
Distinct Signatures of Tumor-Associated Microbiota and Metabolome in Low-Grade vs. High-Grade Dysplastic Colon Polyps: Inference of Their Role in Tumor Initiation and Progression
 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients Enrollment
2.2. Sample Collection
2.3. Histology
2.4. MAM and LAM Analyses
2.5. Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt)
2.6. Mucosa-Associated Metabolome
2.7. Raw Sequence Processing
2.8. Statistical Analysis
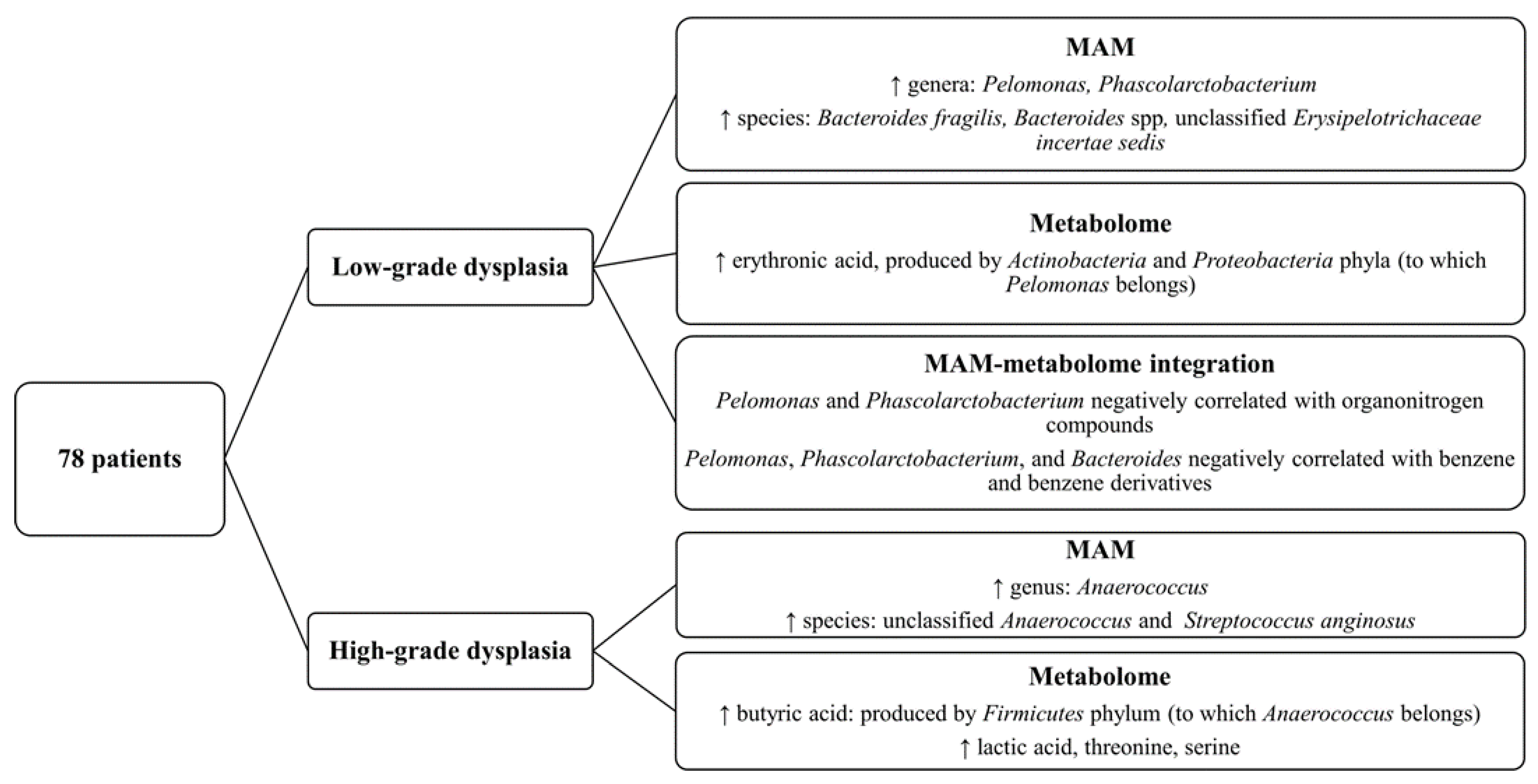
3. Results
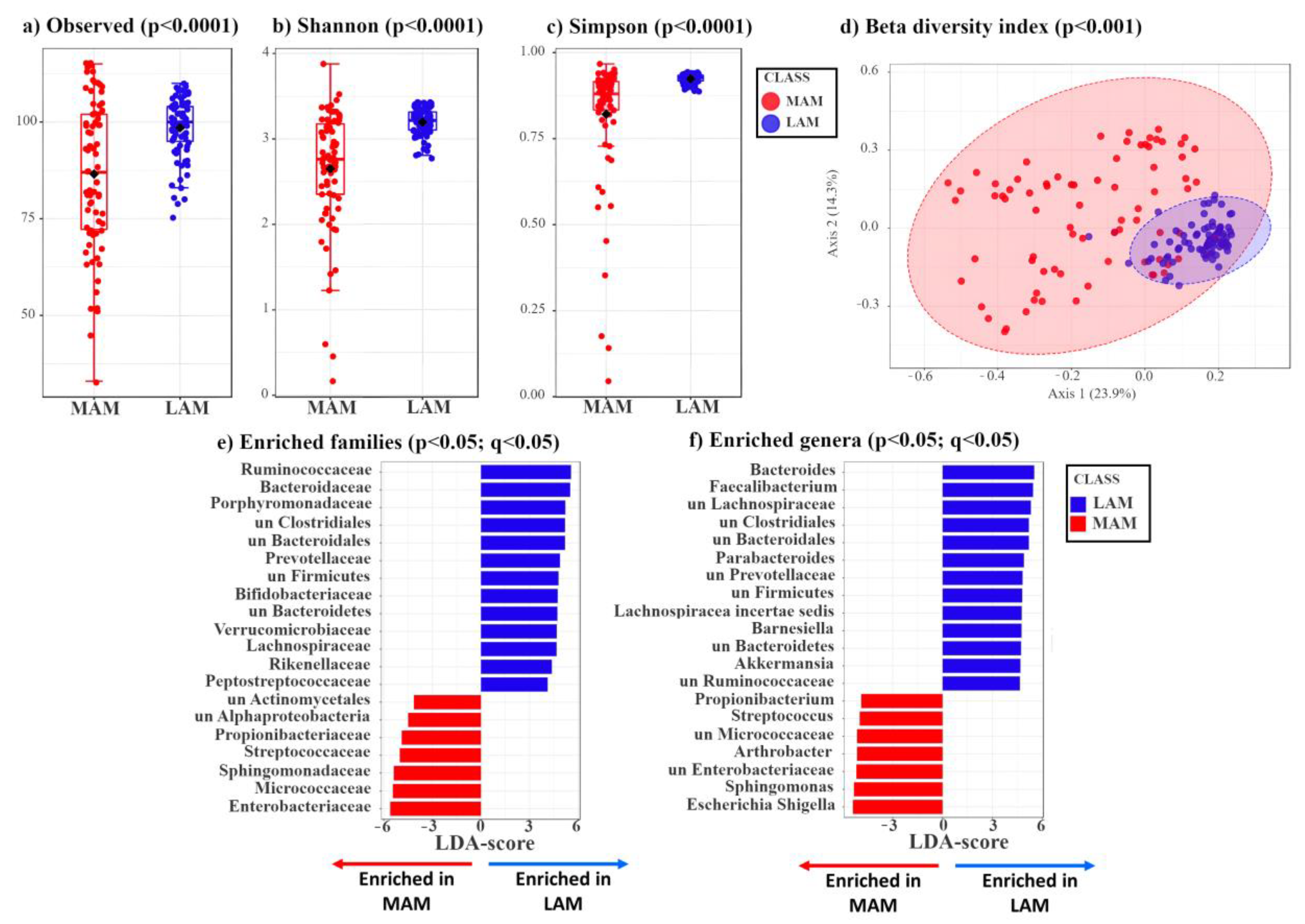
3.1. Characterization of LAM and MAM in Patients with Colon Polyps
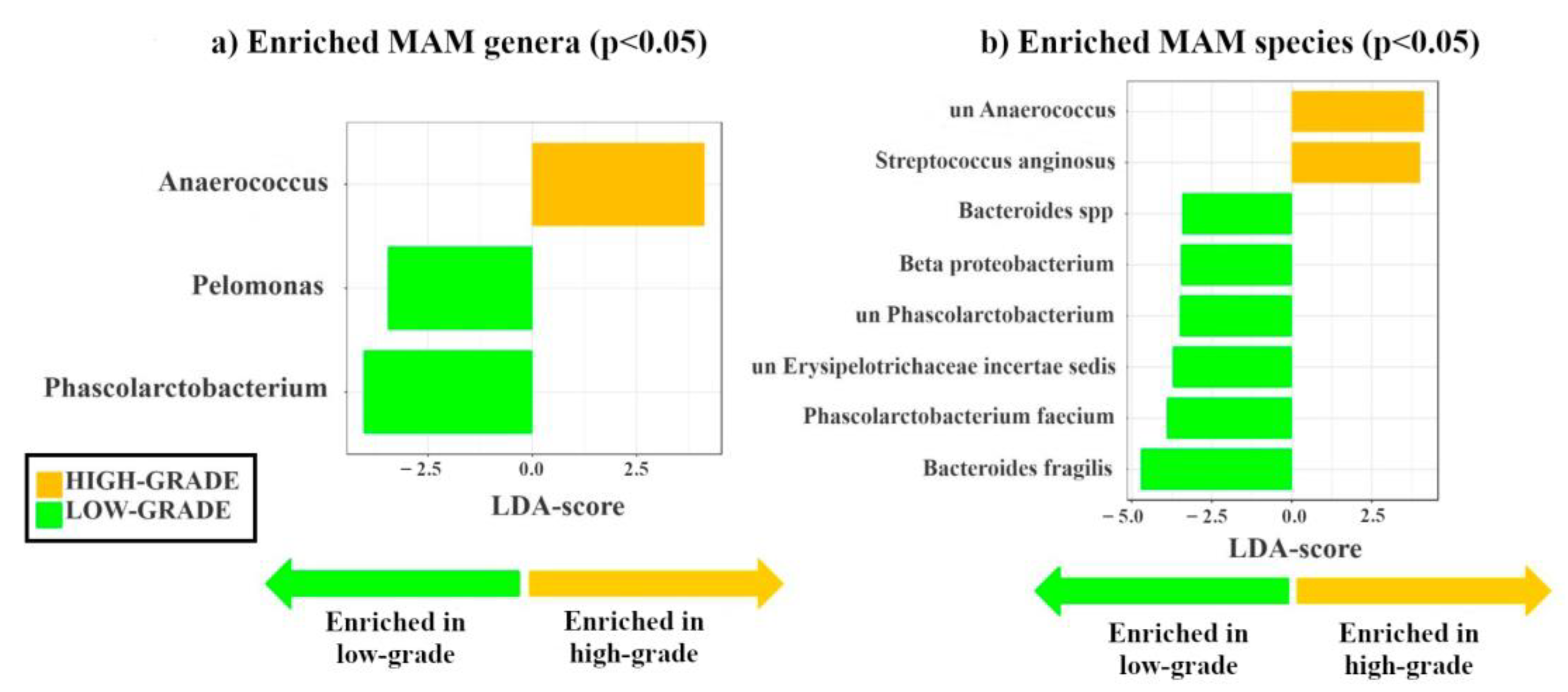
3.2. Identification of Mucosa-Associated Bacterial Signatures Distinguishing Low-Grade from High-Grade Dysplastic Colorectal Polyps
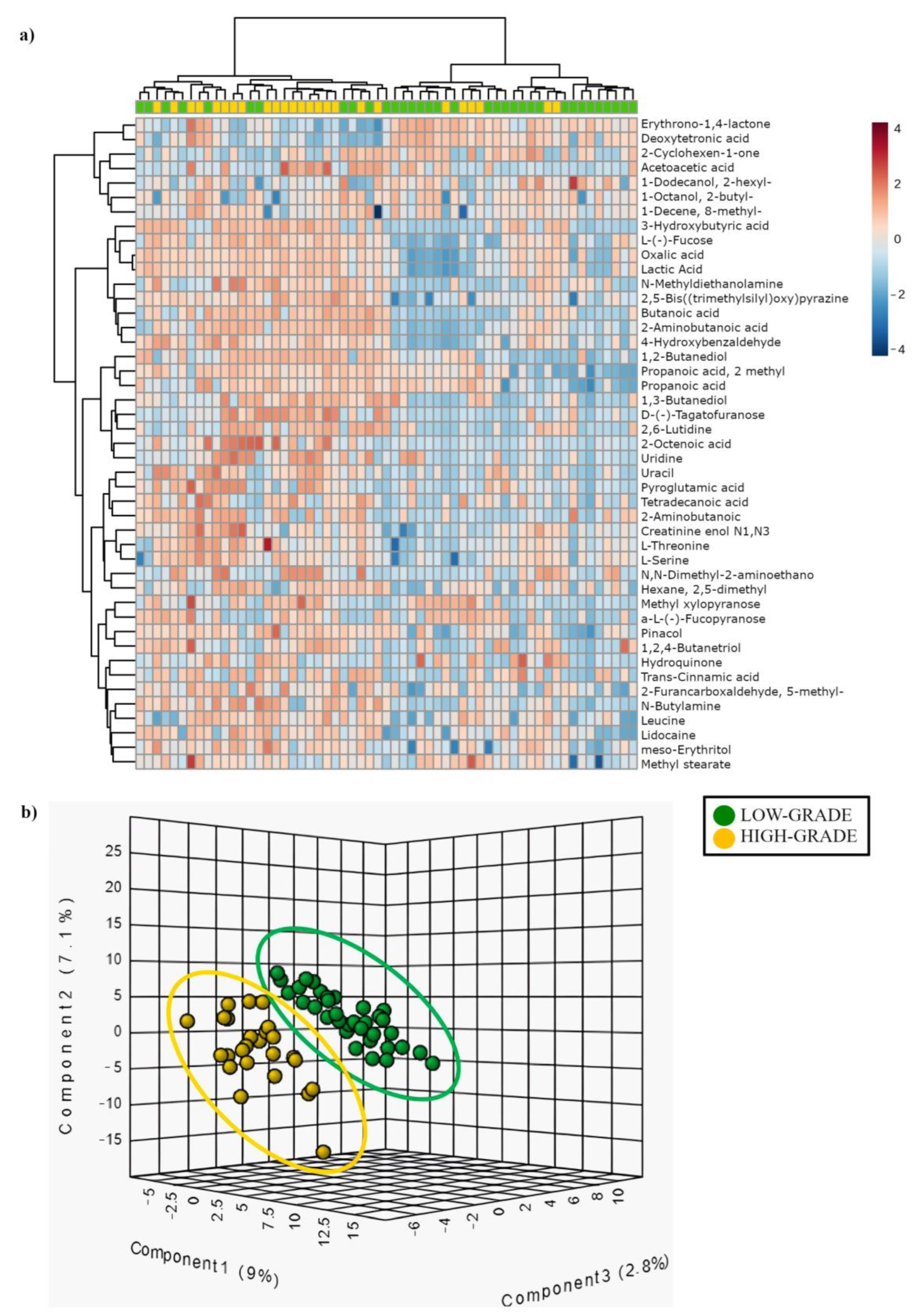
3.3. Comparison of Mucosa-Associated Metabolome between High-Grade and Low-Grade Dysplastic Colorectal Polyps
3.4. Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) in Low-Grade vs. High-Grade
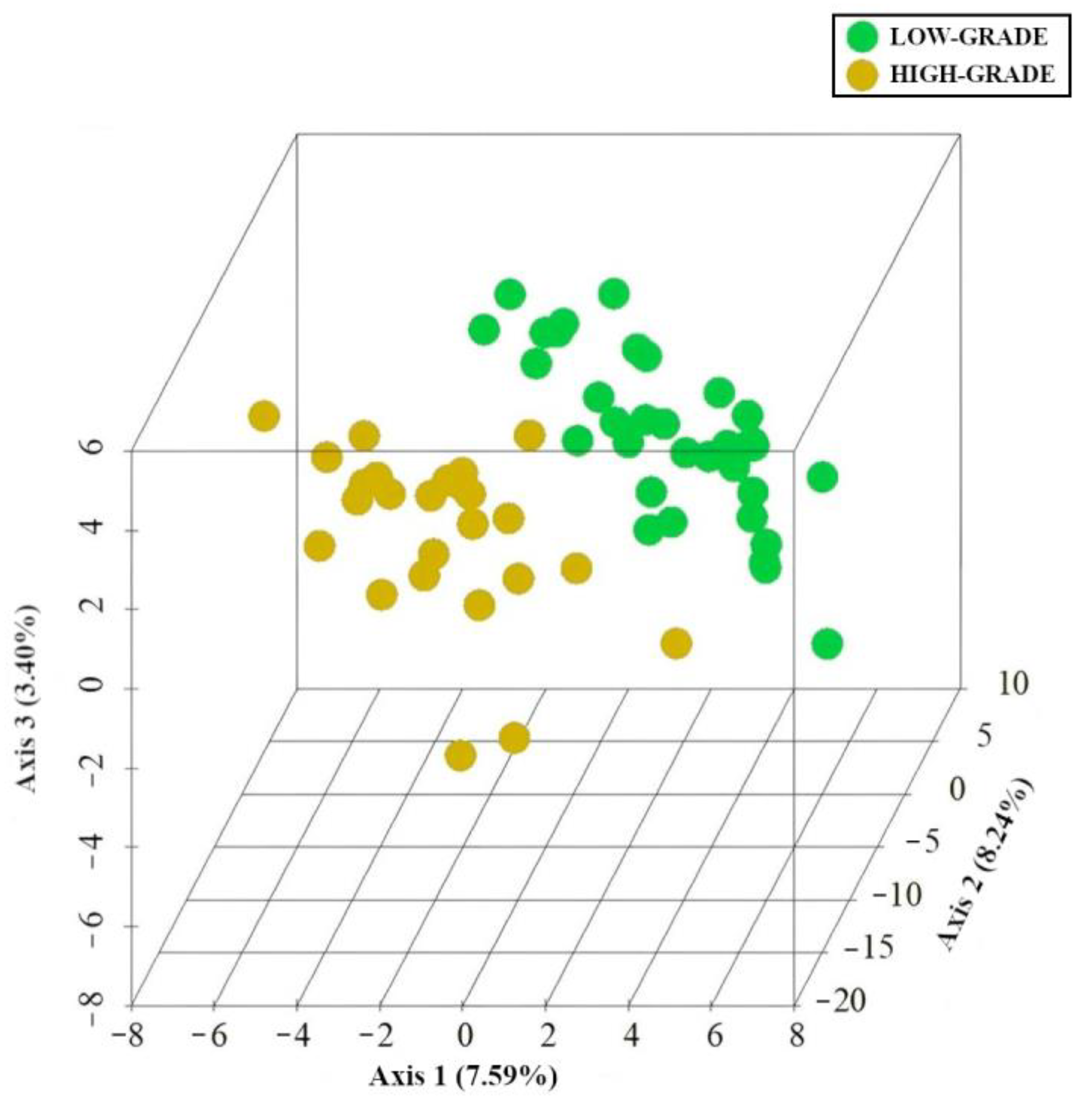
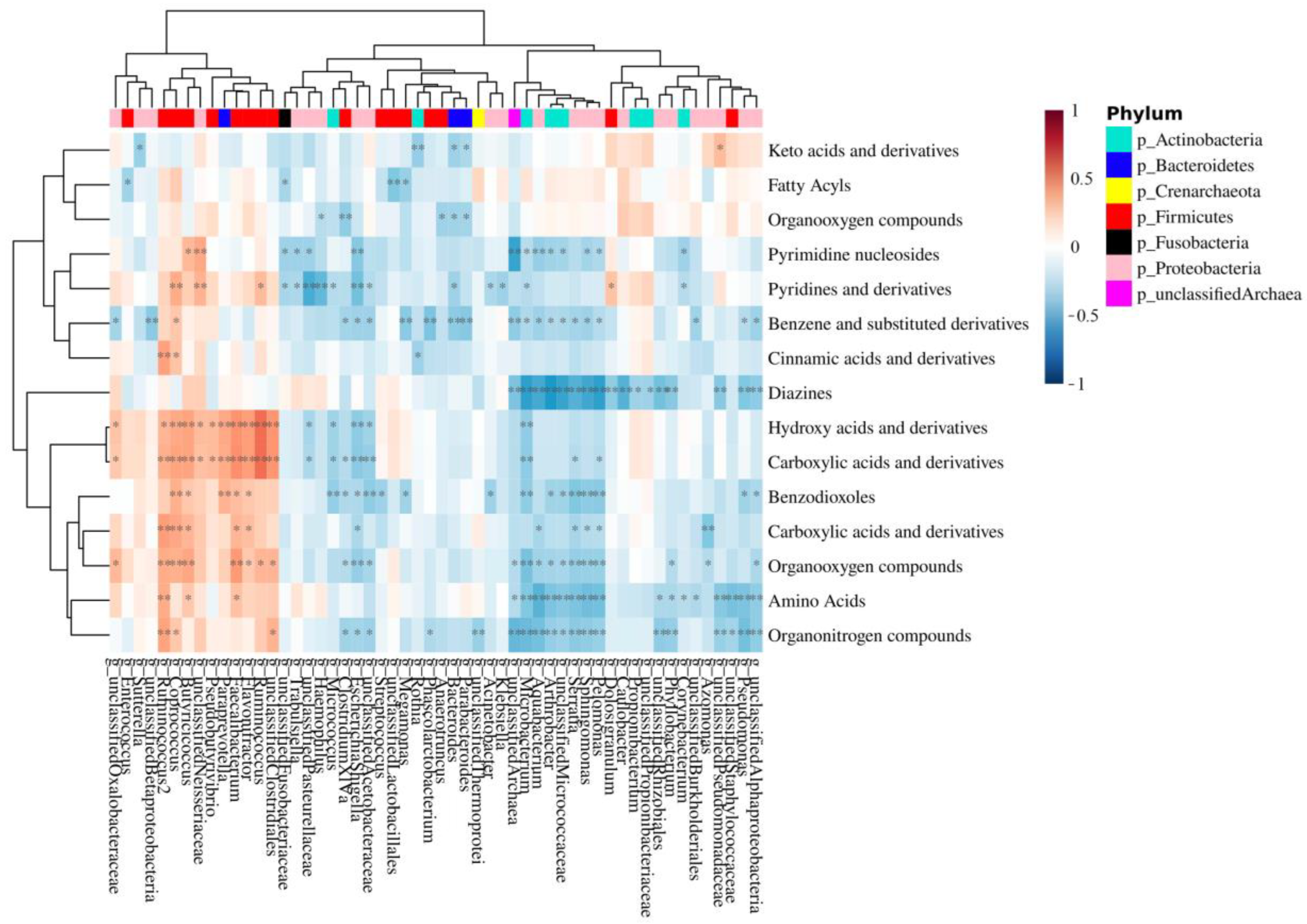
3.5. Integration of MAM and Polyp-Adherent Metabolome Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Chan, A.T.; Sun, J. Influence of the Gut Microbiome, Diet, and Environment on Risk of Colorectal Cancer. Gastroenterology 2020, 158, 322–340. [Google Scholar] [CrossRef] [PubMed]
- Tjalsma, H.; Boleij, A.; Marchesi, J.R.; Dutilh, B.E. A Bacterial Driver-Passenger Model for Colorectal Cancer: Beyond the Usual Suspects. Nat. Rev. Microbiol. 2012, 10, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, C.; Hou, S.; Wu, X.; Liu, J.; Wan, X. Analyses of Potential Driver and Passenger Bacteria in Human Colorectal Cancer. Cancer Manag. Res. 2020, 12, 11553–11561. [Google Scholar] [CrossRef]
- Mori, G.; Pasca, M.R. Gut Microbial Signatures in Sporadic and Hereditary Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 1312. [Google Scholar] [CrossRef] [PubMed]
- Kosumi, K.; Mima, K.; Baba, H.; Ogino, S. Dysbiosis of the Gut Microbiota and Colorectal Cancer: The Key Target of Molecular Pathological Epidemiology. J. Lab. Precis. Med. 2018, 3, 76. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium Nucleatum Potentiates Intestinal Tumorigenesis and Modulates the Tumor-Immune Microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef]
- Wu, S.; Powell, J.; Mathioudakis, N.; Kane, S.; Fernandez, E.; Sears, C.L. Bacteroides Fragilis Enterotoxin Induces Intestinal Epithelial Cell Secretion of Interleukin-8 through Mitogen-Activated Protein Kinases and a Tyrosine Kinase-Regulated Nuclear Factor-KappaB Pathway. Infect. Immun. 2004, 72, 5832–5839. [Google Scholar] [CrossRef]
- Chung, L.; Thiele Orberg, E.; Geis, A.L.; Chan, J.L.; Fu, K.; DeStefano Shields, C.E.; Dejea, C.M.; Fathi, P.; Chen, J.; Finard, B.B.; et al. Bacteroides Fragilis Toxin Coordinates a Pro-Carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host Microbe 2018, 23, 203–214.e5. [Google Scholar] [CrossRef]
- Nougayrède, J.-P.; Homburg, S.; Taieb, F.; Boury, M.; Brzuszkiewicz, E.; Gottschalk, G.; Buchrieser, C.; Hacker, J.; Dobrindt, U.; Oswald, E. Escherichia Coli Induces DNA Double-Strand Breaks in Eukaryotic Cells. Science 2006, 313, 848–851. [Google Scholar] [CrossRef]
- Avril, M.; DePaolo, R.W. “Driver-Passenger” Bacteria and Their Metabolites in the Pathogenesis of Colorectal Cancer. Gut Microbes 2021, 13, 1941710. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Hold, G.L.; Flint, H.J. The Gut Microbiota, Bacterial Metabolites and Colorectal Cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Hassan, C.; Gimeno-García, A.; Kalager, M.; Spada, C.; Zullo, A.; Costamagna, G.; Senore, C.; Rex, D.K.; Quintero, E. Systematic Review with Meta-Analysis: The Incidence of Advanced Neoplasia after Polypectomy in Patients with and without Low-Risk Adenomas. Aliment. Pharmacol. Ther. 2014, 39, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Nie, Y.; Yu, J.; Wong, C.C. Microbial Metabolites in Colorectal Cancer: Basic and Clinical Implications. Metabolites 2021, 11, 159. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Luo, S.; Xu, M.; Zhang, Y.; Song, S.; Wang, S.; Kong, X.; He, N.; Cao, X.; Yan, F.; et al. The Secondary Bile Acid, Deoxycholate Accelerates Intestinal Adenoma-Adenocarcinoma Sequence in ApcMin/+ Mice through Enhancing Wnt Signaling. Fam. Cancer 2014, 13, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Payne, C.M.; Weber, C.; Crowley-Skillicorn, C.; Dvorak, K.; Bernstein, H.; Bernstein, C.; Holubec, H.; Dvorakova, B.; Garewal, H. Deoxycholate Induces Mitochondrial Oxidative Stress and Activates NF-KappaB through Multiple Mechanisms in HCT-116 Colon Epithelial Cells. Carcinogenesis 2007, 28, 215–222. [Google Scholar] [CrossRef]
- Garza, D.R.; Taddese, R.; Wirbel, J.; Zeller, G.; Boleij, A.; Huynen, M.A.; Dutilh, B.E. Metabolic Models Predict Bacterial Passengers in Colorectal Cancer. Cancer Metab. 2020, 8, 3. [Google Scholar] [CrossRef]
- Yachida, S.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Watanabe, H.; Masuda, K.; Nishimoto, Y.; Kubo, M.; et al. Metagenomic and Metabolomic Analyses Reveal Distinct Stage-Specific Phenotypes of the Gut Microbiota in Colorectal Cancer. Nat. Med. 2019, 25, 968–976. [Google Scholar] [CrossRef]
- Bisht, V.; Nash, K.; Xu, Y.; Agarwal, P.; Bosch, S.; Gkoutos, G.V.; Acharjee, A. Integration of the Microbiome, Metabolome and Transcriptomics Data Identified Novel Metabolic Pathway Regulation in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 5763. [Google Scholar] [CrossRef]
- Chen, F.; Dai, X.; Zhou, C.-C.; Li, K.-X.; Zhang, Y.-J.; Lou, X.-Y.; Zhu, Y.-M.; Sun, Y.-L.; Peng, B.-X.; Cui, W. Integrated Analysis of the Faecal Metagenome and Serum Metabolome Reveals the Role of Gut Microbiome-Associated Metabolites in the Detection of Colorectal Cancer and Adenoma. Gut 2022, 71, 1315–1325. [Google Scholar] [CrossRef]
- Sun, J.; Kato, I. Gut Microbiota, Inflammation and Colorectal Cancer. Genes. Dis. 2016, 3, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Ringel, Y.; Maharshak, N.; Ringel-Kulka, T.; Wolber, E.A.; Sartor, R.B.; Carroll, I.M. High Throughput Sequencing Reveals Distinct Microbial Populations within the Mucosal and Luminal Niches in Healthy Individuals. Gut Microbes 2015, 6, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the Human Intestinal Microbial Flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhu, X.; Huang, X.; Murff, H.J.; Ness, R.M.; Seidner, D.L.; Sorgen, A.A.; Blakley, I.C.; Yu, C.; Dai, Q.; et al. On the Robustness of Inference of Association with the Gut Microbiota in Stool, Rectal Swab and Mucosal Tissue Samples. Sci. Rep. 2021, 11, 14828. [Google Scholar] [CrossRef]
- Avelar-Barragan, J.; DeDecker, L.; Lu, Z.N.; Coppedge, B.; Karnes, W.E.; Whiteson, K.L. Distinct Colon Mucosa Microbiomes Associated with Tubular Adenomas and Serrated Polyps. NPJ Biofilms Microbiomes 2022, 8, 69. [Google Scholar] [CrossRef]
- O’Brien, C.L.; Allison, G.E.; Grimpen, F.; Pavli, P. Impact of Colonoscopy Bowel Preparation on Intestinal Microbiota. PLoS ONE 2013, 8, e62815. [Google Scholar] [CrossRef]
- Nagata, N.; Tohya, M.; Fukuda, S.; Suda, W.; Nishijima, S.; Takeuchi, F.; Ohsugi, M.; Tsujimoto, T.; Nakamura, T.; Shimomura, A.; et al. Effects of Bowel Preparation on the Human Gut Microbiome and Metabolome. Sci. Rep. 2019, 9, 4042. [Google Scholar] [CrossRef]
- Riboli, E.; Hunt, K.J.; Slimani, N.; Ferrari, P.; Norat, T.; Fahey, M.; Charrondière, U.R.; Hémon, B.; Casagrande, C.; Vignat, J.; et al. European Prospective Investigation into Cancer and Nutrition (EPIC): Study Populations and Data Collection. Public Health Nutr. 2002, 5, 1113–1124. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for Prediction of Metagenome Functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical Analysis of Taxonomic and Functional Profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Barberis, E.; Joseph, S.; Amede, E.; Clavenna, M.G.; La Vecchia, M.; Sculco, M.; Aspesi, A.; Occhipinti, P.; Robotti, E.; Boldorini, R.; et al. A New Method for Investigating Microbiota-Produced Small Molecules in Adenomatous Polyps. Anal. Chim. Acta 2021, 1179, 338841. [Google Scholar] [CrossRef] [PubMed]
- Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-Calling of Automated Sequencer Traces Using Phred. I. Accuracy Assessment. Genome Res. 1998, 8, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and Tools for High Throughput RRNA Analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef]
- Bona, E.; Massa, N.; Toumatia, O.; Novello, G.; Cesaro, P.; Todeschini, V.; Boatti, L.; Mignone, F.; Titouah, H.; Zitouni, A.; et al. Climatic Zone and Soil Properties Determine the Biodiversity of the Soil Bacterial Communities Associated to Native Plants from Desert Areas of North-Central Algeria. Microorganisms 2021, 9, 1359. [Google Scholar] [CrossRef] [PubMed]
- Torre, E.; Sola, D.; Caramaschi, A.; Mignone, F.; Bona, E.; Fallarini, S. A Pilot Study on Clinical Scores, Immune Cell Modulation, and Microbiota Composition in Allergic Patients with Rhinitis and Asthma Treated with a Probiotic Preparation. Int. Arch. Allergy Immunol. 2022, 183, 186–200. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A Web-Based Tool for Comprehensive Statistical, Visual and Meta-Analysis of Microbiome Data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef]
- Foster, Z.S.L.; Sharpton, T.J.; Grünwald, N.J. Metacoder: An R Package for Visualization and Manipulation of Community Taxonomic Diversity Data. PLoS Comput. Biol. 2017, 13, e1005404. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Agus, A.; Clément, K.; Sokol, H. Gut Microbiota-Derived Metabolites as Central Regulators in Metabolic Disorders. Gut 2021, 70, 1174–1182. [Google Scholar] [CrossRef]
- Lamichhane, S.; Sen, P.; Dickens, A.M.; Orešič, M.; Bertram, H.C. Gut Metabolome Meets Microbiome: A Methodological Perspective to Understand the Relationship between Host and Microbe. Methods 2018, 149, 3–12. [Google Scholar] [CrossRef]
- Yuan, C.; Graham, M.; Staley, C.; Subramanian, S. Mucosal Microbiota and Metabolome along the Intestinal Tract Reveal a Location-Specific Relationship. mSystems 2020, 5, e00055-20. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Kong, C.; Huang, L.; Li, H.; Qu, X.; Liu, Z.; Lan, P.; Wang, J.; Qin, H. Mucosa-Associated Microbiota Signature in Colorectal Cancer. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 2073–2083. [Google Scholar] [CrossRef]
- Wang, Q.; Ye, J.; Fang, D.; Lv, L.; Wu, W.; Shi, D.; Li, Y.; Yang, L.; Bian, X.; Wu, J.; et al. Multi-Omic Profiling Reveals Associations between the Gut Mucosal Microbiome, the Metabolome, and Host DNA Methylation Associated Gene Expression in Patients with Colorectal Cancer. BMC Microbiol. 2020, 20, 83. [Google Scholar] [CrossRef]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut Biogeography of the Bacterial Microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.S.; Poles, J.; Leung, J.M.; Wolff, M.J.; Davenport, M.; Lee, S.C.; Lim, Y.A.; Chua, K.H.; Loke, P.; Cho, I. Inferred Metagenomic Comparison of Mucosal and Fecal Microbiota from Individuals Undergoing Routine Screening Colonoscopy Reveals Similar Differences Observed during Active Inflammation. Gut Microbes 2015, 6, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.; Woltemate, S.; Piazuelo, M.B.; Bravo, L.E.; Yepez, M.C.; Romero-Gallo, J.; Delgado, A.G.; Wilson, K.T.; Peek, R.M.; Correa, P.; et al. Different Gastric Microbiota Compositions in Two Human Populations with High and Low Gastric Cancer Risk in Colombia. Sci. Rep. 2016, 6, 18594. [Google Scholar] [CrossRef]
- Mori, G.; Rampelli, S.; Orena, B.S.; Rengucci, C.; De Maio, G.; Barbieri, G.; Passardi, A.; Casadei Gardini, A.; Frassineti, G.L.; Gaiarsa, S.; et al. Shifts of Faecal Microbiota During Sporadic Colorectal Carcinogenesis. Sci. Rep. 2018, 8, 10329. [Google Scholar] [CrossRef] [PubMed]
- Weir, T.L.; Manter, D.K.; Sheflin, A.M.; Barnett, B.A.; Heuberger, A.L.; Ryan, E.P. Stool Microbiome and Metabolome Differences between Colorectal Cancer Patients and Healthy Adults. PLoS ONE 2013, 8, e70803. [Google Scholar] [CrossRef]
- Zagato, E.; Pozzi, C.; Bertocchi, A.; Schioppa, T.; Saccheri, F.; Guglietta, S.; Fosso, B.; Melocchi, L.; Nizzoli, G.; Troisi, J.; et al. Endogenous Murine Microbiota Member Faecalibaculum Rodentium and Its Human Homologue Protect from Intestinal Tumour Growth. Nat. Microbiol. 2020, 5, 511–524. [Google Scholar] [CrossRef]
- Clos-Garcia, M.; Garcia, K.; Alonso, C.; Iruarrizaga-Lejarreta, M.; D’Amato, M.; Crespo, A.; Iglesias, A.; Cubiella, J.; Bujanda, L.; Falcón-Pérez, J.M. Integrative Analysis of Fecal Metagenomics and Metabolomics in Colorectal Cancer. Cancers 2020, 12, 1142. [Google Scholar] [CrossRef]
- Dinh, D.M.; Volpe, G.E.; Duffalo, C.; Bhalchandra, S.; Tai, A.K.; Kane, A.V.; Wanke, C.A.; Ward, H.D. Intestinal Microbiota, Microbial Translocation, and Systemic Inflammation in Chronic HIV Infection. J. Infect. Dis. 2015, 211, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, L.; Schmid, J.; Ebert, M.; Soucek, P.; Kunicka, T.; Liska, V.; Bruha, J.; Neary, P.; Dezeeuw, N.; Tommasino, M.; et al. Fusobacterium Nucleatum Associates with Stages of Colorectal Neoplasia Development, Colorectal Cancer and Disease Outcome. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 1381–1390. [Google Scholar] [CrossRef]
- Cheng, Y.; Xie, G.; Chen, T.; Qiu, Y.; Zou, X.; Zheng, M.; Tan, B.; Feng, B.; Dong, T.; He, P.; et al. Distinct Urinary Metabolic Profile of Human Colorectal Cancer. J. Proteome Res. 2012, 11, 1354–1363. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Xie, G.; Jia, W. Metabonomics of Human Colorectal Cancer: New Approaches for Early Diagnosis and Biomarker Discovery. J. Proteome Res. 2014, 13, 3857–3870. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.G.; Rao, S.; Weir, T.L.; O’Malia, J.; Bazan, M.; Brown, R.J.; Ryan, E.P. Metabolomics and Metabolic Pathway Networks from Human Colorectal Cancers, Adjacent Mucosa, and Stool. Cancer Metab. 2016, 4, 11. [Google Scholar] [CrossRef]
- Sinha, R.; Ahn, J.; Sampson, J.N.; Shi, J.; Yu, G.; Xiong, X.; Hayes, R.B.; Goedert, J.J. Fecal Microbiota, Fecal Metabolome, and Colorectal Cancer Interrelations. PLoS ONE 2016, 11, e0152126. [Google Scholar] [CrossRef] [PubMed]
- Goedert, J.J.; Sampson, J.N.; Moore, S.C.; Xiao, Q.; Xiong, X.; Hayes, R.B.; Ahn, J.; Shi, J.; Sinha, R. Fecal Metabolomics: Assay Performance and Association with Colorectal Cancer. Carcinogenesis 2014, 35, 2089–2096. [Google Scholar] [CrossRef]
- Wang, X.; Wang, J.; Rao, B.; Deng, L. Gut Flora Profiling and Fecal Metabolite Composition of Colorectal Cancer Patients and Healthy Individuals. Exp. Ther. Med. 2022, 23, 250. [Google Scholar] [CrossRef]
- Zhao, X.; Fu, J.; Hu, B.; Chen, L.; Wang, J.; Fang, J.; Ge, C.; Lin, H.; Pan, K.; Fu, L.; et al. Serine Metabolism Regulates YAP Activity Through USP7 in Colon Cancer. Front. Cell. Dev. Biol. 2021, 9, 639111. [Google Scholar] [CrossRef]
- Qian, J.; Gong, Z.-C.; Zhang, Y.-N.; Wu, H.-H.; Zhao, J.; Wang, L.-T.; Ye, L.-J.; Liu, D.; Wang, W.; Kang, X.; et al. Lactic Acid Promotes Metastatic Niche Formation in Bone Metastasis of Colorectal Cancer. Cell. Commun. Signal. 2021, 19, 9. [Google Scholar] [CrossRef]
- Wei, Y.; Xu, H.; Dai, J.; Peng, J.; Wang, W.; Xia, L.; Zhou, F. Prognostic Significance of Serum Lactic Acid, Lactate Dehydrogenase, and Albumin Levels in Patients with Metastatic Colorectal Cancer. BioMed Res. Int. 2018, 2018, 1804086. [Google Scholar] [CrossRef] [PubMed]
- Cruz, M.D.; Ledbetter, S.; Chowdhury, S.; Tiwari, A.K.; Momi, N.; Wali, R.K.; Bliss, C.; Huang, C.; Lichtenstein, D.; Bhattacharya, S.; et al. Metabolic Reprogramming of the Premalignant Colonic Mucosa Is an Early Event in Carcinogenesis. Oncotarget 2017, 8, 20543–20557. [Google Scholar] [CrossRef] [PubMed]
- Van den Abbeele, P.; Belzer, C.; Goossens, M.; Kleerebezem, M.; De Vos, W.M.; Thas, O.; De Weirdt, R.; Kerckhof, F.-M.; Van de Wiele, T. Butyrate-Producing Clostridium Cluster XIVa Species Specifically Colonize Mucins in an in Vitro Gut Model. ISME J. 2013, 7, 949–961. [Google Scholar] [CrossRef]
- Dalal, N.; Jalandra, R.; Sharma, M.; Prakash, H.; Makharia, G.K.; Solanki, P.R.; Singh, R.; Kumar, A. Omics Technologies for Improved Diagnosis and Treatment of Colorectal Cancer: Technical Advancement and Major Perspectives. BioMed Pharmacother. 2020, 131, 110648. [Google Scholar] [CrossRef]
- Nitter, M.; Norgård, B.; de Vogel, S.; Eussen, S.J.P.M.; Meyer, K.; Ulvik, A.; Ueland, P.M.; Nygård, O.; Vollset, S.E.; Bjørge, T.; et al. Plasma Methionine, Choline, Betaine, and Dimethylglycine in Relation to Colorectal Cancer Risk in the European Prospective Investigation into Cancer and Nutrition (EPIC). Ann. Oncol. 2014, 25, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Simó, C.; García-Cañas, V. Dietary Bioactive Ingredients to Modulate the Gut Microbiota-Derived Metabolite TMAO. New Opportunities for Functional Food Development. Food Funct. 2020, 11, 6745–6776. [Google Scholar] [CrossRef]
- Oellgaard, J.; Winther, S.A.; Hansen, T.S.; Rossing, P.; von Scholten, B.J. Trimethylamine N-Oxide (TMAO) as a New Potential Therapeutic Target for Insulin Resistance and Cancer. Curr. Pharm. Des. 2017, 23, 3699–3712. [Google Scholar] [CrossRef]
- Li, T.; Chen, Y.; Gua, C.; Li, X. Elevated Circulating Trimethylamine N-Oxide Levels Contribute to Endothelial Dysfunction in Aged Rats through Vascular Inflammation and Oxidative Stress. Front. Physiol. 2017, 8, 350. [Google Scholar] [CrossRef]
- Anantharaju, P.G.; Reddy, B.D.; Padukudru, M.A.; Kumari Chitturi, C.M.; Vimalambike, M.G.; Madhunapantula, S.V. Naturally Occurring Benzoic Acid Derivatives Retard Cancer Cell Growth by Inhibiting Histone Deacetylases (HDAC). Cancer Biol. Ther. 2017, 18, 492–504. [Google Scholar] [CrossRef]
- Oren, A.; Garrity, G.M. Valid publication of the names of forty-two phyla of prokaryotes. Int. J. Syst. Evol. Microbiol. 2021, 71, 005056. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Features | Patients n = 78 (%) | Patients with Low-Grade Dysplastic Polyps n = 44 (%) | Patients with High-Grade Dysplastic Polyps n = 34 (%) | p-Value |
|---|---|---|---|---|
| Gender | ||||
| Female | 33 (42.3%) | 21 (47.7%) | 12 (35.3%) | 0.3 |
| Male | 45 (57.7%) | 23 (52.3%) | 22 (64.7%) | |
| BMI (body mass index) | ||||
| Normal weight | 34 (43.6%) | 20 (45.4%) | 14 (41.2%) | 0.7 |
| Overweight or obese | 44 (56.4%) | 24 (54.6%) | 20 (58.8%) | |
| Age | ||||
| Median (IQR) | 61 (58–70) | 61 (58–68) | 62 (56–70) | 0.9 |
| Polyp size mm | ||||
| Median (IQR) | 14 (10–23) | 12 (10–16) | 15 (12–25) | 0.02 |
| Type of polyp | ||||
| Tubular | 28 (35.9%) | 21 (47.7%) | 7 (20.6%) | 0.005 |
| Villous | 3 (3.8%) | 1 (2.3%) | 2 (5.9%) | |
| Tubulo-villous | 40 (51.3%) | 16 (36.4%) | 24 (70.6%) | |
| Others | 7 (9.0%) | 6 (13.6%) | 1 (2.9%) | |
| Previous gastrointestinal conditions | ||||
| Diverticulitis | 26 (33.3%) | 19 (43.2%) | 7 (20.6%) | 0.2 |
| Previous polyp occurrence | 8 (10.2%) | 6 (13.6%) | 2 (5.9%) | |
| IBD | 1 (1.3%) | 0 | 1 (2.9%) | |
| Previous cholecystectomy | 5 (6.4%) | 4 (9.1%) | 1 (2.9%) | |
| Slight mucosal inflammation | 1 (1.3%) | 0 | 1 (2.9%) | |
| Polyp localization | ||||
| Right colon | 18 (23.1%) | 11 (25.0%) | 7 (20.6%) | 0.8 |
| Left colon | 52 (66.7%) | 28 (63.6%) | 24 (70.6%) | |
| Transversal colon | 8 (10.2%) | 5 (11.4%) | 3 (8.8%) |
| Pathway | Description | p-Value |
|---|---|---|
| Superpathway of adenosylcobalamin salvage from cobinamide I (COBALSYN-PWY) | Vitamin biosynthesis | 8.54 × 10−3 |
| Adenosylcobalamin biosynthesis from adenosylcobinamide-GDP I (PWY-5509) | Vitamin biosynthesis | 0.016 |
| Superpathway of adenosylcobalamin salvage from cobinamide II (PWY-6269) | Vitamin biosynthesis | 2.78 × 10−3 |
| Sucrose degradation IV (sucrose phosphorylase) (PWY-5384) | Carbohydrate degradation | 0.035 |
| Mixed acid fermentation (FERMENTATION-PWY) * | Carbohydrate degradation | 5.94 × 10−3 |
| Superpathway of tetrahydrofolate biosynthesis and salvage (FOLSYN-PWY) | Vitamin biosynthesis | 0.036 |
| Superpathway of tetrahydrofolate biosynthesis (PWY-6612) | Vitamin biosynthesis | 0.034 |
| Superpathway of thiamine diphosphate biosynthesis II (PWY-6895) | Vitamin biosynthesis | 0.034 |
| Superpathway of purine nucleotides de novo biosynthesis II (DENOVOPURINE2-PWY) | Nucleotides synthesis | 6.73 × 10−3 |
| Superpathway of guanosine nucleotides de novo biosynthesis II (PWY-6125) | Nucleotides synthesis | 0.028 |
| Superpathway of pyrimidine ribonucleosides salvage (PWY-7196) * | Nucleotides synthesis | 7.02 × 10−3 |
| Superpathway of guanosine nucleotides de novo biosynthesis I (PWY-7228) | Nucleotides synthesis | 0.036 |
| Superpathway of purine nucleotides de novo biosynthesis I (PWY-841) | Nucleotides synthesis | 0.038 |
| Superpathway of pyrimidine ribonucleotides de novo biosynthesis (PWY0-162) | Nucleotides synthesis | 0.031 |
| Incomplete reductive TCA cycle (P42-PWY) | Reductive TCA cycle | 0.017 |
| PreQ0 biosynthesis (PWY-6703) | Secondary metabolite biosynthesis | 0.049 |
| Pyrimidine deoxyribonucleotides de novo biosynthesis II (PWY-7187) | Nucleoside and nucleotide synthesis | 0.019 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clavenna, M.G.; La Vecchia, M.; Sculco, M.; Joseph, S.; Barberis, E.; Amede, E.; Mellai, M.; Brossa, S.; Borgonovi, G.; Occhipinti, P.; et al. Distinct Signatures of Tumor-Associated Microbiota and Metabolome in Low-Grade vs. High-Grade Dysplastic Colon Polyps: Inference of Their Role in Tumor Initiation and Progression. Cancers 2023, 15, 3065. https://doi.org/10.3390/cancers15123065
Clavenna MG, La Vecchia M, Sculco M, Joseph S, Barberis E, Amede E, Mellai M, Brossa S, Borgonovi G, Occhipinti P, et al. Distinct Signatures of Tumor-Associated Microbiota and Metabolome in Low-Grade vs. High-Grade Dysplastic Colon Polyps: Inference of Their Role in Tumor Initiation and Progression. Cancers. 2023; 15(12):3065. https://doi.org/10.3390/cancers15123065
Chicago/Turabian StyleClavenna, Michela Giulia, Marta La Vecchia, Marika Sculco, Soni Joseph, Elettra Barberis, Elia Amede, Marta Mellai, Silvia Brossa, Giulia Borgonovi, Pietro Occhipinti, and et al. 2023. "Distinct Signatures of Tumor-Associated Microbiota and Metabolome in Low-Grade vs. High-Grade Dysplastic Colon Polyps: Inference of Their Role in Tumor Initiation and Progression" Cancers 15, no. 12: 3065. https://doi.org/10.3390/cancers15123065
APA StyleClavenna, M. G., La Vecchia, M., Sculco, M., Joseph, S., Barberis, E., Amede, E., Mellai, M., Brossa, S., Borgonovi, G., Occhipinti, P., Boldorini, R., Robotti, E., Azzimonti, B., Bona, E., Pasolli, E., Ferrante, D., Manfredi, M., Aspesi, A., & Dianzani, I. (2023). Distinct Signatures of Tumor-Associated Microbiota and Metabolome in Low-Grade vs. High-Grade Dysplastic Colon Polyps: Inference of Their Role in Tumor Initiation and Progression. Cancers, 15(12), 3065. https://doi.org/10.3390/cancers15123065