
Cancer Cell-Intrinsic Alterations Associated with an Immunosuppressive Tumor Microenvironment and Resistance to Immunotherapy in Lung Cancer

 , , , , , ,
, , , , , ,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Main Gene Mutations That Shape the Tumor Microenvironment in NSCLC
2.1. KRAS Mutations
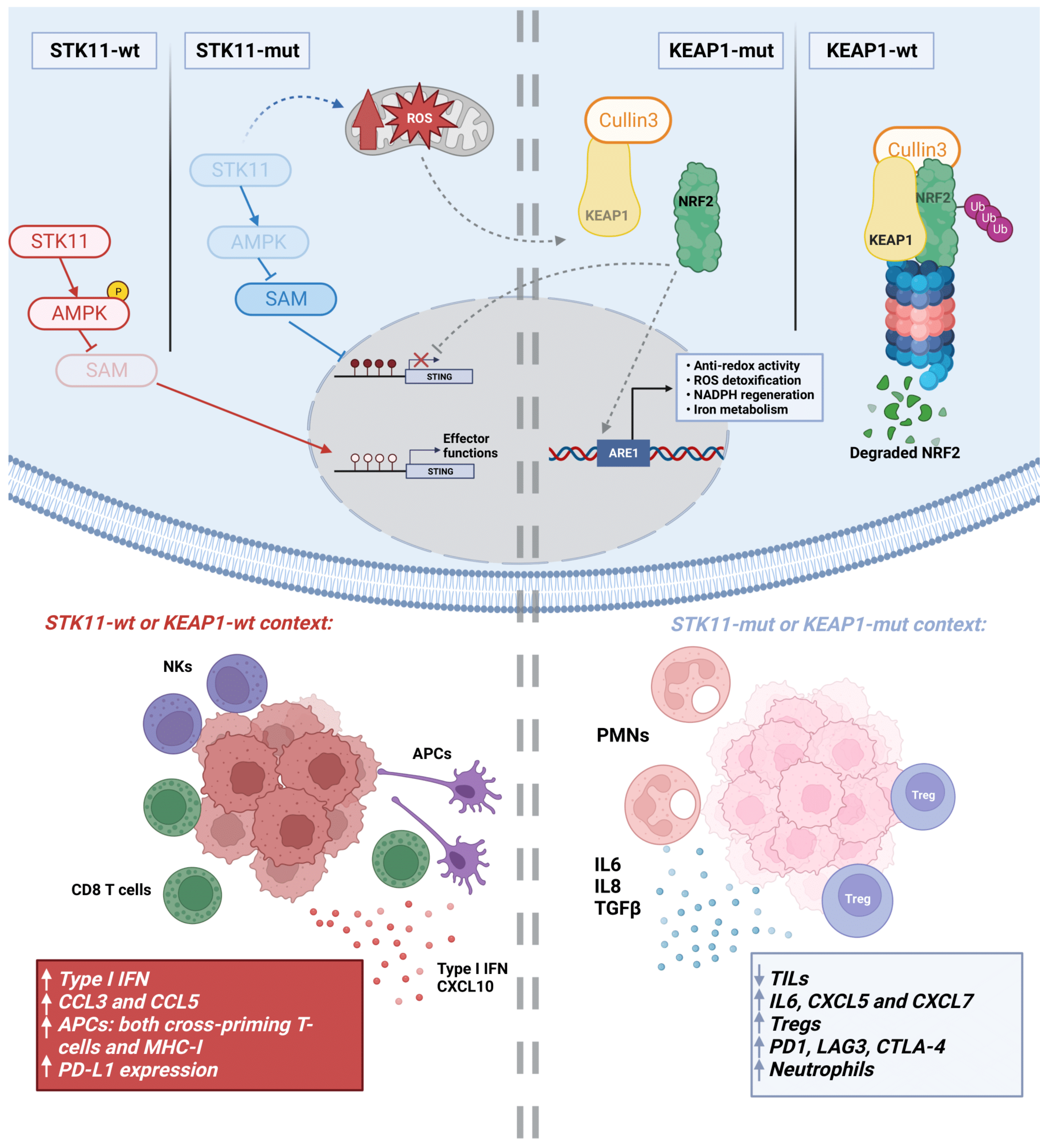
2.2. STK11 Mutations
2.3. TP53 Mutations
2.4. EGFR Mutations
2.5. KEAP1 Mutations
2.6. Mutations in Genes of the Antigen Processing and Presentation Machinery
2.7. Mutations in other Genes with Potential Importance in Immunosuppression
3. Metabolic Rewiring in NSCLC and its Influence on the TME
3.1. Lactate
3.2. Hypoxia
3.3. Amino Acids
3.4. Lipids
3.5. Metabolic Rewiring by STK11 Alterations
3.6. Metabolic Rewiring by KEAP1 Alterations
4. Emerging Altered Cancer Pathways Associated with Immunosuppression in NSCLC
4.1. Wnt/β-Catenin
4.2. YES1
4.3. DSTYK
4.4. MUC1 and Hippo Pathways
4.5. DNA Damage Repair (DDR)
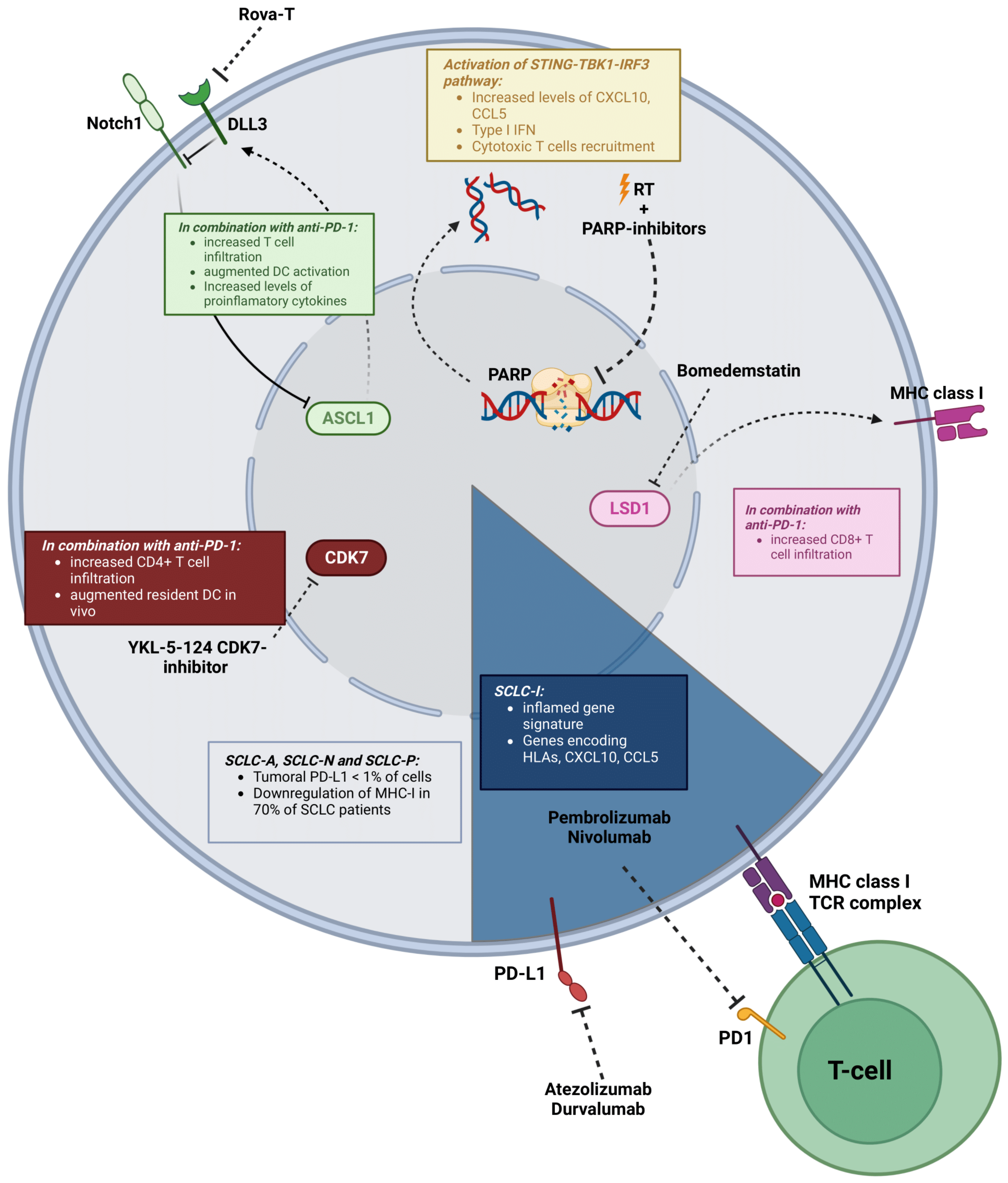
5. Cancer Cell-Intrinsic Genetic Alterations That Contribute to Immunosuppression in SCLC
5.1. Main Genetic Alterations Driving Immunosuppression in SCLC
5.2. Molecular Subtypes
5.3. Notch Signaling
5.4. Targeting DNA Damage Repair (DDR) in SCLC to Reshape the TME
5.5. Other Targets to Reshape the SCLC TME
6. Epigenetic Changes That Modify the TME in Lung Cancer and Immunotherapy Response
6.1. Epigenetic Alterations in NSCLC and SCLC
6.2. Epigenetic Changes in Lung Cancer Related to the Immune TME
7. Personalized Immunotherapy beyond ICIs
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.M.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances since the 2004 Classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The Biology and Management of Non-Small Cell Lung Cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Brambilla, E.; Faivre-Finn, C.; Sage, J. Small-Cell Lung Cancer. Nat. Rev. Dis. Prim. 2021, 7, 3. [Google Scholar] [CrossRef]
- Helin, K.; Holm, K.; Niebuhr, A.; Eiberg, H.; Tommerup, N.; Hougaard, S.; Poulsen, H.S.; Spang-Thomsen, M.; Nøroaard, P. Loss of the Retinoblastoma Protein-Related P130 Protein in Small Cell Lung Carcinoma. Proc. Natl. Acad. Sci. USA 1997, 94, 6933–6938. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretia, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive Genomic Profiles of Small Cell Lung Cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Augert, A.; Rongione, M.; Conkrite, K.; Parazzoli, S.; Nikitin, A.Y.; Ingolia, N.; MacPherson, D. PTEN Is a Potent Suppressor of Small Cell Lung Cancer. Mol. Cancer Res. 2014, 12, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Ibaseta, A.; Fischer, M.M.; Cancilla, B.; O’Young, G.; Cristea, S.; Luca, V.C.; Yang, D.; Jahchan, N.S.; Hamard, C.; et al. Intratumoural Heterogeneity Generated by Notch Signalling Promotes Small-Cell Lung Cancer. Nature 2017, 545, 360–364. [Google Scholar] [CrossRef]
- Peifer, M.; Fernández-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative Genome Analyses Identify Key Somatic Driver Mutations of Small-Cell Lung Cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef]
- Coles, G.L.; Cristea, S.; Webber, J.T.; Levin, R.S.; Moss, S.M.; He, A.; Sangodkar, J.; Hwang, Y.C.; Arand, J.; Drainas, A.P.; et al. Unbiased Proteomic Profiling Uncovers a Targetable GNAS/PKA/PP2A Axis in Small Cell Lung Cancer Stem Cells. Cancer Cell 2020, 38, 129–143.e7. [Google Scholar] [CrossRef]
- Ferone, G.; Song, J.Y.; Krijgsman, O.; van der Vliet, J.; Cozijnsen, M.; Semenova, E.A.; Adams, D.J.; Peeper, D.; Berns, A. FGFR1 Oncogenic Activation Reveals an Alternative Cell of Origin of SCLC in Rb1/P53 Mice. Cell Rep. 2020, 30, 3837–3850.e3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhang, J.; Wang, G.; He, X.; Mi, Y.; Cao, Y.; Yu, X. Predictive Efficacy of Blood-Based Tumor Mutation Burden Assay for Immune Checkpoint Inhibitors Therapy in Non-Small Cell Lung Cancer: A Systematic Review and Meta-Analysis. Front. Oncol. 2022, 12, 206. [Google Scholar] [CrossRef] [PubMed]
- Willis, C.; Fiander, M.; Tran, D.; Korytowsky, B.; Thomas, J.M.; Calderon, F.; Zyczynski, T.M.; Brixner, D.; Stenehjem, D.D. Tumor Mutational Burden in Lung Cancer: A Systematic Literature Review. Oncotarget 2019, 10, 6604–6622. [Google Scholar] [CrossRef]
- Doroshow, D.B.; Sanmamed, M.F.; Hastings, K.; Politi, K.; Rimm, D.L.; Chen, L.; Melero, I.; Schalper, K.A.; Herbst, R.S. Immunotherapy in Non-Small Cell Lung Cancer: Facts and Hopes. Clin. Cancer Res. 2019, 25, 4592–4602. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.M.; Zheng, M.M.; Pan, Y.; Liu, S.Y.; Li, Y.; Wu, Y.L. Emerging Evidence and Treatment Paradigm of Non-Small Cell Lung Cancer. J. Hematol. Oncol. 2023, 16, 40. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Sosman, J.A.; Atkins, M.B.; Leming, P.D.; et al. Five-Year Survival and Correlates Among Patients With Advanced Melanoma, Renal Cell Carcinoma, or Non-Small Cell Lung Cancer Treated With Nivolumab. JAMA Oncol. 2019, 5, 1411–1420. [Google Scholar] [CrossRef]
- Russano, M.; La Cava, G.; Cortellini, A.; Citarella, F.; Galletti, A.; Di Fazio, G.R.; Santo, V.; Brunetti, L.; Vendittelli, A.; Fioroni, I.; et al. Immunotherapy for Metastatic Non-Small Cell Lung Cancer: Therapeutic Advances and Biomarkers. Curr. Oncol. 2023, 30, 2366–2387. [Google Scholar] [CrossRef]
- Anichini, A.; Perotti, V.E.; Sgambelluri, F.; Mortarini, R. Immune Escape Mechanisms in Non Small Cell Lung Cancer. Cancers 2020, 12, 3605. [Google Scholar] [CrossRef]
- Dejima, H.; Hu, X.; Chen, R.; Zhang, J.; Fujimoto, J.; Parra, E.R.; Haymaker, C.; Hubert, S.M.; Duose, D.; Solis, L.M.; et al. Immune Evolution from Preneoplasia to Invasive Lung Adenocarcinomas and Underlying Molecular Features. Nat. Commun. 2021, 12, 2722. [Google Scholar] [CrossRef]
- Mascaux, C.; Angelova, M.; Vasaturo, A.; Beane, J.; Hijazi, K.; Anthoine, G.; Buttard, B.; Rothe, F.; Willard-Gallo, K.; Haller, A.; et al. Immune Evasion before Tumour Invasion in Early Lung Squamous Carcinogenesis. Nature 2019, 571, 570–575. [Google Scholar] [CrossRef]
- Borcoman, E.; Kanjanapan, Y.; Champiat, S.; Kato, S.; Servois, V.; Kurzrock, R.; Goel, S.; Bedard, P.; Le Tourneau, C. Novel Patterns of Response under Immunotherapy. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Horvath, L.; Thienpont, B.; Zhao, L.; Wolf, D.; Pircher, A. Overcoming Immunotherapy Resistance in Non-Small Cell Lung Cancer (NSCLC)-Novel Approaches and Future Outlook. Mol. Cancer 2020, 19, 141. [Google Scholar] [CrossRef] [PubMed]
- Otano, I.; Ucero, A.C.; Zugazagoitia, J.; Paz-Ares, L. At the Crossroads of Immunotherapy for Oncogene-Addicted Subsets of NSCLC. Nat. Rev. Clin. Oncol. 2023, 20, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Aminu, M.; Li, S.; Lu, X.; Petranovic, M.; Saad, M.B.; Chen, P.; Qin, K.; Varghese, S.; Rinsurongkawong, W.; et al. Efficacy and Clinicogenomic Correlates of Response to Immune Checkpoint Inhibitors Alone or with Chemotherapy in Non-Small Cell Lung Cancer. Nat. Commun. 2023, 14, 695. [Google Scholar] [CrossRef]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef]
- Hastings, K.; Yu, H.A.; Wei, W.; Sanchez-Vega, F.; Deveaux, M.; Choi, J.; Rizvi, H.; Lisberg, A.; Truini, A.; Lydon, C.A.; et al. EGFR Mutation Subtypes and Response to Immune Checkpoint Blockade Treatment in Non-Small-Cell Lung Cancer. Ann. Oncol. 2019, 30, 1311–1320. [Google Scholar] [CrossRef]
- Cucurull, M.; Notario, L.; Sanchez-Cespedes, M.; Hierro, C.; Estival, A.; Carcereny, E.; Saigí, M. Targeting KRAS in Lung Cancer Beyond KRAS G12C Inhibitors: The Immune Regulatory Role of KRAS and Novel Therapeutic Strategies. Front. Oncol. 2022, 11, 5768. [Google Scholar] [CrossRef]
- Brooks, G.D.; McLeod, L.; Alhayyani, S.; Miller, A.; Russell, P.A.; Ferlin, W.; Rose-John, S.; Ruwanpura, S.; Jenkins, B.J. IL6 Trans-Signaling Promotes KRAS-Driven Lung Carcinogenesis. Cancer Res. 2016, 76, 866–876. [Google Scholar] [CrossRef]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune Checkpoint Inhibitors for Patients with Advanced Lung Cancer and Oncogenic Driver Alterations: Results from the IMMUNOTARGET Registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef]
- Jeanson, A.; Tomasini, P.; Souquet-Bressand, M.; Brandone, N.; Boucekine, M.; Grangeon, M.; Chaleat, S.; Khobta, N.; Milia, J.; Mhanna, L.; et al. Efficacy of Immune Checkpoint Inhibitors in KRAS-Mutant Non-Small Cell Lung Cancer (NSCLC). J. Thorac. Oncol. 2019, 14, 1095–1101. [Google Scholar] [CrossRef]
- Arbour, K.C.; Rizvi, H.; Plodkowski, A.J.; Hellmann, M.D.; Knezevic, A.; Heller, G.; Yu, H.A.; Ladanyi, M.; Kris, M.G.; Arcila, M.E.; et al. Treatment Outcomes and Clinical Characteristics of Patients with KRAS-G12C-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2021, 27, 2209–2215. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zheng, S.; Wang, Z.; Wang, S.; Wang, X.; Yang, L.; Xu, H.; Cao, Z.; Feng, X.; Xue, Q.; et al. KRAS-G12D Mutation Drives Immune Suppression and the Primary Resistance of Anti-PD-1/PD-L1 Immunotherapy in Non-Small Cell Lung Cancer. Cancer Commun 2022, 42, 828–847. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Heymach, J.V. Co-Occurring Genomic Alterations in Non-Small-Cell Lung Cancer Biology and Therapy. Nat. Rev. Cancer 2019, 19, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Zugazagoitia, J.; Herbertz, S.; John, W.; Paz-Ares, L.; Schmid-Bindert, G. KRAS-Mutant Non-Small Cell Lung Cancer: From Biology to Therapy. Lung Cancer 2018, 124, 53–64. [Google Scholar] [CrossRef]
- Li, C.; Lin, W.Y.; Rizvi, H.; Cai, H.; McFarland, C.D.; Rogers, Z.N.; Yousefi, M.; Winters, I.P.; Rudin, C.M.; Petrov, D.A.; et al. Quantitative in Vivo Analyses Reveal a Complex Pharmacogenomic Landscape in Lung Adenocarcinoma. Cancer Res. 2021, 81, 4570–4580. [Google Scholar] [CrossRef]
- Mazzaschi, G.; Leonetti, A.; Minari, R.; Gnetti, L.; Quaini, F.; Tiseo, M.; Facchinetti, F. Modulating Tumor Microenvironment: A Review on STK11 Immune Properties and Predictive vs Prognostic Role for Non-Small-Cell Lung Cancer Immunotherapy. Curr. Treat. Options Oncol. 2021, 22, 96. [Google Scholar] [CrossRef]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK Pathway: Metabolism and Growth Control in Tumour Suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef]
- Ylikorkala, A.; Rossi, D.J.; Korsisaari, N.; Luukko, K.; Alitalo, K.; Henkemeyer, M.; Mäkelä, T.P. Vascular Abnormalities and Deregulation of VEGF in Lkb1-Deficient Mice. Science 2001, 293, 1323–1326. [Google Scholar] [CrossRef]
- Glantz, M.M.; Chikisheva, T.; Viola, B.; Krivoshapkin, A.I.; Derevianko, A.P.; Islamov, U.I.; Wrinn, P.J.; Ritzman, T.B. Move over Teshik-Tash: New Hominid Remains from Uzbekistan. Nat. Rev. Cancer 2004, 9, A67. [Google Scholar] [CrossRef]
- Della Corte, C.M.; Sen, T.; Gay, C.M.; Ramkumar, K.; Diao, L.; Cardnell, R.J.; Rodriguez, B.L.; Stewart, C.A.; Papadimitrakopoulou, V.A.; Gibson, L.; et al. STING Pathway Expression Identifies NSCLC With an Immune-Responsive Phenotype. J. Thorac. Oncol. 2020, 15, 777–791. [Google Scholar] [CrossRef]
- Kitajima, S.; Ivanova, E.; Guo, S.; Yoshida, R.; Campisi, M.; Sundararaman, S.K.; Tange, S.; Mitsuishi, Y.; Thai, T.C.; Masuda, S.; et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov. 2019, 9, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Aref, A.R.; Skoulidis, F.; Herter-Sprie, G.S.; Buczkowski, K.A.; Liu, Y.; Awad, M.M.; Denning, W.L.; et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-Cell Activity in the Lung Tumor Microenvironment. Cancer Res. 2016, 76, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Luo, Y.; Guo, W.; Niu, Q.; Xue, T.; Yang, F.; Sun, X.; Chen, S.; Liu, Y.; Liu, J.; et al. Lkb1 Maintains Treg Cell Lineage Identity. Nat. Commun. 2017, 8, 15876. [Google Scholar] [CrossRef] [PubMed]
- MacIver, N.J.; Blagih, J.; Saucillo, D.C.; Tonelli, L.; Griss, T.; Rathmell, J.C.; Jones, R.G. The Liver Kinase B1 (LKB1) Is a Central Regulator of T Cell Development, Activation, and Metabolism. J. Immunol. 2011, 187, 4187. [Google Scholar] [CrossRef]
- Wan, L.; Xu, K.; Wei, Y.; Zhang, J.; Han, T.; Fry, C.; Zhang, Z.; Wang, Y.V.; Huang, L.; Yuan, M.; et al. Phosphorylation of EZH2 by AMPK Suppresses PRC2 Methyltransferase Activity and Oncogenic Function. Mol. Cell 2018, 69, 279–291.e5. [Google Scholar] [CrossRef]
- Partanen, J.I.; Nieminen, A.I.; Mäkelä, T.P.; Klefstrom, J. Suppression of Oncogenic Properties of C-Myc by LKB1-Controlled Epithelial Organization. Proc. Natl. Acad. Sci. USA 2007, 104, 14694–14699. [Google Scholar] [CrossRef]
- Deng, J.; Thennavan, A.; Dolgalev, I.; Chen, T.; Li, J.; Marzio, A.; Poirier, J.T.; Peng, D.H.; Bulatovic, M.; Mukhopadhyay, S.; et al. ULK1 Inhibition Overcomes Compromised Antigen Presentation and Restores Antitumor Immunity in LKB1 Mutant Lung Cancer. Nat. Cancer 2021, 2, 503–514. [Google Scholar] [CrossRef]
- Kitajima, S.; Tani, T.; Springer, B.F.; Campisi, M.; Osaki, T.; Haratani, K.; Chen, M.; Knelson, E.H.; Mahadevan, N.R.; Ritter, J.; et al. MPS1 Inhibition Primes Immunogenicity of KRAS-LKB1 Mutant Lung Cancer. Cancer Cell 2022, 40, 1128–1144.e8. [Google Scholar] [CrossRef]
- Collisson, E.A.; Campbell, J.D.; Brooks, A.N.; Berger, A.H.; Lee, W.; Chmielecki, J.; Beer, D.G.; Cope, L.; Creighton, C.J.; Danilova, L.; et al. Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Assoun, S.; Theou-Anton, N.; Nguenang, M.; Cazes, A.; Danel, C.; Abbar, B.; Pluvy, J.; Gounant, V.; Khalil, A.; Namour, C.; et al. Association of TP53 Mutations with Response and Longer Survival under Immune Checkpoint Inhibitors in Advanced Non-Small-Cell Lung Cancer. Lung Cancer 2019, 132, 65–71. [Google Scholar] [CrossRef]
- Sun, H.; Liu, S.Y.; Zhou, J.Y.; Xu, J.T.; Zhang, H.K.; Yan, H.H.; Huan, J.J.; Dai, P.P.; Xu, C.R.; Su, J.; et al. Specific TP53 Subtype as Biomarker for Immune Checkpoint Inhibitors in Lung Adenocarcinoma. EBioMedicine 2020, 60, 102990. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.Y.; Zhong, W.Z.; Zhang, X.C.; Su, J.; Xie, Z.; Liu, S.Y.; Tu, H.Y.; Chen, H.J.; Sun, Y.L.; Zhou, Q.; et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2017, 23, 3012–3024. [Google Scholar] [CrossRef] [PubMed]
- Taki, M.; Abiko, K.; Ukita, M.; Murakami, R.; Yamanoi, K.; Yamaguchi, K.; Hamanishi, J.; Baba, T.; Matsumura, N.; Mandai, M. Tumor Immune Microenvironment during Epithelial-Mesenchymal Transition. Clin. Cancer Res. 2021, 27, 4669–4679. [Google Scholar] [CrossRef] [PubMed]
- Kogan-Sakin, I.; Tabach, Y.; Buganim, Y.; Molchadsky, A.; Solomon, H.; Madar, S.; Kamer, I.; Stambolsky, P.; Shelly, A.; Goldfinger, N.; et al. Mutant P53(R175H) Upregulates Twist1 Expression and Promotes Epithelial-Mesenchymal Transition in Immortalized Prostate Cells. Cell Death Differ. 2011, 18, 271–281. [Google Scholar] [CrossRef]
- Roszkowska, K.A.; Piecuch, A.; Sady, M.; Gajewski, Z.; Flis, S. Gain of Function (GOF) Mutant P53 in Cancer-Current Therapeutic Approaches. Int. J. Mol. Sci. 2022, 23, 13287. [Google Scholar] [CrossRef]
- De Matteis, S.; Canale, M.; Verlicchi, A.; Bronte, G.; Delmonte, A.; Crinò, L.; Martinelli, G.; Ulivi, P. Advances in Molecular Mechanisms and Immunotherapy Involving the Immune Cell-Promoted Epithelial-to-Mesenchymal Transition in Lung Cancer. J. Oncol. 2019, 2019, 7475364. [Google Scholar] [CrossRef]
- Madeddu, C.; Donisi, C.; Liscia, N.; Lai, E.; Scartozzi, M.; Macciò, A. EGFR-Mutated Non-Small Cell Lung Cancer and Resistance to Immunotherapy: Role of the Tumor Microenvironment. Int. J. Mol. Sci. 2022, 23, 6849. [Google Scholar] [CrossRef]
- Lin, A.; Wei, T.; Meng, H.; Luo, P.; Zhang, J. Role of the Dynamic Tumor Microenvironment in Controversies Regarding Immune Checkpoint Inhibitors for the Treatment of Non-Small Cell Lung Cancer (NSCLC) with EGFR Mutations. Mol. Cancer 2019, 18, 139. [Google Scholar] [CrossRef]
- Wang, M.; Zhu, L.; Yang, X.; Li, J.; Liu, Y.; Tang, Y. Targeting Immune Cell Types of Tumor Microenvironment to Overcome Resistance to PD-1/PD-L1 Blockade in Lung Cancer. Front. Pharmacol. 2023, 14, 1132158. [Google Scholar] [CrossRef]
- Chen, S.; Tang, J.; Liu, F.; Li, W.; Yan, T.; Shangguan, D.; Yang, N.; Liao, D. Changes of Tumor Microenvironment in Non-Small Cell Lung Cancer after TKI Treatments. Front. Immunol. 2023, 14, 1094764. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Pan, G.; Cheng, G.; Zhang, F.; Xu, Y.; Huang, Z.; Fan, Y. Immune Microenvironment Features and Efficacy of PD-1/PD-L1 Blockade in Non-Small Cell Lung Cancer Patients with EGFR or HER2 Exon 20 Insertions. Thorac. Cancer 2021, 12, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.G.; Shih, J.Y. Management of Acquired Resistance to EGFR TKI-Targeted Therapy in Advanced Non-Small Cell Lung Cancer. Mol. Cancer 2018, 17, 38. [Google Scholar] [CrossRef] [PubMed]
- Arbour, K.C.; Jordan, E.; Kim, H.R.; Dienstag, J.; Yu, H.A.; Sanchez-Vega, F.; Lito, P.; Berger, M.; Solit, D.B.; Hellmann, M.; et al. Effects of Co-Occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Kerk, S.A.; Papagiannakopoulos, T.; Shah, Y.M.; Lyssiotis, C.A. Metabolic Networks in Mutant KRAS-Driven Tumours: Tissue Specificities and the Microenvironment. Nat. Rev. Cancer 2021, 21, 510–525. [Google Scholar] [CrossRef] [PubMed]
- Best, S.A.; De Souza, D.P.; Kersbergen, A.; Policheni, A.N.; Dayalan, S.; Tull, D.; Rathi, V.; Gray, D.H.; Ritchie, M.E.; McConville, M.J.; et al. Synergy between the KEAP1/NRF2 and PI3K Pathways Drives Non-Small-Cell Lung Cancer with an Altered Immune Microenvironment. Cell Metab. 2018, 27, 935–943.e4. [Google Scholar] [CrossRef] [PubMed]
- Ricciuti, B.; Arbour, K.C.; Lin, J.J.; Vajdi, A.; Vokes, N.; Hong, L.; Zhang, J.; Tolstorukov, M.Y.; Li, Y.Y.; Spurr, L.F.; et al. Diminished Efficacy of Programmed Death-(Ligand)1 Inhibition in STK11- and KEAP1-Mutant Lung Adenocarcinoma Is Affected by KRAS Mutation Status. J. Thorac. Oncol. 2022, 17, 399–410. [Google Scholar] [CrossRef]
- Marinelli, D.; Mazzotta, M.; Scalera, S.; Terrenato, I.; Sperati, F.; D’Ambrosio, L.; Pallocca, M.; Corleone, G.; Krasniqi, E.; Pizzuti, L.; et al. KEAP1-Driven Co-Mutations in Lung Adenocarcinoma Unresponsive to Immunotherapy despite High Tumor Mutational Burden. Ann. Oncol. 2020, 31, 1746–1754. [Google Scholar] [CrossRef]
- Montesion, M.; Murugesan, K.; Jin, D.X.; Sharaf, R.; Sanchez, N.; Guria, A.; Minker, M.; Li, G.; Fisher, V.; Sokol, E.S.; et al. Somatic Hla Class i Loss Is a Widespread Mechanism of Immune Evasion Which Refines the Use of Tumor Mutational Burden as a Biomarker of Checkpoint Inhibitor Response. Cancer Discov. 2021, 11, 282–292. [Google Scholar] [CrossRef]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e11. [Google Scholar] [CrossRef]
- Kalbasi, A.; Tariveranmoshabad, M.; Hakimi, K.; Kremer, S.; Campbell, K.M.; Funes, J.M.; Vega-Crespo, A.; Parisi, G.; Champekar, A.; Nguyen, C.; et al. Uncoupling Interferon Signaling and Antigen Presentation to Overcome Immunotherapy Resistance Due to JAK1 Loss in Melanoma. Sci. Transl. Med. 2020, 12, eabb0152. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, Immunity and Cancer Immunoediting. Nat. Rev. Immunol. 2006, 6, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Three Es of Cancer Immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K. Cancer Immunoediting from Immune Surveillance to Immune Escape. Immunology 2007, 121, 9–27. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Blum, J.S.; Wearsch, P.A.; Cresswell, P. Pathways of Antigen Processing. Annu. Rev. Immunol. 2013, 31, 443–473. [Google Scholar] [CrossRef]
- Seliger, B.; Maeurer, M.J.; Ferrone, S. Antigen-Processing Machinery Breakdown and Tumor Growth. Immunol. Today 2000, 21, 455–464. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to Checkpoint Blockade Therapy through Inactivation of Antigen Presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef]
- Mpakali, A.; Stratikos, E. The Role of Antigen Processing and Presentation in Cancer and the Efficacy of Immune Checkpoint Inhibitor Immunotherapy. Cancers 2021, 13, 134. [Google Scholar] [CrossRef]
- Pereira, C.; Gimenez-Xavier, P.; Pros, E.; Pajares, M.J.; Moro, M.; Gomez, A.; Navarro, A.; Condom, E.; Moran, S.; Gomez-Lopez, G.; et al. Genomic Profiling of Patient-Derived Xenografts for Lung Cancer Identifies B2M Inactivation Impairing Immunorecognition. Clin. Cancer Res. 2017, 23, 3203–3213. [Google Scholar] [CrossRef]
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Miao, D.; Demetri, G.D.; Adeegbe, D.; Rodig, S.J.; Shukla, S.; Lipschitz, M.; Amin-Mansour, A.; Raut, C.P.; Carter, S.L.; et al. Loss of PTEN Is Associated with Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity 2017, 46, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Wise, H.M.; Hermida, M.A.; Leslie, N.R. Prostate Cancer, PI3K, PTEN and Prognosis. Clin. Sci. 2017, 131, 197–210. [Google Scholar] [CrossRef]
- Chen, H.; Chong, W.; Teng, C.; Yao, Y.; Wang, X.; Li, X. The Immune Response-Related Mutational Signatures and Driver Genes in Non-Small-Cell Lung Cancer. Cancer Sci. 2019, 110, 2348–2356. [Google Scholar] [CrossRef]
- Teng, J.; Zhou, K.; Lv, D.; Wu, C.; Feng, H. Case Report: PTEN Mutation Induced by Anti-PD-1 Therapy in Stage IV Lung Adenocarcinoma. Front. Pharmacol. 2022, 13, 714408. [Google Scholar] [CrossRef]
- Janghorban, M.; Xin, L.; Rosen, J.M.; Zhang, X.H.F. Notch Signaling as a Regulator of the Tumor Immune Response: To Target or Not To Target? Front. Immunol. 2018, 9, 1649. [Google Scholar] [CrossRef]
- Li, X.; Wang, Y.; Li, X.; Feng, G.; Hu, S.; Bai, Y. The Impact of NOTCH Pathway Alteration on Tumor Microenvironment and Clinical Survival of Immune Checkpoint Inhibitors in NSCLC. Front. Immunol. 2021, 12, 638763. [Google Scholar] [CrossRef]
- Lv, L.; Huang, R.H.; Li, J.; Xu, J.; Gao, W. Impact of NSCLC Metabolic Remodeling on Immunotherapy Effectiveness. Biomark. Res. 2022, 10, 66. [Google Scholar] [CrossRef]
- Kempkes, R.W.M.; Joosten, I.; Koenen, H.J.P.M.; He, X. Metabolic Pathways Involved in Regulatory T Cell Functionality. Front. Immunol. 2019, 10, 2839. [Google Scholar] [CrossRef]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371.e9. [Google Scholar] [CrossRef] [PubMed]
- Wiel, C.; Le Gal, K.; Ibrahim, M.X.; Jahangir, C.A.; Kashif, M.; Yao, H.; Ziegler, D.V.; Xu, X.; Ghosh, T.; Mondal, T.; et al. BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell 2019, 178, 330–345.e22. [Google Scholar] [CrossRef]
- Bovenzi, C.D.; Hamilton, J.; Tassone, P.; Johnson, J.; Cognetti, D.M.; Luginbuhl, A.; Keane, W.M.; Zhan, T.; Tuluc, M.; Bar-Ad, V.; et al. Prognostic Indications of Elevated MCT4 and CD147 across Cancer Types: A Meta-Analysis. Biomed Res. Int. 2015, 2015, 242437. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.H.; Hu, X.P.; Xiang, X.P.; Fang, L. High Expression of Monocarboxylate Transporter 4 (MCT 4), but Not MCT 1, Predicts Poor Prognosis in Patients with Non-Small Cell Lung Cancer. Transl. Cancer Res. 2021, 10, 1336. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.J.; Pareja, F.; Socci, N.D.; Shen, R.; Doane, A.S.; Schwartz, J.; Khanin, R.; Morris, E.A.; Sutton, E.J.; Blasberg, R.G. Increased Tumor Glycolysis Is Associated with Decreased Immune Infiltration across Human Solid Tumors. Front. Immunol. 2022, 13, 880959. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Zhou, J.; Chen, Q.; Xu, X.; Gao, J.; Li, X.; Shao, Q.; Zhou, B.; Zhou, H.; Wei, S.; et al. Tumor Metabolite Lactate Promotes Tumorigenesis by Modulating MOESIN Lactylation and Enhancing TGF-β Signaling in Regulatory T Cells. Cell Rep. 2022, 39, 110986. [Google Scholar] [CrossRef]
- Kumagai, S.; Koyama, S.; Itahashi, K.; Tanegashima, T.; Lin, Y.-T.; Togashi, Y.; Kamada, T.; Irie, T.; Okumura, G.; Kono, H.; et al. Lactic Acid Promotes PD-1 Expression in Regulatory T Cells in Highly Glycolytic Tumor Microenvironments. Cancer Cell 2022, 40, 201–218.e9. [Google Scholar] [CrossRef]
- Feng, J.; Yang, H.; Zhang, Y.; Wei, H.; Zhu, Z.; Zhu, B.; Yang, M.; Cao, W.; Wang, L.; Wu, Z. Tumor Cell-Derived Lactate Induces TAZ-Dependent Upregulation of PD-L1 through GPR81 in Human Lung Cancer Cells. Oncogene 2017, 36, 5829–5839. [Google Scholar] [CrossRef]
- Qiao, T.; Xiong, Y.; Feng, Y.; Guo, W.; Zhou, Y.; Zhao, J.; Jiang, T.; Shi, C.; Han, Y. Inhibition of LDH-A by Oxamate Enhances the Efficacy of Anti-PD-1 Treatment in an NSCLC Humanized Mouse Model. Front. Oncol. 2021, 11, 1033. [Google Scholar] [CrossRef]
- Gähler, A.; Trufa, D.I.; Chiriac, M.T.; Tausche, P.; Hohenberger, K.; Brunst, A.K.; Rauh, M.; Geppert, C.I.; Rieker, R.J.; Krammer, S.; et al. Glucose-Restricted Diet Regulates the Tumor Immune Microenvironment and Prevents Tumor Growth in Lung Adenocarcinoma. Front. Oncol. 2022, 12, 873293. [Google Scholar] [CrossRef]
- De Saedeleer, C.J.; Copetti, T.; Porporato, P.E.; Verrax, J.; Feron, O.; Sonveaux, P. Lactate Activates HIF-1 in Oxidative but Not in Warburg-Phenotype Human Tumor Cells. PLoS ONE 2012, 7, e46571. [Google Scholar] [CrossRef] [PubMed]
- Roland, C.L.; Arumugam, T.; Deng, D.; Liu, S.H.; Philip, B.; Gomez, S.; Burns, W.R.; Ramachandran, V.; Wang, H.; Cruz-Monserrate, Z.; et al. Cell Surface Lactate Receptor GPR81 Is Crucial for Cancer Cell Survival. Cancer Res. 2014, 74, 5301–5310. [Google Scholar] [CrossRef] [PubMed]
- Ancel, J.; Perotin, J.M.; Dewolf, M.; Launois, C.; Mulette, P.; Nawrocki-Raby, B.; Dalstein, V.; Gilles, C.; Deslée, G.; Polette, M.; et al. Hypoxia in Lung Cancer Management: A Translational Approach. Cancers 2021, 13, 3421. [Google Scholar] [CrossRef]
- Multhoff, G.; Vaupel, P. Hypoxia Compromises Anti-Cancer Immune Responses. Adv. Exp. Med. Biol. 2020, 1232, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Lu, F.T.; Cao, J.X.; Ma, W.J.; Xia, Z.F.; Zhan, J.H.; Zeng, K.M.; Huang, Y.; Zhao, H.Y.; Zhang, L. HIF-1α Inhibition Promotes the Efficacy of Immune Checkpoint Blockade in the Treatment of Non-Small Cell Lung Cancer. Cancer Lett. 2022, 531, 39–56. [Google Scholar] [CrossRef]
- Lieu, E.L.; Nguyen, T.; Rhyne, S.; Kim, J. Amino Acids in Cancer. Exp. Mol. Med. 2020, 52, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative Flux Analysis Reveals Folate-Dependent NADPH Production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef]
- Passarelli, A.; Pisano, C.; Cecere, S.C.; Di Napoli, M.; Rossetti, S.; Tambaro, R.; Ventriglia, J.; Gherardi, F.; Iannacone, E.; Venanzio, S.S.; et al. Targeting Immunometabolism Mediated by the IDO1 Pathway: A New Mechanism of Immune Resistance in Endometrial Cancer. Front. Immunol. 2022, 13, 953115. [Google Scholar] [CrossRef]
- Suzuki, Y.; Suda, T.; Furuhashi, K.; Suzuki, M.; Fujie, M.; Hahimoto, D.; Nakamura, Y.; Inui, N.; Nakamura, H.; Chida, K. Increased Serum Kynurenine/Tryptophan Ratio Correlates with Disease Progression in Lung Cancer. Lung Cancer 2010, 67, 361–365. [Google Scholar] [CrossRef]
- Greene, L.I.; Bruno, T.C.; Christenson, J.L.; D’Alessandro, A.; Culp-Hill, R.; Torkko, K.; Borges, V.F.; Slansky, J.E.; Richer, J.K. A Role for Tryptophan-2,3-Dioxygenase in CD8 T-Cell Suppression and Evidence of Tryptophan Catabolism in Breast Cancer Patient Plasma. Mol. Cancer Res. 2019, 17, 131–139. [Google Scholar] [CrossRef]
- Fallarino, F.; Grohmann, U.; Hwang, K.W.; Orabona, C.; Vacca, C.; Bianchi, R.; Belladonna, M.L.; Fioretti, M.C.; Alegre, M.L.; Puccetti, P. Modulation of Tryptophan Catabolism by Regulatory T Cells. Nat. Immunol. 2003, 4, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Arensman, M.D.; Yang, X.S.; Leahy, D.M.; Toral-Barza, L.; Mileski, M.; Rosfjord, E.C.; Wang, F.; Deng, S.; Myers, J.S.; Abraham, R.T.; et al. Cystine–Glutamate Antiporter XCT Deficiency Suppresses Tumor Growth While Preserving Antitumor Immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 9533–9542. [Google Scholar] [CrossRef]
- Byun, J.K.; Park, M.; Lee, S.; Yun, J.W.; Lee, J.; Kim, J.S.; Cho, S.J.; Jeon, H.J.; Lee, I.K.; Choi, Y.K.; et al. Inhibition of Glutamine Utilization Synergizes with Immune Checkpoint Inhibitor to Promote Antitumor Immunity. Mol. Cell 2020, 80, 592–606.e8. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Gao, L.; Wang, Y.; Xu, B.; Maswikiti, E.P.; Li, H.; Zheng, P.; Tao, P.; Xiang, L.; Gu, B.; et al. A Forgotten Corner in Cancer Immunotherapy: The Role of Lipids. Front. Oncol. 2021, 11, 751086. [Google Scholar] [CrossRef]
- Corn, K.C.; Windham, M.A.; Rafat, M. Lipids in the Tumor Microenvironment: From Cancer Progression to Treatment. Prog. Lipid Res. 2020, 80, 101055. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.R.; Mitchell, D.; Carroll, E.; Li, M.; Schneck, J.; Edidin, M. Differential Effects of a Saturated and a Monounsaturated Fatty Acid on MHC Class I Antigen Presentation. Scand. J. Immunol. 2008, 68, 30–42. [Google Scholar] [CrossRef]
- Coutzac, C.; Jouniaux, J.M.; Paci, A.; Schmidt, J.; Mallardo, D.; Seck, A.; Asvatourian, V.; Cassard, L.; Saulnier, P.; Lacroix, L.; et al. Systemic Short Chain Fatty Acids Limit Antitumor Effect of CTLA-4 Blockade in Hosts with Cancer. Nat. Commun. 2020, 11, 2168. [Google Scholar] [CrossRef]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the Antitumour Response of CD8+ T Cells by Modulating Cholesterol Metabolism. Nature 2016, 531, 651–655. [Google Scholar] [CrossRef]
- Ahmadi, M.; Emery, D.C.; Morgan, D.J. Prevention of Both Direct and Cross-Priming of Antitumor CD8+ T-Cell Responses Following Overproduction of Prostaglandin E2 by Tumor Cells in vivo. Cancer Res. 2008, 68, 7520–7529. [Google Scholar] [CrossRef]
- Yan, D.; Adeshakin, A.O.; Xu, M.; Afolabi, L.O.; Zhang, G.; Chen, Y.H.; Wan, X. Lipid Metabolic Pathways Confer the Immunosuppressive Function of Myeloid-Derived Suppressor Cells in Tumor. Front. Immunol. 2019, 10, 1399. [Google Scholar] [CrossRef]
- Shimizu, K.; Nakata, M.; Hirami, Y.; Yukawa, T.; Maeda, A.; Tanemoto, K. Tumor-Infiltrating Foxp3+ Regulatory T Cells Are Correlated with Cyclooxygenase-2 Expression and Are Associated with Recurrence in Resected Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2010, 5, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Tomić, S.; Joksimović, B.; Bekić, M.; Vasiljević, M.; Milanović, M.; Čolić, M.; Vučević, D. Prostaglanin-E2 Potentiates the Suppressive Functions of Human Mononuclear Myeloid-Derived Suppressor Cells and Increases Their Capacity to Expand IL-10-Producing Regulatory T Cell Subsets. Front. Immunol. 2019, 10, 475. [Google Scholar] [CrossRef] [PubMed]
- Ballav, S.; Biswas, B.; Sahu, V.K.; Ranjan, A.; Basu, S. PPAR-γ Partial Agonists in Disease-Fate Decision with Special Reference to Cancer. Cells 2022, 11, 3215. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.; Cai, Y.; Chen, D.; Jiang, G.; Xu, Y.; Chen, R.; Wang, F.; Wang, X.; Zheng, M.; Zhao, X.; et al. Statin Shapes Inflamed Tumor Microenvironment and Enhances Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. JCI Insight 2022, 7, e161940. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer Metabolism: Fatty Acid Oxidation in the Limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Svensson, R.U.; Parker, S.J.; Eichner, L.J.; Kolar, M.J.; Wallace, M.; Brun, S.N.; Lombardo, P.S.; Van Nostrand, J.L.; Hutchins, A.; Vera, L.; et al. Inhibition of Acetyl-CoA Carboxylase Suppresses Fatty Acid Synthesis and Tumor Growth of Non-Small-Cell Lung Cancer in Preclinical Models. Nat. Med. 2016, 22, 1108–1119. [Google Scholar] [CrossRef]
- Kim, J.; Lee, H.M.; Cai, F.; Ko, B.; Yang, C.; Lieu, E.L.; Muhammad, N.; Rhyne, S.; Li, K.; Haloul, M.; et al. The Hexosamine Biosynthesis Pathway Is a Targetable Liability in KRAS/LKB1 Mutant Lung Cancer. Nat. Metab. 2020, 2, 1401–1412. [Google Scholar] [CrossRef]
- Kerr, E.M.; Martins, C.P. Metabolic Rewiring in Mutant Kras Lung Cancer. FEBS J. 2018, 285, 28–41. [Google Scholar] [CrossRef]
- Biton, J.; Mansuet-Lupo, A.; Pécuchet, N.; Alifano, M.; Ouakrim, H.; Arrondeau, J.; Boudou-Rouquette, P.; Goldwasser, F.; Leroy, K.; Goc, J.; et al. TP53, STK11, and EGFR Mutations Predict Tumor Immune Profile and the Response to Anti-PD-1 in Lung Adenocarcinoma. Clin. Cancer Res. 2018, 24, 5710–5723. [Google Scholar] [CrossRef]
- Jure-Kunkel, M.; Wu, S.; Xiao, F.; Abdullah, S.E.; Gao, G.; Englert, J.M.; Hsieh, H.-J.; Mukhopadhyay, P.; Gupta, A.K.; Dennis, P.A.; et al. Somatic STK11/LKB1 Mutations to Confer Resistance to Immune Checkpoint Inhibitors as Monotherapy or in Combination in Advanced NSCLC. J. Clin. Oncol. 2018, 36, 3028. [Google Scholar] [CrossRef]
- Rizvi, N.; Cho, B.C.; Reinmuth, N.; Lee, K.H.; Luft, A.; Ahn, M.; Papadimitrakopoulou, V.; Heymach, J.; Scheuring, U.; Higgs, B.; et al. OA04.07 Mutations Associated with Sensitivity or Resistance to Immunotherapy in MNSCLC: Analysis from the MYSTIC Trial. J. Thorac. Oncol. 2019, 14, S217. [Google Scholar] [CrossRef]
- Zhao, J.; Lin, X.; Meng, D.; Zeng, L.; Zhuang, R.; Huang, S.; Lv, W.; Hu, J. Nrf2 Mediates Metabolic Reprogramming in Non-Small Cell Lung Cancer. Front. Oncol. 2020, 10, 578315. [Google Scholar] [CrossRef] [PubMed]
- Denicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-Induced Nrf2 Transcription Promotes ROS Detoxification and Tumorigenesis. Nature 2011, 475, 106. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Ma, J.; Hall, S.R.R.; Peng, R.W.; Yang, H.; Yao, F. Battles against Aberrant KEAP1-NRF2 Signaling in Lung Cancer: Intertwined Metabolic and Immune Networks. Theranostics 2023, 13, 704–723. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining Glutaminolysis Bolsters Chemotherapy While NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res. 2020, 80, 1630–1643. [Google Scholar] [CrossRef]
- Best, S.A.; Gubser, P.M.; Sethumadhavan, S.; Kersbergen, A.; Negrón Abril, Y.L.; Goldford, J.; Sellers, K.; Abeysekera, W.; Garnham, A.L.; McDonald, J.A.; et al. Glutaminase Inhibition Impairs CD8 T Cell Activation in STK11-/Lkb1-Deficient Lung Cancer. Cell Metab. 2022, 34, 874–887.e6. [Google Scholar] [CrossRef]
- Oh, M.H.; Sun, I.H.; Zhao, L.; Leone, R.D.; Sun, I.M.; Xu, W.; Collins, S.L.; Tam, A.J.; Blosser, R.L.; Patel, C.H.; et al. Targeting Glutamine Metabolism Enhances Tumor-Specific Immunity by Modulating Suppressive Myeloid Cells. J. Clin. Investig. 2020, 130, 3865–3884. [Google Scholar] [CrossRef]
- Olagnier, D.; Brandtoft, A.M.; Gunderstofte, C.; Villadsen, N.L.; Krapp, C.; Thielke, A.L.; Laustsen, A.; Peri, S.; Hansen, A.L.; Bonefeld, L.; et al. Nrf2 Negatively Regulates STING Indicating a Link between Antiviral Sensing and Metabolic Reprogramming. Nat. Commun. 2018, 9, 3506. [Google Scholar] [CrossRef]
- Lemos, H.; Mohamed, E.; Huang, L.; Ou, R.; Pacholczyk, G.; Arbab, A.S.; Munn, D.; Mellor, A.L. STING Promotes the Growth of Tumors Characterized by Low Antigenicity via IDO Activation. Cancer Res. 2016, 76, 2076–2081. [Google Scholar] [CrossRef]
- Fahrmann, J.F.; Tanaka, I.; Irajizad, E.; Mao, X.; Dennison, J.B.; Murage, E.; Casabar, J.; Mayo, J.; Peng, Q.; Celiktas, M.; et al. Mutational Activation of the NRF2 Pathway Upregulates Kynureninase Resulting in Tumor Immunosuppression and Poor Outcome in Lung Adenocarcinoma. Cancers 2022, 14, 2543. [Google Scholar] [CrossRef] [PubMed]
- Galan-Cobo, A.; Sitthideatphaiboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.H.; Boroughs, L.K.; Rodriguez, M.L.M.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 Pathways Cooperatively Promote Metabolic Reprogramming with Enhanced Glutamine Dependence in KRAS-Mutant Lung Adenocarcinoma. Cancer Res. 2019, 79, 3251–3267. [Google Scholar] [CrossRef] [PubMed]
- Muto, S.; Enta, A.; Maruya, Y.; Inomata, S.; Yamaguchi, H.; Mine, H.; Takagi, H.; Ozaki, Y.; Watanabe, M.; Inoue, T.; et al. Wnt/β-Catenin Signaling and Resistance to Immune Checkpoint Inhibitors: From Non-Small-Cell Lung Cancer to Other Cancers. Biomedicines 2023, 11, 190. [Google Scholar] [CrossRef] [PubMed]
- Muto, S.; Ozaki, Y.; Yamaguchi, H.; Mine, H.; Takagi, H.; Watanabe, M.; Inoue, T.; Yamaura, T.; Fukuhara, M.; Okabe, N.; et al. Tumor β-Catenin Expression Is Associated with Immune Evasion in Non-Small Cell Lung Cancer with High Tumor Mutation Burden. Oncol. Lett. 2021, 21, 203. [Google Scholar] [CrossRef] [PubMed]
- Garmendia, I.; Redin, E.; Montuenga, L.M.; Calvo, A. YES1: A Novel Therapeutic Target and Biomarker in Cancer. Mol. Cancer Ther. 2022, 21, 1371–1380. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, J.; Zheng, R.; Song, B.; Huang, L.; Liu, Y.; Hao, Y.; Bai, X. MiR-133 Targets YES1 and Inhibits the Growth of Triple-Negative Breast Cancer Cells. Technol. Cancer Res. Treat. 2020, 19, 1533033820927011. [Google Scholar] [CrossRef]
- Chatterji, T.; Varkaris, A.S.; Parikh, N.U.; Song, J.H.; Cheng, C.J.; Schweppe, R.E.; Alexander, S.; Davis, J.W.; Troncoso, P.; Friedl, P.; et al. Yes-Mediated Phosphorylation of Focal Adhesion Kinase at Tyrosine 861 Increases Metastatic Potential of Prostate Cancer Cells. Oncotarget 2015, 6, 10175–10194. [Google Scholar] [CrossRef]
- Fang, Z.; Yin, S.; Sun, R.; Zhang, S.; Fu, M.; Wu, Y.; Zhang, T.; Khaliq, J.; Li, Y. MiR-140-5p Suppresses the Proliferation, Migration and Invasion of Gastric Cancer by Regulating YES1. Mol. Cancer 2017, 16, 139. [Google Scholar] [CrossRef]
- Guo, L.; Zheng, J.; Luo, J.; Zhang, Z.; Shao, G. Targeting Yes1 Associated Transcriptional Regulator Inhibits Hepatocellular Carcinoma Progression and Improves Sensitivity to Sorafenib: An in Vitro and in Vivo Study. Onco Targets Ther. 2020, 13, 11071–11087. [Google Scholar] [CrossRef]
- Garmendia, I.; Pajares, M.J.; Hermida-Prado, F.; Ajona, D.; Bértolo, C.; Sainz, C.; Lavín, A.; Remírez, A.B.; Valencia, K.; Moreno, H.; et al. YES1 Drives Lung Cancer Growth and Progression and Predicts Sensitivity to Dasatinib. Am. J. Respir. Crit. Care Med. 2019, 200, 888–899. [Google Scholar] [CrossRef]
- Redin, E.; Garrido-Martin, E.M.; Valencia, K.; Redrado, M.; Solorzano, J.L.; Carias, R.; Echepare, M.; Exposito, F.; Serrano, D.; Ferrer, I.; et al. YES1 Is a Druggable Oncogenic Target in SCLC. J. Thorac. Oncol. 2022, 17, 1387–1403. [Google Scholar] [CrossRef] [PubMed]
- Serrels, A.; Lund, T.; Serrels, B.; Byron, A.; McPherson, R.C.; Von Kriegsheim, A.; Gómez-Cuadrado, L.; Canel, M.; Muir, M.; Ring, J.E.; et al. Nuclear FAK Controls Chemokine Transcription, Tregs, and Evasion of Anti-Tumor Immunity. Cell 2015, 163, 160. [Google Scholar] [CrossRef]
- Murakami, S.; Shahbazian, D.; Surana, R.; Zhang, W.; Chen, H.; Graham, G.T.; White, S.M.; Weiner, L.M.; Yi, C. Yes-Associated Protein Mediates Immune Reprogramming in Pancreatic Ductal Adenocarcinoma. Oncogene 2017, 36, 1232–1244. [Google Scholar] [CrossRef]
- Redin, E.; Garmendia, I.; Lozano, T.; Serrano, D.; Senent, Y.; Redrado, M.; Villalba, M.; De Andrea, C.E.; Exposito, F.; Ajona, D.; et al. SRC Family Kinase (SFK) Inhibitor Dasatinib Improves the Antitumor Activity of Anti-PD-1 in NSCLC Models by Inhibiting Treg Cell Conversion and Proliferation. J. Immunother. Cancer 2021, 9, e001496. [Google Scholar] [CrossRef] [PubMed]
- Valencia, K.; Echepare, M.; Teijeira, Á.; Pasquier, A.; Bértolo, C.; Sainz, C.; Tamayo, I.; Picabea, B.; Bosco, G.; Thomas, R.; et al. DSTYK Inhibition Increases the Sensitivity of Lung Cancer Cells to T Cell-Mediated Cytotoxicity. J. Exp. Med. 2022, 219, e20220726. [Google Scholar] [CrossRef] [PubMed]
- Bouillez, A.; Rajabi, H.; Jin, C.; Samur, M.; Tagde, A.; Alam, M.; Hiraki, M.; Maeda, T.; Hu, X.; Adeegbe, D.; et al. MUC1-C Integrates PD-L1 Induction with Repression of Immune Effectors in Non-Small-Cell Lung Cancer. Oncogene 2017, 36, 4037–4046. [Google Scholar] [CrossRef] [PubMed]
- Van Rensburg, H.J.J.; Azad, T.; Ling, M.; Hao, Y.; Snetsinger, B.; Khanal, P.; Minassian, L.M.; Graham, C.H.; Rauh, M.J.; Yang, X. The Hippo Pathway Component TAZ Promotes Immune Evasion in Human Cancer through PD-L1. Cancer Res. 2018, 78, 1457–1470. [Google Scholar] [CrossRef]
- Miao, J.; Hsu, P.C.; Yang, Y.L.; Xu, Z.; Dai, Y.; Wang, Y.; Chan, G.; Huang, Z.; Hu, B.; Li, H.; et al. YAP Regulates PD-L1 Expression in Human NSCLC Cells. Oncotarget 2017, 8, 114576–114587. [Google Scholar] [CrossRef]
- Li, L.; Zou, B.J.; Zhao, J.Z.; Liang, J.B.; She, Z.Y.; Zhou, W.Y.; Lin, S.X.; Tian, L.; Luo, W.J.; He, F.Z. A Novel DNA Damage Repair-Related Signature for Predicting Prognositc and Treatment Response in Non-Small Lung Cancer. Front. Oncol. 2022, 12, 961274. [Google Scholar] [CrossRef]
- Cortellini, A.; Giusti, R.; Filetti, M.; Citarella, F.; Adamo, V.; Santini, D.; Buti, S.; Nigro, O.; Cantini, L.; Di Maio, M.; et al. High Familial Burden of Cancer Correlates with Improved Outcome from Immunotherapy in Patients with NSCLC Independent of Somatic DNA Damage Response Gene Status. J. Hematol. Oncol. 2022, 15, 9. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, G.; Li, J.; Huang, Y.Y.; Li, Y.; Lin, J.; Chen, L.Z.; Lu, J.P.; Wang, Y.Q.; Wang, C.X.; et al. Association of Tumor Protein P53 and Ataxia-Telangiectasia Mutated Comutation With Response to Immune Checkpoint Inhibitors and Mortality in Patients With Non-Small Cell Lung Cancer. JAMA Netw. Open 2019, 2, e1911895. [Google Scholar] [CrossRef] [PubMed]
- Remon, J.; Aldea, M.; Besse, B.; Planchard, D.; Reck, M.; Giaccone, G.; Soria, J.C. Small Cell Lung Cancer: A Slightly Less Orphan Disease after Immunotherapy. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, 32, 698–709. [Google Scholar] [CrossRef] [PubMed]
- Gazdar, A.F.; Bunn, P.A.; Minna, J.D. Small-Cell Lung Cancer: What We Know, What We Need to Know and the Path Forward. Nat. Rev. Cancer 2017, 17, 725–737. [Google Scholar] [CrossRef]
- Mahadevan, N.R.; Knelson, E.H.; Wolff, J.O.; Vajdi, A.; Saigí, M.; Campisi, M.; Hong, D.; Thai, T.C.; Piel, B.; Han, S.; et al. Intrinsic Immunogenicity of Small Cell Lung Carcinoma Revealed by Its Cellular Plasticity. Cancer Discov. 2021, 11, 1952–1969. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, Y.; Ozasa, H.; Kim, Y.H. PD-L1 Expression in Small Cell Lung Cancer. J. Thorac. Oncol. 2018, 13, e40–e41. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Callahan, M.K.; Awad, M.M.; Calvo, E.; Ascierto, P.A.; Atmaca, A.; Rizvi, N.A.; Hirsch, F.R.; Selvaggi, G.; Szustakowski, J.D.; et al. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer Cell 2018, 33, 853–861.e4. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, E.M.; Taniguchi, H.; Chan, J.M.; Zhan, Y.A.; Chen, X.; Qiu, J.; de Stanchina, E.; Allaj, V.; Shah, N.S.; Uddin, F.; et al. Targeting Lysine-Specific Demethylase 1 Rescues Major Histocompatibility Complex Class I Antigen Presentation and Overcomes Programmed Death-Ligand 1 Blockade Resistance in SCLC. J. Thorac. Oncol. 2022, 17, 1014–1031. [Google Scholar] [CrossRef]
- Hiatt, J.B.; Sandborg, H.; Garrison, S.M.; Arnold, H.U.; Liao, S.Y.; Norton, J.P.; Friesen, T.J.; Wu, F.; Sutherland, K.D.; Rienhoff, H.Y.; et al. Inhibition of LSD1 with Bomedemstat Sensitizes Small Cell Lung Cancer to Immune Checkpoint Blockade and T-Cell Killing. Clin. Cancer Res. 2022, 28, 4551–4564. [Google Scholar] [CrossRef]
- Poirier, J.T.; George, J.; Owonikoko, T.K.; Berns, A.; Brambilla, E.; Byers, L.A.; Carbone, D.; Chen, H.J.; Christensen, C.L.; Dive, C.; et al. New Approaches to SCLC Therapy: From the Laboratory to the Clinic. J. Thorac. Oncol. 2020, 15, 520–540. [Google Scholar] [CrossRef]
- Longo, V.; Catino, A.; Montrone, M.; Pizzutilo, P.; Annese, T.; Pesola, F.; Marech, I.; Cassiano, S.; Ribatti, D.; Galetta, D. What Are the Biomarkers for Immunotherapy in SCLC? Int. J. Mol. Sci. 2021, 22, 11123. [Google Scholar] [CrossRef]
- Iams, W.T.; Porter, J.; Horn, L. Immunotherapeutic Approaches for Small-Cell Lung Cancer. Nat. Rev. Clin. Oncol. 2020, 17, 300–312. [Google Scholar] [CrossRef]
- Liu, J.; He, D.; Cheng, L.; Huang, C.; Zhang, Y.; Rao, X.; Kong, Y.; Li, C.; Zhang, Z.; Liu, J.; et al. P300/CBP Inhibition Enhances the Efficacy of Programmed Death-Ligand 1 Blockade Treatment in Prostate Cancer. Oncogene 2020, 39, 3939–3951. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lu, D.; Yin, Y.; Song, J.; Liu, Y.; Hao, W.; Qi, F.; Zhang, G.; Zhang, X.; Liu, L.; et al. PTENα Functions as an Immune Suppressor and Promotes Immune Resistance in PTEN-Mutant Cancer. Nat. Commun. 2021, 12, 5147. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Poirier, J.T.; Byers, L.A.; Dive, C.; Dowlati, A.; George, J.; Heymach, J.V.; Johnson, J.E.; Lehman, J.M.; MacPherson, D.; et al. Molecular Subtypes of Small Cell Lung Cancer: A Synthesis of Human and Mouse Model Data. Nat. Rev. Cancer 2019, 19, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Owonikoko, T.K.; Dwivedi, B.; Chen, Z.; Zhang, C.; Barwick, B.; Ernani, V.; Zhang, G.; Gilbert-Ross, M.; Carlisle, J.; Khuri, F.R.; et al. YAP1 Expression in SCLC Defines a Distinct Subtype With T-Cell-Inflamed Phenotype. J. Thorac. Oncol. 2021, 16, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Gay, C.M.; Stewart, C.A.; Park, E.M.; Diao, L.; Groves, S.M.; Heeke, S.; Nabet, B.Y.; Fujimoto, J.; Solis, L.M.; Lu, W.; et al. Patterns of Transcription Factor Programs and Immune Pathway Activation Define Four Major Subtypes of SCLC with Distinct Therapeutic Vulnerabilities. Cancer Cell 2021, 39, 346–360.e7. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, Q.; Yang, Z.; Zhang, S.; Xu, J.; Wang, Z.; Bai, H.; Duan, J.; Zheng, B.; Li, W.; et al. Single-Cell Transcriptomic Profiling Reveals the Tumor Heterogeneity of Small-Cell Lung Cancer. Signal Transduct. Target. Ther. 2022, 7, 346. [Google Scholar] [CrossRef]
- Fasoulakis, Z.; Daskalakis, G.; Theodora, M.; Antsaklis, P.; Sindos, M.; Diakosavvas, M.; Angelou, K.; Loutradis, D.; Kontomanolis, E.N. The Relevance of Notch Signaling in Cancer Progression. Adv. Exp. Med. Biol. 2021, 1287, 169–181. [Google Scholar] [CrossRef]
- Leonetti, A.; Facchinetti, F.; Minari, R.; Cortellini, A.; Rolfo, C.D.; Giovannetti, E.; Tiseo, M. Notch Pathway in Small-Cell Lung Cancer: From Preclinical Evidence to Therapeutic Challenges. Cell. Oncol. 2019, 42, 261–273. [Google Scholar] [CrossRef]
- Radojcic, V.; Maillard, I. Notch Signaling and Alloreactivity. Transplantation 2016, 100, 2593–2600. [Google Scholar] [CrossRef]
- Murakami, N.; Maillard, I.; Riella, L.V. Notch Signaling and Immune Regulation in Alloimmunity. Curr. Transplant. Rep. 2016, 3, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Roper, N.; Velez, M.J.; Chiappori, A.; Kim, Y.S.; Wei, J.S.; Sindiri, S.; Takahashi, N.; Mulford, D.; Kumar, S.; Ylaya, K.; et al. Notch Signaling and Efficacy of PD-1/PD-L1 Blockade in Relapsed Small Cell Lung Cancer. Nat. Commun. 2021, 12, 3880. [Google Scholar] [CrossRef] [PubMed]
- Vitorino, P.; Chuang, C.H.; Iannello, A.; Zhao, X.; Anderson, W.; Ferrando, R.; Zhang, Z.; Madhavan, S.; Karsunky, H.; Saunders, L.R. Rova-T Enhances the Anti-Tumor Activity of Anti-PD1 in a Murine Model of Small Cell Lung Cancer with Endogenous Dll3 Expression. Transl. Oncol. 2021, 14, 100883. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Amar, N.; Zhu, Y.; Wang, C.; Xia, C.; Yang, X.; Wu, D.; Feng, M. Combined DLL3-Targeted Bispecific Antibody with PD-1 Inhibition Is Efficient to Suppress Small Cell Lung Cancer Growth. J. Immunother. Cancer 2020, 8, e000785. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Gay, C.M.; Byers, L.A. Targeting DNA Damage Repair in Small Cell Lung Cancer and the Biomarker Landscape. Transl. Lung Cancer Res. 2018, 7, 50–68. [Google Scholar] [CrossRef]
- Byers, L.A.; Wang, J.; Nilsson, M.B.; Fujimoto, J.; Saintigny, P.; Yordy, J.; Giri, U.; Peyton, M.; Fan, Y.H.; Diao, L.; et al. Proteomic Profiling Identifies Dysregulated Pathways in Small Cell Lung Cancer and Novel Therapeutic Targets Including PARP1. Cancer Discov. 2012, 2, 798–811. [Google Scholar] [CrossRef]
- Sen, T.; Tong, P.; Stewart, C.A.; Cristea, S.; Valliani, A.; Shames, D.S.; Redwood, A.B.; Fan, Y.H.; Li, L.; Glisson, B.S.; et al. CHK1 Inhibition in Small-Cell Lung Cancer Produces Single-Agent Activity in Biomarker-Defined Disease Subsets and Combination Activity with Cisplatin or Olaparib. Cancer Res. 2017, 77, 3870–3884. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Della Corte, C.M.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-Cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef]
- Zhang, N.; Gao, Y.; Huang, Z.; Dai, P.; Luo, Y.; Wu, Q.; Jiang, X.; Sun, W.; Zhang, J.; Han, L.; et al. PARP Inhibitor plus Radiotherapy Reshapes an Inflamed Tumor Microenvironment That Sensitizes Small Cell Lung Cancer to the Anti-PD-1 Immunotherapy. Cancer Lett. 2022, 545, 215852. [Google Scholar] [CrossRef]
- Thomas, A.; Vilimas, R.; Trindade, C.; Erwin-Cohen, R.; Roper, N.; Xi, L.; Krishnasamy, V.; Levy, E.; Mammen, A.; Nichols, S.; et al. Durvalumab in Combination with Olaparib in Patients with Relapsed SCLC: Results from a Phase II Study. J. Thorac. Oncol. 2019, 14, 1447–1457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Christensen, C.L.; Dries, R.; Oser, M.G.; Deng, J.; Diskin, B.; Li, F.; Pan, Y.; Zhang, X.; Yin, Y.; et al. CDK7 Inhibition Potentiates Genome Instability Triggering Anti-Tumor Immunity in Small Cell Lung Cancer. Cancer Cell 2020, 37, 37–54.e9. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.L.; Groves, S.M.; Zhang, Y.K.; Li, J.; Gonzalez-Ericsson, P.; Sivagnanam, S.; Betts, C.B.; Chen, H.C.; Liu, Q.; Lowe, C.; et al. Beyond Programmed Death-Ligand 1: B7-H6 Emerges as a Potential Immunotherapy Target in SCLC. J. Thorac. Oncol. 2021, 16, 1211–1223. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, K.; Jahchan, N.S.; Schnorr, P.J.; Cristea, S.; Ring, A.M.; Maute, R.L.; Volkmer, A.K.; Volkmer, J.P.; Liu, J.; Lim, J.S.; et al. CD47-Blocking Immunotherapies Stimulate Macrophage-Mediated Destruction of Small-Cell Lung Cancer. J. Clin. Investig. 2016, 126, 2610–2620. [Google Scholar] [CrossRef]
- Perrier, A.; Didelot, A.; Laurent-Puig, P.; Blons, H.; Garinet, S. Epigenetic Mechanisms of Resistance to Immune Checkpoint Inhibitors. Biomolecules 2020, 10, 1061. [Google Scholar] [CrossRef]
- Dai, E.; Zhu, Z.; Wahed, S.; Qu, Z.; Storkus, W.J.; Guo, Z.S. Epigenetic Modulation of Antitumor Immunity for Improved Cancer Immunotherapy. Mol. Cancer 2021, 20, 171. [Google Scholar] [CrossRef]
- Bajbouj, K.; Al-ali, A.; Ramakrishnan, R.K.; Saber-ayad, M.; Hamid, Q. Histone Modification in NSCLC: Molecular Mechanisms and Therapeutic Targets. Int. J. Mol. Sci. 2021, 22, 11701. [Google Scholar] [CrossRef]
- Khan, P.; Siddiqui, J.A.; Maurya, S.K.; Lakshmanan, I.; Jain, M.; Ganti, A.K.; Salgia, R.; Batra, S.K.; Nasser, M.W. Epigenetic Landscape of Small Cell Lung Cancer: Small Image of a Giant Recalcitrant Disease. Semin. Cancer Biol. 2022, 83, 57–76. [Google Scholar] [CrossRef]
- Yang, S.; Huang, Y.; Zhao, Q. Epigenetic Alterations and Inflammation as Emerging Use for the Advancement of Treatment in Non-Small Cell Lung Cancer. Front. Immunol. 2022, 13, 1610. [Google Scholar] [CrossRef]
- Luo, H.; Shan, J.; Zhang, H.; Song, G.; Li, Q.; Xu, C.X. Targeting the Epigenetic Processes to Enhance Antitumor Immunity in Small Cell Lung Cancer. Semin. Cancer Biol. 2022, 86, 960–970. [Google Scholar] [CrossRef]
- Wang, Y.; Tong, C.; Dai, H.; Wu, Z.; Han, X.; Guo, Y.; Chen, D.; Wei, J.; Ti, D.; Liu, Z.; et al. Low-Dose Decitabine Priming Endows CAR T Cells with Enhanced and Persistent Antitumour Potential via Epigenetic Reprogramming. Nat. Commun. 2021, 12, 409. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Buchlis, G.; Kapoor, V.; Cheng, G.; Sun, J.; Singhal, S.; Crisanti, C.; Wang, L.C.S.; Heitjan, D.; Snyder, L.A.; et al. CCL2 Blockade Augments Cancer Immunotherapy. Cancer Res. 2010, 70, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157.e19. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.R.; Chasalow, S.D.; Wang, L.; Hamid, O.; Schmidt, H.; Cogswell, J.; Alaparthy, S.; Berman, D.; Jure-Kunkel, M.; Siemers, N.O.; et al. An Immune-Active Tumor Microenvironment Favors Clinical Response to Ipilimumab. Cancer Immunol. Immunother. 2012, 61, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Zhao, W.; Yan, C.; Watson, C.C.; Massengill, M.; Xie, M.; Massengill, C.; Noyes, D.R.; Martinez, G.V.; Afzal, R.; et al. HDAC Inhibitors Enhance T-Cell Chemokine Expression and Augment Response to PD-1 Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2016, 22, 4119–4132. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Towatari, M.; Kosugi, H.; Saito, H. Up-Regulation of Costimulatory/Adhesion Molecules by Histone Deacetylase Inhibitors in Acute Myeloid Leukemia Cells. Blood 2000, 96, 3847–3856. [Google Scholar] [CrossRef]
- Magner, W.J.; Kazim, A.L.; Stewart, C.; Romano, M.A.; Catalano, G.; Grande, C.; Keiser, N.; Santaniello, F.; Tomasi, T.B. Activation of MHC Class I, II, and CD40 Gene Expression by Histone Deacetylase Inhibitors. J. Immunol. 2000, 165, 7017–7024. [Google Scholar] [CrossRef]
- Briere, D.; Sudhakar, N.; Woods, D.M.; Hallin, J.; Engstrom, L.D.; Aranda, R.; Chiang, H.; Sodré, A.L.; Olson, P.; Weber, J.S.; et al. The Class I/IV HDAC Inhibitor Mocetinostat Increases Tumor Antigen Presentation, Decreases Immune Suppressive Cell Types and Augments Checkpoint Inhibitor Therapy. Cancer Immunol. Immunother. 2018, 67, 381–392. [Google Scholar] [CrossRef]
- Miyanaga, A.; Gemma, A.; Noro, R.; Kataoka, K.; Matsuda, K.; Nara, M.; Okano, T.; Seike, M.; Yoshimura, A.; Kawakami, A.; et al. Antitumor Activity of Histone Deacetylase Inhibitors in Non-Small Cell Lung Cancer Cells: Development of a Molecular Predictive Model. Mol. Cancer Ther. 2008, 7, 1923–1930. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Maitland, M.L.; Frankel, P.; Argiris, A.E.; Koczywas, M.; Gitlitz, B.; Thomas, S.; Espinoza-Delgado, I.; Vokes, E.E.; Gandara, D.R.; et al. Carboplatin and Paclitaxel in Combination with Either Vorinostat or Placebo for First-Line Therapy of Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2010, 28, 56–62. [Google Scholar] [CrossRef]
- Boivin, A.J.; Momparler, L.F.; Hurtubise, A.; Momparler, R.L. Antineoplastic Action of 5-Aza-2′-Deoxycytidine and Phenylbutyrate on Human Lung Carcinoma Cells. Anticancer Drugs 2002, 13, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Duruisseaux, M.; Martínez-Cardús, A.; Calleja-Cervantes, M.E.; Moran, S.; Castro de Moura, M.; Davalos, V.; Piñeyro, D.; Sanchez-Cespedes, M.; Girard, N.; Brevet, M.; et al. Epigenetic Prediction of Response to Anti-PD-1 Treatment in Non-Small-Cell Lung Cancer: A Multicentre, Retrospective Analysis. Lancet Respir. Med. 2018, 6, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Kim, H.S.; Kim, J.Y.; Sun, J.M.; Ahn, J.S.; Ahn, M.J.; Park, K.; Esteller, M.; Lee, S.H.; Choi, J.K. DNA Methylation Loss Promotes Immune Evasion of Tumours with High Mutation and Copy Number Load. Nat. Commun. 2019, 10, 4278. [Google Scholar] [CrossRef] [PubMed]
- Burr, M.L.; Sparbier, C.E.; Chan, K.L.; Chan, Y.C.; Kersbergen, A.; Lam, E.Y.N.; Azidis-Yates, E.; Vassiliadis, D.; Bell, C.C.; Gilan, O.; et al. An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer Cell 2019, 36, 385–401.e8. [Google Scholar] [CrossRef] [PubMed]
- Murai, F.; Koinuma, D.; Shinozaki-Ushiku, A.; Fukayama, M.; Miyaozono, K.; Ehata, S. EZH2 Promotes Progression of Small Cell Lung Cancer by Suppressing the TGF-β-Smad-ASCL1 Pathway. Cell Discov. 2015, 1, 15026. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; Vansteenkiste, J.F.; Moon, E. Immunotherapy: Beyond Anti-PD-1 and Anti-PD-L1 Therapies. Am. Soc. Clin. Oncol. Educ. Book Am. Soc. Clin. Oncol. Annu. Meet. 2016, 35, e450–e458. [Google Scholar] [CrossRef]
- Hou, A.J.; Chen, L.C.; Chen, Y.Y. Navigating CAR-T Cells through the Solid-Tumour Microenvironment. Nat. Rev. Drug Discov. 2021, 20, 531–550. [Google Scholar] [CrossRef]
- Qu, J.; Mei, Q.; Chen, L.; Zhou, J. Chimeric Antigen Receptor (CAR)-T-Cell Therapy in Non-Small-Cell Lung Cancer (NSCLC): Current Status and Future Perspectives. Cancer Immunol. Immunother. 2021, 70, 619–631. [Google Scholar] [CrossRef]
- Abdin, S.M.; Paasch, D.; Morgan, M.; Lachmann, N. CARs and beyond: Tailoring Macrophage-Based Cell Therapeutics to Combat Solid Malignancies. J. Immunother. Cancer 2021, 9, e002741. [Google Scholar] [CrossRef]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human Chimeric Antigen Receptor Macrophages for Cancer Immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- Hu, B.; Sajid, M.; Lv, R.; Liu, L.; Sun, C. A Review of Spatial Profiling Technologies for Characterizing the Tumor Microenvironment in Immuno-Oncology. Front. Immunol. 2022, 13, 996721. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Subtype | Clinical Trial Identifier | Phase and Status | Epigenetic Agent | Target | Combined Agent |
|---|---|---|---|---|---|
| NSCLC | NCT01928576 | Phase II; Ongoing | Azacitidine Entinostat | DNMT1 HDAC | Nivolumab |
| NSCLC | NCT03233724 | Phase I/II; Completed | Decitabine Tetrahydrouridine | DNMT1 CDA | Pembrolizumab |
| NSCLC | NCT03220477 | Phase I; Ongoing | Guadecitabine Mocetinostat | DNMT1 | Pembrolizumab |
| NSCLC | NCT01209520 | Pilot study; Completed | Azacitidine | DNMT1 | Cisplatin Carboplatin Paclitaxel Vinorelbine Docetaxel Pemetrexed |
| SCLC NSCLC | NCT02489903 | Phase II; Completed | RRx-001 | CD47 and SIRP-α | Cisplatin Etoposide Carboplatin Irinotecan Vinorelbine Doxil Gemcitabine Taxane Paclitaxel Nab-paclitaxel Pemetrexed |
| NSCLC | NCT02959437 | Phase I/II; Terminated | Azacitidine INCB057643 INCB059872 | DNMT1 BET LSD1 | Pembrolizumab Epacadostat |
| NSCLC | NCT02664181 | Phase II; Ongoing | Oral decitabine Tetrahdrouridine | DNMT1 CDA | Nivolumab |
| NSCLC | NCT02638090 | Phase I/II; Ongoing | Vorinostat | HDAC | Pembrolizumab |
| NSCLC | NCT02437136 | Phase IB/II; Ongoing | Entinostat | HDAC | Pembrolizumab |
| SCLC | NCT02446704 | Phase I/II; Ongoing | Temozolomide | m5C | Olaparib |
| SCLC | NCT01638546 | Phase II; Completed | Temozolomide | m5C | Veliparib |
| NSCLC | NCT00423150 | Phase II; Terminated | Temozolomide | m5C | - |
| SCLC | NCT01222936 | Phase II; Completed | LBH589 Panobinostat | HDAC | - |
| SCLC | NCT02034123 | Phase I; Terminated | GSK2879552 | LSD1/KMD1A | - |
| Metastatic solid tumor | NCT05268666 | Phase I/II; Ongoing | JBI-802 | LSD1/HDAC6 | - |
| SCLC NSCLC | NCT04350463 | Phase II; Ongoing | CC-90011 | LSD1 | Nivolumab |
| NSCLC | NCT05467748 | Phase I/II; Ongoing | Tazemetostat | EZH2 | Pembrolizumab |
| SCLC | NCT03460977 | Phase I; Ongoing | PF-06821497 | EZH2 | - |
| Lung neoplasms | NCT00978250 | Phase II; Completed | FdCyd Tetrahydrouridine | DNMT CDA | - |
| NSCLC | NCT02546986 | Phase II; Ongoing | CC-486 | DNMT1 | Pembrolizumab |
| NSCLC | NCT01478685 | Phase I; Completed | CC-486 | DNMT1 | Carboplatin Nab-paclitaxel |
| NSCLC | NCT02250326 | Phase II; Ongoing | CC-486 | DNMT1 | Durvalumab Nab-paclitaxel |
| NSCLC | NCT05573035 | Phase I; Ongoing | LYL845 | TILs | - |
| NSCLC | NCT05607108 | Phase II; Ongoing | ZEN003694 | BET | - |
| NSCLC | NCT01207726 | Phase II; Terminated | Azacitidine Entinostat | DNMT1 HDAC | - |
| SCLC | NCT05191797 | Phase I/II; Ongoing | Bomedemstat | LSD1 | Atezolizumab |
| NSCLC | NCT02635061 | Phase IB; Ongoing | ACY-241 | HDAC6 | Nivolumab |
| NSCLC | NCT01059552 | Phase I; Completed | Vorinostat | HDAC | Cisplatin Pemetrexed Radiation 70Gy |
| NSCLC | NCT00821951 | Phase I; Completed | Vorinostat | HDAC | Radiotherapy |
| NSCLC | NCT02728492 | Phase I; Completed | Quisinostat | HDAC1 | Paclitaxel Carboplatin Gemcitabine Cisplatin |
| NSCLC | NCT00667082 | Phase I; Completed | Vorinostat | HDAC | NPI-0052 Marizomib |
| NSCLC | NCT00005093 | Phase III; Completed | Tacedinaline | HDAC | Gemcitabine |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Otegui, N.; Houry, M.; Arozarena, I.; Serrano, D.; Redin, E.; Exposito, F.; Leon, S.; Valencia, K.; Montuenga, L.; Calvo, A. Cancer Cell-Intrinsic Alterations Associated with an Immunosuppressive Tumor Microenvironment and Resistance to Immunotherapy in Lung Cancer. Cancers 2023, 15, 3076. https://doi.org/10.3390/cancers15123076
Otegui N, Houry M, Arozarena I, Serrano D, Redin E, Exposito F, Leon S, Valencia K, Montuenga L, Calvo A. Cancer Cell-Intrinsic Alterations Associated with an Immunosuppressive Tumor Microenvironment and Resistance to Immunotherapy in Lung Cancer. Cancers. 2023; 15(12):3076. https://doi.org/10.3390/cancers15123076
Chicago/Turabian StyleOtegui, Nerea, Maeva Houry, Imanol Arozarena, Diego Serrano, Esther Redin, Francisco Exposito, Sergio Leon, Karmele Valencia, Luis Montuenga, and Alfonso Calvo. 2023. "Cancer Cell-Intrinsic Alterations Associated with an Immunosuppressive Tumor Microenvironment and Resistance to Immunotherapy in Lung Cancer" Cancers 15, no. 12: 3076. https://doi.org/10.3390/cancers15123076
APA StyleOtegui, N., Houry, M., Arozarena, I., Serrano, D., Redin, E., Exposito, F., Leon, S., Valencia, K., Montuenga, L., & Calvo, A. (2023). Cancer Cell-Intrinsic Alterations Associated with an Immunosuppressive Tumor Microenvironment and Resistance to Immunotherapy in Lung Cancer. Cancers, 15(12), 3076. https://doi.org/10.3390/cancers15123076