
Genomic and Transcriptomic Analyses of Malignant Pleural Mesothelioma (MPM) Samples Reveal Crucial Insights for Preclinical Testing

 ,
,  , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Characteristics
2.2. Human Commercially Available Mesothelioma Cell Lines
2.3. Generation of Patient-Derived Cell Lines
2.4. Immunocytochemistry
2.5. Immunohistochemistry (IHC)
2.6. Western Blot
2.7. Whole Exome Sequencing
2.8. Murine RN5 Model
2.9. Tumor Cell Isolation from Human and Mouse Tumors for RNA Sequencing
2.10. RNA Sequencing
2.11. RNA Sequencing Analysis and Statistics
2.12. Deconvolution of Bulk RNA Sequencing Data Using Granulator
2.13. Gene Set Enrichment Analysis (GSEA)
3. Results
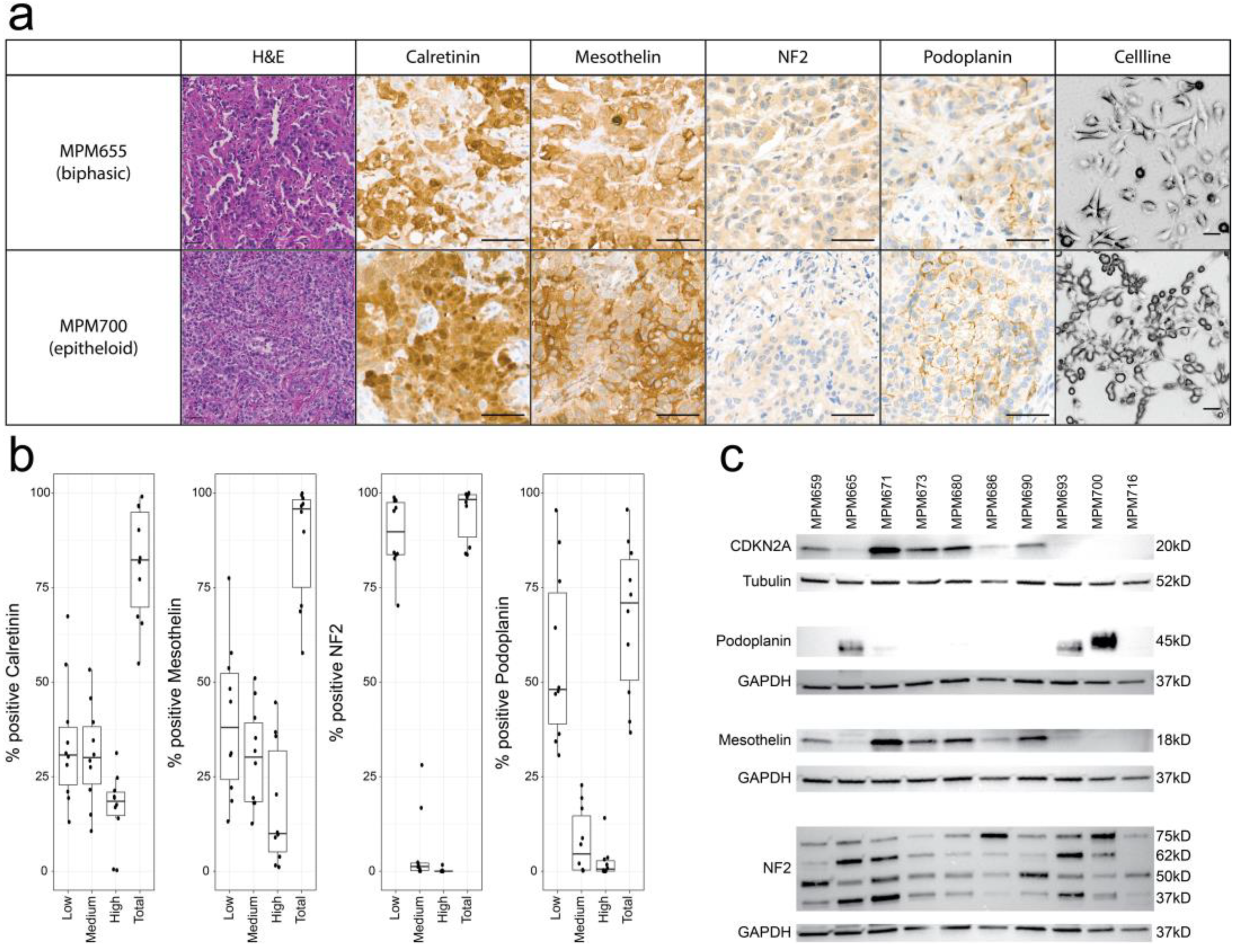
3.1. Characterization of MPM Tumors
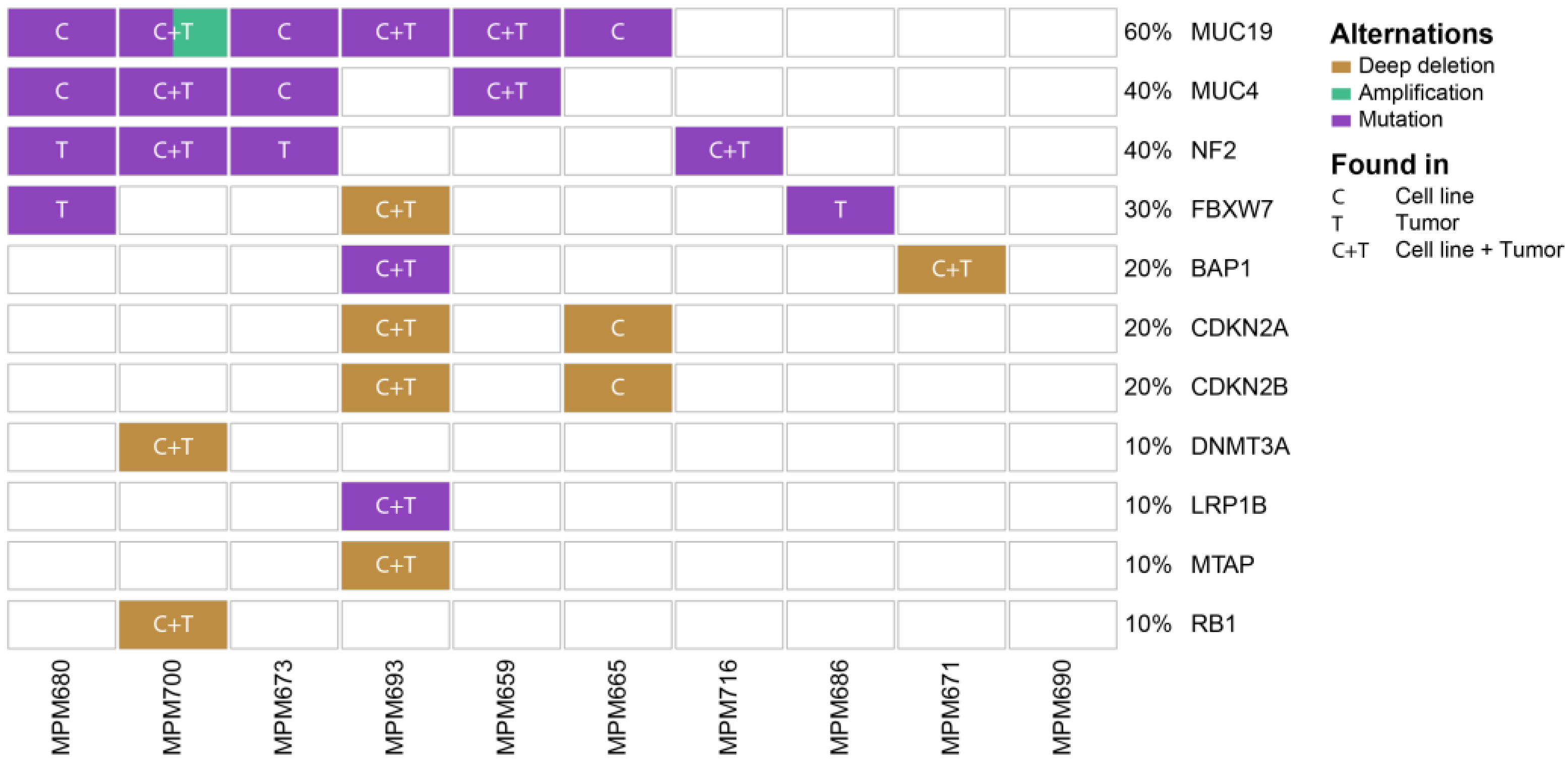
3.2. Genomic Profiling Identifies MPM Specific Overlapping Genetic Mutations in Patient-Derived Cell Lines and Originating Tumors
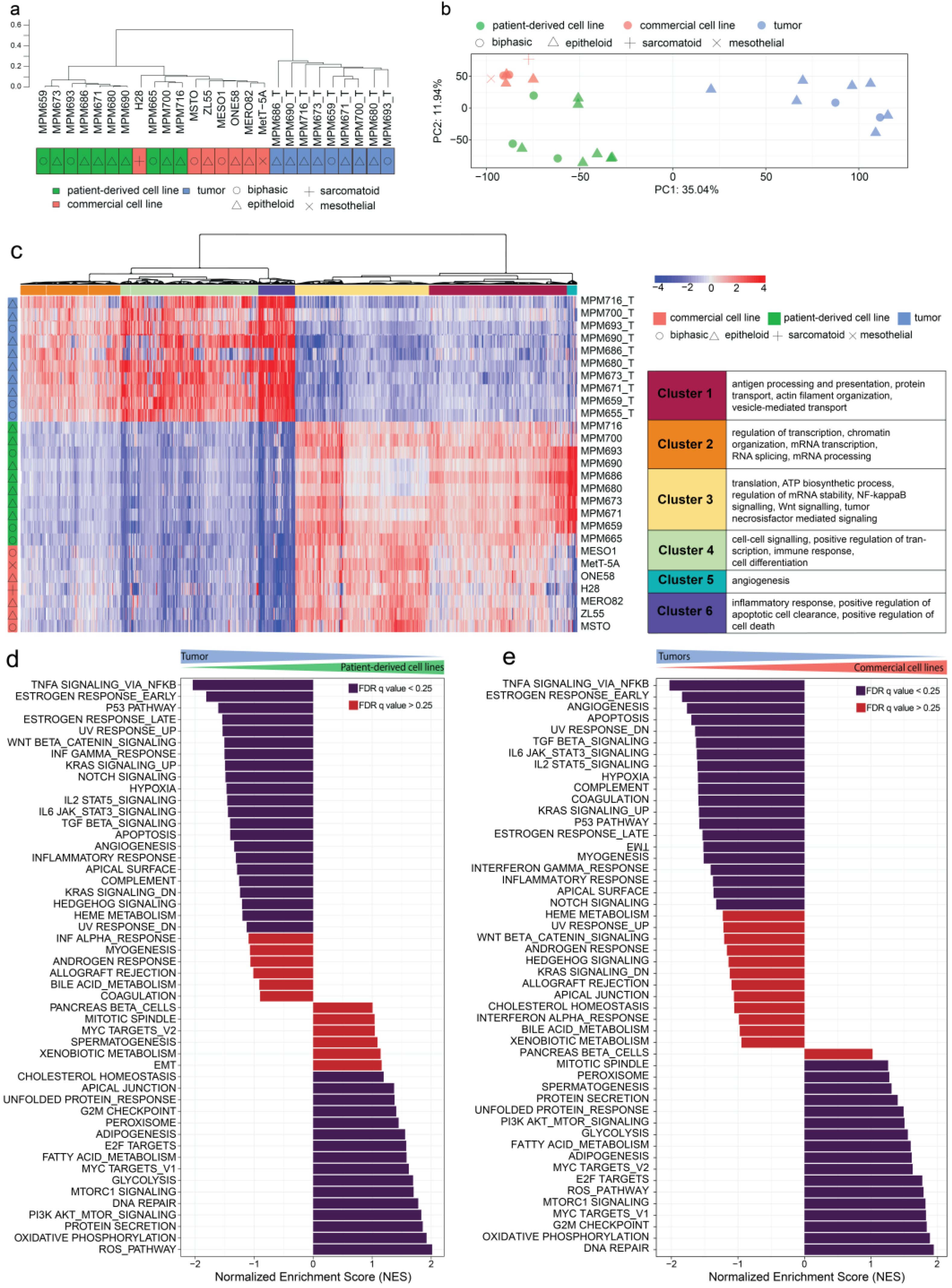
3.3. Gene Expression Profiles of MPM Tumors, Patient-Derived Cell Lines, and Commercial Cell Lines form Distinct Clusters
3.4. Differential Gene Expression Analysis Reveals Upregulation of Metabolic and Cell-Cycle-Related Processes in Cell Lines Compared to an Upregulation of Genes Involved in Transcription, EMT, and Immune System Response in Tumors
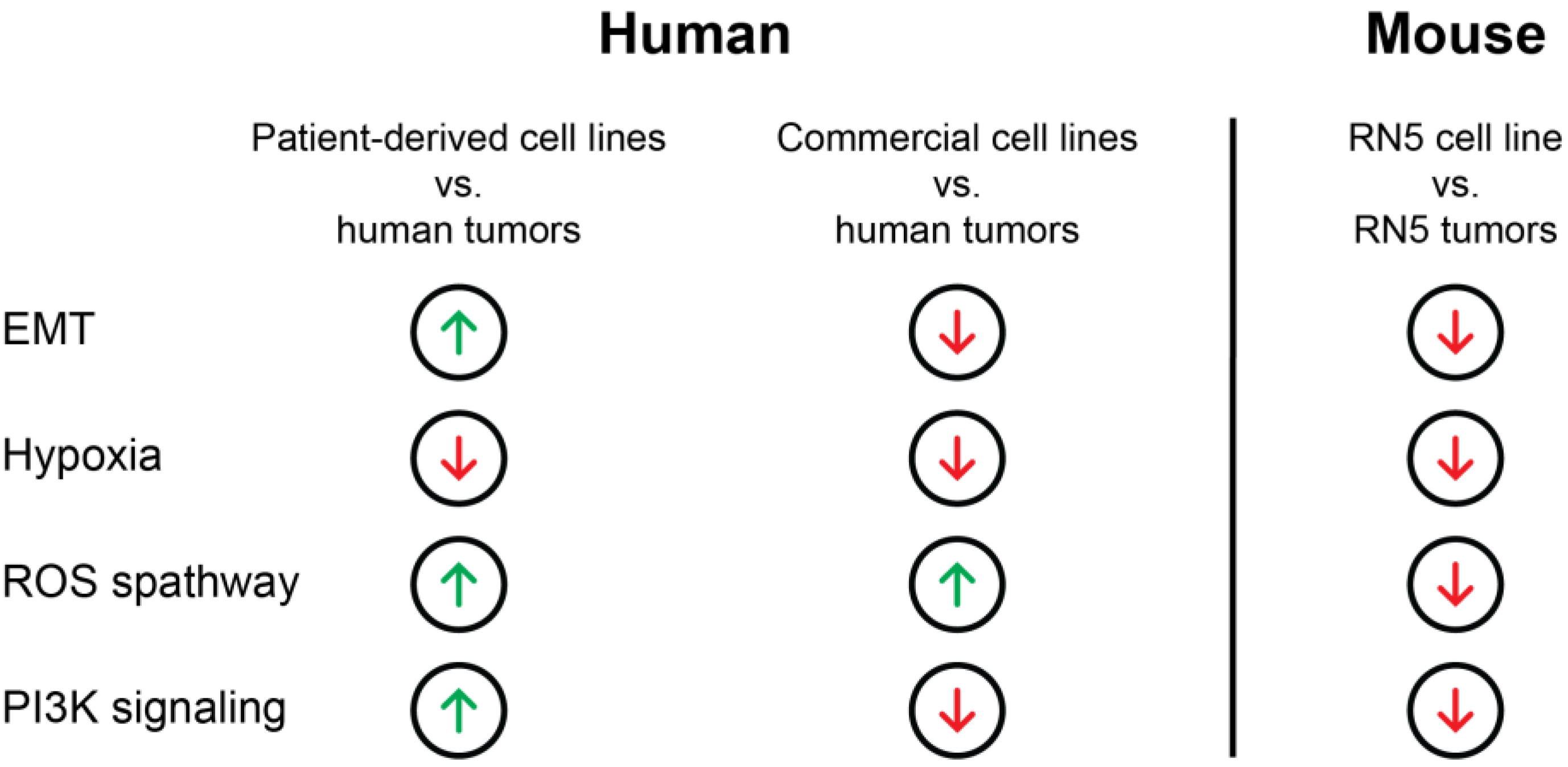
3.5. Transcriptomic Regulation of EMT, Apoptosis, UV-Response Inhibition, Myogenesis, and Angiogenesis Are More Comparable to Tumors in Patient-Derived Cell Lines Than Commercial Cell Lines Based on Gene Set Enrichment Analysis (GSEA)
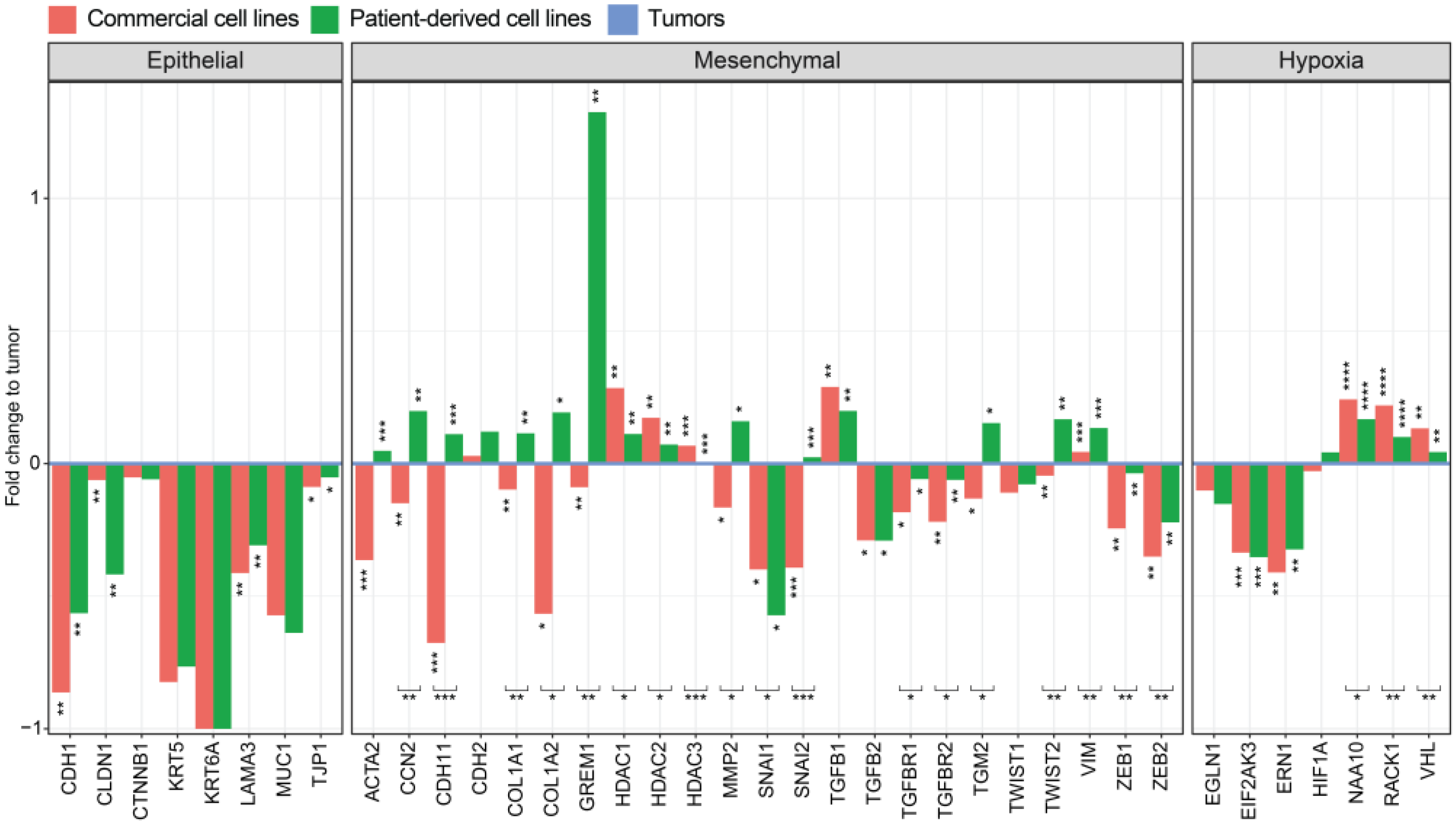
3.6. Genes of the EMT Pathway Are Upregulated in Patient-Derived Cell Lines Compared to Commercial Cell Lines Displaying Similarities to Tumors
3.7. Genes Involved in Oxidative Folding of Proteins Are Downregulated and Negative Regulators of Hypoxia Are Upregulated in Cell Lines Suggesting a Highly Hypoxic State in MPM Tumors
3.8. Gene Sets of the P53 Pathway, EMT, and TGFβ Signaling Are Upregulated in RN5 Tumors Compared to the RN5 Cell Line
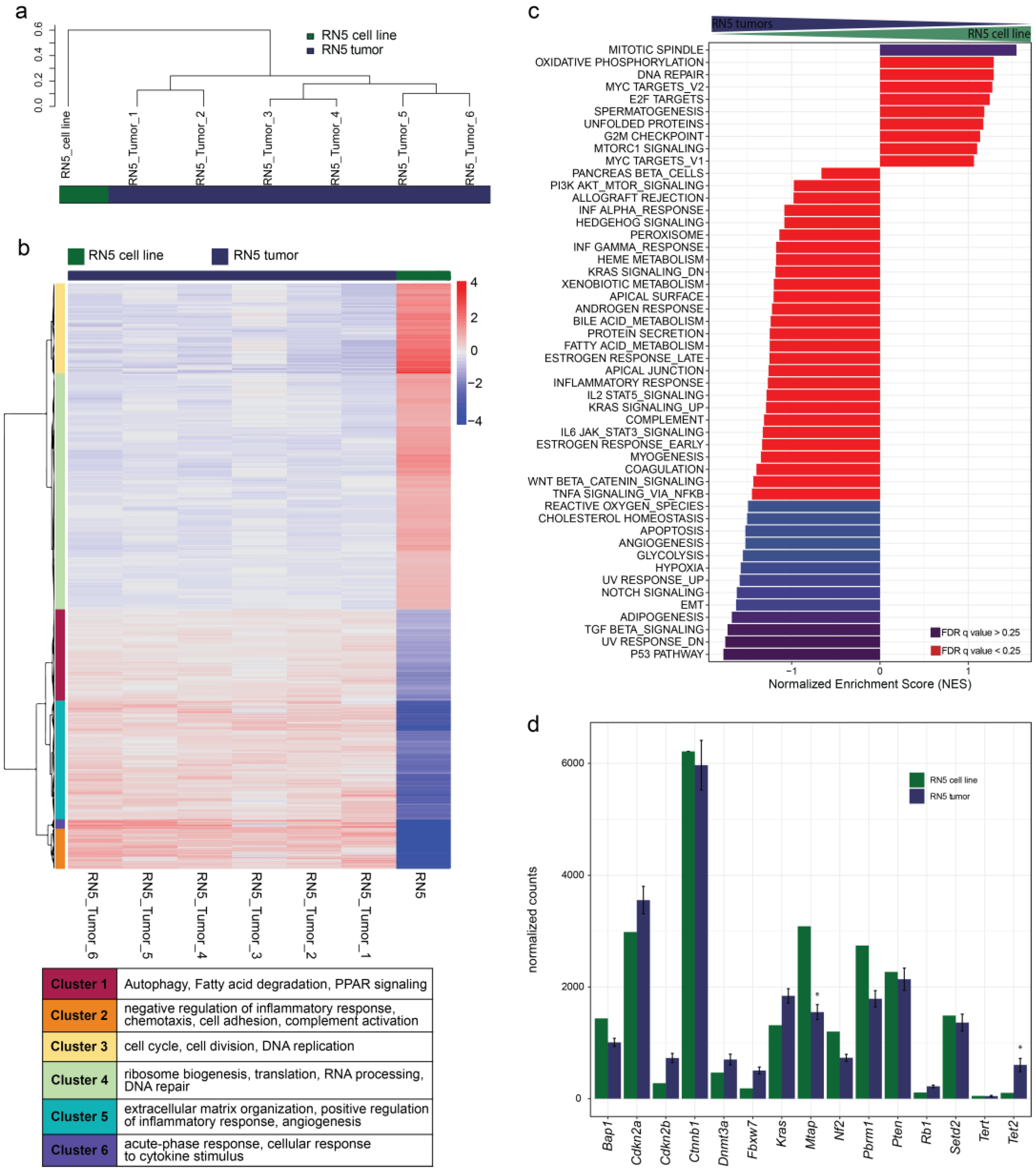
3.9. Genomic Alterations of the RN5 Model
3.10. Gene Expression Patterns Differ from Human to Murine MPM Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baas, P.; Scherpereel, A.; Nowak, A.K.; Fujimoto, N.; Peters, S.; Tsao, A.S.; Mansfield, A.S.; Popat, S.; Jahan, T.; Antonia, S.; et al. First-Line Nivolumab Plus Ipilimumab in Unresectable Malignant Pleural Mesothelioma (Checkmate 743): A Multicentre, Randomised, Open-Label, Phase 3 Trial. Lancet 2021, 397, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Scherpereel, A.; Mazieres, J.; Greillier, L.; Lantuejoul, S.; Do, P.; Bylicki, O.; Monnet, I.; Corre, R.; Audigier-Valette, C.; Locatelli-Sanchez, M.; et al. Nivolumab or Nivolumab Plus Ipilimumab in Patients with Relapsed Malignant Pleural Mesothelioma (Ifct-1501 Maps2): A Multicentre, Open-Label, Randomised, Non-Comparative, Phase 2 Trial. Lancet Oncol. 2019, 20, 239–253. [Google Scholar] [CrossRef] [PubMed]
- de Gooijer, C.J.; Borm, F.J.; Scherpereel, A.; Baas, P. Immunotherapy in Malignant Pleural Mesothelioma. Front. Oncol. 2020, 10, 187. [Google Scholar] [CrossRef] [PubMed]
- Shamseddin, M.; Obacz, J.; Garnett, M.J.; Rintoul, R.C.; Francies, H.E.; Marciniak, S.J. Use of preclinical models for malignant pleural mesothelioma. Thorax 2021, 76, 1154–1162. [Google Scholar] [CrossRef]
- JJohnson, B.W.; Takahashi, K.; Cheng, Y.Y. Preclinical Models and Resources to Facilitate Basic Science Research on Malignant Mesothelioma—A Review. Front. Oncol. 2021, 11, 748444. [Google Scholar] [CrossRef]
- Gillet, J.-P.; Varma, S.; Gottesman, M.M. The Clinical Relevance of Cancer Cell Lines. J. Natl. Cancer Inst. 2013, 105, 452–458. [Google Scholar] [CrossRef]
- Gazdar, A.F.; Gao, B.; Minna, J.D. Lung cancer cell lines: Useless artifacts or invaluable tools for medical science? Lung Cancer 2010, 68, 309–318. [Google Scholar] [CrossRef]
- Vincent, K.M.; Findlay, S.D.; Postovit, L.M. Assessing breast cancer cell lines as tumour models by comparison of mRNA expression profiles. Breast Cancer Res. 2015, 17, 114. [Google Scholar] [CrossRef]
- Jiang, G.; Zhang, S.; Yazdanparast, A.; Li, M.; Pawar, A.V.; Liu, Y.; Inavolu, S.M.; Cheng, L. Comprehensive comparison of molecular portraits between cell lines and tumors in breast cancer. BMC Genom. 2016, 17 (Suppl. S7), 525. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Sirota, M.; Fan-Minogue, H.; Hadley, D.; Butte, A.J. Relating hepatocellular carcinoma tumor samples and cell lines using gene expression data in translational research. BMC Med. Genom. 2015, 8 (Suppl. S2), S5. [Google Scholar] [CrossRef]
- Gordon, G.J.; Jensen, R.V.; Hsiao, L.-L.; Gullans, S.R.; Blumenstock, J.E.; Ramaswamy, S.; Richards, W.G.; Sugarbaker, D.J.; Bueno, R. Translation of microarray data into clinically relevant cancer diagnostic tests using gene expression ratios in lung cancer and mesothelioma. Cancer Res. 2002, 62, 4963–4967. [Google Scholar] [PubMed]
- Gordon, G.J.; Rockwell, G.N.; Jensen, R.V.; Rheinwald, J.G.; Glickman, J.N.; Aronson, J.P.; Pottorf, B.J.; Nitz, M.D.; Richards, W.G.; Sugarbaker, D.J.; et al. Identification of Novel Candidate Oncogenes and Tumor Suppressors in Malignant Pleural Mesothelioma Using Large-Scale Transcriptional Profiling. Am. J. Pathol. 2005, 166, 1827–1840. [Google Scholar] [CrossRef] [PubMed]
- Nair, N.U.; Jiang, Q.; Wei, J.S.; Misra, V.A.; Morrow, B.; Kesserwan, C.; Hermida, L.C.; Lee, J.S.; Mian, I.; Zhang, J.; et al. Genomic and transcriptomic analyses identify a prognostic gene signature and predict response to therapy in pleural and peritoneal mesothelioma. Cell Rep. Med. 2023, 4, 100938. [Google Scholar] [CrossRef] [PubMed]
- Chernova, T.; Sun, X.M.; Powley, I.R.; Galavotti, S.; Grosso, S.; Murphy, F.A.; Miles, G.J.; Cresswell, L.; Antonov, A.V.; Bennett, J.; et al. Molecular profiling reveals primary mesothelioma cell lines recapitulate human disease. Cell Death Differ. 2016, 23, 1152–1164. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Garcia, M.; Juhos, S.; Larsson, M.; Olason, P.I.; Martin, M.; Eisfeldt, J.; DiLorenzo, S.; Sandgren, J.; De Stahl, T.D.; Ewels, P.; et al. Sarek: A Portable Workflow for Whole-Genome Sequencing Analysis of Germline and Somatic Variants. F1000Research 2020, 9, 63. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef]
- Kurtzer, G.M.; Sochat, V.; Bauer, M.W. Singularity: Scientific containers for mobility of compute. PLoS ONE 2017, 12, e0177459. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with Bwa-Mem. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef] [PubMed]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-FREEC: A tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 2012, 28, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Blum, W.; Pecze, L.; Felley-Bosco, E.; Worthmüller-Rodriguez, J.; Wu, L.; Vrugt, B.; De Perrot, M.; Schwaller, B. Establishment of immortalized murine mesothelial cells and a novel mesothelioma cell line. Vitr. Cell. Dev. Biol.-Anim. 2015, 51, 714–721. [Google Scholar] [CrossRef]
- Picelli, S. Full-Length Single-Cell Rna Sequencing with Smart-Seq2. Methods Mol. Biol. 2019, 1979, 25–44. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Alaoui, A.; Olengoba, B.; Ettaki, B.; Zerouaoui, J. A Generic Methodology for Clustering to Maximises Inter-Cluster Inertia. Int. J. Adv. Comput. Sci. Appl. 2017, 8, 187–191. [Google Scholar] [CrossRef]
- Kuettel, V.; Pfister, S.; Ferrero, E. Deconvolution of Bulk Rna-Seq Data with Granulator. Available online: http://bioconductor.jp/packages/release/bioc/vignettes/granulator/inst/doc/granulator.html (accessed on 17 May 2023).
- Monaco, G.; Lee, B.; Xu, W.; Mustafah, S.; Hwang, Y.Y.; Carré, C.; Burdin, N.; Visan, L.; Ceccarelli, M.; Poidinger, M.; et al. RNA-Seq Signatures Normalized by mRNA Abundance Allow Absolute Deconvolution of Human Immune Cell Types. Cell Rep. 2019, 26, 1627–1640.e7. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (Msigdb) Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Vasaikar, S.V.; Deshmukh, A.P.; Hollander, P.D.; Addanki, S.; Kuburich, N.A.; Kudaravalli, S.; Joseph, R.; Chang, J.T.; Soundararajan, R.; Mani, S.A. EMTome: A resource for pan-cancer analysis of epithelial-mesenchymal transition genes and signatures. Br. J. Cancer 2020, 124, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Hiltbrunner, S.; Fleischmann, Z.; Sokol, E.S.; Zoche, M.; Felley-Bosco, E.; Curioni-Fontecedro, A. Genomic landscape of pleural and peritoneal mesothelioma tumours. Br. J. Cancer 2022, 127, 1997–2005. [Google Scholar] [CrossRef] [PubMed]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Kallergi, G.; Papadaki, M.A.; Politaki, E.; Mavroudis, D.; Georgoulias, V.; Agelaki, S. Epithelial to mesenchymal transition markers expressed in circulating tumour cells of early and metastatic breast cancer patients. Breast Cancer Res. 2011, 13, R59. [Google Scholar] [CrossRef]
- Spaderna, S.; Schmalhofer, O.; Hlubek, F.; Berx, G.; Eger, A.; Merkel, S.; Jung, A.; Kirchner, T.; Brabletz, T. A Transient, EMT-Linked Loss of Basement Membranes Indicates Metastasis and Poor Survival in Colorectal Cancer. Gastroenterology 2006, 131, 830–840. [Google Scholar] [CrossRef]
- Rehrauer, H.; Wu, L.; Blum, W.; Pecze, L.; Henzi, T.; Serre-Beinier, V.; Aquino, C.; Vrugt, B.; de Perrot, M.; Schwaller, B.; et al. How Asbestos Drives the Tissue towards Tumors: Yap Activation, Macrophage and Mesothelial Precursor Recruitment, Rna Editing, and Somatic Mutations. Oncogene 2018, 37, 2645–2659. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Tissari, M.; Tamminen, J.; Ylivinkka, I.; Rönty, M.; von Nandelstadh, P.; Lehti, K.; Hyytiäinen, M.; Myllärniemi, M.; Koli, K. Gremlin-1 is a key regulator of the invasive cell phenotype in mesothelioma. Oncotarget 2017, 8, 98280–98297. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, G.; Grun, D.; Alexander, H.R.; Friedberg, J.S.; Xu, W.; Keillor, J.W.; Kandasamy, S.; Eckert, R.L. Transglutaminase is a mesothelioma cancer stem cell survival protein that is required for tumor formation. Oncotarget 2018, 9, 34495–34505. [Google Scholar] [CrossRef] [PubMed]
- Wawruszak, A.; Kalafut, J.; Okon, E.; Czapinski, J.; Halasa, M.; Przybyszewska, A.; Miziak, P.; Okla, K.; Rivero-Muller, A.; Stepulak, A. Histone Deacetylase Inhibitors and Phenotypical Transformation of Cancer Cells. Cancers 2019, 11, 148. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood-Flow, Oxygen and Nutrient Supply, and Metabolic Microenvironment of Human-Tumors—A Review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar]
- Wouters, B.G.; Koritzinsky, M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat. Rev. Cancer 2008, 8, 851–864. [Google Scholar] [CrossRef]
- Koumenis, C.; Naczki, C.; Koritzinsky, M.; Rastani, S.; Diehl, A.; Sonenberg, N.; Koromilas, A.; Wouters, B.G. Regulation of Protein Synthesis by Hypoxia via Activation of the Endoplasmic Reticulum Kinase Perk and Phosphorylation of the Translation Initiation Factor Eif2 Alpha. Mol. Cell. Biol. 2002, 22, 7405–7416. [Google Scholar] [CrossRef]
- Cobos, F.A.; Alquicira-Hernandez, J.; Powell, J.E.; Mestdagh, P.; De Preter, K. Benchmarking of cell type deconvolution pipelines for transcriptomics data. Nat. Commun. 2020, 11, 5650. [Google Scholar] [CrossRef]
- Jin, H.; Liu, Z. A benchmark for RNA-seq deconvolution analysis under dynamic testing environments. Genome Biol. 2021, 22, 102. [Google Scholar] [CrossRef]
- Kanellakis, N.I.; Asciak, R.; Hamid, M.A.; Yao, X.; McCole, M.; McGowan, S.; Seraia, E.; Hatch, S.; Hallifax, R.J.; Mercer, R.M.; et al. Patient-derived malignant pleural mesothelioma cell cultures: A tool to advance biomarker-driven treatments. Thorax 2020, 75, 1004–1008. [Google Scholar] [CrossRef]
- Sinha, R.; Luna, A.; Schultz, N.; Sander, C. A pan-cancer survey of cell line tumor similarity by feature-weighted molecular profiles. Cell Rep. Methods 2021, 1, 100039. [Google Scholar] [CrossRef]
- Kresoja-Rakic, J.; Kapaklikaya, E.; Ziltener, G.; Dalcher, D.; Santoro, R.; Christensen, B.C.; Johnson, K.C.; Schwaller, B.; Weder, W.; Stahel, R.A.; et al. Identification of cis- and trans-acting elements regulating calretinin expression in mesothelioma cells. Oncotarget 2016, 7, 21272–21286. [Google Scholar] [CrossRef] [PubMed]
- Kresoja-Rakic, J.; Sulemani, M.; Kirschner, M.B.; Ronner, M.; Reid, G.; Kao, S.; Schwaller, B.; Weder, W.; Stahel, R.A.; Felley-Bosco, E. Posttranscriptional Regulation Controls Calretinin Expression in Malignant Pleural Mesothelioma. Front. Genet. 2017, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Zhou, Z.; Su, W.; Wang, Z.; Wu, Q. A novel alternative splicing isoform of NF2 identified in human Schwann cells. Oncol. Lett. 2016, 12, 977–982. [Google Scholar] [CrossRef][Green Version]
- MMartin, S.D.; Cheung, S.; Churg, A. Immunohistochemical Demonstration of Merlin/NF2 Loss in Mesothelioma. Mod. Pathol. 2023, 36, 100036. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, S.-R.; Chen, Y.-B.; Adusumilli, P.S.; Bialik, A.; Bodd, F.M.; Ladanyi, M.; Lopardo, J.; Offin, M.D.; Rusch, V.W.; et al. Neurofibromatosis Type 2-Yes-Associated Protein and Transcriptional Coactivator with PDZ-Binding Motif Dual Immunohistochemistry Is a Reliable Marker for the Detection of Neurofibromatosis Type 2 Alterations in Diffuse Pleural Mesothelioma. Mod. Pathol. 2023, 36, 100030. [Google Scholar] [CrossRef]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R.; et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325–330. [Google Scholar] [CrossRef]
- Meiller, C.; Montagne, F.; Hirsch, T.Z.; Caruso, S.; De Wolf, J.; Bayard, Q.; Assié, J.B.; Meunier, L.; Blum, Y.; Quetel, L.; et al. Multi-Site Tumor Sampling Highlights Molecular Intra-Tumor Heterogeneity in Malignant Pleural Mesothelioma. Genome Med. 2021, 13, 113. [Google Scholar] [CrossRef]
- Jeong, M.; Park, H.J.; Celik, H.; Ostrander, E.L.; Reyes, J.M.; Guzman, A.; Rodriguez, B.; Lei, Y.; Lee, Y.; Ding, L.; et al. Loss of Dnmt3a Immortalizes Hematopoietic Stem Cells In Vivo. Cell Rep. 2018, 23, 1–10. [Google Scholar] [CrossRef]
- Beer, A.G.; Zenzmaier, C.; Schreinlechner, M.; Haas, J.; Dietrich, M.F.; Herz, J.; Marschang, P. Expression of a recombinant full-length LRP1B receptor in human non-small cell lung cancer cells confirms the postulated growth-suppressing function of this large LDL receptor family member. Oncotarget 2016, 7, 68721–68733. [Google Scholar] [CrossRef]
- McLoughlin, K.C.; Kaufman, A.S.; Schrump, D.S. Targeting the epigenome in malignant pleural mesothelioma. Transl. Lung Cancer Res. 2017, 6, 350–365. [Google Scholar] [CrossRef]
- Komatsu, M.; Jepson, S.; Arango, M.E.; Carraway, C.A.C.; Carraway, K.L. Muc4/sialomucin complex, an intramembrane modulator of ErbB2/HER2/Neu, potentiates primary tumor growth and suppresses apoptosis in a xenotransplanted tumor. Oncogene 2001, 20, 461–470. [Google Scholar] [CrossRef]
- Gautam, S.K.; Kumar, S.; Dam, V.; Ghersi, D.; Jain, M.; Batra, S.K. MUCIN-4 (MUC4) is a novel tumor antigen in pancreatic cancer immunotherapy. Semin. Immunol. 2020, 47, 101391. [Google Scholar] [CrossRef] [PubMed]
- Ponnusamy, M.P.; Seshacharyulu, P.; Vaz, A.; Dey, P.; Batra, S.K. MUC4 stabilizes HER2 expression and maintains the cancer stem cell population in ovarian cancer cells. J. Ovarian Res. 2011, 4, 7–10. [Google Scholar] [CrossRef]
- van der Meer, D.; Barthorpe, S.; Yang, W.; Lightfoot, H.; Hall, C.; Gilbert, J.; Francies, H.E.; Garnett, M.J. Cell Model Passports—A hub for clinical, genetic and functional datasets of preclinical cancer models. Nucleic Acids Res. 2019, 47, D923–D929. [Google Scholar] [CrossRef]
- Singh, A.; Busacca, S.; Gaba, A.; Sheaff, M.; Poile, C.; Nakas, A.; Dzialo, J.; Bzura, A.; Dawson, A.G.; Fennell, D.A.; et al. BAP1 loss induces mitotic defects in mesothelioma cells through BRCA1-dependent and independent mechanisms. Oncogene 2023, 42, 572–585. [Google Scholar] [CrossRef]
- Sneddon, S.; Dick, I.; Lee, Y.G.; Musk, A.; Patch, A.-M.; Pearson, J.V.; Waddell, N.; Allcock, R.J.; Holt, R.; Robinson, B.W.; et al. Malignant cells from pleural fluids in malignant mesothelioma patients reveal novel mutations. Lung Cancer 2018, 119, 64–70. [Google Scholar] [CrossRef]
- Garman, B.; Anastopoulos, I.N.; Krepler, C.; Brafford, P.; Sproesser, K.; Jiang, Y.; Wubbenhorst, B.; Amaravadi, R.; Bennett, J.; Beqiri, M.; et al. Genetic and Genomic Characterization of 462 Melanoma Patient-Derived Xenografts, Tumor Biopsies, and Cell Lines. Cell Rep. 2017, 21, 1936–1952. [Google Scholar] [CrossRef]
- Ramundo, V.; Zanirato, G.; Aldieri, E. The Epithelial-to-Mesenchymal Transition (EMT) in the Development and Metastasis of Malignant Pleural Mesothelioma. Int. J. Mol. Sci. 2021, 22, 12216. [Google Scholar] [CrossRef]
- Marzo, A.L.; Fitzpatrick, D.R.; Robinson, B.W.; Scott, B. Antisense oligonucleotides specific for transforming growth factor beta2 inhibit the growth of malignant mesothelioma both in vitro and in vivo. Cancer Res. 1997, 57, 3200–3207. [Google Scholar]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Dev. Ther. 2015, 9, 4479–4499. [Google Scholar]
- Yin, T.; Wang, G.; He, S.; Shen, G.; Su, C.; Zhang, Y.; Wei, X.; Ye, T.; Li, L.; Yang, S.; et al. Malignant Pleural Effusion and Ascites Induce Epithelial-Mesenchymal Transition and Cancer Stem-Like Cell Properties via the Vascular Endothelial Growth Factor (Vegf)/Phosphatidylinositol 3-Kinase (Pi3k)/Akt/Mechanistic Target of Rapamycin (Mtor) Pathway*. J. Biol. Chem. 2016, 291, 26750–26761. [Google Scholar] [CrossRef]
- Roscilli, G.; De Vitis, C.; Ferrara, F.F.; Noto, A.; Cherubini, E.; Ricci, A.; Mariotta, S.; Giarnieri, E.; Giovagnoli, M.R.; Torrisi, M.R.; et al. Human lung adenocarcinoma cell cultures derived from malignant pleural effusions as model system to predict patients chemosensitivity. J. Transl. Med. 2016, 14, 61. [Google Scholar] [CrossRef]
- Basak, S.K.; Veena, M.S.; Oh, S.; Huang, G.; Srivatsan, E.; Huang, M.; Sharma, S.; Batra, R.K. The Malignant Pleural Effusion as a Model to Investigate Intratumoral Heterogeneity in Lung Cancer. PLoS ONE 2009, 4, e5884. [Google Scholar] [CrossRef]
- Mancini, R.; Giarnieri, E.; de Vitis, C.; Malanga, N.; Roscilli, G.; Noto, A.; Marra, E.; Laudanna, C.; Zoppoli, P.; De Luca, P.; et al. Spheres Derived from Lung Adenocarcinoma Pleural Effusions: Molecular Characterization and Tumor Engraftment. PLoS ONE 2011, 6, e21320. [Google Scholar] [CrossRef]
- Cao, X.X.; Mohuiddin, I.; Ece, F.; McConkey, D.J.; Smythe, W.R. Histone Deacetylase Inhibitor Downregulation of bcl-xl Gene Expression Leads to Apoptotic Cell Death in Mesothelioma. Am. J. Respir. Cell Mol. Biol. 2001, 25, 562–568. [Google Scholar] [CrossRef]
- Symanowski, J.; Vogelzang, N.; Zawel, L.; Atadja, P.; Pass, H.; Sharma, S. A Histone Deacetylase Inhibitor LBH589 Downregulates XIAP in Mesothelioma Cell Lines Which is Likely Responsible for Increased Apoptosis with TRAIL. J. Thorac. Oncol. 2009, 4, 149–160. [Google Scholar] [CrossRef]
- Vandermeers, F.; Hubert, P.; Delvenne, P.; Mascaux, C.; Grigoriu, B.; Burny, A.; Scherpereel, A.; Willems, L. Valproate, in Combination with Pemetrexed and Cisplatin, Provides Additional Efficacy to the Treatment of Malignant Mesothelioma. Clin. Cancer Res. 2009, 15, 2818–2828. [Google Scholar] [CrossRef]
- Kelly, W.K.; O’Connor, O.A.; Krug, L.M.; Chiao, J.H.; Heaney, M.; Curley, T.; MacGregore-Cortelli, B.; Tong, W.; Secrist, J.P.; Schwartz, L.; et al. Phase I Study of an Oral Histone Deacetylase Inhibitor, Suberoylanilide Hydroxamic Acid, in Patients with Advanced Cancer. J. Clin. Oncol. 2005, 23, 3923–3931. [Google Scholar] [CrossRef]
- Rangarajan, A.; Weinberg, R.A. Comparative biology of mouse versus human cells: Modelling human cancer in mice. Nat. Rev. Cancer 2003, 3, 952–959. [Google Scholar] [CrossRef]
- Wu, L.; Amjad, S.; Yun, H.; Mani, S.; de Perrot, M. A panel of emerging EMT genes identified in malignant mesothelioma. Sci. Rep. 2022, 12, 1007. [Google Scholar] [CrossRef] [PubMed]
- Pardo-Sánchez, J.M.; Mancheño, N.; Cerón, J.; Jordá, C.; Ansotegui, E.; Juan, Ó.; Palanca, S.; Cremades, A.; Gandía, C.; Farràs, R. Increased Tumor Growth Rate and Mesenchymal Properties of Nsclc-Patient-Derived Xenograft Models During Serial Transplantation. Cancers 2021, 13, 2980. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, M.L.; Francescangeli, F.; Nicolazzo, C.; Xhelili, E.; La Torre, F.; Colace, L.; Bruselles, A.; Macchia, D.; Vitale, S.; Gazzaniga, P.; et al. An Orthotopic Patient-Derived Xenograft (PDX) Model Allows the Analysis of Metastasis-Associated Features in Colorectal Cancer. Front. Oncol. 2022, 12, 869485. [Google Scholar] [CrossRef] [PubMed]
- Chahinian, A.P.; Beranek, J.T.; Suzuki, Y.; Bekesi, J.G.; Wisniewski, L.; Selikoff, I.J.; Holland, J.F. Transplantation of Human Malignant Mesothelioma into Nude Mice. Cancer Res. 1980, 40, 181–185. [Google Scholar]
- Wu, L.; Allo, G.; John, T.; Li, M.; Tagawa, T.; Opitz, I.; Anraku, M.; Yun, Z.; Pintilie, M.; Pitcher, B.; et al. Patient-Derived Xenograft Establishment from Human Malignant Pleural Mesothelioma. Clin. Cancer Res. 2017, 23, 1060–1067. [Google Scholar] [CrossRef]
- Fujiwara, S. Humanized mice: A brief overview on their diverse applications in biomedical research. J. Cell. Physiol. 2018, 233, 2889–2901. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laure, A.; Rigutto, A.; Kirschner, M.B.; Opitz, L.; Grob, L.; Opitz, I.; Felley-Bosco, E.; Hiltbrunner, S.; Curioni-Fontecedro, A. Genomic and Transcriptomic Analyses of Malignant Pleural Mesothelioma (MPM) Samples Reveal Crucial Insights for Preclinical Testing. Cancers 2023, 15, 2813. https://doi.org/10.3390/cancers15102813
Laure A, Rigutto A, Kirschner MB, Opitz L, Grob L, Opitz I, Felley-Bosco E, Hiltbrunner S, Curioni-Fontecedro A. Genomic and Transcriptomic Analyses of Malignant Pleural Mesothelioma (MPM) Samples Reveal Crucial Insights for Preclinical Testing. Cancers. 2023; 15(10):2813. https://doi.org/10.3390/cancers15102813
Chicago/Turabian StyleLaure, Alexander, Angelica Rigutto, Michaela B. Kirschner, Lennart Opitz, Linda Grob, Isabelle Opitz, Emanuela Felley-Bosco, Stefanie Hiltbrunner, and Alessandra Curioni-Fontecedro. 2023. "Genomic and Transcriptomic Analyses of Malignant Pleural Mesothelioma (MPM) Samples Reveal Crucial Insights for Preclinical Testing" Cancers 15, no. 10: 2813. https://doi.org/10.3390/cancers15102813
APA StyleLaure, A., Rigutto, A., Kirschner, M. B., Opitz, L., Grob, L., Opitz, I., Felley-Bosco, E., Hiltbrunner, S., & Curioni-Fontecedro, A. (2023). Genomic and Transcriptomic Analyses of Malignant Pleural Mesothelioma (MPM) Samples Reveal Crucial Insights for Preclinical Testing. Cancers, 15(10), 2813. https://doi.org/10.3390/cancers15102813