ma, H. Metformin Synergizes with PD-L1 Monoclonal Antibody Enhancing Tumor Immune Response in Treating Non-Small Cell Lung Cancer and Its Molecular Mechanism Investigation. Evid.-Based Complement. Altern. Med. 2022, 2022, 5983959. [Google Scholar] [CrossRef] [PubMed]
- Seabloom, D.E.; Galbraith, A.R.; Haynes, A.M.; Antonides, J.D.; Wuertz, B.R.; Miller, W.A.; Miller, K.A.; Steele, V.E.; Miller, M.S.; Clapper, M.L.; et al. Fixed-Dose Combinations of Pioglitazone and Metformin for Lung Cancer Prevention. Cancer Prev. Res. 2017, 10, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Dwyer-Nield, L.D.; McArthur, D.G.; Hudish, T.M.; Hudish, L.I.; Mirita, C.; Sompel, K.; Smith, A.J.; Alavi, K.; Ghosh, M.; Merrick, D.T.; et al. PPARgamma agonism inhibits progression of premalignant lesions in a murine lung squamous cell carcinoma model. Int. J. Cancer 2022, 151, 2195–2205. [Google Scholar] [CrossRef] [PubMed]
- Mal, S.; Dwivedi, A.R.; Kumar, V.; Kumar, N.; Kumar, B.; Kumar, V. Role of peroxisome proliferator-activated receptor gamma (PPARγ) in different disease states: Recent updates. Curr. Med. Chem. 2021, 28, 3193–3215. [Google Scholar] [CrossRef]
- To, K.K.; Wu, W.K.; Loong, H.H. PPARgamma agonists sensitize PTEN-deficient resistant lung cancer cells to EGFR tyrosine kinase inhibitors by inducing autophagy. Eur. J. Pharmacol. 2018, 823, 19–26. [Google Scholar] [CrossRef]
- Fukushiro-Lopes, D.; Hegel, A.D.; Russo, A.; Senyuk, V.; Liotta, M.; Beeson, G.C.; Burdette, J.; Potkul, R.K.; Gentile, S. Repurposing Kir6/SUR2 channel activator minoxidil to arrests growth of gynecologic cancers. Front. Pharmacol. 2020, 11, 577. [Google Scholar] [CrossRef]
- Zhao, S.; Fan, S.; Shi, Y.; Ren, H.; Hong, H.; Gao, X.; Zhang, M.; Qin, Q.; Li, H. Propranolol induced apoptosis and autophagy via the ROS/JNK signaling pathway in Human Ovarian Cancer. J. Cancer 2020, 11, 5900–5910. [Google Scholar] [CrossRef]
- Suganuma, T.; Ino, K.; Shibata, K.; Kajiyama, H.; Nagasaka, T.; Mizutani, S.; Kikkawa, F. Functional Expression of the Angiotensin II Type1 Receptor in Human Ovarian Carcinoma Cells and Its Blockade Therapy Resulting in Suppression of Tumor Invasion, Angiogenesis, and Peritoneal Dissemination. Clin. Cancer Res. 2005, 11, 2686–2694. [Google Scholar] [CrossRef] [PubMed]
- Pu, Z.; Zhu, M.; Kong, F. Telmisartan prevents proliferation and promotes apoptosis of human ovarian cancer cells through upregulating PPARγ and downregulating MMP-9 expression. Mol. Med. Rep. 2015, 13, 555–559. [Google Scholar] [CrossRef]
- Mani, E.; A Medina, L.; Isaac-Olivé, K.; Dueñas-González, A. Radiosensitization of cervical cancer cells with epigenetic drugs hydralazine and valproate. Eur. J. Gynaecol. Oncol. 2014, 35, 140–142. [Google Scholar]
- Coronel, J.; Cetina, L.; Pacheco, I.; Trejo-Becerril, C.; González-Fierro, A.; De La Cruz-Hernandez, E.; Perez-Cardenas, E.; Taja-Chayeb, L.; Arias-Bofill, D.; Candelaria, M.; et al. A double-blind, placebo-controlled, randomized phase III trial of chemotherapy plus epigenetic therapy with hydralazine valproate for advanced cervical cancer. Preliminary results. Med. Oncol. 2010, 28, 540–546. [Google Scholar] [CrossRef]
- Candelaria, M.; Herrera, A.; Labardini, J.; González-Fierro, A.; Trejo-Becerril, C.; Taja-Chayeb, L.; Pérez-Cárdenas, E.; de la Cruz-Hernández, E.; Arias-Bofill, D.; Vidal, S.; et al. Hydralazine and magnesium valproate as epigenetic treatment for myelodysplastic syndrome. Preliminary results of a phase-II trial. Ann. Hematol. 2010, 90, 379–387. [Google Scholar] [CrossRef]
- Candelaria, M.; De La Cruz-Hernandez, E.; Taja-Chayeb, L.; Perez-Cardenas, E.; Trejo-Becerril, C.; Gonzalez-Fierro, A.; Chavez-Blanco, A.; Soto-Reyes, E.; Dominguez, G.; Trujillo, J.E.; et al. DNA Methylation-Independent Reversion of Gemcitabine Resistance by Hydralazine in Cervical Cancer Cells. PLoS ONE 2012, 7, e29181. [Google Scholar] [CrossRef]
- Bao, X.-X.; Xie, B.-S.; Li, Q.; Li, X.-P.; Wei, L.-H.; Wang, J.-L. Nifedipine induced autophagy through Beclin1 and mTOR pathway in endometrial carcinoma cells. Chin. Med. J. 2012, 125, 3120–3126. [Google Scholar]
- Koyama, N.; Nishida, Y.; Ishii, T.; Yoshida, T.; Furukawa, Y.; Narahara, H. Telmisartan Induces Growth Inhibition, DNA Double-Strand Breaks and Apoptosis in Human Endometrial Cancer Cells. PLoS ONE 2014, 9, e93050. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.C.; Chiyoda, T.; Liu, X.; Weigert, M.; Curtis, M.; Chiang, C.Y.; Loth, R.; Lastra, R.; McGregor, S.M.; Locasale, J.W.; et al. SPHK1 Is a Novel Target of Metformin in Ovarian CancerMetformin Targets SPHK1 in Ovarian Cancer. Mol. Cancer Res. 2019, 17, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Wei, J.; Wan, J.; Wang, W.; Wang, L.; Yuan, Y.; Yang, Z.; Liu, X.; Ming, L. Low glucose and metformin-induced apoptosis of human ovarian cancer cells is connected to ASK1 via mitochondrial and endoplasmic reticulum stress-associated pathways. J. Exp. Clin. Cancer Res. 2019, 38, 77. [Google Scholar] [CrossRef]
- Lu, C.-C.; Chiang, J.-H.; Tsai, F.-J.; Hsu, Y.-M.; Juan, Y.-N.; Yang, J.-S.; Chiu, H.-Y. Metformin triggers the intrinsic apoptotic response in human AGS gastric adenocarcinoma cells by activating AMPK and suppressing mTOR/AKT signaling. Int. J. Oncol. 2019, 54, 1271–1281. [Google Scholar] [CrossRef]
- Shigeto, T.; Yokoyama, Y.; Xin, B.; Mizunuma, H. Peroxisome proliferator-activated receptor α and γ ligands inhibit the growth of human ovarian cancer. Oncol. Rep. 2007, 18, 833–840. [Google Scholar] [CrossRef]
- Sulzberger, L.; Baillie, R.; Itinteang, T.; de Jong, S.; Marsh, R.; Leadbitter, P.; Tan, S. Serum levels of renin, angiotensin-converting enzyme and angiotensin II in patients treated by surgical excision, propranolol and captopril for problematic proliferating infantile haemangioma. J. Plast. Reconstr. Aesthetic Surg. 2015, 69, 381–386. [Google Scholar] [CrossRef]
- Pinheiro, L.; Perdomo-Pantoja, A.; Casaos, J.; Huq, S.; Paldor, I.; Vigilar, V.; Mangraviti, A.; Wang, Y.; Witham, T.F.; Brem, H.; et al. Captopril inhibits Matrix Metalloproteinase-2 and extends survival as a temozolomide adjuvant in an intracranial gliosarcoma model. Clin. Neurol. Neurosurg. 2021, 207, 106771. [Google Scholar] [CrossRef]
- Wolter, J.K.; E Wolter, N.; Blanch, A.; Partridge, T.; Cheng, L.; Morgenstern, D.A.; Podkowa, M.; Kaplan, D.R.; Irwin, M.S. Anti-tumor activity of the beta-adrenergic receptor antagonist propranolol in neuroblastoma. Oncotarget 2013, 5, 161–172. [Google Scholar] [CrossRef]
- Wolter, N.E.; Wolter, J.K.; Enepekides, D.J.; Irwin, M.S. Propranolol as a novel adjunctive treatment for head and neck squamous cell carcinoma. J. Otolaryngol. Head Neck Surg. 2012, 41, 334–344. [Google Scholar] [PubMed]
- Pasquier, E.; Street, J.; Pouchy, C.; Carre, M.; Gifford, A.; Murray, J.; Norris, M.D.; Trahair, T.; Andre, N.; Kavallaris, M. β-blockers increase response to chemotherapy via direct antitumour and anti-angiogenic mechanisms in neuroblastoma. Br. J. Cancer 2013, 108, 2485–2494. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Yin, D.; Morimura, T.; Kubo, H.D.; Nakatsu, S.; Takeuchi, J. Combination therapy with cisplatin and nifedipine induces apoptosis in cisplatin-sensitive and cisplatin-resistant human glioblastoma cells. Br. J. Cancer 1995, 71, 282–289. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rivera, E.; Arrieta, O.; Guevara, P.; Duarte-Rojo, A.; Sotelo, J. AT1 receptor is present in glioma cells; its blockage reduces the growth of rat glioma. Br. J. Cancer 2001, 85, 1396–1399. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Raj, G.M.; Wyawahare, M. Dapagliflozin for heart failure: Is it a class effect? Future Cardiol. 2020, 17, 355–361. [Google Scholar] [CrossRef]
- Guarnaccia, L.; Marfia, G.; Masseroli, M.M.; Navone, S.E.; Balsamo, M.; Caroli, M.; Valtorta, S.; Moresco, R.M.; Campanella, R.; Garzia, E.; et al. Frontiers in Anti-Cancer Drug Discovery: Challenges and Perspectives of Metformin as Anti-Angiogenic Add-On Therapy in Glioblastoma. Cancers 2021, 14, 112. [Google Scholar] [CrossRef] [PubMed]
- Afshari, A.R.; Sahebkar, A.; Sanati, M.; Aminyavari, S.; Mollazadeh, H.; Motamed-Sanaye, A.; Bibak, B.; Mohtashami, E.; Teng, Y. The potential therapeutic impact of metformin in glioblastoma multiforme. Curr. Med. Chem. 2022, 30, 7. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Shin, D.; Lee, J.; Jung, A.R.; Roh, J.-L. CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett. 2018, 432, 180–190. [Google Scholar] [CrossRef]
- Sobol, N.T.; Solernó, L.M.; Beltrán, B.; Vásquez, L.; Ripoll, G.V.; Garona, J.; Alonso, D.F. Anticancer activity of repurposed hemostatic agent desmopressin on AVPR2-expressing human osteosarcoma. Exp. Ther. Med. 2021, 21, 566. [Google Scholar] [CrossRef]
- Feng, L.-H.; Sun, H.-C.; Zhu, X.-D.; Zhang, S.-Z.; Li, X.-L.; Li, K.-S.; Liu, X.-F.; Lei, M.; Li, Y.; Tang, Z.-Y. Irbesartan inhibits metastasis by interrupting the adherence of tumor cell to endothelial cell induced by angiotensin II in hepatocellular carcinoma. Ann. Transl. Med. 2021, 9, 207. [Google Scholar] [CrossRef] [PubMed]
- Oura, K.; Tadokoro, T.; Fujihara, S.; Morishita, A.; Chiyo, T.; Samukawa, E.; Yamana, Y.; Fujita, K.; Sakamoto, T.; Nomura, T.; et al. Telmisartan inhibits hepatocellular carcinoma cell proliferation in vitro by inducing cell cycle arrest. Oncol. Rep. 2017, 38, 2825–2835. [Google Scholar] [CrossRef]
- Júnior, R.F.D.A.; Oliveira, A.L.C.L.; Silveira, R.F.D.M.; Rocha, H.A.D.O.; Cavalcanti, P.D.F.; Araújo, A. Telmisartan induces apoptosis and regulates Bcl-2 in human renal cancer cells. Exp. Biol. Med. 2014, 240, 34–44. [Google Scholar] [CrossRef]
- Matsuyama, M.; Funao, K.; Kuratsukuri, K.; Tanaka, T.; Kawahito, Y.; Sano, H.; Chargui, J.; Touraine, J.-L.; Yoshimura, N.; Yoshimura, R. Telmisartan inhibits human urological cancer cell growth through early apoptosis. Exp. Ther. Med. 2010, 1, 301–306. [Google Scholar] [CrossRef]
- Hii, S.I.; Nicol, D.L.; Gotley, D.; Thompson, L.; Green, M.; Jonsson, J.R. Captopril inhibits tumour growth in a xenograft model of human renal cell carcinoma. Br. J. Cancer 1998, 77, 880–883. [Google Scholar] [CrossRef]
- McIver, L.A.; Preuss, C.V.; Tripp, J. Acarbose; StatPearls: Treasure Island, FL, USA, 2023.
- Orlandella, R.M.; Turbitt, W.J.; Gibson, J.T.; Boi, S.K.; Li, P.; Smith, D.L.; Norian, L.A. The Antidiabetic Agent Acarbose Improves Anti-PD-1 and Rapamycin Efficacy in Preclinical Renal Cancer. Cancers 2020, 12, 2872. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ghoshal, S.; Sojoodi, M.; Arora, G.; Masia, R.; Erstad, D.J.; Lanuti, M.; Hoshida, Y.; Baumert, T.F.; Tanabe, K.K.; et al. Pioglitazone Reduces Hepatocellular Carcinoma Development in Two Rodent Models of Cirrhosis. J. Gastrointest. Surg. 2018, 23, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhao, L.-H.; Huang, B.; Wang, R.-Y.; Yuan, S.-X.; Tao, Q.-F.; Xu, Y.; Sun, H.-Y.; Lin, C.; Zhou, W.-P. Pioglitazone, a PPARγ agonist, inhibits growth and invasion of human hepatocellular carcinoma via blockade of the rage signaling. Mol. Carcinog. 2014, 54, 1584–1595. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, Z.; Wang, H.; Chen, X.; Jin, C. Activation of AMPK by metformin promotes renal cancer cell proliferation under glucose deprivation through its interaction with PKM2. Int. J. Biol. Sci. 2019, 15, 617–627. [Google Scholar] [CrossRef]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brûlé, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, Independent of AMPK, Inhibits mTORC1 in a Rag GTPase-Dependent Manner. Cell Metab. 2010, 11, 390–401. [Google Scholar] [CrossRef]
- Wu, L.; Zhou, B.; Oshiro-Rapley, N.; Li, M.; Paulo, J.A.; Webster, C.M.; Mou, F.; Kacergis, M.C.; Talkowski, M.E.; Carr, C.E.; et al. An Ancient, Unified Mechanism for Metformin Growth Inhibition in C. elegans and Cancer. Cell 2016, 167, 1705–1718.e13. [Google Scholar] [CrossRef]
- Jin, Y.Y.; Han, C.; Geng, N.; Li, Y.R.; Zheng, L.Y.; Zhu, W.J.; Bai, H. AKR1B10 inhibitor enhances the inhibitory effect of sorafenib on liver cancer xenograft. Zhonghua Gan Zang Bing Za Zhi = Zhonghua Ganzangbing Zazhi = Chin. J. Hepatol. 2019, 27, 39–44. [Google Scholar]
- Zhong, L.; Shen, H.; Huang, C.; Jing, H.; Cao, D. AKR1B10 induces cell resistance to daunorubicin and idarubicin by reducing C13 ketonic group. Toxicol. Appl. Pharmacol. 2011, 255, 40–47. [Google Scholar] [CrossRef]
- Bailly, C. Moving toward a new horizon for the aldose reductase inhibitor epalrestat to treat drug-resistant cancer. Eur. J. Pharmacol. 2022, 931, 175191. [Google Scholar] [CrossRef]
- Tanagala, K.K.; Baba, A.B.; Kowshik, J.; Reddy, G.B.; Nagini, S.; Gedunin, A. Neem Limonoid in Combination with Epalrestat Inhibits Cancer Hallmarks by Attenuating Aldose Reductase-Driven Oncogenic Signaling in SCC131 Oral Cancer Cells. Anti-Cancer Agents Med. Chem. 2019, 18, 2042–2052. [Google Scholar] [CrossRef]
- Ji, J.; Xu, M.-X.; Qian, T.-Y.; Zhu, S.-Z.; Jiang, F.; Liu, Z.-X.; Xu, W.-S.; Zhou, J.; Xiao, M.-B. The AKR1B1 inhibitor epalrestat suppresses the progression of cervical cancer. Mol. Biol. Rep. 2020, 47, 6091–6103. [Google Scholar] [CrossRef]
- Geng, N.; Jin, Y.Y.; Zhu, S.X.; Li, Y.R.; Zheng, L.Y.; Zhu, W.J.; Li, Y.W.; Han, C.; Dou, X.G.; Bai, H. Aldo-keto reductase family 1 B10 participates in the regulation of hepatoma cell cycle through p27/p-Rb signaling pathway. Zhonghua Gan Zang Bing Za Zhi = Zhonghua Ganzangbing Zazhi = Chin. J. Hepatol. 2020, 28, 861–867. [Google Scholar]
- Geng, N.; Jin, Y.; Li, Y.; Zhu, S.; Bai, H. AKR1B10 Inhibitor Epalrestat Facilitates Sorafenib-Induced Apoptosis and Autophagy Via Targeting the mTOR Pathway in Hepatocellular Carcinoma. Int. J. Med. Sci. 2020, 17, 1246–1256. [Google Scholar] [CrossRef]
- Koh, M.; Takahashi, T.; Kurokawa, Y.; Kobayashi, T.; Saito, T.; Ishida, T.; Serada, S.; Fujimoto, M.; Naka, T.; Wada, N.; et al. Propranolol suppresses gastric cancer cell growth by regulating proliferation and apoptosis. Gastric Cancer 2021, 24, 1037–1049. [Google Scholar] [CrossRef]
- Hu, Q.; Liao, P.; Li, W.; Hu, J.; Chen, C.; Zhang, Y.; Wang, Y.; Chen, L.; Song, K.; Liu, J.; et al. Clinical Use of Propranolol Reduces Biomarkers of Proliferation in Gastric Cancer. Front. Oncol. 2021, 11, 628613. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Y.; Wang, Z.; Li, H.; Wang, Y. Clinical value of propranolol combined with oxaliplatin and tigio in concurrent chemoradiotherapy for locally advanced gastric cancer. Pak. J. Med. Sci. 2022, 38, 1316. [Google Scholar] [CrossRef]
- Williams, R.; Parsons, S.; Morris, T.; Rowlands, B.; Watson, S. Inhibition of matrix metalloproteinase activity and growth of gastric adenocarcinoma cells by an angiotensin converting enzyme inhibitor in in vitro and murine models. Eur. J. Surg. Oncol. (EJSO) 2005, 31, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Fujihara, S.; Morishita, A.; Ogawa, K.; Tadokoro, T.; Chiyo, T.; Kato, K. The angiotensin II type 1 receptor antagonist telmisartan inhibits cell proliferation and tumor growth of esophageal adenocarcinoma via the AMPKa/mTOR pathway in vitro and in vivo. Oncotarget 2017, 8, 8536. [Google Scholar] [CrossRef]
- Matsui, T.; Chiyo, T.; Kobara, H.; Fujihara, S.; Fujita, K.; Namima, D.; Nakahara, M.; Kobayashi, N.; Nishiyama, N.; Yachida, T.; et al. Telmisartan Inhibits Cell Proliferation and Tumor Growth of Esophageal Squamous Cell Carcinoma by Inducing S-Phase Arrest In Vitro and In Vivo. Int. J. Mol. Sci. 2019, 20, 3197. [Google Scholar] [CrossRef]
- Samukawa, E.; Fujihara, S.; Oura, K.; Iwama, H.; Yamana, Y.; Tadokoro, T.; Chiyo, T.; Kobayashi, K.; Morishita, A.; Nakahara, M.; et al. Angiotensin receptor blocker telmisartan inhibits cell proliferation and tumor growth of cholangiocarcinoma through cell cycle arrest. Int. J. Oncol. 2017, 51, 1674–1684. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Y. Telmisartan inhibits NSCLC A549 cell proliferation and migration by regulating the PI3K/AKT signaling pathway. Oncol. Lett. 2018, 15, 5859–5864. [Google Scholar] [CrossRef] [PubMed]
- Shuai, Y.; Li, C.; Zhou, X. The effect of metformin on gastric cancer in patients with type 2 diabetes: A systematic review and meta-analysis. Clin. Transl. Oncol. 2020, 22, 1580–1590. [Google Scholar] [CrossRef]
- Seo, H.S.; Jung, Y.J.; Kim, J.H.; Lee, H.H.; Park, C.H. The Effect of Metformin on Prognosis in Patients With Locally Advanced Gastric Cancer Associated With Type 2 Diabetes Mellitus. Am. J. Clin. Oncol. 2019, 42, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Júnior, A.D.C.; Bragagnoli, A.C.; Costa, F.O.; Carvalheira, J.B.C. Repurposing metformin for the treatment of gastrointestinal cancer. World J. Gastroenterol. 2021, 27, 1883–1904. [Google Scholar] [CrossRef]
- Wang, L.; Li, K.; Lin, X.; Yao, Z.; Wang, S.; Xiong, X.; Zhang, H. Metformin induces human esophageal carcinoma cell pyroptosis by targeting the miR-497/PELP1 axis. Cancer Lett. 2019, 450, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Grahovac, J.; Srdić-Rajić, T.; Santibañez, J.F.; Pavlović, M.; Čavić, M.; Radulović, S. Telmisartan induces melanoma cell apoptosis and synergizes with vemurafenib in vitro by altering cell bioenergetics. Cancer Biol. Med. 2019, 16, 247. [Google Scholar]
- Benish, M.; Bartal, I.; Goldfarb, Y.; Levi, B.; Avraham, R.; Raz, A.; Ben-Eliyahu, S. Perioperative Use of β-blockers and COX-2 Inhibitors May Improve Immune Competence and Reduce the Risk of Tumor Metastasis. Ann. Surg. Oncol. 2008, 15, 2042–2052. [Google Scholar] [CrossRef]
- Goldfarb, Y.; Sorski, L.; Benish, M.; Levi, B.; Melamed, R.; Ben-Eliyahu, S. Improving postoperative immune status and resistance to cancer metastasis: A combined perioperative approach of immunostimulation and prevention of excessive surgical stress responses. Ann. Surg. 2011, 253, 798–810. [Google Scholar] [CrossRef]
- Glasner, A.; Avraham, R.; Rosenne, E.; Benish, M.; Zmora, O.; Shemer, S.; Meiboom, H.; Ben-Eliyahu, S. Improving Survival Rates in Two Models of Spontaneous Postoperative Metastasis in Mice by Combined Administration of a β-Adrenergic Antagonist and a Cyclooxygenase-2 Inhibitor. J. Immunol. 2010, 184, 2449–2457. [Google Scholar] [CrossRef]
- Wrobel, L.; Le Gal, F.A. Inhibition of Human Melanoma Growth by a Non-Cardioselective β-Blocker. J. Investig. Dermatol. 2015, 135, 525–531. [Google Scholar] [CrossRef]
- Dal Monte, M.; Casini, G.; Filippi, L.; Nicchia, G.P.; Svelto, M.; Bagnoli, P. Functional involvement of β3-adrenergic receptors in melanoma growth and vascularization. J. Mol. Med. 2013, 91, 1407–1419. [Google Scholar] [CrossRef]
- Lv, Z.; Guo, Y. Metformin and Its Benefits for Various Diseases. Front. Endocrinol. 2020, 11, 191. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zhang, T.-T.; Wang, F.; Cui, B.; Zhao, C.-X.; Yu, J.-J.; Lv, X.-X.; Zhang, X.-W.; Yang, Z.-N.; Huang, B.; et al. Metformin suppresses melanoma progression by inhibiting KAT5-mediated SMAD3 acetylation, transcriptional activity and TRIB3 expression. Oncogene 2018, 37, 2967–2981. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.-W.; Li, S.-C.; Tsai, K.-W. Metformin Treatment Suppresses Melanoma Cell Growth and Motility through Modulation of microRNA Expression. Cancers 2019, 11, 209. [Google Scholar] [CrossRef]
- Deng, C.; Lu, Q.; Zhang, Z.; Rao, T.; Attwood, J.; Yung, R.; Richardson, B. Hydralazine may induce autoimmunity by inhibiting extracellular signal-regulated kinase pathway signaling. Arthritis Rheum. 2003, 48, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Kozako, T.; Soeda, S.; Yoshimitsu, M.; Arima, N.; Kuroki, A.; Hirata, S.; Tanaka, H.; Imakyure, O.; Tone, N.; Honda, S.; et al. Angiotensin II type 1 receptor blocker telmisartan induces apoptosis and autophagy in adult T-cell leukemia cells. FEBS Open Bio 2016, 6, 442–460. [Google Scholar] [CrossRef]
- Purclutepe, O.; Iskender, G.; Kiper, H.D.; Tezcanli, B.; Selvi, N.; Avci, C.B.; Kosova, B.; Gokbulut, A.A.; Sahin, F.; Baran, Y.; et al. Enalapril-induced apoptosis of acute promyelocytic leukaemia cells involves STAT5A. Anticancer Res. 2012, 32, 2885–2893. [Google Scholar]
- Corey, S.J.; Jha, J.; McCart, E.A.; Rittase, W.B.; George, J.; Mattapallil, J.J.; Day, R.M. Captopril mitigates splenomegaly and myelofibrosis in the Gata1low murine model of myelofibrosis. J. Cell. Mol. Med. 2018, 22, 4274–4282. [Google Scholar] [CrossRef]
- Hanusova, V.; Skálová, L.; Králová, V.; Matouskova, P. Potential Anti-cancer Drugs Commonly Used for Other Indications. Curr. Cancer Drug Targets 2015, 15, 35–52. [Google Scholar] [CrossRef]
- Papanagnou, P.; Stivarou, T.; Tsironi, M. Unexploited Antineoplastic Effects of Commercially Available Anti-Diabetic Drugs. Pharmaceuticals 2016, 9, 24. [Google Scholar] [CrossRef]
- Vella, V.; Nicolosi, M.L.; Giuliano, S.; Bellomo, M.; Belfiore, A.; Malaguarnera, R. PPAR-γ Agonists As Antineoplastic Agents in Cancers with Dysregulated IGF Axis. Front. Endocrinol. 2017, 8, 31. [Google Scholar] [CrossRef]
- Prost, S.; Relouzat, F.; Spentchian, M.; Ouzegdouh, Y.; Saliba, J.; Massonnet, G.; Beressi, J.-P.; Verhoeyen, E.; Raggueneau, V.; Maneglier, B.; et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARγ agonists. Nature 2015, 525, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, F.; Guan, J.; Zhou, L.; Chen, B. Action Mechanism of Metformin and Its Application in Hematological Malignancy Treatments: A Review. Biomolecules 2023, 13, 250. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Wu, L.; Zou, L.; Wang, M.; Liu, X. Metformin induces myeloma cells necrosis and apoptosis and it is considered for therapeutic use. J. Chemother. 2022, 35, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, S.R.; Agusala, V.; Sher, D.J. Financial toxicity and cancer therapy: A primer for radiation oncologists. Hematol./Oncol. Clin. 2019, 33, 1117–1128. [Google Scholar] [CrossRef]
- Petrie, J.; Guzik, T.J.; Touyz, R.M. Diabetes, Hypertension, and Cardiovascular Disease: Clinical Insights and Vascular Mechanisms. Can. J. Cardiol. 2017, 34, 575–584. [Google Scholar] [CrossRef]
- A Cameron, H.; E Ramsay, L. The lupus syndrome induced by hydralazine: A common complication with low dose treatment. BMJ 1984, 289, 410–412. [Google Scholar] [CrossRef]
- Battistoni, A.; Volpe, M. Recent warnings about antihypertensive drugs and cancer risk: Where do they come from? Eur. Cardiol. Rev. 2020, 15, e21. [Google Scholar] [CrossRef]
- Pan, E.; Bogumil, D.; Cortessis, V.; Yu, S.; Nieva, J. A Systematic Review of the Efficacy of Preclinical Models of Lung Cancer Drugs. Front. Oncol. 2020, 10, 591. [Google Scholar] [CrossRef]
- Emptage, N.P.; A Koster, M.; E Schottinger, J.; Petitti, D.B. Critical Appraisal of Clinical Studies: An Example from Computed Tomography Screening for Lung Cancer. Perm. J. 2007, 11, 81–85. [Google Scholar] [CrossRef]
- Chen, K.; Li, Y.; Guo, Z.; Zeng, Y.; Zhang, W.; Wang, H. Metformin: Current clinical applications in nondiabetic patients with cancer. Aging 2020, 12, 3993–4009. [Google Scholar] [CrossRef]
- Dulskas, A.; Patasius, A.; Linkeviciute-Ulinskiene, D.; Zabuliene, L.; Smailyte, G. A cohort study of antihyperglycemic medication exposure and survival in patients with gastric cancer. Aging 2019, 11, 7197–7205. [Google Scholar] [CrossRef]
- Saraei, P.; Asadi, I.; Kakar, M.A.; Moradi-Kor, N. The beneficial effects of metformin on cancer prevention and therapy: A comprehensive review of recent advances. Cancer Manag. Res. 2019, 11, 3295–3313. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Daley, B.; Klubo-Gwiezdzinska, J. The role of the antidiabetic drug metformin in the treatment of endocrine tumors. J. Mol. Endocrinol. 2019, 63, R17–R35. [Google Scholar] [CrossRef] [PubMed]
- Lord, S.R.; Harris, A.L. Is it still worth pursuing the repurposing of metformin as a cancer therapeutic? Br. J. Cancer 2023, 128, 958–966. [Google Scholar] [CrossRef]
- Kian, W.; Zemel, M.; Levitas, D.; Alguayn, W.; Remilah, A.A.; Rahman, N.A.; Peled, N. Lung cancer screening: A critical appraisal. Curr. Opin. Oncol. 2021, 34, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Kaczor, M.; Wójcik, R.; Połowinczak-Przybyłek, J.; Potemski, P. Critical appraisal of clinical trials in oncology—Part I. Oncol. Clin. Pract. 2019, 15, 89–103. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hijazi, M.A.; Gessner, A.; El-Najjar, N. Repurposing of Chronically Used Drugs in Cancer Therapy: A Chance to Grasp. Cancers 2023, 15, 3199. https://doi.org/10.3390/cancers15123199
Hijazi MA, Gessner A, El-Najjar N. Repurposing of Chronically Used Drugs in Cancer Therapy: A Chance to Grasp. Cancers. 2023; 15(12):3199. https://doi.org/10.3390/cancers15123199
Chicago/Turabian StyleHijazi, Mohamad Ali, André Gessner, and Nahed El-Najjar. 2023. "Repurposing of Chronically Used Drugs in Cancer Therapy: A Chance to Grasp" Cancers 15, no. 12: 3199. https://doi.org/10.3390/cancers15123199
APA StyleHijazi, M. A., Gessner, A., & El-Najjar, N. (2023). Repurposing of Chronically Used Drugs in Cancer Therapy: A Chance to Grasp. Cancers, 15(12), 3199. https://doi.org/10.3390/cancers15123199