
Potential of Synthetic and Natural Compounds as Novel Histone Deacetylase Inhibitors for the Treatment of Hematological Malignancies

 ,
,  ,
,  ,
,  and
and 
Abstract
Simple Summary
Abstract
1. Introduction
- Cancer cells express changes in the global pattern of acetylation
- Levels of HDACs are increased in lymphoma cells
- HDACs are abnormally attracted to the promoter of a target gene, where they suppress transcription and prevent differentiation, thereby contributing to the generation of acute promyelocytic leukemia.
2. Classification of HDACs and Inhibitors
3. Induction of DNA Damage by HDAC Inhibitors
4. DNA Double-Strand Breaks
4.1. Homology-Directed Repair
4.2. Nonhomologous End-Joining
5. Downregulation of DSB Repair
6. Phenomenon of Chromatin Remodeling
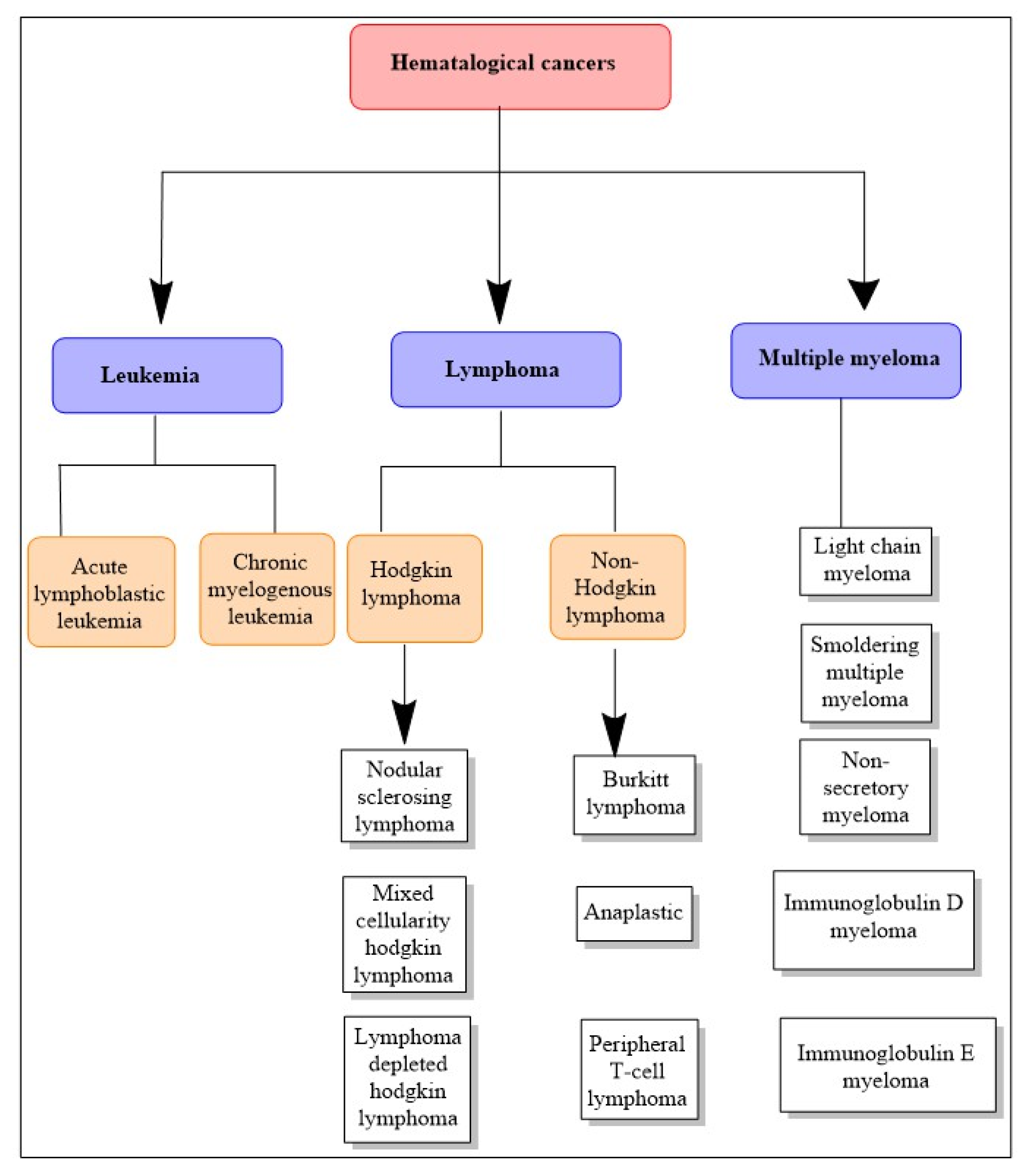
7. Hematological Cancers
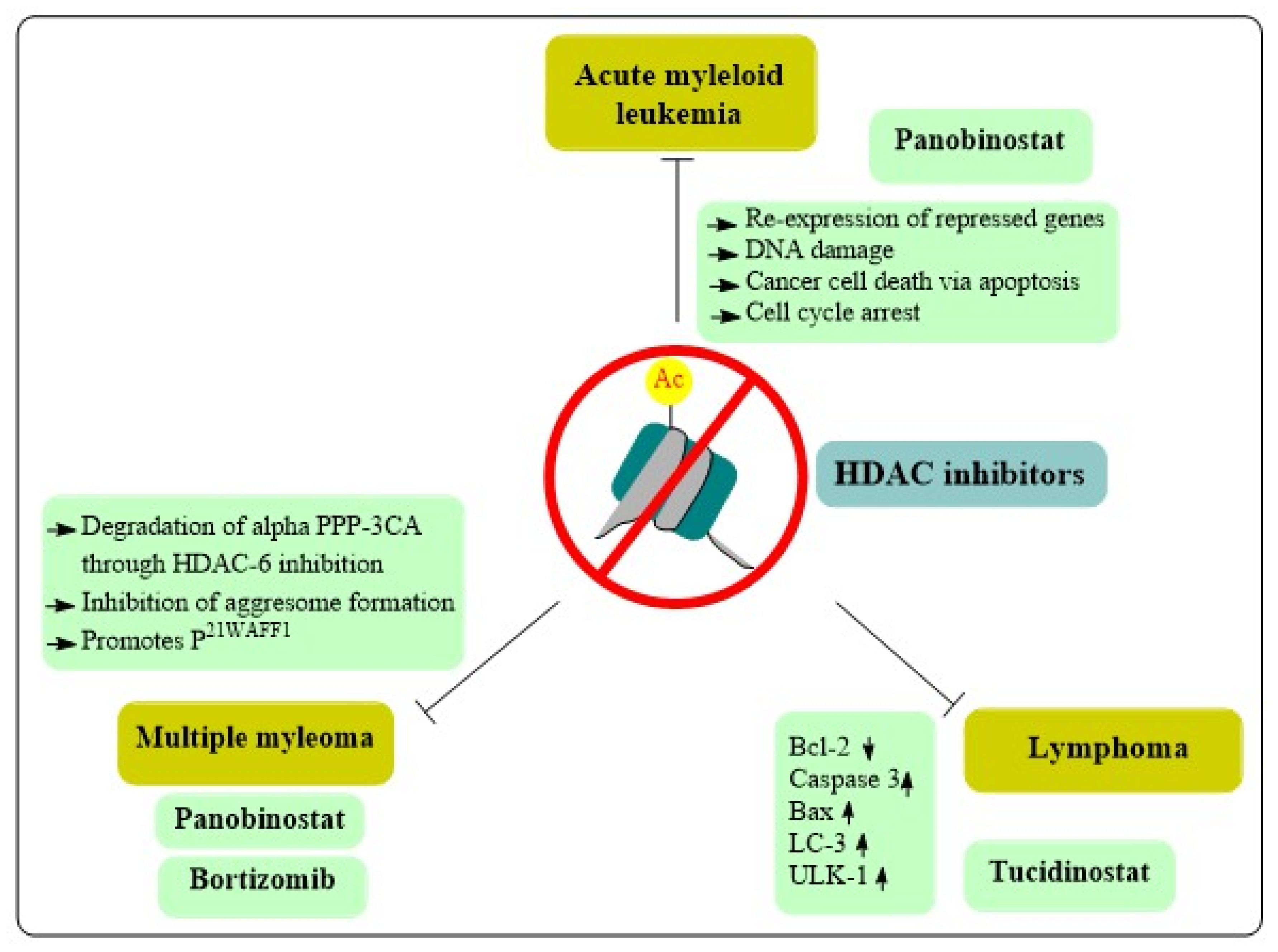
8. Role of HDAC Inhibitors in Hematological Cancers
8.1. Acute Myeloid Leukemia
8.2. Lymphomas
8.3. Multiple Myeloma
9. Development of HDAC Inhibitors for Hematological Cancer Treatment
10. Natural Products as HDAC Inhibitors in Hematological Malignancies
10.1. Pure Compounds
10.1.1. Berberine
10.1.2. Chrysin
10.1.3. Cowaxanthone and Cowain

10.1.4. Curcumin
10.1.5. Cyclostellettamines
10.1.6. Ginsenoside 20(s)-Rh2
10.1.7. Halenaquinones
10.1.8. Largazole
10.1.9. Oleacein
10.1.10. Phenylhexyl Isothiocyanate
10.1.11. Pterostilbene
10.1.12. Quercetin
10.1.13. Thujaplicin
10.1.14. Ursolic Acid
10.1.15. Xestoquinone
10.2. Plant Products and Extracts
10.2.1. Antrodia camphorate
10.2.2. Feijoa sellowiana
10.2.3. Olive Oil

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Active Constituents | Source | Mechanism(s) of Action | References |
|---|---|---|---|
| Berberine | Berberis aristate | Inhibited protein synthesis, HDACs, and Akt/mTOR pathways | [101,102,103] |
| Chrysin | Oroxylum indicum and Pelargonium crispum | Derepressed the epigenetically silenced genes and inhibited HDAC8 both directly and indirectly by reducing its protein concentration | [104] |
| Cowaxanthone and Cowain | Garcinia fusca | Induced apoptosis and autophagy in leukemic T-cells | [109,110] |
| Curcumin | Curcuma longa | Increased histone acetylation on gene promoters of the proapoptotic BAX gene due to inhibition of HDAC1, HDAC3, and HDAC8 activity and expression in leukemic cells | [113] |
| Cyclostellettamine G and dehydrocyclostellettamines D and E | Haliclona and Xestospongia | Induced apoptosis | [114,115] |
| Ginsenoside 20(s)-Rh2 | Panax ginseng | Induced apoptosis | [118] |
| Halenaquinone | Xestospongia vansoesti and Paracheilinus alfiani | Induced apoptosis | [120] |
| Hydroxytyrosol [3,4-dyhydroxyphenyl-ethanol (3,4-DHPEA)] | Virgin olive oil and olive mill wastewater | Induced DNA damage | [138] |
| Largazole | Symploca sp. | Induced apoptosis | [121] |
| Oleacein | Olea europaea | Exhibited epigenetic modulation in multiple myeloma cells | [122] |
| Phenylhexyl isothiocyanate | Cruciferous vegetables | Promoted G1 arrest and apoptosis | [124] |
| Pterostilbene | Blueberries and grapes | Cell cycle arrest, autophagy | [127] |
| Quercetin | Apples, onions, parsley, and sage | Induced apoptosis | [129] |
| Thujaplicin | Troplone | Promoted cell cycle arrest and induced apoptosis | [130] |
| Ursolic acid | Panax ginseng, Rosmarinus officinalis, and Prunus domestica | Promoted cell cycle arrest and induced apoptosis | [132] |
| Xestoquinone | Petrosia sp. | Induced apoptosis | [115] |
| Acetonic extract and flavone | Feijoa sellowiana | Induced apoptosis | [136] |
| Antrodia camphorate | Antrodia camphorate | Induced apoptosis | [135] |
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Adams, H.; Fritzsche, F.R.; Dirnhofer, S.; Kristiansen, G.; Tzankov, A. Class I histone deacetylases 1, 2 and 3 are highly expressed in classical Hodgkin’s lymphoma. Expert Opin. Ther. Targets 2010, 14, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, T.; Kiweler, N.; Krämer, O.H. Interstrand crosslink repair as a target for HDAC inhibition. Trends Pharmacol. Sci. 2017, 38, 822–836. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Bai, L.; Zhou, Y.; Lu, Y.; Jiang, Z.; Shi, J. Recent study of dual HDAC/PARP inhibitor for the treatment of tumor. Curr. Top Med. Chem. 2019, 19, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and Mechanisms of Non-Histone Protein Acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Rassool, F.V. HDAC inhibitors: Roles of DNA damage and repair. Adv. Cancer Res. 2012, 116, 87–129. [Google Scholar]
- Butler, L.M.; Zhou, X.; Xu, W.S.; Scher, H.I.; Rifkind, R.A.; Marks, P.A.; Richon, V.M. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc. Natl. Acad. Sci. USA 2002, 99, 11700–11707. [Google Scholar] [CrossRef]
- Singh, A.K.; Bishayee, A.; Pandey, A.K. Targeting histone deacetylases with natural and synthetic agents: An emerging anti-cancer strategy. Nutrients 2018, 10, 731. [Google Scholar] [CrossRef]
- Bouyahya, A.; El Hachlafi, N.; Aanniz, T.; Bourais, I.; Mechchate, H.; Benali, T.; Shariati, M.A.; Burkov, P.; Lorenzo, J.M.; Wilairatana, P.; et al. Natural Bioactive Compounds Targeting Histone Deacetylases in Human Cancers: Recent Updates. Molecules 2022, 27, 2568. [Google Scholar] [CrossRef]
- Ruzic, D.; Djoković, N.; Srdić-Rajić, T.; Echeverria, C.; Nikolic, K.; Santibanez, J.F. Targeting histone deacetylases: Opportunities for cancer treatment and chemoprevention. Pharmaceutics 2022, 14, 209. [Google Scholar] [CrossRef]
- De Felice, F.; Tombolini, V.; Marampon, F.; Musella, A.; Marchetti, C. Defective DNA repair mechanisms in prostate cancer: Impact of olaparib. Drug Des Devel Ther. 2017, 11, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Shilatifard, A. Epigenetics of hematopoiesis and hematological malignancies. Genes Dev. 2016, 30, 2021–2041. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, Q.; Wang, X.; Gou, S. Design, synthesis, and biological evaluation of novel HDAC inhibitors with a 3-(benzazol-2-yl) quinoxaline framework. Bioorg. Med. Chem. 2023, 88, 129305. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Lu, J.J.; Ding, J. Natural products in cancer therapy: Past, present and future. Nat. Prod. Bioprospect. 2021, 11, 5–13. [Google Scholar] [CrossRef]
- Lecumberri, E.; Dupertuis, Y.M.; Miralbell, R.; Pichard, C. Green tea polyphenol epigallocatechin-3-gallate (EGCG) as adjuvant in cancer therapy. Clin. Nutr. 2013, 32, 894–903. [Google Scholar] [CrossRef]
- Pramanik, S.D.; Kumar Halder, A.; Mukherjee, U.; Kumar, D.; Dey, Y.N. Potential of histone deacetylase inhibitors in the control and regulation of prostate, breast and ovarian cancer. Front. Chem. 2022, 10, 847. [Google Scholar] [CrossRef]
- Feng, J.; Meng, X. Histone modification and histone modification-targeted anti-cancer drugs in breast cancer: Fundamentals and beyond. Front. Pharmacol. 2022, 13, 946811. [Google Scholar] [CrossRef]
- Hieu, D.T.; Anh, D.T.; Tuan, N.M.; Hai, P.T.; Kim, J.; Kang, J.S.; Vu, T.K.; Dung, P.T.; Han, S.B.; Nam, N.H.; et al. Design, synthesis and evaluation of novel N-hydroxybenzamides/N-hydroxypropenamides incorporating quinazolin-4 (3H)-ones as histone deacetylase inhibitors and antitumor agents. Bioorg. Chem. 2018, 76, 258–267. [Google Scholar] [CrossRef]
- Saha, S.; Pal, D.; Kumar, S. Design, synthesis and antiproliferative activity of hydroxyacetamide derivatives against HeLa cervical carcinoma cell and breast cancer cell line. Trop. J. Pharm. Res. 2016, 15, 1401–1411. [Google Scholar] [CrossRef]
- García, S.; Mercado-Sánchez, I.; Bahena, L.; Alcaraz, Y.; García-Revilla, M.A.; Robles, J.; Santos-Martínez, N.; Ordaz-Rosado, D.; García-Becerra, R.; Vazquez, M.A. Design of fluorescent coumarin-hydroxamic acid derivatives as inhibitors of HDACs: Syn-thesis, anti-proliferative evaluation and docking studies. Molecules 2020, 25, 5134. [Google Scholar] [CrossRef]
- Moseley, V.R.; Morris, J.; Knackstedt, R.W.; Wargovich, M.J. Green Tea Polyphenol Epigallocatechin 3-Gallate, Contributes to the Degradation of DNMT3A and HDAC3 in HCT 116 Human Colon Cancer Cells. Anticancer Res. 2013, 33, 5325–5333. [Google Scholar] [PubMed]
- Meeran, S.M.; Patel, S.N.; Chan, T.-H.; Tollefsbol, T.O. A Novel Prodrug of Epigallocatechin-3-Gallate: Differential Epigenetic HTERT Repression in Human Breast Cancer Cells. Cancer Prev. Res. 2011, 4, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.; Kaur, P.; Shukla, S.; Abbas, A.; Fu, P.; Gupta, S. Plant Flavone Apigenin Inhibits HDAC and Remodels Chromatin to Induce Growth Arrest and Apoptosis in Human Prostate Cancer Cells: In Vitro and in Vivo Study. Mol. Carcinog. 2012, 51, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Huang, P.; Wang, X.; Wu, J.; Wu, M.; Huang, J. Galangin-Induced down-Regulation of BACE1 by Epigenetic Mechanisms in SH-SY5Y Cells. Neuroscience 2015, 294, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, K.; Grønbæk, K. Epigenetic therapy in hematological cancers. Apmis 2019, 127, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Serrano, M.; Winkler, R.; Santos, J.C.; Le Pannérer, M.M.; Buschbeck, M.; Roué, G. Histone modifications and their targeting in lymphoid malignancies. Int. J. Mol. Sci. 2022, 23, 253. [Google Scholar] [CrossRef]
- Rathinavelu, A.; Natarajan, U. HDAC inhibition in cancer. In Epigenetics in Organ Specific Disorders; Academic Press: Cambridge, MA, USA, 2023; pp. 63–97. [Google Scholar]
- Andrade, M.B. The Role of Epigenetics in Glioblastoma—A Meta Analysis. 2020. Available online: https://repositorio-aberto.up.pt/bitstream/10216/130224/2/429915.pdf (accessed on 29 April 2023).
- Wang, L.; Syn, N.L.; Subhash, V.V.; Any, Y.; Thuya, W.L.; Cheow, E.S.; Kong, L.; Yu, F.; Peethala, P.C.; Wong, A.L.; et al. Pan-HDAC inhibition by panobino-stat medi-ates chemosensi-tization to carboplatin in non-small cell lung cancer via at-tenuation of EGFR signaling. Cancer Lett. 2018, 417, 152–160. [Google Scholar] [CrossRef]
- Khot, A.; Dickinson, M.; Prince, H.M. Panobinostat in lymphoid and myeloid malignancies. Expert Opin. Investig. Drugs 2013, 22, 1211–1223. [Google Scholar] [CrossRef]
- Giaccone, G.; Rajan, A.; Berman, A.; Kelly, R.J.; Szabo, E.; Lopez-Chavez, A.; Trepel, J.; Lee, M.J.; Cao, L.; Espinoza-Delgado, I.; et al. Phase II study of belinostat in patients with recurrent or refractory advanced thymic epithelial tumors. Clin. Oncol. 2011, 29, 2052. [Google Scholar] [CrossRef]
- Tavakoli-Yaraki, M.; Karami-Tehrani, F.; Salimi, V.; Sirati-Sabet, M. Induction of apoptosis by Trichostatin A in human breast cancer cell lines: Involvement of 15-Lox-1. Tumor Biol. 2013, 34, 241–249. [Google Scholar] [CrossRef]
- Liu, Y.; He, G.; Wang, Y.; Guan, X.; Pang, X.; Zhang, B. MCM-2 is a therapeutic target of Trichostatin A in colon cancer cells. Toxicol. Lett. 2013, 221, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Reid, T.; Valone, F.; Lipera, W.; Irwin, D.; Paroly, W.; Natale, R.; Sreedharan, S.; Keer, H.; Lum, B.; Scappaticci, F.; et al. Phase II trial of the histone deacetylase inhibitor pivaloyloxymethyl butyrate (Pivanex, AN-9) in advanced non-small cell lung cancer. Clin. Lung Cancer 2004, 45, 381–386. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, J.; Moral, M.A.; Bozzo, J. Combination therapy with valproic acid in cancer: Initial clinical approach. Drugs Future 2007, 32, 45. [Google Scholar] [CrossRef]
- Connolly, R.M.; Rudek, M.A.; Piekarz, R. Entinostat: A promising treatment option for patients with advanced breast cancer. Future Oncol. 2017, 13, 1137–1148. [Google Scholar] [CrossRef]
- Saito, Y.; Mizokami, A.; Izumi, K.; Naito, R.; Goto, M.; Nakagawa-Goto, K. α-Trifluoromethyl chalcones as potent anticancer agents for androgen receptor-independent prostate cancer. Molecules 2021, 26, 2812. [Google Scholar] [CrossRef] [PubMed]
- Slingerland, M.; Guchelaar, H.J.; Gelderblom, H. Histone deacetylase inhibitors: An overview of the clinical studies in solid tumors. Anticancer. Drugs 2014, 25, 140–149. [Google Scholar] [CrossRef]
- Jang, K.Y.; Hwang, S.H.; Kwon, K.S.; Kim, K.R.; Choi, H.N.; Lee, N.R.; Kwak, J.Y.; Park, B.H.; Park, H.S.; Chung, M.J.; et al. SIRT1 expression is associated with poor prognosis of diffuse large B-cell lymphoma. Am. J. Surg. Pathol. 2008, 32, 1523–1531. [Google Scholar] [CrossRef]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissocia-tion. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Gaymes, T.J.; Padua, R.A.; Pla, M.; Orr, S.; Omidvar, N.; Chomienne, C.; Mufti, G.J.; Rassool, F.V. Histone deacetylase inhibitors (HDI) cause DNA damage in leukemia cells: A mechanism for leukemia-specific HDI-dependent apoptosis? Mol. Cancer 2006, 4, 563–573. [Google Scholar] [CrossRef]
- Lukas, J.; Lukas, C.; Bartek, J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 2011, 13, 1161–1169. [Google Scholar] [CrossRef]
- Daśko, M.; de Pascual-Teresa, B.; Ortín, I.; Ramos, A. HDAC inhibitors: Innovative strategies for their design and applications. Molecules 2022, 27, 715. [Google Scholar] [CrossRef] [PubMed]
- Misteli, T.; Soutoglou, E. The emerging role of nuclear architecture in DNA repair and genome maintenance. Nat. Rev. Mol. Cell Biol. 2009, 10, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, G.; Rakshit, S.; Sarkar, K. HDAC inhibitors: Targets for tumor therapy, immune modulation and lung diseases. Transl. Oncol. 2022, 16, 101312. [Google Scholar] [CrossRef]
- Lanzi, C.; Cassinelli, G. Combinatorial strategies to potentiate the efficacy of HDAC inhibitors in fusion-positive sarcomas. Biochem. Pharmacol. 2022, 198, 114944. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Kawaguchi, Y.; Lai, C.H.; Kovacs, J.J.; Higashimoto, Y.; Appella, E.; Yao, T.P. MDM2–HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002, 21, 6236–6245. [Google Scholar] [CrossRef]
- Johnstone, R.W. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 2002, 1, 287–299. [Google Scholar] [CrossRef]
- Jiang, X.; Sun, Y.; Chen, S.; Roy, K.; Price, B.D. The FATC domains of PIKK proteins are functionally equivalent and participate in the Tip60-dependent activation of DNA-PKcs and ATM. J. Biol. Chem. 2006, 281, 15741–15746. [Google Scholar] [CrossRef]
- Lee, S.F.; Pervaiz, S. Assessment of oxidative stress-induced DNA damage by immunoflourescent analysis of 8-oxodG. Methods Cell Biol. 2011, 103, 99–113. [Google Scholar]
- Dasmahapatra, G.; Lembersky, D.; Son, M.P.; Attkisson, E.; Dent, P.; Fisher, R.I.; Friedberg, J.W.; Grant, S. Carfilzomib Interacts Synergistically with Histone Deacetylase Inhibitors in Mantle Cell Lymphoma Cells In Vitro and In VivoCarfilzomib and Vorinostat in Mantle Cells. Mol. Cancer Ther. 2011, 10, 1686–1697. [Google Scholar] [CrossRef]
- McClure, J.J.; Li, X.; Chou, C.J. Advances and Challenges of HDAC inhibitors in cancer therapeutics. Adv. Cancer Res. 2018, 138, 183–211. [Google Scholar]
- Dai, Y.; Rahmani, M.; Dent, P.; Grant, S. Blockade of histone deacetylase inhibitor-induced RelA/p65 acetylation and NF-κB activation potentiates apoptosis in leukemia cells through a process mediated by oxidative damage, XIAP downregulation, and c-Jun N-terminal kinase 1 activation. Mol. Cell. Biol. 2005, 25, 5429–5444. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P.; Jackson, S.P. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem. 2009, 284, 9558–9565. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Hong, Q.; Wei, R.; Li, H.; Wan, C.; Chen, X.; Zhao, S.; Bu, H.; Zhang, B.; Yang, D.; et al. Design and synthesis of HDAC inhibitors to enhance the therapeutic effect of diffuse large B-cell lymphoma by improving metabolic stability and pharmacokinetic characteristics. Eur. J. Med. Chem. 2022, 22, 114049. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.C.; Chan, A.H.; Ganesan, A. Thirty years of HDAC inhibitors: 2020 insight and hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Dong, H.; Xu, Q.; Zhang, Y. Histone deacetylase (HDAC) inhibitors in cancer: A patent review (2017-present). Expert Opin. Ther. Pat. 2020, 30, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Kukita, A.; Kukita, T.; Shobuike, T.; Nakamura, T.; Kohashi, O. Two histone deacetylase inhibitors, trichostatin A and sodium butyrate, suppress differentiation into osteoclasts but not into macrophages. Blood J. Am. Soc. Hematol. 2003, 101, 3451–3459. [Google Scholar] [CrossRef]
- Chang, J.; Varghese, D.S.; Gillam, M.C.; Peyton, M.; Modi, B.; Schiltz, R.L.; Girard, L.; Martinez, E.D. Differential response of cancer cells to HDAC inhibitors trichostatin A and depsipeptide. BJC 2012, 106, 116–125. [Google Scholar] [CrossRef]
- Schäker-Hübner, L.; Haschemi, R.; Büch, T.; Kraft, F.B.; Brumme, B.; Schöler, A.; Jenke, R.; Meiler, J.; Aigner, A.; Bendas, G.; et al. Balancing Histone Deacetylase (HDAC) Inhibition and Drug-likeness: Biological and Physicochemical Evaluation of Class I Selective HDAC Inhibitors. Chem. Med. Chem. 2022, 17, e202100755. [Google Scholar] [CrossRef]
- Basile, V.; Mantovani, R.; Imbriano, C. DNA damage promotes histone deacetylase 4 nuclear localization and repression of G2/M promoters, via p53 C-terminal lysines. J. Biol. Chem. 2006, 281, 2347–2357. [Google Scholar] [CrossRef]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Kutil, Z.; Novakova, Z.; Meleshin, M.; Mikesova, J.; Schutkowski, M.; Barinka, C. Histone deacetylase 11 is a fatty-acid deac-ylase. ACS Chem. Biol. 2018, 13, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.R.; Corpina, R.A.; Goldberg, J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 2001, 412, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, T.M.; Jackson, S.P. The DNA-dependent protein kinase: Requirement for DNA ends and association with Ku antigen. Cell 1993, 72, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, T.C.; Kn, H.; El-Osta, A. The epigenetic modifier, valproic acid, enhances radiation sensitivity. Epigenetics 2006, 1, 131–137. [Google Scholar] [CrossRef]
- Lee, J.H.; Choy, M.L.; Ngo, L.; Foster, S.S.; Marks, P.A. Histone deacetylase inhibitor induces DNA damage, which normal but nottransformed cells can repair. Proc. Natl. Acad. Sci. USA 2010, 107, 14639–14644. [Google Scholar] [CrossRef]
- Kachhap, S.K.; Rosmus, N.; Collis, S.J.; Kortenhorst, M.S.; Wissing, M.D.; Hedayati, M.; Shabbeer, S.; Mendonca, J.; Deangelis, J.; Marchionni, L.; et al. Downregulation of homologous recombination DNA repair genes by HDAC inhibition in prostate cancer is mediated through the E2F1 transcription factor. PLoS ONE 2010, 5, e11208. [Google Scholar] [CrossRef]
- Sun, Y.; Jiang, X.; Price, B.D. Tip60: Connecting chromatin to DNA damage signaling. Cell Cycle 2010, 9, 930–936. [Google Scholar] [CrossRef]
- Sharma, G.G.; So, S.; Gupta, A.; Kumar, R.; Cayrou, C.; Avvakumov, N.; Bhadra, U.; Pandita, R.K.; Porteus, M.H.; Chen, D.J.; et al. MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Mol. Cell. Biol. 2010, 30, 3582–3595. [Google Scholar] [CrossRef]
- Polo, S.E.; Kaidi, A.; Baskcomb, L.; Galanty, Y.; Jackson, S.P. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBOJ 2010, 29, 3130–3139. [Google Scholar] [CrossRef]
- Petruccelli, L.A.; Dupéré-Richer, D.; Pettersson, F.; Retrouvey, H.; Skoulikas, S.; Miller, W.H., Jr. Vorinostat induces reactive oxygen species and DNA damage in acute myeloid leukemia cells. PLoS ONE 2011, 6, e20987. [Google Scholar] [CrossRef]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.C.; Li, H.; Van Neste, L.; Cai, Y.; Robert, C.; Rassool, F.V.; Shin, J.J.; Harbom, K.M.; Beaty, R.; Pappou, E.; et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell 2012, 21, 430–446. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Sun, Y. Epigenetic regulators as promising targets in acute myeloid leukemia therapy. Signal Transduct. Target The.r 2020, 5, 1–20. [Google Scholar]
- Lawson, M.; Uciechowska, U.; Schemies, J.; Rumpf, T.; Jung, M.; Sippl, W. Inhibitors to understand molecular mechanisms of NAD+-dependent deacetylases (sirtuins). Biochim. Et Biophys. Acta (BBA)-Gene Regul. Mech. 2010, 1799, 726–739. [Google Scholar] [CrossRef]
- San José-Enériz, E.; Gimenez-Camino, N.; Agirre, X.; Prosper, F. HDAC inhibitors in acute myeloid leukemia. Cancers 2019, 11, 1794. [Google Scholar] [CrossRef] [PubMed]
- Maiso, P.; Colado, E.; Ocio, E.M.; Garayoa, M.; Martin, J.; Atadja, P.; Pandiella, A.; San-Miguel, J.F. The synergy of panobinostat plus doxorubicin in acute myeloid leukemia suggests a role for HDAC inhibitors in the control of DNA repair. Leukemia 2009, 23, 2265–2274. [Google Scholar] [CrossRef] [PubMed]
- Ramaiah, M.J.; Tangutur, A.D.; Manyam, R.R. Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy. Life Sci. 2021, 277, 119504. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hoshino, T.; Redner, R.L.; Kajigaya, S.; Liu, J.M. ETO, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3/HDAC1 complex. Proc. Natl. Acad. Sci. USA 1998, 95, 10860–10865. [Google Scholar] [CrossRef] [PubMed]
- Zullo, K.M.; Guo, Y.; Cooke, L.; Jirau-Serrano, X.; Mangone, M.; Scotto, L.; Amengual, J.E.; Mao, Y.; Nandakumar, R.; Cremers, S.; et al. Aurora A Kinase Inhibition Selectively Synergizes with Histone Deacetylase Inhibitor through Cytokinesis Failure in T-cell LymphomaAlisertib Synergizes with Romidepsin in TCL. Clin. Cancer Res. 2015, 21, 4097–4109. [Google Scholar] [CrossRef]
- Palomero, T.; Couronne, L.; Khiabanian, H. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat. Genet. 2014, 46, 166–170. [Google Scholar] [CrossRef]
- Vader, G.; Lens, S.M. The Aurora kinase family in cell division and cancer. Biochim. Biophys. Acta 2008, 1786, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Marchi, E.; Zullo, K.M.; Amengual, J.E. The combination of hypomethylating agents and histone deacetylase inhibitors produce marked synergy in preclinical models of T-cell lymphoma. Br. J. Haematol. 2015, 171, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.; Rahman, F.; Piekarz, R.; Peer, C.; Frye, R.; Robey, R.W.; Gardner, E.R.; Figg, W.D.; Bates, S.E. Romidepsin: A new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev. Anticancer Ther. 2010, 10, 997–1008. [Google Scholar] [CrossRef]
- Patra, S.; Praharaj, P.P.; Klionsky, D.J.; Bhutia, S.K. Vorinostat in autophagic cell death: A critical insight into autopha-gy-mediated,-associated and-dependent cell death for cancer prevention. Drug Discov. Today 2022, 27, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Mrakovcic, M.; Bohner, L.; Hanisch, M.; Fröhlich, L.F. Epigenetic targeting of autophagy via HDAC inhibition in tumor cells: Role of p53. Int. J. Mol. Sci. 2018, 19, 3952. [Google Scholar] [CrossRef]
- Heltweg, B.; Gatbonton, T.; Schuler, A.D.; Posakony, J.; Li, H.; Goehle, S.; Kollipara, R.; DePinho, R.A.; Gu, Y.; Simon, J.A.; et al. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 2006, 66, 4368–4377. [Google Scholar] [CrossRef]
- Grozinger, C.M.; Chao, E.D.; Blackwell, H.E.; Moazed, D.; Schreiber, S.L. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J. Biol. Chem. 2001, 276, 38837–38843. [Google Scholar] [CrossRef]
- Bedalov, A.; Gatbonton, T.; Irvine, W.P.; Gottschling, D.E.; Simon, J.A. Identification of a small molecule inhibitor of Sir2p. Proc. Natl. Acad. Sci. USA 2001, 98, 15113–15118. [Google Scholar] [CrossRef]
- Huang, J.; Chan, S.C.; Lok, V.; Zhang, L.; Lucero-Prisno, D.E.; Xu, W.; Zheng, Z.J.; Elcarte, E.; Withers, M.; Wong, M.C. The epidemiological landscape of multiple myeloma: A global cancer registry estimate of disease burden, risk factors, and temporal trends. Lancet Haematol. 2022, 9, e670–e677. [Google Scholar] [CrossRef]
- Pei, X.Y.; Dai, Y.; Grant, S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin. Cancer Res. 2004, 10, 3839–3852. [Google Scholar] [CrossRef]
- Duvic, M.; Dimopoulos, M. The safety profile of vorinostat (suberoylanilide hydroxamic acid) in hematologic malignancies: A review of clinical studies. Cancer Treat. Rev. 2016, 43, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, N.; Mitsiades, C.S.; Richardson, P.G. Molecular sequelae of histone deacetylase inhibition in human malignant B cells. Blood 2003, 101, 4055–4062. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Vogl, D.T.; Raje, N.; Jagannath, S.; Richardson, P.; Hari, P.; Orlowski, R.; Supko, J.G.; Tamang, D.; Yang, M.; Jones, S.S.; et al. Ricolinostat, the First Selective Histone Deacetylase 6 Inhibitor, in Combination with Bortezomib and Dexamethasone for Relapsed or Refractory Multiple MyelomaRicolinostat, Bortezomib, and Dexamethasone for Mye-loma. Clin. Cancer Res. 2017, 23, 3307–3315. [Google Scholar] [CrossRef]
- Amengual, J.E.; Lue, J.K.; Ma, H.; Lichtenstein, R.; Shah, B.; Cremers, S.; Jones, S.; Sawas, A. First-in-Class Selective HDAC6 Inhibitor (ACY-1215) Has a Highly Favorable Safety Profile in Patients with Relapsed and Refractory Lymphoma. J. Oncol. 2021, 26, 184-e366. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Ohta, E.; Takeda, S.; Sunamura, S.; Ishibashi, M.; Tamura, H.; Wang, Y.H.; Deguchi, A.; Tanaka, J.; Maru, Y.; et al. Histone deacetylase inhibitor panobinostat induces calcineurin degradation in multiple myeloma. JCI Insight 2016, 1, e85061. [Google Scholar] [CrossRef]
- Di Bello, E.; Noce, B.; Fioravanti, R.; Mai, A. Current HDAC Inhibitors in Clinical Trials. Chimia 2022, 76, 448. [Google Scholar] [CrossRef]
- Ghosh, S.K.; Perrine, S.P.; Williams, R.M.; Faller, D.V. Histone deacetylaseinhibitors are potent inducers of gene expression in latent EBVand sensitize lymphoma cells to nucleoside antiviral agents. Blood 2012, 119, 1008–1017. [Google Scholar] [CrossRef]
- Park, S.H.; Sung, J.H.; Chung, N. Berberine diminishes side population and down regulates stem cell-associated genes in the pancreatic cancer cell lines PANC-1 and MIA sssPaCa-2. Mol. Mol. Cell. Biochem. 2014, 394, 209–215. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Y.; Xue, Y.; Hu, H.; Ye, J.; Li, X.; Lu, Z.; Meng, F.; Liang, S. Berberine Acts as a Putative Epigenetic Modulator by Affecting the Histone Code. Toxicol. In Vitro 2016, 36, 10–17. [Google Scholar] [CrossRef]
- Li, C.; Su, R.; Wang, X.; Huang, G.; Liu, Y.; Yang, J.; Yin, Z.; Gu, C.; Fei, J. Discovery of the oncogenic MDM2, a direct binding target of berberine and a potential therapeutic, in multiple myeloma. Funct. Integr. Genom. 2022, 22, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Moghadam, E.R.; Ang, H.L.; Asnaf, S.E.; Zabolian, A.; Saleki, H.; Yavari, M.; Esmaeili, H.; Zarrabi, A.; Ashrafizadeh, M.; Kumar, A.P. Broad-spectrum preclinical antitumor activity of chrysin: Current trends and future perspectives. Biomolecules 2020, 10, 1374. [Google Scholar] [CrossRef] [PubMed]
- Talebi, M.; Farkhondeh, T.; Simal-Gandara, J.; Kopustinskiene, D.M.; Bernatoniene, J.; Pourbagher-Shahri, A.M.; Sa-marghandian, S. Promising protective effects of chrysin in cardiometabolic diseases. Curr. Drug Targets 2022, 23, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Ganai, S.A.; Sheikh, F.A.; Baba, Z.A. Plant flavone Chrysin as an emerging histone deacetylase inhibitor for prosperous epigenetic-based anticancer therapy. Phytother. Res. 2021, 35, 823–834. [Google Scholar] [CrossRef]
- Ganai, S.A. Designing isoform-selective inhibitors against classical HDACs for effective anticancer therapy: Insight and perspectives from in Silico. Curr. Drug Targets 2018, 19, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Murthy, H.N.; Yadav, G.G. Chemistry and Biological Activities of Garcinia Resin. In Gums, Resins and Latexes of Plant Origin: Chemistry, Biological Activities and Uses; Springer International Publishing: Cham, Switzerland, 2021; pp. 1–38. [Google Scholar]
- Punpai, S.; Saenkham, A.; Jarintanan, F.; Jongrungruangchok, S.; Choowongkomon, K.; Suksamrarn, S.; Tanechpongtamb, W. HDAC inhibitor cowanin extracted from G. fusca induces apoptosis and autophagy via inhibition of the PI3K/Akt/mTOR pathways in Jurkat cells. Biomed. Pharmacother 2022, 147, 112577. [Google Scholar] [CrossRef]
- Wahyuni, F.S.; Shaari, K.; Stanslas, J.; Lajis, N.H.; Hamidi, D. Cytotoxic properties and complete nuclear magnetic resonance assignment of isolated xanthones from the root of Garcinia cowa Roxb. Pharmacogn. Mag. 2016, 12 (Suppl. 1), S52. [Google Scholar]
- Kasi, P.D.; Tamilselvam, R.; Skalicka-Woźniak, K.; Nabavi, S.F.; Daglia, M.; Bishayee, A.; Pazoki-Toroudi, H.; Nabavi, S.M. Molecular targets of curcumin for cancer therapy: An updated review. Tumor Biol. 2016, 37, 13017–13028. [Google Scholar] [CrossRef]
- Arslan, A.K.; Uzunhisarcıklı, E.; Yerer, M.B.; Bishayee, A. The golden spice curcumin in cancer: A perspective on finalized clinical trials during the last 10 years. J. Cancer Ther. 2022, 18, 19–26. [Google Scholar]
- Liu, H.L.; Chen, Y.; Cui, G.H.; Zhou, J.F. Curcumin, a potent anti-tumor reagent, is a novel histone deacetylase inhibitor regulating B-NHL cell line Raji proliferation. Acta Pharmacol. Sin. 2005, 26, 603–609. [Google Scholar] [CrossRef]
- Oku, N.; Nagai, K.; Shindoh, N.; Terada, Y.; van Soest, R.W.; Matsunaga, S.; Fusetani, N. Three new cyclostellettamines, which inhibit histone deacetylase, from a marine sponge of the genus Xestospongia. Bioorg. Med. Chem. Lett. 2004, 14, 2617–2620. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Lu, M.C.; Hsu, K.C.; El-Shazly, M.; Shih, S.P.; Lien, S.T.; Kuo, F.W.; Yang, S.C.; Chen, C.L.; Yang, Y.C. The an-tileukemic effect of xestoquinone, a marine-derived polycyclic quinone-type metabolite, is mediated through ros-induced inhibition of hsp-90. Molecules 2021, 26, 7037. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.H.; Li, J.; Xia, J.; Jiang, R.; Zuo, G.W.; Li, X.P.; Chen, Y.; Xiong, W.; Chen, D.L. Ginsenoside 20 (s)-Rh2 as potent natural histone deacetylase inhibitors suppressing the growth of human leukemia cells. Chem. Biol. Interact. 2015, 242, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H. Pharmacological and medical applications of Panax ginseng and ginsenosides: A review for use in cardiovascular diseases. JGR 2018, 42, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Sun, S.; Fang, W.; Jin, J. 20(S)-Ginsenoside Rh2 induces apoptosis and differentiation of K562 leukemia cells. J. Ethnopharmacol. 2016, 194, 126–133. [Google Scholar]
- Shih, S.P.; Lee, M.G.; El-Shazly, M.; Juan, Y.S.; Wen, Z.H.; Du, Y.C.; Su, J.H.; Sung, P.J.; Chen, Y.C.; Yang, J.C. Tackling the Cytotoxic Effect of a Marine Polycyclic Quinone-Type Metabolite: Halenaquinone Induces Molt 4 Cells Apoptosis via Oxi-dative Stress Combined with the Inhibition of HDAC and Topoisomerase Activities. Mar. Drugs 2015, 13, 3132–3153. [Google Scholar] [CrossRef]
- Luparello, C.; Mauro, M.; Arizza, V.; Vazzana, M. Histone deacetylase inhibitors from marine invertebrates. Biology 2020, 9, 429. [Google Scholar] [CrossRef]
- Souto, J.A.; Vaz, E.; Lepore, I.; Pöppler, A.C.; Franci, G.; Alvarez, R.; Altucci, L.; de Lera, A.R. Synthesis and biological char-acterization of the histone deacetylase inhibitor largazole and C7-modified analogues. J. Med. Chem. 2010, 53, 4654–4667. [Google Scholar] [CrossRef]
- Emma, M.R.; Augello, G.D.; Stefano, V.; Azzolina, A.; Giannitrapani, L.; Montalto, G.; Cervello, M.; Cusimano, A. Potential uses of olive oil secoiridoids for the prevention and treatment of cancer: A narrative review of preclinical studies. Int. J. Mol. Sci. 2021, 22, 1234. [Google Scholar] [CrossRef]
- Juli, G.; Oliverio, M.; Bellizzi, D.; Gallo Cantafio, M.E.; Grillone, K.; Passarino, G.; Colica, C.; Nardi, M.; Rossi, M.; Procopio, A.; et al. Anti-tumor activity and epigenetic impact of the polyphenol oleacein in multiple myeloma. Cancers 2019, 11, 990. [Google Scholar] [CrossRef]
- Ma, X.; Fang, Y.; Beklemisheva, A.; Dai, W.; Feng, J.; Ahmed, T.; Liu, D.; Chiao, J.W. Phenylhexyl isothiocyanate inhibits histone deacetylases and remodels chromatins to induce growth arrest in human leukemia cells. Int. J. Oncol. 2006, 28, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Xu, Z.; Chang, G.; Hou, J.; Hu, L.; Zhang, Y.; Yu, D.; Li, B.; Chang, S.; Xie, Y.; et al. The blueberry component pterostilbene has potent anti-myeloma activity in bortezomib-resistant cells. Oncol. Rep. 2017, 38, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.J.; Ho, C.T.; Wang, Y.J. Pterostilbene induces autophagy and apoptosis in sensitive and chemoresistant human bladder cancer cells. Mol. Nutr. Food Res. 2010, 54, 1819–1832. [Google Scholar] [CrossRef] [PubMed]
- McCormack, D.; McFadden, D. Pterostilbene and cancer: Current review. J. Surg Res. 2012, 173, e53–e61. [Google Scholar] [CrossRef] [PubMed]
- Maurya, P.K. Health benefits of quercetin in age-related diseases. Molecules 2022, 27, 2498. [Google Scholar]
- Alvarez, M.C.; Maso, V.; Torello, C.O.; Ferro, K.P.; Saad, S.T. The polyphenol quercetin induces cell death in leukemia by targeting epigenetic regulators of pro-apoptotic genes. Clin. Epigenetics 2018, 10, 139. [Google Scholar] [CrossRef] [PubMed]
- Ononye, S.N.; VanHeyst, M.D.; Giardina, C.; Wright, D.L.; Anderson, A.C. Studies on the antiproliferative effects of tropolone derivatives in Jurkat T-lymphocyte cells. Bioorg. Med. Chem. 2014, 22, 2188–2193. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Dai, X.; Kumar, A.P.; Tan, B.K.; Sethi, G.; Bishayee, A. Ursolic acid in cancer prevention and treatment: Molecular targets, pharmacokinetics and clinical studies. Biochem. Pharmacol. 2013, 85, 1579–1587. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, C.; Li, W.; Wu, R.; Guo, Y.; Cheng, D.; Yang, Y.; Androulakis, I.P.; Kong, A.N. Pharmacokinetics and Pharmacodynamics of the Triterpenoid Ursolic Acid in Regulating the Antioxidant, Anti-Inflammatory, and Epigenetic Gene Responses in Rat Leukocytes. Mol. Pharm. 2017, 14, 3709–3717. [Google Scholar] [CrossRef]
- Dung, D.T.; Hang, D.T.; Yen, P.H.; Quang, T.H.; Nhiem, N.X.; Tai, B.H.; Minh, C.V.; Kim, Y.C.; Kim, D.C.; Oh, H.; et al. Macrocyclic bis-quinolizidine alkaloids from Xestospongia muta. Nat. Prod. Res. 2019, 33, 400–406. [Google Scholar] [CrossRef]
- Yang, H.L.; Kumar, K.S.; Kuo, Y.T.; Chang, H.C.; Liao, J.W.; Hsu, L.S.; Hseu, Y.C. Antrodia camphorata induces G 1 cell-cycle arrest in human premyelocytic leukemia (HL-60) cells and suppresses tumor growth in athymic nude mice. Food Funct. 2014, 5, 2278–2288. [Google Scholar] [CrossRef] [PubMed]
- Hseu, Y.C.; Yang, H.L.; Lai, Y.C.; Lin, J.G.; Chen, G.W.; Chang, Y.H. Induction of apoptosis by Antrodia camphorata in human premyelocytic leukemia HL-60 cells. Nutr. Cancer 2004, 48, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Bontempo, P.; Mita, L.; Miceli, M.; Doto, A.; Nebbioso, A.; De Bellis, F.; Conte, M.; Minichiello, A.; Manzo, F.; Carafa, V.; et al. Feijoa sellowiana derived natural Flavone exerts anti-cancer action displaying HDAC inhibitory activities. Int. J. Biochem. 2007, 39, 1902–1914. [Google Scholar] [CrossRef]
- Gawlik-Dziki, U.; Świeca, M.; Dziki, D. Comparison of Phenolic Acids Profile and Antioxidant Potential of Six Varieties of Spelt (Triticum spelta L.). J. Agric. Food Chem. 2012, 60, 4603–4612. [Google Scholar] [CrossRef] [PubMed]
- Fabiani, R.; Rosignoli, P.; De Bartolomeo, A.; Fuccelli, R.; Servili, M.; Montedoro, G.F.; Morozzi, G. Oxidative DNA damage is prevented by extracts of olive oil, hydroxytyrosol, and other olive phenolic compounds in human blood mononuclear cells and HL60 cells. J. Nutr. 2008, 138, 1411–1416. [Google Scholar] [CrossRef] [PubMed]





| Class | HDAC | Localization | % Similarity to Human (Nucleic Acid/Amino Acid) | Associated Cancer(s) | Physiological Functions |
|---|---|---|---|---|---|
| I | HDAC 1 | Nucleus | 90.8/99.4 | Prostate, gastric, colorectal, pancreas, and esophageal | Cell survival and proliferation |
| HDAC 2 | Nucleus | 91.1/98.6 | Colorectal, gastric, cervical dysplasia, and invasive carcinoma | Cell proliferation; insulin resistance | |
| HDAC 3 | Nucleus | 92.5/99.6 | Lung, prostate, and colon | Cell survival and proliferation | |
| HDAC 8 | Nucleus | 90.9/96.3 | Poor outcomes in pediatric neuroblastoma | Cell proliferation | |
| IIa | HDAC 4 | Nucleus/cytoplasm | 86.3/94.2 | Breast | Regulation of skeletogenesis and gluconeogenesis |
| HDAC 5 | Nucleus/cytoplasm | 91.1/95.6 | Colon, AML, and poor outcomes in lung cancer | Cardiovascular growth and function; gluconeogenesis; cardiac myocyte and endothelial cell function | |
| HDAC 7 | Nucleus/cytoplasm | 86.8/90.3 | Colon | Thymocyte differentiations; endothelial function; gluconeogenesis; homologous recombination | |
| HDAC 9 | Nucleus/cytoplasm | 90.394.8 | Medulloblastoma and astrocytoma | Thymocyte differentiation; cardiovascular growth and function | |
| IIb | HDAC 6 | Cytoplasm | 81.1/78.7 | Ovarian and AML | Cell motility; control of cytoskeletal dynamics |
| HDAC 10 | Cytoplasm | 78.1/76.4 | Hepatocellular carcinoma | Homologous recombination; autophagy-mediated cell survival | |
| IV | HDAC 11 | Nucleus/cytoplasm | 87.3/91.9 | Breast | Immunomodulators DNA replications |
| III | SIRT1 | Nucleus | Not available | AML, colon, prostate, skin, and BCLL | Aging; redox regulation; cell survival; autoimmune system regulation |
| SIRT2 | Cytoplasm | Not available | Glioma | Cell survival-cell migration and invasion | |
| SIRT3 | Nucleus/Mitochondria | Not available | Breast, prostate, head and neck, and glioblastoma | Urea cycle; redox balance; ATP regulation; metabolism; apoptosis; cell signaling | |
| SIRT4 | Mitochondria | Not available | Breast | Energy metabolism; ATP regulation; metabolism; apoptosis; cell signaling | |
| SIRT5 | Mitochondria | Not available | Pancreas and breast | Urea cycle; energy metabolism; ATP regulation; metabolism; apoptosis; cell signaling | |
| SIRT6 | Nucleus | Not available | Colon and breast | Metabolism | |
| SIRT7 | Nucleus | Not available | Breast | Apoptosis |
| Class | HDACis | Target | Structure | Associated Cancer(s) | References |
|---|---|---|---|---|---|
| Hydroxamates | SAHA (vorinostat) | HDAC1-11 |  | Glioblastoma multiforme and brain metastasis | [28] |
| Panobinostat | HDAC1-11 |  | Small cell lung cancer, myelofibrosis, and cutaneous T-cell lymphoma | [29,30] | |
| Belinostat | HDAC1-11 |  | Thymic epithelial tumor and ovarian cancer | [31] | |
| Trichostatin | HDAC1-11 |  | Colon and breast | [32,33] | |
| Aliphatic acids | Pivanex | HDAC1-9 |  | Liver carcinoma, Lung carcinoma and melanoma | [34] |
| Valproic acid | HDAC1-9 |  | Prostate, breast and melanoma | [35] | |
| Benzamides | Entinostat | HDAC1-3 |  | Breast cancer and advanced solid tumors | [36] |
| Electrophilic ketone | Trifluromethyl Ketones | HDAC4-9 |  | Prostate cancer | [37] |
| Cyclic tetrapeptides | Romidepsin | HDAC1-2 |  | Thyroid, ovarian, pancreatic and breast cancer | [38] |
| Sirtuin inhibitors | Cambinol | SIRT1 |  | Sarcomas and lymphomas | [39] |
| Classifications | Drug Name | Role in Treatment of AML | References |
|---|---|---|---|
| Hydroximates | Trichostatin A | Inhibits the pathway for DNA repair (NHEJ). Acetylation of repair factors and trapping of PARP1 at DNA double-strand brakes in chromatin, inducing leukemic toxicity. Possible synergistic effect of TSA with an inhibitor of PARP1. | [32,33] |
| Vorinostat | Apoptosis and inhibition of cell growth. Increases differentiation induced by retinoic acid in acute promyelocytic leukemia cells. Induction of double-strand breaks and oxidative DNA damage. | [28] | |
| Panobinostat | Promotes apoptosis and inhibition of cell growth. | [29,30] | |
| Belinostat | Promotes cell cycle arrest, inhibits cell proliferation, and induces apoptosis. | [31] | |
| Benzamides | Entinostat | Induces growth arrest and apoptosis. Downregulates antiapoptotic molecules BCL-2 and MCL-1, increases p21, and induces acetylation of H3. | [36] |
| Cyclic peptides | Romidepsin | Promotes apoptosis of chemo-resistant malignant cells and reversed their gene expression profile. | [38] |
| Apicidin | Induces selective changes in P21WAF1/Cip1 and gelsolin gene expression, which control cell cycle and cell morphology. | [38] | |
| Aliphatic acids | Valproic acid | Induces differentiation and inhibits proliferation and apoptosis of AML cells. No clinical effect when used as a single-agent therapy for AML. Synergistic effects with ATRA, decitabine, gemtuzumab ozogamicin, curcumin, hydroxyurea, 6-mercaptopurine, dasatinib, bortezomib, cytarabine | [35] |
| Hydroxamate Family | Non-Hydroxamate Family | ||||
|---|---|---|---|---|---|
| Drug Name | Investigated Malignancy | Clinical Trial Phase | Drug Name | Investigated Malignancy | Clinical Trial Phase |
| Abexinostat | Skin cancers, non-small lung cancer, mantle cell lymphoma, acute myeloid leukemia | Phase-3 | Tacedinaline | Solid and hematological cancers | Phase 3 |
| Fimepinostat | lymphomas, brain tumors | Phase1/2 | Entinostat | Gynecological cancers, CNS tumors, pancreatic cancer, non-small lung cancer | Phase 2 |
| Quisinostat | JNJ26481585 ovarian cancer, non-small lung cancer | Phase 2 | Domatinostat | Cutaneous T-cell lymphoma | Phase1 |
| Ricolinostat | ACY-1215 lymphomas, Breast cancer, gynecological cancers | Phase 2 | Givinostat | Polycythemia vera | Phase 2 |
| Trichostatin A | Hematological cancers | Phase 1 | KA2507 | Melanoma | Phase 1 |
| Nanatinostat | VRx-3996, EBV-associated cancers | Phase 1 | Mocetinostat | Leiomyosarcoma and melanoma | Phase 2 |
| CG200745 | Myelodysplastic syndrome, pancreatic cancer | Phase1/2 | OBP-801 | Lung cancer | Phase 1a |
| Pracinostat | Prostate cancer, sarcoma, myelofibrosis, myelodysplastic syndrome | Phase 3 | AR-42 | Sarcoma and meningioma | Phase1 |
| Resminostat | Pancreatic cancer, non-small lung cancer, colorectal carcinoma, Hodgkin’s lymphoma | Phase 2 | Pivanex | Melanoma and lymphoblastic leukemia | Phase 2 |
| CUDC-101 | Advanced solid tumors | Phase 1 | Givinostat | Polycythemia vera | Phase 2 |
| MPT0E028 | Advanced solid tumors | Phase 1 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pal, D.; Raj, K.; Nandi, S.S.; Sinha, S.; Mishra, A.; Mondal, A.; Lagoa, R.; Burcher, J.T.; Bishayee, A. Potential of Synthetic and Natural Compounds as Novel Histone Deacetylase Inhibitors for the Treatment of Hematological Malignancies. Cancers 2023, 15, 2808. https://doi.org/10.3390/cancers15102808
Pal D, Raj K, Nandi SS, Sinha S, Mishra A, Mondal A, Lagoa R, Burcher JT, Bishayee A. Potential of Synthetic and Natural Compounds as Novel Histone Deacetylase Inhibitors for the Treatment of Hematological Malignancies. Cancers. 2023; 15(10):2808. https://doi.org/10.3390/cancers15102808
Chicago/Turabian StylePal, Dilipkumar, Khushboo Raj, Shyam Sundar Nandi, Surajit Sinha, Abhishek Mishra, Arijit Mondal, Ricardo Lagoa, Jack T. Burcher, and Anupam Bishayee. 2023. "Potential of Synthetic and Natural Compounds as Novel Histone Deacetylase Inhibitors for the Treatment of Hematological Malignancies" Cancers 15, no. 10: 2808. https://doi.org/10.3390/cancers15102808
APA StylePal, D., Raj, K., Nandi, S. S., Sinha, S., Mishra, A., Mondal, A., Lagoa, R., Burcher, J. T., & Bishayee, A. (2023). Potential of Synthetic and Natural Compounds as Novel Histone Deacetylase Inhibitors for the Treatment of Hematological Malignancies. Cancers, 15(10), 2808. https://doi.org/10.3390/cancers15102808